

Abstract
C-type inactivation underlies important roles played by voltage-gated K+ (Kv) channels. Functional studies have provided strong evidence that a common underlying cause of this type of inactivation is an alteration near the extracellular end of the channel's ion selectivity filter. Unlike N-type inactivation, which is known to reflect occlusion of the channel's intracellular end, the structural mechanism of C-type inactivation remains controversial and may have many detailed variations. Here, we report that in voltage-gated Shaker K+ channels lacking N-type inactivation, a mutation enhancing inactivation disrupts the outermost K+ site in the selectivity filter. Furthermore, in a crystal structure of the Kv1.2-2.1 chimeric channel bearing the same mutation, the outermost K+ site, which is formed by eight carbonyl oxygen atoms, appears to be slightly too small to readily accommodate a K+ ion and in fact exhibits little ion density; this structural finding is consistent with the functional hallmark characteristic of C-type inactivation.
Kv channels underlie the repolarization phase of the action potential in excitable cells including nerve and heart cells. The channel activation gate is controlled by membrane voltage such that it opens upon membrane depolarization and closes on hyperpolarization1, 2. However, even when depolarization is maintained and the activation gate remains open, most Kv channels still enter a nonconducting state, a process called inactivation. Two mechanistically distant types of inactivation are commonly recognized3-5: N-type inactivation, which results from occlusion of the channel's ion pore by the N-terminus of either the channel protein itself or its (auxiliary) β subunit4-6, and C-type inactivation, whose mechanistic interpretation presently remains controversial.
C-type inactivation enables Kv channels to perform important tasks such as shaping cardiac action potentials to allow sufficient Ca2+ influx to trigger effective myocyte contraction and accelerating repolarization. This phenomenon was originally discovered in voltage-gated Shaker K+ channels, on the basis that in the absence of the N-terminal inactivation-causing sequence, the channels still undergo inactivation, albeit with a much slower rate5. Furthermore, the inactivation kinetics differs between two splice variants; a variation of residue type (hereafter simply called mutation) at position 463 (in the ShB variant) has been identified as the cause5. This residue is located in the sixth transmembran segment (S6), which was then considered to be a part of the C-terminal region (S6). This segment was later shown to form the intracellular part of the pore and the activation gate7-9. A subsequent study revealed that increasing the concentration of extracellular permeant ions antagonizes C-type inactivation and that mutations in the extracellular vestibule of the ion pore can also affect the inactivation10. These and other findings strongly suggest that conformational changes in the extracellular part of the selectivity filter are the primary cause of C-type inactivation, whereas the mutation in S6 allosterically influences the inactivation process.
Here, we adopt the following operational definition of C-type inactivation, which is based primarily on the original series of studies of Aldrich and colleagues5, 10. C-type inactivation reflects a non-conducting state that occurs even in the absence of N-type inactivation, exhibits apparent dependence on activation gating, is allosterically influenced by the conformation of S6, and primarily involves changes in the extracellular part of the K+-selective filter. Given that C-type inactivation is affected by many mutations around the selectivity filter itself and in S6 5, 10, 11, it is likely a common functional manifestation of multiple forms of the channel's overall structure.
One hypothesis regarding the mechanism of C-type inactivation is that the extracellular end of the ion pore becomes markedly constricted, thus preventing ion conduction. It is one of two original interpretations of the observation that in a mutant Shaker channel, whose Thr449 located in the selectivity filter's external vestibule is replaced with cysteine, Cd2+ promotes C-type inactivation with very high affinity (Kd = 0.2 μ M)12. As such strong Cd2+ binding would typically require coordination by multiple cysteine residues ∼4 Å apart, one interpretation is that the extracellular part of the ion pore becomes markedly constricted in the inactivated state. This interpretation was called into question after the crystal structures of (wild-type and mutant) Kv1.2 channels were solved13, 14. In these structures, Val377 (corresponding to Shaker's Thr449) in a given one of the four subunits is 12 - 18 Å away from its counterpart (Cβ – Cβ) in another subunit15. The second original interpretation of the above finding is that the accessibility of the mutant cysteine differs between conducting and C-type inactivated states. An alternative hypothesis, based on a different line of reasoning, posits that C-type inactivation reflects dilation of the ion filter at the outermost K+ ion site (IS1), which removes its ability to bind a K+ ion and the filter's ability to conduct K+ current15, 16.
In Shaker channels, C-type inactivation causes up to an ∼10-fold reduction of current in the presence of a low concentration (2 mM) of extracellular K+ 5. In theory, a 10-fold reduction in a channel's steady-state open probability (Po) corresponds to a free-energy change (ΔG) of ∼0.5 kT per subunit, which, for reference, is on the magnitude of the average inherent energy of an ideal monatomic gas molecule in each degree of freedom. In the framework of Monod, Wyman and Changeux's allosteric model17, one plausible view is that the selectivity filter spontaneously switches between conducting and non-conducting states, a process reflecting some bond activity of low free energy that gives rise to subtle structural changes. Energy from alteration, formation or elimination of other nearby or distant bonds may propagate to the filter and thereby alter the relative probability of the conducting and non-conducting states. Again for reference, if formation of a single hydrogen bond results in a ΔG of 2 - 7 kT, four such bonds in a tetrametric Kv channel would give rise to a ΔG that could lower the channel's Po by about 103 to 1012 fold. The above view is consistent with the observation that C-type inactivation varies markedly among Kv subtypes and can be influenced by mutations outside the selectivity filter. For example, besides the already mentioned mutation in S6, the W434F mutation near the filter also dramatically enhances C-type inactivation, rendering the channel practically nonconducting11, 18. Intriguingly, the Swartz and colleagues have showed that V478W in S6 also renders the Shaker channel nonconducting19, 20. The Po of a mutant channel, in which two of four symmetric 478 residues are replaced by tryptophan, is drastically lower even though the voltage sensors remain functional20. The single-channel conductance of this mutant channel is essentially unaffected. These findings suggest that V478W allosterically stabilizes the selectivity filter in a non-conducting state.
Results
The V478W mutation favors inactivation in Shaker Kv channels
As previously shown19, 20, in Shaker channels with N-type inactivation removed, the V478W mutation eliminates the ionic but not the gating current21 (Fig. 1a), effectively rendering them inactive. To investigate further the effect of the V478W mutation, we made a concatemeric cDNA expressing all four subunits as a single protein, which allows replacement of fewer than four V478 residues per channel, thus lessening the mutation's impact. This approach was previously used to show that the apparent non-conducting behavior of W434F-mutant channels reflects enhanced C-type inactivation11. The concatemer channel itself acts like the wild type (Fig. S1). However, when three of the four V478 residues in the concatemer channel were replaced with tryptophan, this mutant construct (denoted 3/4V478W) exhibited robust gating current but only modest ionic current at strongly depolarized potentials; the ratio of ionic and gating currents of 3/4V478W was drastically lower (Fig. 1b, c) than the wild type. Over a 10-s sustained depolarization, the ionic current of 3/4V478W declined more and faster than that of the wild type (Fig. 1d-f). As previously reported for wild-type channels11, accumulation of K+ around the extracellular end of the pore, which stems from outward K+ flux during a 10-s long depolarization pulse needed to elicit sizable inactivation, precludes meaningful examination of the effect of extracellular K+.
Figure 1.

V478W mutation enhances C-type inactivation in Shaker channels. (a-c) Current traces of homotetrameric V478W (a) or 3/4V478W (b, c) mutant channels. Panel b shows 3/4V478W gating currents whereas panel c shows both gating and ionic currents, where the ionic currents were elicited by stronger depolarization. Currents shown in panels a-c were recorded from three different oocytes in the presence of 100 mM extracellular K+, as membrane voltage was stepped from the -100 mV holding potential to between -90 and -10 mV (a, b) or between -90 and 50 mV (c) in 10 mV increments and back to -100 mV. The dashed line indicates the zero current level. (d) Time courses of wild-type (wt) and 3/4V478W-mutant (mut) channel ionic currents normalized to peak currents, elicited by stepping the voltage from the -80 mV holding potential to 50 mV. (e, f) Mean time constants (e) or currents (normalized to peak currents) remaining at the end of the 10-s voltage pulse (f), obtained from data of the kind shown in panel d; data in e and f are shown as mean ± s.e.m. (n = 16). (g) Normalized current (mean ± s.e.m.; n = 3) of wild-type (circles) or 3/4V478W-mutant (squares) channels (Ch) against concentration of wild-type (closed symbols) or K27M mutant (open symbols) AgTx2 (Tx). Superimposed curves are fits of a monomolecular inhibition model with Kd (mean ± s.e.m.) values of 2.5 ± 0.5 nM for wild-type AgTx2 and wild-type channels, 9.2 ± 1.1 nM for wild-type AgTx2 and mutant channels, 0.58 ± 0.11 μM for mutant AgTx2 and wild-type channels, and 0.67 ± 0.06 μM for mutant AgTx2 and mutant channels.
To determine whether the perturbation caused by V478W mutation is limited to the S6-lined intracellular part of the pore or whether it propagates to the extracellular K+-selective part, we employed the pore-blocking scorpion toxin AgTx2 as a probe22. When the toxin is bound to the pore from the extracellular side, the ε amine of its Lys27 residue “displaces” K+ from the outermost IS1 of the four ion sites (IS1-IS4) in the selectivity filter and directly interacts with the site23-26. Replacing Lys27 with a neutral residue may affect the toxin-channel interactions in three ways. First, it inevitably eliminates the interaction between the amine and IS1 site, likely a modest effect because ammonium ions traverse the selectivity filter as rapidly as K+ ions and thus do not strongly interact with the site. Second, the difference between Lys27's long alkyl chain and the mutant side chain can also perturb the channel-toxin interaction. Finally, the replacement may indirectly alter interactions elsewhere along the channel-toxin interface. It is therefore not surprising that the reduction of channel-toxin affinity caused by a neutral mutation at the toxin's Lys27 varies greatly with both K+ channel type and substituting residue type, ranging from five- to over 10,000-fold24, 25. If the lower bound of this range (fivefold) primarily reflects the amine-IS1 interaction common to all cases, then the variable additional reduction in affinity will reflect the two other types of perturbation. Consistent with this scenario, the 3/4V478W mutation, suspected to perturb the IS1 site, lowers the channel-toxin affinity about fourfold, whereas, as previously shown 25, replacement of Lys27 by methionine lowered the affinity much more (Fig. 1g). Furthermore, if a channel mutation had largely eliminated IS1's interaction with Lys27's amine but left other toxin-interacting loci unaffected, the affinities of a mutant toxin (with a neutral residue replacing Lys27) for both wild-type and mutant channels would be decreased to comparable levels. Here, the K27M-mutant AgTx2 indeed inhibited both wild-type and 3/4V478W-mutant channels, with affinities decreased to comparable levels, a finding strongly implying that the channel's tryptophan mutation alllosterically perturbs the Lys27-IS1 interaction (Fig. 1g). This perturbation in turn explains why the V478W mutation enhances inactivation. Thus, a Kv channel with all four valine residues replaced by tryptophan may provide useful structural insight into the inactivation mechanism.
Crystallization of the V406W mutant Kv1.2-2.1 chimera
Kv1.2, a mammalian Shaker ortholog, has successfully served in crystallographic studies of mammalian Kv channels. One of its mutants, the so-called Kv1.2-2.1 (chimeric) channel, i.e., Kv1.2 containing Kv2.1's voltage-sensor paddle sequence, forms crystals that diffract to higher resolution than Kv1.2 itself14. In crystals, the pore of Kv1.2 and its mutants tends to adopt an open conformation13, 14, 26, 27. The above functional studies motivated us to examine the structure of Kv1.2-2.1 containing the V406W (equivalent to Shaker's V478W) in all four subunits. As in the case of the Shaker channel, Kv1.2 bearing the same mutation, co-expressed with a β subunit, carried no detectable ionic current (Fig. S2). In our hands, Kv1.2 expresses less current than the Shaker channel, thus thwarting examination of its mutant's gating current. Nevertheless, the V406W construct did yield enough protein for crystallographic studies. In the presence of 150 mM K+, the mutant protein crystallized in the P4212 space group, the same space group as that of the original Kv1.2-2.1 construct (hereafter called wild type; PDB: 2R9R), each asymmetric unit also containing two molecules14. We solved the structures of both molecules (I and II) with a crystal diffracting to 3.3 Å Bragg spacing (table 1). The wild-type structure (PDB 2R9R) obtained at 2.4 Å resolution was used as initial model and provided more accurate phase information than would be from at 3.3 Å. As in the wild-type structure, the S6 activation gate of both mutant molecules appears to be open.
Table 1. Data collection and refinement statistics (molecular replacement).
| Kv1.2-2.1 V406W (PDB 5WIE) |
|
|---|---|
| Data collection | |
| Space group | P 4 21 2 |
| Cell dimensions | |
| a, b, c (Å) | 143.12, 143.12, 284.51 |
| α, β, γ (°) | 90, 90,90 |
| Resolution (Å) | 3.3(3.42-3.30)a |
| Rmerge | 0.33(>1) |
| I/σ (I) | 7.9(1.7) |
| CC1/2 | 0.985(0.604) |
| Completeness (%) | 99.5(97.2) |
| Redundancy | 11.7(9.0) |
| Refinement | |
| Resolution (Å) | 3.3 |
| No. reflections | 45103 |
| Rwork / Rfree | 23.6/24.4 |
| No. atoms | 10955 |
| Protein | 10771 |
| Ligand/ion | NADP 96, PGW 80, K ion 8 |
| Water | |
| B factors | |
| Protein | 64.1 |
| Ligand/ion | NADP 50.1, PGW 66.3, K ion 62.6 |
| Water | |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.017 |
| Bond angles (°) | 1.6 |
Values in parentheses are for highest-resolution shell.
Structural characteristics of mutant molecule I
Fig. 2a shows views of molecule I's crystal structure around the V406W residue, superimposed on the corresponding 2Fo-Fc composite-omit electron-density map. Four V406W side chains, one from each subunit, point toward the pore's central axis. The closest atom-atom distance between the side chains of V406W in two diagonally opposed subunits is 5.5 Å (left panel, Fig. 2b), which is wider than the diameter of a K+ ion of ∼3 Å but is narrower than a fully hydrated K+ of ∼8 Å. The V406W mutation clearly causes structural perturbations in S6 and consequently in S5 (Fig. 2b). Conformational changes to the local backbone are evidently necessary to accommodate the V406W side chain. In the mutant structure, the V406W side chain is sandwiched between those of Val402 in the same subunit and Pro403 in an adjacent one (right panels, Fig. 2a). The nitrogen atom of the V406W side chain is 2.8 Å away from the carbonyl oxygen of Ala399 in the adjacent subunit. Judging from this distance, the V406W side chain appears to attach to the main chain of the adjacent subunit via a hydrogen bond, which would help stabilize the altered conformation. The residue triplet Pro401-Val402-Pro403 appears to act as a responsive joint, enabling S6 to adjust its conformation. This adjustment also shortens the distance between the Pro401 carbonyl oxygen and the Ile405 amide from 3.6 Å (PDB: 2R9R) to 3.2 Å, again allowing formation of a hydrogen bond. These two new hydrogen bonds together would in turn help stabilize the joint. In the Shaker channel, the Pro-Val-Pro residue triplet has been suggested to form a kink site in the region of the activation gate28.
Figure 2.
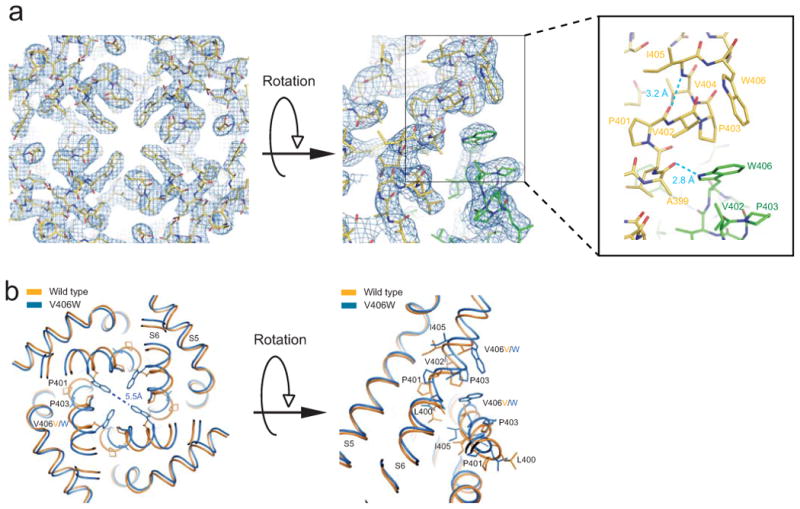
Structure and electron density map of the region around residue 406 in mutant molecule I. (a) Crystal structure of S5 and S6 viewed from the inside of the cell, superimposed on the corresponding 2Fo-Fc composite-omit map generated with CNS and contoured at 1.5 σ. At right is a side view of two contiguous subunits (green and yellow). Dashed blue lines in the enlarged view of the boxed region suggest new hydrogen bonds as judged from distances. (b) Comparison of wild type (PDB: 2R9R; orange) and V406W mutant (blue) structures around residue 406; same views as in a.
Structural perturbations caused by the V406W mutation are clearly evident in the region from residues 398 to 408 in S6 but are not apparent in the more extracellular region toward the selectivity filter. In this latter region, perturbation may have been prevented by more extensive interactions of S6 with neighboring structures, including the so-called pore helix (Ser356-Thr369) to which the ion filter is attached (Fig. S3). Although these, and possibly other, damping mechanisms effectively minimize propagation of visible perturbations to the more extracellular region, structurally minimal yet energetically impactful perturbations to the filter may still exist. Even if only a fraction of the free energy change from formation of the two new hydrogen bonds (per subunit) propagated to the selectivity filter, it could have a consequential effect on the filter.
The selectivity filter of the V406W mutant is generally similar to that of the wild type. The Cβ of Val377, which correspond to Shaker's Thr449, is little affected (Fig. S4; recall that the T449C Shaker mutant is strongly inhibited by Cd2+). However, in the mutant structure, the outermost set of K+-coordinating carbonyl oxygen atoms of Tyr373 appear to move slightly in the intracellular direction (Figs. 3a-c). Fig. S5 compares the structures of Gly372-Tyr-373-Gly374 in wild-type (orange) and mutant (blue) channels, superimposed on a Fo-Fc omit map contoured at 5 σ and 8 σ; the map was calculated with a model where the three residues are omitted. The distances from the hydroxyl oxygen atom of Tyr373 to a nitrogen atom of Trp363 and to the hydroxyl oxygen atom of Ser367 in a neighboring subunit remain in a hydrogen-bond range. Thus, Tyr373's side chain is held in place so that its carbonyl oxygen atom cannot rotate much further. In the Shaker channel, a mutation of this tyrosine markedly enhances C-type inactivation29. As it stands, the rotation of the carbonyl oxygen atom of Tyr373 in one given subunit reduces its distance to that in the diagonally opposed subunit and its distance to the carbonyl oxygen atom of Gly372 in the same subunit, which seems to make IS1 a bit too small to readily accommodate a K+ ion (Figs. 3c). If this structural change is genuine and consequential, the electron density at IS1 should largely vanish, which, as shown below, is indeed the case. The mutant tryptophan-induced free energy change in S6 propagates to the filter's extracellular region most probably through the structures around its intracellular region. If so, these structures must be relatively rigid as they exhibit no observable changes. Supporting this view, the filter's intracellular region is attached to the pore helix almost exclusively via backbone hydrogen bonds. In contrast, its extracellular region is primarily attached via side chains and some of these side-chain interactions involve water molecules (Fig. S3). These features suggest that the extracellular region of the filter is inherently more flexible than the intracellular region and therefore more susceptible to energetic perturbations.
Figure 3.
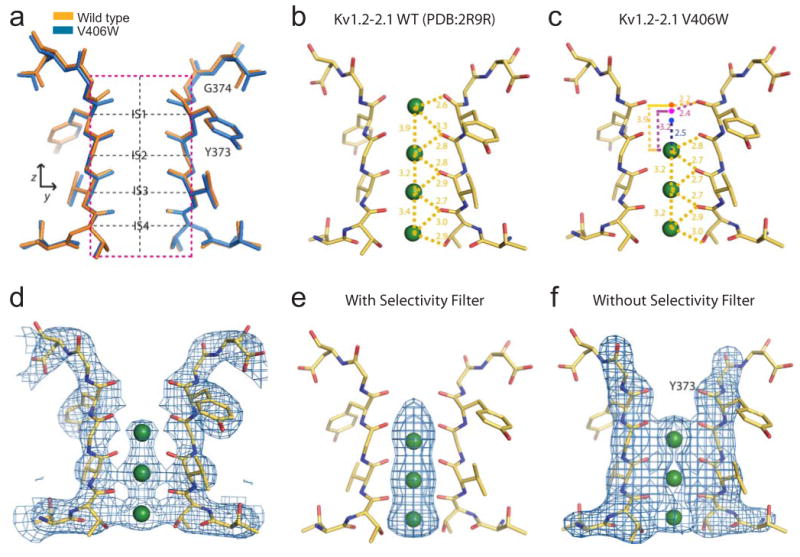
Structure and electron density of the selectivity filter in mutant molecule I. (a) Superposition of the structure (blue) and its wild-type counterpart (orange; PDB: 2R9R); the black dashed vertical line indicated the central axis of the selectivity filter whereas the four black dashed horizontal lines traverse IS1 - IS4. (b, c) Distances between K+ ions and carbonyl oxygens in the selectivity filter of wild-type (b) and V406W mutant (c) channels are indicated. Green spheres indicate the experimentally observed K+ positions. Three dots in panel c indicate hypothetical IS1 positions in the mutant structures, located according to its position (i) as in the wild-type (orange dot), (ii) 3.2 Å from IS2 (magenta dot), and (iii) centered among all coordinating carbonyl oxygen atoms (blue dot). (d) Selectivity filter structure of two diagonally opposed subunits superimposed on the corresponding 2Fo-Fc composite-omit map generated with CNS and contoured at 1.7 σ; K+ ions are shown as green spheres. (e, f) Filter structure superimposed on the corresponding Fo-Fc maps at 5 σ, calculated using CNS with models omitting either only the K+ ions (e) or both the filter and the ions (f).
In the filter of wild-type Kv1.2-2.1 (PDB: 2R9R) all four ion sites exhibit clear electron densities14. In contrast, the IS1 density of the V406W mutant is invisible on a 2FO–FC composite-omit map contoured at 1.7 σ, despite the presence of 150 mM K+ in the crystallization solution (Fig. 3d). On a Fo-Fc map contoured at 5 σ, visible density extends from the intracellular direction to the center of IS1 (Fig. 3e). Evaluation of electron density within the selectivity filter on this map can be seriously confounded by local structures. To demonstrate this effect, we removed the filter in the model by omitting atoms within a cylinder of 5 Å radius centered on the filter's axis (henceforth in all cases, filter removal is done as described here). In a Fo-Fc map recalculated with this filter-omitted model (contoured also at 5 σ) the electron density at IS1 vanished (Fig. 3f versus 3e).
Using one-dimensional electron density profiles along the central axis of the selectivity filter (vertical dashed line, Fig. 3a), we further show that removal of the narrow selectivity filter was in fact critical for uniform bulk solvent correction. As a control, Fig. 4a (magenta curve) shows the one-dimensional density profile extracted from the Fo-Fc map for a model containing the selectivity filter. As expected from the Fo-Fc map itself (Fig. 3e), the plot exhibits three distinct density humps at the loci expected for IS2-IS4 and some modest density at IS1. To assess bulk solvent correction, we placed a string of dummy ions of zero occupancy along the selectivity filter (extending beyond both ends) to create a virtual vacuum in the filter, a maneuver intended to artificially prevent application of bulk solvent correction there. The occupancy of these dummy ions being set at zero, they did not contribute to the phase. We then recalculated an Fo-Fc map, from which we again extracted the one-dimensional electron density profile along the filter (black curve, Fig. 4a). The difference between the two one-dimensional density plots with and without solvent correction gives the solvent correction profile along the filter (blue curve, Fig. 4a). This profile is noticeably non-uniform, most probably because the selectivity filter is only a few angstroms wide, which is too close to the 1.0 - 1.1 Å probe size used in the various solvent correction programs. Solvent correction is not applied to a 1.0 - 1.1 Å zone from any atom surface or to any space smaller than the probe size. If narrowness of the filter space causes non-uniform solvent correction, removing the filter should mitigate the problem. To test this possibility, we recalculated the Fo-Fc map but using the filter-less model, again with or without dummy ions. The difference between the one-dimensional density plots extracted from these two maps (black and magenta curves, Fig. 4b) now shows a nearly uniform solvent correction along the filter (blue curve, Fig. 4b). That the electron density at IS1 appears on the Fo-Fc map (Fig. 3e) not on the filter-omitted Fo-Fc map (Fig. 3f) results from inappropriate solvent correction and possibly a phase bias of the selectivity filter. We conclude that correct evaluation of the filter's K+ densities on a Fo-Fc map should be carried out after omitting not just K+ ions but also the filter.
Figure 4.
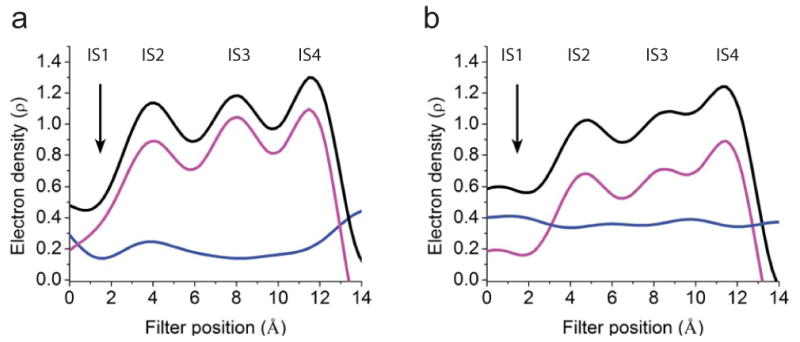
One-dimensional electron density profiles (electrons/Å3) sampled from Fo-Fc maps along the central axis of the filter of V406W mutant molecule I. (a, b) The maps were calculated for four conditions: without (a) or with (b) the filter removed and without (magenta curve) or with (black curve) dummy ions added in the filter to prevent the bulk solvent correction. The blue curve in a or b represents the difference between the corresponding black and magenta curves. The origin of the horizontal axes corresponds to G374's Cα.
Following appropriate solvent correction and elimination of local model biases, the electron density at IS1 is insignificant in one-dimensional plots (Fig. 5a, b) not just along the central axis but also perpendicular to it (vertical and horizontal black dashed lines, Fig 3a). In the view perpendicular to the central axis (Fig. 5b), the density bumps at sites IS2-IS4 are distinct from the densities of the filter structure proper. In this particular case, a two-dimensional contoured (filter-omitted) Fo-Fc map (Fig. 5c) is arguably visually more informative and captures more completely all characteristics discussed above, as compared with a (filter-omitted) Fo-Fc map displayed at a single selected density level (Fig. 3f).
Figure 5.
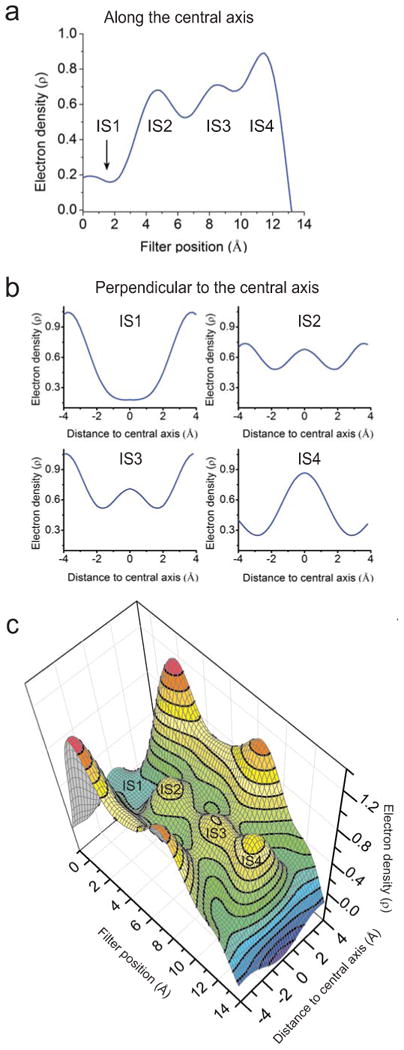
Electron density profiles within the selectivity filter of V406W mutant molecule I. (a, b) One-dimensional profiles of electron density (electrons/Å3) sampled from the filter-omitted Fo-Fc map along (a; taken from Fig. 4b for comparison) or perpendicular to (b) the central axis at the levels of four individual K+ sites, as indicated by the vertical and four horizontal black dashed lines, respectively, in Fig. 3a. The origin of the horizontal axis in panel a corresponds to G374's Cα whereas those in panel b are at the filter's central axis. (c) Contour representation of a filter-omitted Fo-Fc electron density map within the plane of the mutant channel defined by the dashed magenta box in Fig. 3a; highest density is red, and lowest is blue.
Structural features of mutant molecule II
In molecule II, the V406W mutant residue also causes some structural perturbations in S6 and S5, although its side-chain is not well ordered (Fig. 6a). The selectivity filter structure in this molecule is practically indistinguishable from that in the wild type (Fig. 6b). Molecule II is generally less ordered than molecule I, a feature inherited from the wild-type crystal14. Unlike the case of molecule I, abundant electron density is present at the filter's IS1 (Fig. 6c, d). Thus, in molecule II, the extracellular part of the selectivity filter must be stabilized by some structural factors that are not present in molecule I. Searching for possible causes that might stabilize the selectivity filter in molecule II, we found that molecules I and II differ in crystal contacts. In molecule I, the extracellular end of the pore faces a solvent cavity, whereas in molecule II, the so-called turret region, which surrounds the extracellular end of the selectivity filter, is involved in crystal contacts. Specifically, Arg350 in the turret region of molecule II is within hydrogen-bonding distance from Asn148 in a β subunit of molecule I, and Asp351 is within bonding distance from Lys104 in the β subunit (Fig. 7). These two hydrogen bonds appear to be the factors that counteract the energetic impact of V478W and thereby stabilize the selectivity filter in the conformation of the wild type.
Figure 6.
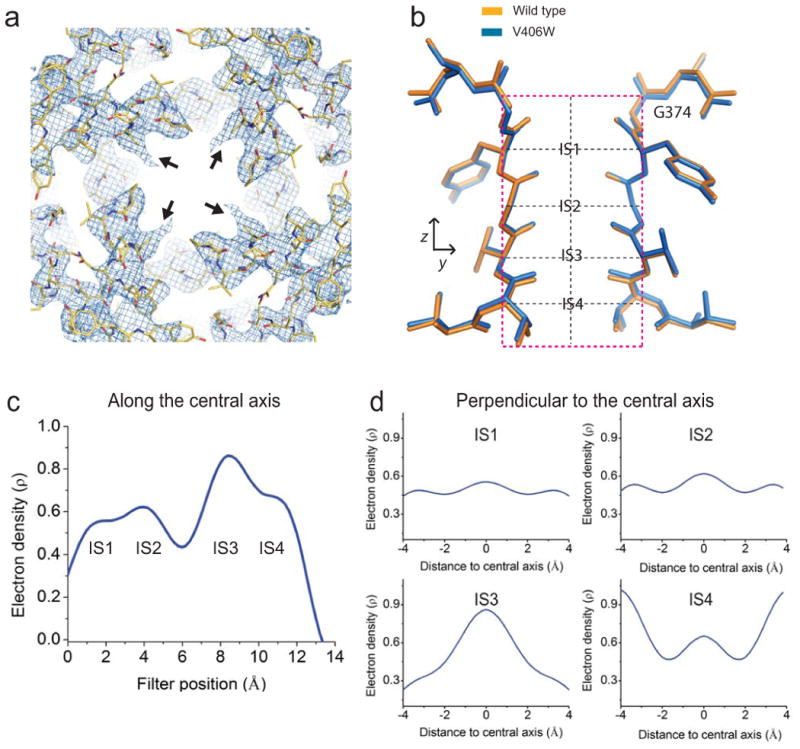
Structure and density of V406W mutant molecule II. (a) Crystal structure of S5 and S6 of molecule II, viewed from inside the cell, superimposed on the corresponding 2Fo-Fc composite-omit map generated with CNS and contoured at 1.5 σ. All symmetric V406W side chains of molecule II were truncated at the Cβ atoms indicated by black arrows. (b) Selectivity filter of the V406W mutant (blue) superimposed on its wild-type counterpart (orange; PDB: 2R9R). (c,d) One-dimensional electron-density profiles (electrons/Å3) sampled from a filter-omitted Fo-Fc map along (c) and perpendicular to (d) the central axis at the levels of four individual K+ sites indicated by the vertical and four horizontal dashed black lines in panel b. Origin of the horizontal axis for panel c corresponds to G374's Cα; those for the plots in panel d are at the filter's central axis.
Figure 7.
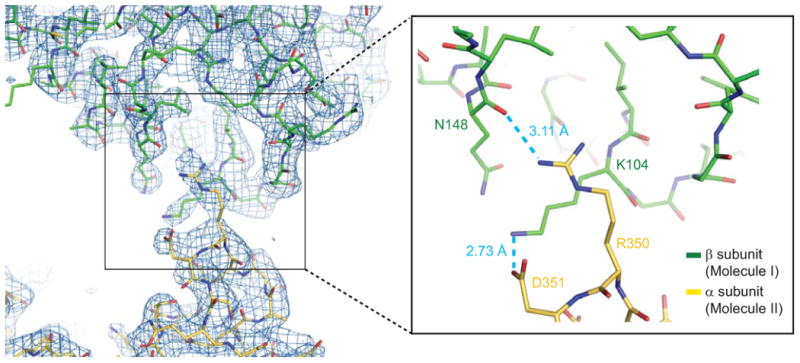
Crystal contacts between the turret region of the α-subunit of molecule II and the β-subunit of molecule I. An interface region between molecule II's α-subunit (yellow; part of the turret region) and molecule I's β-subunit (carbon atoms in green), superimposed on the corresponding 2Fo-Fc composite-omit map contoured at 1.8 σ. Two pairs of atoms within hydrogen-bonding distance are identified by dashed blue lines in the enlarged view of the boxed region in the left panel.
Discussion
While the structural mechanism underlying C-type inactivation is presently controversial, its functional characteristics are well documented. For example, raising the extracellular concentration of permeant ions antagonizes inactivation with the potency that correlates well with the filter's ion selectivity sequence10, whereas reducing K+ supply to the filter promotes inactivation30, 31. A Ba2+ ion, bound in the intracellular part of the selectivity filter in the C-type inactivated state, cannot access the extracellular solution32. These and other findings have led to the view that C-type inactivation in Kv channels is related to some local structural changes and reduced K+ occupancy in the extracellular part of the filter.
Here, we find that the V478W mutation in S6 enhances inactivation of Shaker channels lacking N-type inactivation. Furthermore, using AgTx2 as a probe, we show that this distant mutation also allosterically perturbs the outermost K+ site in the channel's selectivity filter. This latter effect is not surprising, given that a mutation in S6 is already known to affect C-type inactivation and thus the selectivity filter5, and that S6 and the filter are known to be energetically coupled33. Like homotetrameric V478W-mutant Shaker channels, Kv1.2 channels with the equivalent mutation also carry no measureable ionic current upon depolarization. We have obtained crystal structures of mutant Kv1.2-2.1 in two states. In state I, four symmetric V406W residues generate a modest constriction of 5.5 Å in the S6 region, and the outermost set of K+-coordinating atoms, i.e., carbonyl oxygens of the Tyr373 resides, in the selectivity filter moves slightly in the intracellular direction. This alteration of the carbonyl oxygen atoms results in a smaller IS1 (defined by eight carbonyl oxygen atoms) with little density. In contrast, state II appears to have a conducting filter similar to that of the wild type. This difference between the two states most probably reflects that the extracellular end of the pore in state I faces a solvent cavity, whereas in state II it is involved in crystal packing, which stabilizes the selectivity filter in a conducting state (Fig. 7).
In the first of two landmark studies, MacKinnon and colleagues obtained a crystal structure of KcsA at 3.2 Å resolution (here dubbed the original 3.2 Å structure) with only three resolvable ion density peaks in the selectivity filter9. A second structure at 2 Å resolution (dubbed the 2 Å structure) displays a fourth one34, which raises the issue of whether at 3.3 Å resolution we can partially resolve individual K+ ions. To examine this question, we used the phase information from the 2 Å structure34 (omitting K+ ions) to generate a one-dimensional electron density profile along the filter from the 2.0 Å diffraction data34 either extended fully (i.e., to 2.0 Å) or truncated at 3.3 or 3.5 Å (Fig. 8a). At 2.0 Å resolution, the four K+ ions appear, as expected, as fully resolved peaks, whereas at 3.3 Å or 3.5 Å, they appear as partially resolved peaks (bumps). This findings, plus that the four K+ ions in the filter are separated by 3.2 - 3.4 Å, offer the hope that the more accurate phase information provided by the 2.0 Å structure34 might allows us, in retrospect, to partially resolve all four K+ density peaks present in the original 3.2 Å data9.
Figure 8.
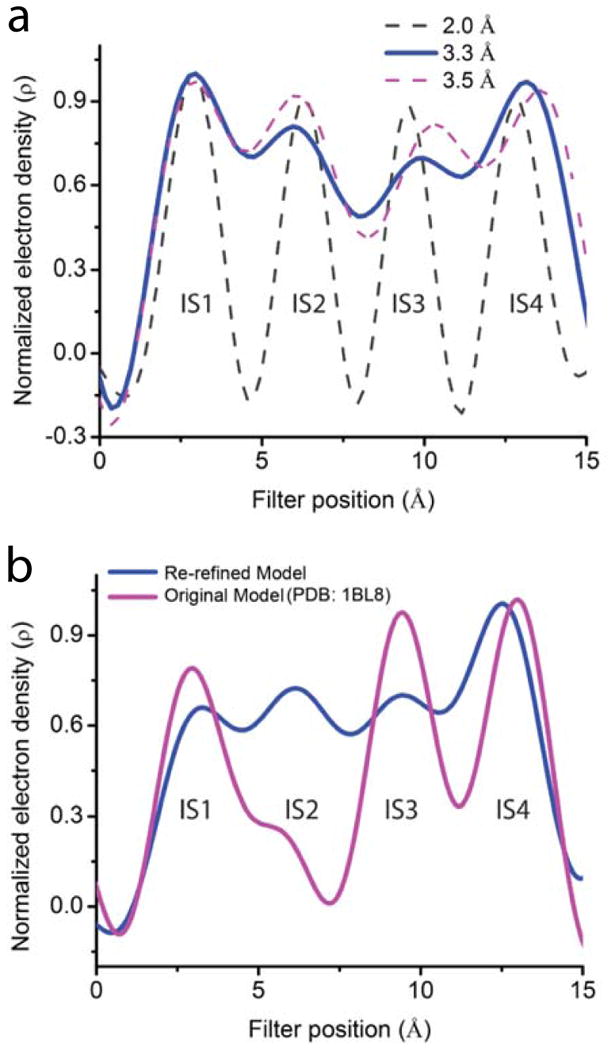
Electron density profiles within KcsA's selectivity filter. (a) Normalized one-dimensional electron density profiles sampled from Fo-Fc maps calculated from the previously published 2.0 Å diffraction data and structure34 (PDB: 1K4C) to three different resolutions: 2.0 Å (black dashed curve), 3.3 Å (solid blue curve) and 3.5 Å (magenta dashed curve). (b) Normalized one-dimensional electron density profiles sampled from Fo-Fc maps calculated with the previously published 3.2 Å data and the original model9 (magenta curve; PDB: 1BL8) or the model re-refined here (blue curve).
As a control, we first calculated from these 3.2 Å data9 the one-dimensional density profile along the filter using the phase information from the original 3.2 Å structure9 (see Methods). Consistent with the originally reported electron density map9, this one-dimensional plot displays three partially resolved peaks (magenta curve, Fig. 8b). We then re-refined a structure against the original 3.2 Å data9 using the phase information provided by the 2.0 Å structure34 (see Methods). The resulting structure (here dubbed the re-refined 3.2 Å structure) has Rfree = 25.6% and Rwork = 24.7% (refinement statistics in supplementary table 1; PDB in supplementary information; refinement procedures in Methods). Using this re-refined 3.2 Å structure, we recalculated from the 3.2 Å data9 the one-dimensional density profile (see Methods). This profile now exhibits four partially resolved density peaks along the filter (the blue curve, Fig. 8b). The above exercise shows that with the 3.2 Å diffraction data, partial resolution of all four ion density peaks in the selectivity filter is practically achievable, provided sufficiently accurate phase information is available, e.g., from a high-resolution structure. In the present study, we partially resolved K+ density peaks from the 3.3 Å diffraction data of V406W-mutant Kv1.2-2.1 by using the phase information from the existing 2.4 Å wild-type structure14.
KcsA, which architecturally resembles Kv's pore module but lacks voltage-sensing modules, has been used as a model to study C-type inactivation. KcsA activated by low intracellular pH (or a constitutively active mutant) undergoes inactivation35. In a crystal structure of KcsA obtained at low (3 mM) K+ concentration, the peptide bond between Val76 and Gly77, which lines the middle part of the selectivity filter, is rotated relative to the structure obtained at a high (200 mM) K+ concentration34. These conformational changes constrict the filter to ∼2 Å and eliminate the middle two of the filter's four ion sites (IS1 – IS4). Similar changes were observed in a mutant constitutively active at neutral pH, which were suggested to be a structural basis for C-type inactivation36. Subsequent studies showed that a maneuver that prevents the rotation of the Val76-Gly77 bond does not prevent inactivation, and that K+ occupancy at IS2-IS4, but not at IS1, affects inactivation kinetics37, 38. The difference between our present structural findings in a Kv channel and those pertaining to inactivation in KcsA reflects the likely case that multiple types of structural changes in the selectivity filter can give rise to non-conduction. The different observations may also stem from these channels' different characteristics. For example, Kv and KcsA differ in how their selectivity filters are attached to the pore-helix.
The human ether-à-go-g-related (HERG) K+ channel, which naturally undergoes profound C-type inactivation39, 40, has recently been determined at 3.7 Å resolution with cryo-EM41. The study shows that the orientation of Phe627's side chain slightly differs from that of a mutant with reduced inactivation42, and the orientation of this side chain in the mutant is more similar to those of other K+ channels41. It was suggested that the difference in the orientation of Phe627's side-chain might be related to C-type inactivation. Residue Phe627 in HERG corresponds to Tyr 373 in Kv1.2-2.1.
To summarize, in a Shaker Kv channel that lacks N-type inactivation, a valine-to-tryptophan mutation in S6 allosterically disrupts the outermost K+ site of the selectivity filter and enhances inactivation. We have obtained one structure of the corresponding Kv1.2-2.1 mutant that exhibits a smaller outermost K+ site with little density. These structural findings are consistent with previously established functional hallmark characteristics of C-type inactivation in eukaryotic Kv channels. The present structure of the K+ selectivity filter may represent an elementary non-conducting conformation whose probability of occurrence is dependent on the conformation energy of S6, where the activation gate is located, and can be modified by input energy to the filter from elsewhere in the channel.
Online Methods
Materials
Detergents were purchased from Anatrace, lipids from Avanti Polar Lipid Inc., anti-Strep-tag antibodies from EMD Millipore or GenScript, N-[N-(L-3-transcarboxyirane-2-carbonyl)-L-Leucyl]-agmatine (E64) from Roche, Superose 6 resin from GE Healthcare, Strep-tactin resin from IBA, and Zeocin from Invitrogen Life Technologies. Other reagents from Sigma, Thermo Fisher Scientific, G-Bioscience or EMD Millipore. Culture media for Pichia pastoris were prepared as described in “Pichia Expression Kit” manual (Invitrogen Life Technologies). cDNA of the rat Kv1.2 α subunit in the pPIZC-C plasmid and cDNA of the rat Kv β2-subunit in the PCR3A+ plasmid were gifts of Drs. Roderick MacKinnon (Rockefeller University) and Carol Deutsch (University of Pennsylvania), respectively. DNA sequencing revealed that residue 15 in the Kv1.2 cDNA used in our study is histidine, compared to leucine in the sequence deposited in NCBI (NP-037102). This residue was not resolved crystallographically.
Molecular biology
For expression in Xenopus oocytes, wild-type and mutant Shaker channels (with N-type inactivation removed)4 including the concatemer construct, as well as wild-type and mutant Kv1.2, were all subcloned in the pGEMHE plasmid43. The concatemer construct was designed as previously described44, with minor changes in the linker sequence. In our construct, the sequence is GQQQQQQQSGYPYDVPDYA(TR/AR)KLQL. Mutant cDNAs were produced through PCR-based mutagenesis and confirmed by DNA sequencing. The cRNAs were synthesized with T7 polymerase using the corresponding cDNAs (linearized with Nhe I) as templates.
For expression in Pichia pastoris, the Kv2.1-1.2 chimera cDNA was constructed as previously described14 with some modifications. The chimeric cDNA, ligated into the multiple cloning site of the pPICZ-B vector (Invitrogen Life Technologies), contained five point mutations (N207Q, C31S, C32S, C431S and C478S), an N-terminal double Strep-tag, and a thrombin protease recognition site (MSAWSHPQFEKGGGSGGGSGGSAWSHPQFEKLVPRGS). The V406W mutation was created on this background. The construct with this mutation were combined with a pPICZ-B vector that already contained the cDNA of the β2 subunit (GI:8393646). Following transformation, P. pastoris clones with successful genomic integration of the PmeI-linearized pPICZ-B plasmid containing the channel cDNAs were selected on agar plates containing yeast extract, peptone, dextrose (YPD), sorbitol and Zeocin (100 – 800 μg/ml). Selected colonies were grown in liquid culture containing 400 μg/ml Zeocin and were screened for protein expression with anti-Strep-tag Western blotting.
Protein expression and purification
The clone of P. pastoris identified to express an adequate amount of protein was grown and induced as described below. All cells were grown using unbaffled flasks with a 1:3 medium to air ratio in a 30°C shaker (200 rpm) incubator (Innova 44, New Brunswick). A single colony was inoculated into 100 ml YPD medium containing 200 μg/ml Zeocin and grown for 36 hours. A ∼40 mL aliquot was diluted with 2L of Buffered Glycerol-complex Medium (BMGY) to an optical density (OD) of ∼0.5, and grown for 6-8 hours until OD reached 4-6. Cells were collected by centrifugation at 3,000 × g for 10 min at 20°C, resuspended (OD of ∼1) and grown in 8 L Buffered Methanol-complex Medium (BMMY). 24 hours later, an additional dose of 0.5% methanol was added to sustain protein induction. The culture was harvested at the 48th hour with centrifugation (3,000 × g, 4°C, 10 min), and the pellet was immediately frozen in liquid nitrogen and stored at −80°C.
To purify the channel protein, frozen cells were broken with a Retsch MM 400 mixer mill or PM 100 planetary ball mill. In the former case, 3 minute milling at 20 Hz was repeated 5 times with intervening 3 minute liquid nitrogen cooling. In the latter case, 3 minute milling at 400 rpm, followed by 1 minute of reverse rotation, was repeated 10 times with 3 minute liquid nitrogen cooling in between. The resulting frozen cell powder was thawed at room temperature in Lysis Buffer (0.8 g/ml). This and other buffers are all defined below. All subsequent steps were carried out at 4°C. To isolate cell membrane, the thawed cell lysate was centrifuged at 2,000 × g for 10 minutes and the resulting supernatant was ultra-centrifuged at 100,000 × g for 100 minutes. The pellet containing cell membranes was resuspended in Lysis Buffer (0.25 g/ml) and homogenized on ice using a Dounce tissue homogenizer followed by a probe sonicator. After addition of dodecyl-maltoside (DDM; 0.2 g/g membranes), the homogenized sample was stirred for 3 hours in a cold room and centrifuged at 20,000 × g for 45 minutes. The supernatant was loaded onto a Strep-tactin column pre-equilibrated with Lysis Buffer containing 5 mM DDM. The column was washed with the wash buffer (40 × bed volume) and eluted with wash buffer containing, in addition, 10 mM d-desthiobiotin (6 × bed volume). The elution fractions identified to contain channel protein (complexed with the β subunit) were combined and reduced to 0.5 mL using a Millipore Amicon Ultra 100K device and further purified on a Superose-6 (10/300) gel filtration column pre-equilibrated in Gel-filtration Buffer. Gel-filtration fractions identified to contain the channel protein were pooled and concentrated to ∼10 mg/mL using a Millipore Amicon Ultra 100K device. The concentrated sample was diluted with a buffer solution, which had the same compositions as Gel-filtration Buffer but without the detergent or lipids, and then re-concentrated. This process was repeated until the DDM concentration reaches 20 - 40 mM while the protein concentration remained at ∼10 mg/mL. The detergent concentration of the sample solution was estimated on thin layer chromatography against a set of standards. A detergent mixture was added to the channel protein sample solutions; the resulting solutions contained approximately 8 mM 3-[(3-cholamidopropyl) dimethylammonio]-1-propanesulfonate (CHAPS) as well as 0.5 - 5 mM 6-Cyclohexyl-1-hexyl-β-D-maltoside (Cymal-6) and 7-Cyclohexyl-1-heptyl-β-D-maltoside (Cymal-7) (1:1 molar ratio). The sample solutions were incubated at 20°C for one hour prior to crystallization trials.
The lysis buffer consisted of 150 mM KCl, 50 mM Tris(hydroxymethyl)aminomethane-HCl (Tris-HCl, pH 8.0), 5 mM beta-mercaptoethanol, 2 mM tris(2-carboxyethyl)phosphine (TCEP, pH 8.0 titrated with NaOH), 2 mM 1,10-phenanthroline, 1 mM ethylenediaminetetraacetic acid (EDTA, pH 8.0), 1 μg/mL aprotinin, 1 μg/mL leupeptin, 1 μg/ml pepstatin A, 0.1 mg/mL soy bean trypsin inhibitor, 0.24 mg/ml 4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride (AEBSF), 5 μg/ml E64, and 1 mM phenylmethylsulfonyl fluoride (PMSF).
The column wash buffer consisted of 150 mM KCl, 50 mM Tris-HCl (pH 8), 5 mM beta-mercaptoethanol, 2 mM TCEP-NaOH, 1 mM EDTA (pH 8), 5 mM DDM, and 0.1 mg/ml lipid mixture (3:1:1 ratio) of 1-Palmitoyl-2-Oleoyl-sn-Glycero-3-Phosphocholine (POPC), 1-Palmitoyl-2-Oleoyl-sn-Glycero-3-Phosphoethanolamine (POPE), and 1-Palmitoyl-2-Oleoyl-sn-Glycero-3-[Phospho-rac-(1-glycerol)] (POPG).
The gel-filtration buffer consisted of 150 mM KCl, 50 mM Tris-HCl (pH 8), 10 mM dithiothreitol (DDT), 2 mM TCEP-NaOH, 1 mM EDTA (pH 8), 2.5 mM DDM, and 0.05 mg/ml lipid mixture (3:1:1 ratio) of POPC, POPE and POPG; pH readjusted to 8.0 with NaOH.
Protein crystallization
All trials were performed using a Mosquito Crystal system (TTP) with a hanging drop method by equilibrating a 1:1 mixture of protein and reservoir solutions against the reservoir solution. The reservoir solution contained 100 μl of 50 mM Tris-Cl at pH 8.3 and 29-31% PEG400. Crystals appeared within 2-3 days following incubation at 20°C, and the selected crystals were protected by raising the PEG400 concentration of the reservoir solution to 35% one day before being frozen in liquid propane and then stored.
Crystallographic analysis of the mutant Kv1.2-2.1 chimera
Channel protein crystals were diffracted at the temperature of 100K and the wavelength of 1 Å on beamline 8.2.1 or 8.2.2 at Advanced Light Source (ALS) in Lawrence Berkeley National Laboratory or on beamline 23-IDB at Advanced Photon Source (APS) in Argonne National Laboratory. Images were processed using HKL-2000 and data reduction were performed with the CCP4 suite45, 46. The Rfree flags were inherited from Long et al.14 The original Kv1.2-2.1 structure (PDB: 2R9R) was used to obtain phase information and as an initial model for structure building. Models were alternately refined in reciprocal space with CNS47 (which includes rigid body refinement, simulated annealing, energy minimization, and individual B refinement) and in real space with Coot48. Statistics of both diffraction data and refinement are shown in table 1. The angles of all peptide bonds are within the favorable or allowable regions in the Ramachandran plot.
All maps were generated using CNS. The 2Fo-Fc composite-omit maps were generated using a model lacking K+ ions. Figures were generated with Pymol49. One-dimensional electron density profiles were obtained using MAPMAN46 to sample the Fo-Fc maps (calculated to 3.3 Å). These maps were generated with annealed models that lack K+ ions, with or without removal of any atoms in the selectivity filter within 5 Å from the central axis. Profiles were obtained along the central axis of the selectivity filter or perpendicularly to it at the level of each of the four ion sites. To obtain a two-dimensional electron density profile of the selectivity filter region, a planar array probe template with 0.2 Å step sizes was created. The probe section is at the level of the origin of the x-axis (x = 0) and spans 8 Å along the y-axis centered on the pore axis and 16 Å along the z-axis. Electron densities at individual probe positions were sampled using MAPMAN and plotted using Origin 8 (OriginLab Corp.).
KcsA re-refinement and calculation of one-dimensional electron density maps
All reciprocal space refinements and map calculations were performed with Phenix50 whereas real space refinements were performed with Coot48. The KcsA structure was re-refined against the 3.2 Å resolution diffraction data (PDB: 1BL8) using the 2.0 Å structure (PDB: 1K4C) for phase information and as the template of an initial model. The initial model contained no water, lipid molecules or K+ ions. After cycles of rigid body refinement, simulated annealing, minimization, b-factor refinement, and manual building, four K+ ions were built (with fixed coordinates) into the selectivity filter to occupy the space, but their occupancy was set to zero to eliminate their bias. A limited number of well-defined water molecules as well as the so-called cavity K+ ion were also built into the model. The resulting model has Rfree of 26.4% and Rwork of 24.6%. The final model, where occupancy of K+ ions was set to 0.5 and their B factors are refined, has Rfree of 25.6% and Rwork of 24.7%. PBD and refinement statistics of this final model are presented in supplementary information.
To calculate all one-dimensional electron density maps of KcsA, K+ ion in all models were effectively removed from the structure by setting their occupancies to zero. The original 3.2 Å low-resolution model (PDB: 1BL8) without K+ ions (but not the 2.0 Å structure) was also subjected to refinements to remove the bias from K+ ions. Following the refinements including cycles of simulated annealing and one cycle of TLS, the model, where occupancy of K+ ions were set to zero, has Rfree of 30.9% and Rwork of 26.9%. It is noteworthy that the low resolution data of the 3.2 Å data set (PDB: 1BL8) were truncated to 10 Å, a maneuver that was typically used at the time to minimize bulk solvent contribution. In the absence of adequate low resolution data, the current bulk solvent correction method is not applicable, and the procedure, which we implemented to achieve a uniform bulk solvent correction in the selectivity filter of the Kv1.2-2.1 mutant structure, is therefore unnecessary here.
Electrophysiological recordings
Xenopus oocytes injected with cRNAs were stored at 18°C in an incubator with ND-96 solution and studied at least 16 hours later51. Channel currents were recorded from oocytes using a two-electrode voltage clamp amplifier (Warner OC-725C; Harvard Apparatus), filtered at 1 kHz, and sampled at 10 kHz using an analog-to-digital converter (Digidata 1322A; MDS Analytical Technologies) interfaced to a PC. pClamp8 software (Molecular Devices) was used for amplifier control and data acquisition. The bath solution contained (in mM): 100 KCl, 0.3 CaCl2, 1 MgCl2 and 10 HEPES; pH was adjusted to 7.6 with KOH unless otherwise specified. The resistance of electrodes filled with 3 M KCl was about 0.2 MΩ. Gating currents were isolated with the P/4 protocol21. Synthetic wild-type and the mutant K27M AgTx2 were folded by means of air oxidation and purified with a C18 HPLC column. For all toxin titration experiments, the extracellular solution contained (in mM): 2 KCl, 0.3 CaCl2, 1 MgCl2 and 10 HEPES, 50μg/ml of BSA; pH was adjusted to 7.6 with KOH. Data analysis and curve fitting were performed with OriginPro 8 (OriginLab Corp.). The figures were made using OriginPro 8 and CorelDRAW X4 (Corel Corp.).
Supplementary Material
Acknowledgments
We thank P. De Weer and T. Hoshi (University of Pennsylvania) for critical review of our manuscript; S. Stayrook (University of Pennsylvania) for his technical assistance on in-house X-ray source; R. MacKinnon (Rockefeller University) for cDNA of the rat Kv1.2 α subunit in the pPIZC-C plasmid and C. Deutsch (University of Pennsylvania) for cDNA of the rat Kv β2-subunit in the PCR3A+ plasmid; S. Long (Memorial Sloan Kettering Cancer Center) for information on Kv expression and cryo-milling; the staff at beamline 8.2.1 & 8.2.2, ALS, Lawrence Berkeley National Laboratory and the staff at beamline 23-IDB, APS, Argonne National Laboratory for their support in data collection. Research reported in this publication was supported by the National Institute of Diabetes and Digestive and Kidney Diseases (DK109919) and the National Institute of General Medical Sciences (GM055560) of the National Institutes of Health.
Footnotes
Contribution: All authors designed research, performed experiment, and analyzed data; V.P., Y.Z., R.Y. and Z.L. wrote the manuscript.
Atomic coordinates and structure factors for chimera Kv1.2-2.1 V406W construct has been deposited with the Protein Data Bank under accession code 5WIE.
Competing financial interest: The authors declare no competing financial interest.
A Life Sciences Reporting Summary is available online.
Data availability: Coordinates and structural factors for the Kv1.2-2.1 V406W mutant have been deposited in the Protein Data Bank under accession code PDB 5WIE. All other data are available from the authors upon reasonable request.
References
- 1.Hodgkin AL, Huxley AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol. 1952;117:500–544. doi: 10.1113/jphysiol.1952.sp004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Armstrong CM. Interaction of tetraethylammonium ion derivatives with the potassium channels of giant axons. J Gen Physiol. 1971;58:413–437. doi: 10.1085/jgp.58.4.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hoshi T, Zagotta WN, Aldrich RW. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science. 1990;250:533–538. doi: 10.1126/science.2122519. [DOI] [PubMed] [Google Scholar]
- 4.Zagotta WN, Hoshi T, Aldrich RW. Restoration of inactivation in mutants of Shaker potassium channels by a peptide derived from ShB. Science. 1990;250:568–571. doi: 10.1126/science.2122520. [DOI] [PubMed] [Google Scholar]
- 5.Hoshi T, Zagotta WN, Aldrich RW. Two types of inactivation in Shaker K+ channels: effects of alterations in the carboxy-terminal region. Neuron. 1991;7:547–556. doi: 10.1016/0896-6273(91)90367-9. [DOI] [PubMed] [Google Scholar]
- 6.Rettig J, et al. Inactivation properties of voltage-gated K+ channels altered by presence of beta-subunit. Nature. 1994;369:289–294. doi: 10.1038/369289a0. [DOI] [PubMed] [Google Scholar]
- 7.Choi KL, Mossman C, Aube J, Yellen G. The internal quaternary ammonium receptor site of Shaker potassium channels. Neuron. 1993;10:533–541. doi: 10.1016/0896-6273(93)90340-w. [DOI] [PubMed] [Google Scholar]
- 8.Liu Y, Holmgren M, Jurman ME, Yellen G. Gated access to the pore of a voltage-dependent K+ channel. Neuron. 1997;19:175–184. doi: 10.1016/s0896-6273(00)80357-8. [DOI] [PubMed] [Google Scholar]
- 9.Doyle DA, et al. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 10.Lopez-Barneo J, Hoshi T, Heinemann SH, Aldrich RW. Effects of external cations and mutations in the pore region on C-type inactivation of Shaker potassium channels. Receptors Channels. 1993;1:61–71. [PubMed] [Google Scholar]
- 11.Yang Y, Yan Y, Sigworth FJ. How does the W434F mutation block current in Shaker potassium channels? J Gen Physiol. 1997;109:779–789. doi: 10.1085/jgp.109.6.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yellen G, Sodickson D, Chen TY, Jurman ME. An engineered cysteine in the external mouth of a K+ channel allows inactivation to be modulated by metal binding. Biophys J. 1994;66:1068–1075. doi: 10.1016/S0006-3495(94)80888-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Long SB, Campbell EB, MacKinnon R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science. 2005;309:897–903. doi: 10.1126/science.1116269. [DOI] [PubMed] [Google Scholar]
- 14.Long SB, Tao X, Campbell EB, MacKinnon R. Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature. 2007;450:376–382. doi: 10.1038/nature06265. [DOI] [PubMed] [Google Scholar]
- 15.Hoshi T, Armstrong CM. C-type inactivation of voltage-gated K+ channels: pore constriction or dilation? J Gen Physiol. 2013;141:151–160. doi: 10.1085/jgp.201210888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Armstrong CM, Hoshi T. K+ channel gating: C-type inactivation is enhanced by calcium or lanthanum outside. J Gen Physiol. 2014;144:221–230. doi: 10.1085/jgp.201411223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Monod J, Wyman J, Changeux JP. On the nature of allosteric transitions: a plausible model. J Mol Biol. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- 18.Perozo E, MacKinnon R, Bezanilla F, Stefani E. Gating currents from a nonconducting mutant reveal open-closed conformations in Shaker K+ channels. Neuron. 1993;11:353–358. doi: 10.1016/0896-6273(93)90190-3. [DOI] [PubMed] [Google Scholar]
- 19.Hackos DH, Chang TH, Swartz KJ. Scanning the intracellular S6 activation gate in the shaker K+ channel. J Gen Physiol. 2002;119:521–532. doi: 10.1085/jgp.20028569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kitaguchi T, Sukhareva M, Swartz KJ. Stabilizing the closed S6 gate in the Shaker Kv channel through modification of a hydrophobic seal. J Gen Physiol. 2004;124:319–332. doi: 10.1085/jgp.200409098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Armstrong CM, Bezanilla F. Currents related to movement of the gating particles of the sodium channels. Nature. 1973;242:459–461. doi: 10.1038/242459a0. [DOI] [PubMed] [Google Scholar]
- 22.Garcia ML, Garcia-Calvo M, Hidalgo P, Lee A, MacKinnon R. Purification and characterization of three inhibitors of voltage-dependent K+ channels from Leiurus quinquestriatus var. hebraeus venom Biochemistry. 1994;33:6834–6839. doi: 10.1021/bi00188a012. [DOI] [PubMed] [Google Scholar]
- 23.MacKinnon R, Miller C. Mechanism of charybdotoxin block of the high-conductance, Ca2+-activated K+ channel. J Gen Physiol. 1988;91:335–349. doi: 10.1085/jgp.91.3.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park CS, Miller C. Interaction of charybdotoxin with permeant ions inside the pore of a K+ channel. Neuron. 1992;9:307–313. doi: 10.1016/0896-6273(92)90169-e. [DOI] [PubMed] [Google Scholar]
- 25.Hidalgo P, MacKinnon R. Revealing the architecture of a K+ channel pore through mutant cycles with a peptide inhibitor. Science. 1995;268:307–310. doi: 10.1126/science.7716527. [DOI] [PubMed] [Google Scholar]
- 26.Banerjee A, Lee A, Campbell E, MacKinnon R. Structure of a pore-blocking toxin in complex with a eukaryotic voltage-dependent K+ channel. Elife. 2013;2:e00594. doi: 10.7554/eLife.00594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tao X, Lee A, Limapichat W, Dougherty DA, MacKinnon R. A gating charge transfer center in voltage sensors. Science. 2010;328:67–73. doi: 10.1126/science.1185954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Webster SM, Del CD, Dekker JP, Yellen G. Intracellular gate opening in Shaker K+ channels defined by high-affinity metal bridges. Nature. 2004;428:864–868. doi: 10.1038/nature02468. [DOI] [PubMed] [Google Scholar]
- 29.Heginbotham L, Lu Z, Abramson T, MacKinnon R. Mutations in the K+ channel signature sequence. Biophys J. 1994;66:1061–1067. doi: 10.1016/S0006-3495(94)80887-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baukrowitz T, Yellen G. Modulation of K+ current by frequency and external K+: a tale of two inactivation mechanisms. Neuron. 1995;15:951–960. doi: 10.1016/0896-6273(95)90185-x. [DOI] [PubMed] [Google Scholar]
- 31.Baukrowitz T, Yellen G. Use-dependent blockers and exit rate of the last ion from the multi-ion pore of a K+ channel. Science. 1996;271:653–656. doi: 10.1126/science.271.5249.653. [DOI] [PubMed] [Google Scholar]
- 32.Harris RE, Larsson HP, Isacoff EY. A permanent ion binding site located between two gates of the Shaker K+ channel. Biophys J. 1998;74:1808–1820. doi: 10.1016/s0006-3495(98)77891-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Panyi G, Deutsch C. Cross talk between activation and slow inactivation gates of Shaker potassium channels. J Gen Physiol. 2006;128:547–559. doi: 10.1085/jgp.200609644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou Y, Morais-Cabral JH, Kaufman A, MacKinnon R. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 Å resolution. Nature. 2001;414:43–48. doi: 10.1038/35102009. [DOI] [PubMed] [Google Scholar]
- 35.Cordero-Morales JF, et al. Molecular determinants of gating at the potassium-channel selectivity filter. Nat Struct Mol Biol. 2006;13:311–318. doi: 10.1038/nsmb1069. [DOI] [PubMed] [Google Scholar]
- 36.Cuello LG, Jogini V, Cortes DM, Perozo E. Structural mechanism of C-type inactivation in K+ channels. Nature. 2010;466:203–208. doi: 10.1038/nature09153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matulef K, Komarov AG, Costantino CA, Valiyaveetil FI. Using protein backbone mutagenesis to dissect the link between ion occupancy and C-type inactivation. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:17866–17891. doi: 10.1073/pnas.1314356110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Matulef K, Annen AW, Nix JC, Valiyaveetil FI. Individual ion biinding sites in the K+ channel play distict roles in C-type inactivation and recovery from inactivation. Structure. 2016;24:750–761. doi: 10.1016/j.str.2016.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Trudeau MC, Warmke JW, Ganetzky B, Robertson GA. HERG, a human inward rectifier in the voltage-gated potassium channel family. Science. 1995;269:92–95. doi: 10.1126/science.7604285. [DOI] [PubMed] [Google Scholar]
- 40.Smith PL, Baukrowitz T, Yellen G. The inward rectification mechanism of the HERG cardiac potassium channel. Nature. 1996;379:833–836. doi: 10.1038/379833a0. [DOI] [PubMed] [Google Scholar]
- 41.Wang W, MacKinnon R. Cryo-EM strcuture of the open human ether-à-go-go-realted K+ channel hERG. Cell. 2017;169:422–430. doi: 10.1016/j.cell.2017.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schonherr R, Heinemann SH. Molecular determinants for activation and inactivation of HERG, a human inward rectifier potassium channel. J Physiol. 1996;493:635–642. doi: 10.1113/jphysiol.1996.sp021410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liman ER, Tytgat J, Hess P. Subunit stoichiometry of a mammalian K+ channel determined by construction of multimeric cDNAs. Neuron. 1992;9:861–871. doi: 10.1016/0896-6273(92)90239-a. [DOI] [PubMed] [Google Scholar]
- 44.McCormack K, Lin L, Iverson LE, Tanouye MA, Sigworth FJ. Tandem linkage of Shaker K+ channel subunits does not ensure the stoichiometry of expressed channels. Biophys J. 1992;63:1406–1411. doi: 10.1016/S0006-3495(92)81703-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 46.Collaborative Computational Project, N.4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 47.Brunger AT, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 48.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallographica Section D-Biological Crystallography. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 49.Schrödinger, LLC; 2002. The PyMOL molecular graphics system. Version 1.5.0.4. http://www.pymol.org. [Google Scholar]
- 50.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Biological crystallography Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Spassova M, Lu Z. Coupled ion movement underlies rectification in an inward-rectifier K+ channel. J Gen Physiol. 1998;112:211–221. doi: 10.1085/jgp.112.2.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.