

Abstract
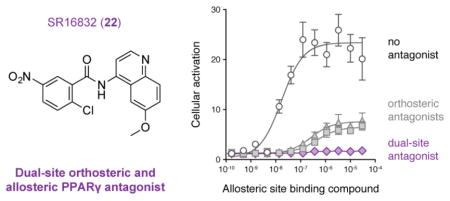
GW9662 and T0070907 are widely used commercially available irreversible antagonists of peroxisome proliferator-activated receptor gamma (PPARγ). These antagonists covalently modify Cys285 located in an orthosteric ligand-binding pocket embedded in the PPARγ ligand-binding domain and are used to block binding of other ligands. However, we recently identified an alternate/allosteric ligand-binding site in the PPARγ LBD to which ligand binding is not inhibited by these orthosteric covalent antagonists. Here, we developed a series of analogs based on the orthosteric covalent antagonist scaffold with the goal of inhibiting both orthosteric and allosteric cellular activation of PPARγ by MRL20, an orthosteric agonist that also binds to an allosteric site. Our efforts resulted in the identification of SR16832 (compound 22), which functions as a dual-site covalent inhibitor of PPARγ transcription by PPARγ-binding ligands. Molecular modeling, protein NMR spectroscopy structural analysis, and biochemical assays indicate the inhibition of allosteric activation occurs in part through expansion of the 2-chloro-5-nitrobenzamidyl orthosteric covalent antagonist towards the allosteric site, weakening of allosteric ligand binding affinity, and inducing conformational changes not competent for cellular PPARγ activation. Furthermore, SR16832 better inhibits binding of rosiglitazone, a thiazolidinedione (TZD) that weakly activates PPARγ when cotreated with orthosteric covalent antagonists, and may better inhibit binding of endogenous PPARγ ligands such as docosahexaenoic acid (DHA) compared to orthosteric covalent antagonists. Compounds such as SR16832 may be useful chemical tools to use as a dual-site bitopic orthosteric and allosteric covalent inhibitor of ligand binding to PPARγ.
Graphical Abstract
INTRODUCTION
Peroxisome proliferator-activated receptor gamma (PPARγ) is a lipid-binding nuclear receptor and molecular target for the FDA-approved thiazolidinedione (TZD) or glitazone class of antidiabetic drugs used clinically in patients with type 2 diabetes mellitus.1–4 TZDs are PPARγ agonists that activate transcription through binding to a canonical orthosteric pocket, the binding site for endogenous ligands such as docosahexaenoic acid (DHA) and other lipids, located in the ligand-binding domain (LBD).5 Nuclear receptors have dynamic ligand-binding pockets with sizes typically in the range of 300–1000 Å3 that expand by residue motions to accommodate ligands.6 Comparatively, the PPARγ pocket is large (>1200 Å3) and composed of three subpockets within distinct regions of the LBD core.5 Agonist binding stabilizes the activation function-2 (AF-2) coactivator interaction surface in the PPARγ LBD, facilitates recruitment of coactivator proteins to PPARγ target gene promoters, which influences chromatin remodeling and increases expression of PPARγ target genes. However, TZDs display adverse side effects,7,8 and considerable effort has been made to discover new antidiabetic compounds with better side effect profiles and determine their mechanism of action. Many pharmacologically distinct PPARγ ligands have been developed with activities ranging from full and partial agonism that robustly or weakly activate transcription, respectively, and favor coactivator recruitment; non-agonists or non-covalent passive antagonists that compete with endogenous ligands to bind PPARγ but do not perturb basal activation when bound to endogenous ligands; and inverse agonists that repress transcription and favor corepressor recruitment. Studies using these newer compounds have shown that transcriptional agonism is not required for antidiabetic efficacy, and although these compounds have not translated into the clinic they display more favorable side effect profiles in cellular and animal models.9,10
Mechanistic studies to determine ligand mechanism of action have shown that in addition to binding PPARγ, TZDs also bind and/or activate other proteins7 including AMP kinase,11 mitochondrial pyruvate carrier proteins,12,13 and mitoNEET,14 which may contribute to the beneficial antidiabetic and adverse effects of TZDs. The notion that PPARγ-binding ligands can display non-PPARγ “off-target” effects has also been suggested in studies where cells treated with PPARγ-binding ligands are cotreated with one of two commercially available irreversible PPARγ antagonists, GW9662 and T0070907 (Figure 1A). These compounds derived from the 2-chloro-5-nitrobenzamidyl scaffold covalently bind to a reactive cysteine (Cys285) in the orthosteric pocket.15,16 In crystal structures, endogenous PPARγ ligands and most synthetic ligands fully or partially occupy the three orthosteric subpockets with binding modes that overlap with covalently bound GW9662.5,17 GW9662 and T0070907 are therefore thought to inhibit the binding of other ligands to PPARγ15 and frequently used as chemical tools to inhibit cellular activation of PPARγ by other PPARγ-binding ligands.
Figure 1. Allosteric activation of PPARγ in the presence of orthosteric covalent antagonists.

(A) Chemical structures of GW9662 (1) and T0070907 (2). (B) Chemical structure of MRL20. (C) Crystal structure of PPARγ LBD bound to GW9662 (PDB 3B0R) compared to MRL20 docked into the allosteric site; * denotes the phenyl (GW9662) to pyridyl (T0070907) change. (D) Schematic of the components of the TR-FRET assay that detects how orthosteric or allosteric ligand binding affects the interaction between PPARγ and a coregulator peptide. (E) TR-FRET assay dose response of MRL20 using FITC-labeled TRAP220 coactivator peptide and His-PPARγ LBD ± commercially available covalent antagonists (n=2). (F) Cell-based transactivation assay dose response of MRL20 using a Gal4-PPARγ LBD/5xUAS-luciferase reporter assay in HEK293T cells. Data plotted as the average ± s.e.m. (n=6; normalized to the vehicle control) and fit to a sigmoidal dose response equation.
In addition to binding to the PPARγ orthosteric pocket, we and others have shown that PPARγ-binding compounds can also bind to an alternate site at pharmacologically relevant concentrations and affect PPARγ function.18–21 Alternate site ligand binding occurs when the orthosteric pocket is either blocked by GW9662 and T0070907 or bound to an endogenous ligand, indicating this alternate site can be classified as an allosteric site because it does not compete with endogenous ligand binding.18 Notably, alternate or allosteric ligand-binding sites have been reported for other nuclear receptors,22–31 and the site we identified was structurally visualized by x-ray crystallography for PPARγ and PPARα (Supplementary Figure 1).20,21,31 Structure-function studies have shown that ligands that form hydrogen bond contacts within the orthosteric subpocket near the AF-2 surface are associated with AF-2 stabilization and classical agonism.32,33 However, this is not a strict rule as AF-2 stabilization and transcriptional activation can also occur upon ligand binding to the allosteric site,18 or be reduced/inhibited when ligands occupy the orthosteric subpocket near the AF-2 surface without hydrogen bond contacts.9,10,32,34 The importance of allosteric site modulation is best exemplified in GPCRs, where the discovery of allosteric GPCR ligands led to the development of ligand “bias”.35,36 This provides a means to pharmacologically regulate receptor activity that permits fine-tuning of a functional response without competing with an endogenous ligand. On the molecular level, an allosteric ligand can “trap” an orthosteric ligand to afford a higher on-target residence time and/or also impart conformational changes that impact function. Although the pharmacological utility of the PPARγ allosteric site and related sites in nuclear receptors remain unclear relative to GPCRs, it is possible that allosteric ligands can synergize with endogenous orthosteric nuclear receptor ligands to provide unique functional activity profiles.
Because GW9662 and T0070907 are both ineffective at inhibiting allosteric ligand binding, they are not ideal chemical tools to explore off-target or PPARγ-independent effects of other PPARγ ligands. Based on the structural proximity of the orthosteric pocket and allosteric site, we hypothesized that expanding the 2-chloro-5-nitrobenzamidyl scaffold of GW9662 and T0070907 towards the allosteric site could provide a bitopic dual-site inhibitor—a covalent antagonist capable of inhibiting both orthosteric and allosteric ligand binding. This concept is different than other dual-site nuclear receptor antagonists such as tamoxifen, a competitive non-covalent orthosteric antagonist of estrogen receptors with an extension that protrudes out of the orthosteric pocket, thereby disrupting the position of the helix 12/AF-2 surface and inhibiting coregulator interaction to antagonize ER activity.37 Here, we report modifications resulting in the identification of SR16832 (compound 22), which inhibits both orthosteric and allosteric transcriptional activation of PPARγ by other PPARγ-binding ligands.
RESULTS AND DISCUSSION
Orthosteric Antagonists Allow Activation of PPARγ
Using NMR spectroscopy, we previously showed the PPARγ LBD can bind two equivalents of the agonist MRL20 (Figure 1B);18 one molecule binds and occupies all three orthosteric subpockets in the previously crystallized binding mode,32 and a second molecule binds to an alternate site located near one of the orthosteric subpockets at the β-sheet surface and flexible Ω-loop region preceding helix 3 (Figure 1C). We also showed that GW9662, T0070907, and oxidized fatty acids that covalently bind to Cys285 do not block the second MRL20 molecule from binding to the allosteric site. In a TR-FRET assay that measures the effect of MRL20 binding to PPARγ on the interaction between PPARγ and a peptide derived from the TRAP220 coactivator (Figure 1D), MRL20 increased the interaction between PPARγ and the TRAP220 peptide with an EC50 of 10 ± 1 nM (Figure 1E). However, when PPARγ protein was pretreated with GW9662 or T0070907 to block the orthosteric pocket, MRL20 still increased the interaction between PPARγ and the TRAP220 peptide with EC50 values of 176 ± 1 nM and 440 ± 1 nM, respectively. In a cell-based luciferase reporter assay, GW9662 and T0070907 reduced the potency and efficacy of MRL20 but did not completely inhibit cellular activation of PPARγ (Figure 1F).
Analysis of the GW9662-PPARγ LBD co-crystal structure showed that the phenyl (GW9662) to pyridyl (T0070907) substitution is situated near the allosteric site (Figure 1C), providing a structural rationale that changes in this region of the 2-chloro-5-nitrobenzamidyl scaffold may weaken in allosteric MRL20 potency. Because of the proximity of the orthosteric pocket and allosteric site, we hypothesized that we could develop orthosteric covalent antagonist analogs that protrude into the allosteric site (i.e., a dual-site bitopic covalent antagonist) and inhibit both orthosteric and allosteric activation of PPARγ. A recent study demonstrated that attaching a large lipophilic moiety to GW9662, which can occupy more space in the large orthosteric pocket, can convert the orthosteric covalent antagonist scaffold into a covalent agonist that activates PPARγ transcription.38 Another study expanded the orthosteric covalent antagonist scaffold into the allosteric site to demonstrate that occupancy of the allosteric site can block phosphorylation of PPARγ.21 Here, we desired to develop a covalent dual-site PPARγ antagonist that induces no or low transcriptional activation on its own and inhibits both orthosteric and allosteric activation by other PPARγ-binding ligands.
GW9662 Analogs Inhibit Allosteric Cellular Activation
We synthesized 22 compound analogs derived from the 2-chloro-5-nitrobenzamidyl scaffold of GW9662 and T0070907 (Figure 2) and characterized their effects in inhibiting allosteric activation of PPARγ in a cell-based transcriptional reporter assay. To screen compounds for their ability to inhibit allosteric PPARγ activation by MRL20, we transfected HEK293T cells with an expression plasmid containing the PPARγ LBD fused to the Gal4 DNA-binding domain along with a 5xUAS-luciferase reporter plasmid. Cells were then treated with vehicle control or a covalent analog, with or without 5 μM of MRL20 (Figure 3A). Simple methyl, ethyl, methoxy, and hydroxyl substitutions provided a minor enhancement of inhibiting allosteric activation (3–8), as shown by the difference in luciferase activity vs. DMSO control (Figure 3B), compared to GW9662 (1) and T0070907 (2). In contrast, compound 7, containing a 2-methylpyridine moiety, exhibited a higher allosteric efficacy. Analogs containing an additional phenyl group ortho (9) or meta (10), but not para (11), to the amide bond showed an improvement in inhibiting allosteric efficacy, as did the addition of an ether linkage between the phenyl moieties (12, 13). We also explored fused ring substitutions (14–24), several of which significantly inhibited cellular allosteric PPARγ activation, including compounds 14, 16, 19–22, and 24 (i.e. no statistical difference, two-way ANOVA, multiple comparisons, Figure 3B). In addition, compared to cells not treated with a covalent analog in the absence of MRL20 all but three analogs (10, 22, and 23) showed a significant increase in luciferase activity in the Gal4 assay (P < 0.05, one-way ANOVA, multiple comparisons, Figure 3A). This indicates that GW9662, T0070907, and many of the analogs increase transcriptional activation of Gal4-PPARγ LBD over the basal constitutive activity of PPARγ.
Figure 2.
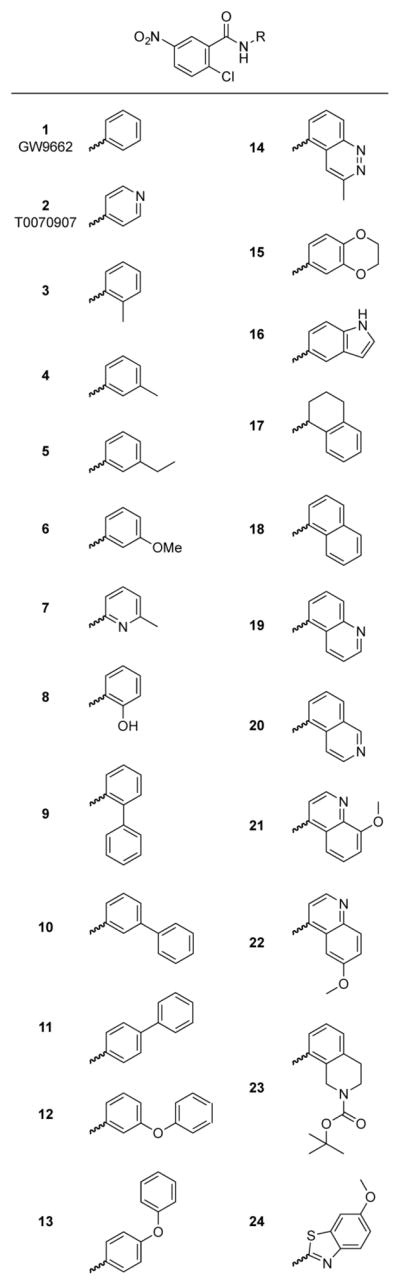
Compound analogs based on the 2-chloro-5-nitrobenzamidyl scaffold.
Figure 3. Efficacy of analogs on inhibiting allosteric cellular activation by MRL20.

(A,B) HEK293T cells transfected with Gal4-PPAR LBD and 5xUAS-luciferase reporter plasmids and treated with 5 μM MRL20 shown as (A) raw luciferase activity or (B) difference in luciferase activity ± MRL20; basal activity marked with a dotted line. (C,D) HEK293T cells transfected with full-length PPARγ and 5xPPRE-luciferase reporter plasmids and treated with 5 μM MRL20 shown as (C) raw luciferase activity or (D) difference in luciferase activity ± MRL20; basal activity marked with a dotted line. Data plotted as the average ± s.e.m. (n=6).
We next tested the analogs on allosteric activation of full-length PPARγ (Figure 3C), where HEK293T cells were transfected with an expression plasmid containing full-length PPARγ along with a luciferase reporter plasmid containing five copies of the PPARγ monomeric DNA-response element (5xPPRE). Full-length PPARγ showed much higher basal transactivation compared to Gal4-PPARγ LBD, which could be due to an influence of the N-terminal activation function-1 (AF-1) region present in full-length PPARγ. In contrast to Gal4-PPARγ LBD, most analogs did not increase activation of full-length PPARγ on their own, and in fact several compounds decreased basal level activation, including compounds 4, 18, 21–23 (P < 0.05, one-way ANOVA, multiple comparisons, Figure 3C). This suggests these analogs might more effectively inhibit binding of an endogenous ligand to PPARγ, which we further verified below (vide infra). When cells were treated with MRL20, several analogs again showed inhibition of allosteric PPARγ activation, including compounds 5, 9, 12, 13, 15, 17, 18, 21, 22, (i.e. no statistical difference, two-way ANOVA, multiple comparisons, Figure 3D).
Effect of Analogs on Coregulator Affinity
We used a fluorescence polarization assay to determine if the analogs affect the ability of PPARγ LBD to interact with FITC-labeled peptides derived from two nuclear receptor coregulator proteins that interact with PPARγ,39–41 including the coactivator TRAP220 (Figure 4A) and the corepressor NCoR (Figure 4B). Apo-protein exhibited weaker affinity for TRAP220 (2.2 ± 0.3 μM) and stronger affinity for NCoR (0.85 ± 0.27 μM). Covalent modification by GW9662 resulted in a slight decrease in TRAP220 binding affinity (2.8 ± 0.3 μM), whereas T0070907 more significantly decreased TRAP220 binding affinity (7.7 ± 0.8 μM). In a similar trend but an opposite effect, T0070907 more significantly increased NCoR binding affinity (0.12 ± 0.06 μM) compared to GW9662 (0.38 ± 0.12 μM). These trends are indicative of inverse agonist activity and suggest that covalent modification by T0070907 in particular, and GW9662 to a lesser degree, may perturb the PPARγ AF-2 coregulator binding surface and affect coregulator interaction. The 22 analogs perturbed affinity for TRAP220 or NCoR to different degrees relative to apo-PPARγ (Figure 4C). Similar to GW9662 and T0070907, many of the compounds showed inverse agonist-like activity by weakening TRAP220 binding and strengthening NCoR binding. However, compounds 7, 8, 10, 15, 15–22, and 24 showed either negligible changes in coregulator peptide affinity or a slight weakening of affinity for both peptides, a profile that may be expected of a true neutral antagonist. We compared the coregulator binding affinities to basal cellular transactivation levels from the luciferase assays, but no correlation could be made. This indicates the transcriptional activity of PPARγ observed in a cellular context upon covalent modification of the analogs may not be related simply to coregulator affinity; there may be additional contributions due to allosteric ligand binding.
Figure 4. Fluorescence polarization coregulator interaction assay.

PPARγ LBD titrated into FITC-labeled (A) TRAP220 coactivator peptide and (B) NCoR corepressor peptide, with or without GW9662 (1) or T0070907 (2). (C) Kd values derived from the assay with or without covalent analogs. Data plotted as the average ± s.e.m. from the fit of the data to a sigmoidal dose-response equation.
Effect of Analogs on Allosteric Potency of MRL20
To gain additional insight into structure-activity relationships, we tested the ability of the analogs to reduce the potency (EC50 value) of recruitment of the TRAP220 coactivator peptide that occurs upon allosteric binding of MRL20 to PPARγ using the TR-FRET assay (Figure 5). Analogs with small changes (3–8) generally exhibited modest or similar improvements in the ability to block or weaken TRAP220 recruitment by allosteric MRL20 binding relative to GW9662 and T0070907. Placement of the methyl group ortho (3), and to a lesser degree meta (4), to the amide bond resulted in decreased allosteric potency, as did meta ethyl (5) or methoxy (6) substitutions. However, a pyridyl ring with the nitrogen ortho and the methyl meta to the amide bond (7) resulted in an increase in allosteric potency. Of these simple changes, the methyl ortho placement (3) exhibited the largest decrease in allosteric potency, whereas a hydroxyl ortho substitution (8) reduced the effect. This suggested that bulkier hydrophobic substitutions at this position could be more favorable in decreasing allosteric potency. Addition of a phenyl substitution (9, 10, and 11) showed a slight improvement over GW9662 (1), with the meta substitution to the amide bond (10) again showing the best improvement. These phenyl substituents, as well as phenoxy substitutions (12 and 13) did not show a significant improvement relative to methyl substitutions; molecular modeling suggested the rotatable bonds contained in the phenyl substitutions can associate near the allosteric site or bury into the orthosteric pocket away from the allosteric site. For the fused ring analogs (14–24), a fairly robust improvement was observed with a 2,3-dihydrobenzo[1,4]dioxine group (15) and an even larger improvement resulted from the addition of an indole group (16). Other compounds tested with increased aromaticity (18, 19, and 20), with the exception of (17), showed improved inhibition. Addition of methoxyquinoline moieties (21 and 22) also showed improved inhibition, whereas other substitutions did not show a strong improvement (23 and 24). In general, all analogs showed decreased allosteric MRL20 potency relative to apo-PPARγ, demonstrating all are active in modifying Cys285, block ligand binding to the orthosteric site and weaken ligand binding to the allosteric site. Relative to GW9662 (1), two compounds (7 and 24) showed decreased ability to inhibit allosteric recruitment of TRAP220 (lower EC50 values) and fourteen compounds (3–6, 8, 10, 11, 15, 16, 18–22) showed an improvement over T0070907.
Figure 5. Analogs weaken allosteric potency of MRL20.

EC50 values from a TR-FRET assay using TRAP220 coactivator peptide and His-PPARγ LBD ± covalent antagonist analogs. Data plotted as the average ± s.e.m. from the fit of the data to a sigmoidal dose-response equation.
SR16832 as a Candidate Dual-Site Covalent Antagonist
Of the covalent antagonist analogs tested, cellular treatment with SR16832 (22) showed little to no allosteric cellular activation by MRL20 (Figures 6A). Additionally, SR16832 only caused a minimal effect on the basal activity of Gal4-PPARγ LBD and lowered the basal activity of full-length PPARγ. Endogenous lipids present in cellular assays are known to activate PPARγ, and TR-FRET showed that SR16832 reduced binding of DHA to PPARγ better than GW9662 and T0070907 (Figure 6B,C). Compared to other analogs, SR16832 only slightly weakened the binding affinity of coregulator peptides, whereas many of the other analogs displayed inverse agonist profiles by weakening affinity for TRAP220 and strengthening affinity for NCoR. Finally, SR16832 is among the best of the fused ring analogs that weaken allosteric potency of MRL20 in the TR-FRET assay with TRAP220, including in the presence of RXRα LBD (Supplementary Figure 2), and a TR-FRET assay with NCoR (Figure 6D,E). These observations indicate that SR16832 is a suitable candidate for selection as a dual-site PPARγ antagonist.
Figure 6. SR16832 as a candidate dual-site inhibitor.

(A) Gal4-PPAR LBD/5xUAS-luciferase reporter assay in HEK293T cells. Data plotted as the average ± s.e.m. (n=4) and fit to a sigmoidal dose response equation. MRL20 was tested up to 30 μM due to cellular toxicity at higher concentrations. (B,C) SR16832 reduces binding of DHA to PPARγ in a TR-FRET assay using TRAP220 coactivator peptide and His-PPARγ LBD ± covalent antagonist analogs; (B) dose response format fit to a sigmoidal dose response equation, (C) endpoint format comparing vehicle control (DMSO) and 30 μM DHA treated conditions. (D) TR-FRET assay using NCoR corepressor peptide and His-PPARγ LBD ± covalent antagonist analogs, plotted as the average ± s.e.m; and (E) the corresponding EC50 values from the fit of the data to a sigmoidal dose-response equation. TR-FRET data plotted as the average ± s.e.m. (n=2).
SR16832 Inhibits Allosteric Activation by Rosiglitazone
We previously showed that rosiglitazone can activate PPARγ in the presence of GW9662.18 To test the effect of SR16832 on inhibiting allosteric activation by rosiglitazone, we performed the TR-FRET assay (Figure 7A). Titration of rosiglitazone into apo/ligand-free PPARγ LBD resulted in a concentration dependent increase in TRAP220 recruitment. The TR-FRET response was lower but not blocked by GW9662 and T0070907. In contrast, SR16832 did not show any detectable increase in TR-FRET response, indicating no binding of rosiglitazone (Figure 7B). We also tested the effect of SR16832 on inhibiting cellular allosteric activation of PPARγ by rosiglitazone. HEK293T cells were transfected with Gal4-PPARγ LBD and 5xUAS-luciferase reporter plasmid, treated with vehicle control or a covalent inhibitor, followed by titration of rosiglitazone (Figure 7C). Compared to GW9662 or T0070907 treated cells, relatively no activation was observed for rosiglitazone in the presence of SR16832 further indicating it is a better allosteric inhibitor.
Figure 7. SR16832 inhibits allosteric potency and activation by rosiglitazone.

(A,B) TR-FRET assay using TRAP220 coactivator peptide and His-PPARγ LBD ± covalent antagonist analogs; (A) dose response format fit to a sigmoidal dose response equation, (B) endpoint format comparing vehicle control (DMSO) and 300 μM rosiglitazone treated conditions. Data plotted as the average ± s.e.m. (n=2). (C) Cell-based transactivation assay dose response of rosiglitazone using a Gal4-PPARγ LBD/5xUAS-luciferase reporter assay in HEK293T cells. Data plotted as the average ± s.e.m. (n=4; normalized to the vehicle control).
Structural Analysis of SR16832 Bound to PPARγ
To gain structural insight into how SR16832 blocks allosteric activation of PPARγ, we performed molecular modeling by modifying the GW9662-bound PPARγ LBD crystal structure (PDB 3B0R). Compared to the binding pose of GW9662 (Figure 8A), the 6-methoxyquinoline group in SR16832 is predicted to protrude into the allosteric site between helix 3 and the β-strand region (Figure 8B). Notably, the binding pose of GW9662 does not clash with MRL20 docked into the allosteric site (Figure 1B),18 whereas the 6-methoxyquinoline group of SR16832 clashes with MRL20 (Figure 8C). To confirm the modeled binding pose of SR16832, we compared 2D [1H,15N]-TROSY HSQC NMR spectroscopy data on 15N-labeled PPARγ LBD bound to GW9662 or SR16832 (Figure 8D). The change from the phenyl group in GW9662 to the 6-methoxyquinoline group in SR16832, resulting in an extension of the analog into the allosteric site, causes NMR chemical shift perturbations for well resolved peaks in the 2D NMR data for residues near the allosteric site, as well as surrounding regions indicating a conformation changes propagated to other parts of the protein (Figure 8E). We also compared 2D [1H,15N]-TROSY HSQC NMR data on SR16832 and 21, where the sole difference is the placement of the methoxy moiety, 6-methoxyquinoline versus 8-methoxyquinoline, respectively, which are located near the β-strand surface (Figure 8F). NMR chemical shift perturbations are primarily localized around the β-strand region near the allosteric site in addition to residues that were previously shown to be protected by HDX exchange by allosteric binding (Figure 8G).14 Thus, the NMR data are consistent with the modeled binding pose of SR16832.
Figure 8. Modeling and structural analysis of SR16832-bound PPARγ LBD.

(A–C) Comparison of the binding poses of (A) GW9662 (PDB 3B0R) and (B) SR16832 (compound 22; modeled from PDB 3B0R). Residues in close proximity to the extensions in SR16832, relative to GW9662, that protrude towards the allosteric site are shown as green sticks and labeled; Cys285 atoms remaining after covalent attachment are shown as yellow spheres. (C) The 6-methoxyquinoline group in SR16832 clashes with MRL20 docked into the allosteric site. (D–G) Differential 2D [1H,15N]-TROSY HSQC NMR spectroscopy data on 15N-labeled PPARγ LBD bound to (D,E) GW9662 (black) or SR16832 (compound 22; green); and (F,G) 21 (magenta) or SR16832 (compound 22; green). NMR chemical shift changes are mapped onto the crystal structure of PPARγ LBD bound to GW9662 (PDB 3B0R) with modeled covalent analogs (E,G). Arrows highlight the respective differences between covalent analogs; (E) extension of the phenyl (1) to 6-methoxyquinoline (22), and (G) placement of the methoxy group at the 8- (21) or 6- (22) position.
Conclusions
Compared to commercially available orthosteric covalent antagonists GW9662 and T0070907, our best compound, SR16832 (compound 22), was effective at inhibiting both orthosteric and allosteric cellular activation by the PPARγ agonists MRL20 and rosiglitazone. In the TR-FRET assay, SR16832 was among the best analogs in weakening allosteric binding of MRL20. However, while SR16832 completely abolished allosteric cellular activation of PPARγ, it did not completely inhibit binding of the TRAP220 peptide to purified PPARγ protein in the TR-FRET assay. There are several possible reasons for this. First, the TR-FRET biochemical assay uses simple components (purified protein and a peptide derived from the TRAP220 coactivator), whereas the cellular reporter assay is representative of PPARγ function in cells with the full cellular complement of full-length nuclear receptor coactivator and corepressor proteins. Furthermore, cellular potency values can differ from biochemical potency due to competition with endogenous ligands in cells. Second, whereas T0070907 and SR16832 both weaken MRL20 EC50 values in the TR-FRET biochemical assay relative to apo-protein, MRL20 caused significant cellular activation when cells are pretreated with T0070907 but no cellular activation when cells are pretreated with SR16832. This observation indicates that in addition to inhibiting allosteric ligand binding, the analogs may induce specific structural conformations that can also influence allosteric activation of PPARγ. It is noteworthy that SR16832 blocks allosteric binding of rosiglitazone and cellular activation caused by rosiglitazone up to 100 μM. Such high pharmacological ligand concentrations have been used in some cellular studies of PPARγ, despite the potent orthosteric binding affinity of rosiglitazone. Because commercially available orthosteric covalent antagonists GW9662 and T0070907 do not inhibit allosteric activation by rosiglitazone and other PPARγ-binding compounds, dual-site antagonists such as SR16832 may be useful, complementary chemical tools for researchers to use to simultaneously inhibit both orthosteric and allosteric ligand-induced cellular activation of PPARγ.
METHODS
Protein, Ligands, and Covalent Ligand Attachment
Human PPARγ LBD (residues 203–477; isoform 1 numbering) was expressed in Escherichia coli BL21(DE3) cells as a Tobacco Etch Virus (TEV) cleavable N-terminal hexahistidine-tagged fusion protein (His-PPARγ LBD) and purified using previously described protocols,18,33 and purity was verified by SDS-PAGE. DHA and rosiglitazone were obtained from Cayman Chemical. For covalent binding of compounds, PPARγ LBD was incubated at 4 °C overnight with 2x molar excess of compound prepared in DMSO-d6; additional detail is provided in the Supporting Information document.
Chemical Synthesis
To a solution of amine (1.2 eq., 1.2 mmol) and triethylamine (209 μL, 1.5 mmol) in dichloromethane (2 mL) was added 2-chloro-5-nitrobenzoyl chloride (219 mg, 1.0 mmol) at 0 °C. The solution was allowed to warm to room temperature overnight with stirring. The reaction mixture was quenched with saturated sodium bicarbonate solution and extracted with dichloromethane. The organic layer was washed with HCl solution (1 N), brine, dried (Na2SO4) and concentrated under reduced pressure. The crude residue was purified by flash chromatography on silica gel (EtOAc/hexanes) to give the target compound. The identity and purity of the compounds (>95%) was confirmed by NMR, mass spectrometry, and reverse-phase analytical HPLC analysis. The reaction scheme, 1H NMR, and masses of compounds are provided in the Supporting Information document. MRL20 was previously synthesized18,33 using established methods;42 solubility issues18 limited use of MRL20 concentrations to 30 μM in assays.
Biochemical and Cellular Assays
The fluorescence polarization (FP) coregulator interaction biochemical assay, time resolved-fluorescence resonance energy transfer (TR-FRET) biochemical assay, and cell-based luciferase reporter assays were performed as described previously;18,19,33,34 details are provided in the Supporting Information document.
NMR Spectroscopy
Experiments were performed as described previously;18,19,33,34 details are provided in the Supporting Information document.
Supplementary Material
Acknowledgments
We thank P. Griffin (Scripps Florida) for mammalian expression and reporter plasmids, and B. Shen (Scripps Florida) for providing access to a high-resolution mass spectrometer. This work was supported by National Institutes of Health (NIH) National Institute of Diabetes and Digestive Kidney Diseases (NIDDK) grant awards R01DK101871 (DK) and F32DK108442 (RB); and American Heart Association (AHA) fellowship award 16POST27780018 (RB).
Footnotes
Supporting Information Available: This material is available free of charge via the Internet.
Supplementary Figures 1 and 2; Experimental Procedures; Supplementary References; Spectral Characterization for Final Compounds; NMR Spectra for Compounds (PDF)
References
- 1.Lehrke M, Lazar MA. The many faces of PPARgamma. Cell. 2005;123:993–999. doi: 10.1016/j.cell.2005.11.026. [DOI] [PubMed] [Google Scholar]
- 2.Ahmadian M, Suh JM, Hah N, Liddle C, Atkins AR, Downes M, Evans RM. PPARgamma signaling and metabolism: the good, the bad and the future. Nat Med. 2013;19:557–566. doi: 10.1038/nm.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma) J Biol Chem. 1995;270:12953–12956. doi: 10.1074/jbc.270.22.12953. [DOI] [PubMed] [Google Scholar]
- 4.Willson TM, Cobb JE, Cowan DJ, Wiethe RW, Correa ID, Prakash SR, Beck KD, Moore LB, Kliewer SA, Lehmann JM. The structure-activity relationship between peroxisome proliferator-activated receptor gamma agonism and the antihyperglycemic activity of thiazolidinediones. J Med Chem. 1996;39:665–668. doi: 10.1021/jm950395a. [DOI] [PubMed] [Google Scholar]
- 5.Kroker AJ, Bruning JB. Review of the structural and dynamic mechanisms of PPARγ partial agonism. PPAR Res. 2015;816856:1–15. doi: 10.1155/2015/816856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gallastegui N, Mackinnon JA, Fletterick RJ, Estebanez-Perpina E. Advances in our structural understanding of orphan nuclear receptors. Trends Biochem Sci. 2015;40:25–35. doi: 10.1016/j.tibs.2014.11.002. [DOI] [PubMed] [Google Scholar]
- 7.Soccio RE, Chen ER, Lazar MA. Thiazolidinediones and the promise of insulin sensitization in type 2 diabetes. Cell Metab. 2014;20:573–591. doi: 10.1016/j.cmet.2014.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guan Y, Hao C, Cha DR, Rao R, Lu W, Kohan DE, Magnuson MA, Redha R, Zhang Y, Breyer MD. Thiazolidinediones expand body fluid volume through PPARgamma stimulation of ENaC-mediated renal salt absorption. Nat Med. 2005;11:861–866. doi: 10.1038/nm1278. [DOI] [PubMed] [Google Scholar]
- 9.Choi JH, Banks AS, Estall JL, Kajimura S, Bostrom P, Laznik D, Ruas JL, Chalmers MJ, Kamenecka TM, Bluher M, Griffin PR, Spiegelman BM. Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARgamma by Cdk5. Nature. 2010;466:451–456. doi: 10.1038/nature09291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choi JH, Banks AS, Kamenecka TM, Busby SA, Chalmers MJ, Kumar N, Kuruvilla DS, Shin Y, He Y, Bruning JB, Marciano DP, Cameron MD, Laznik D, Jurczak MJ, Schurer SC, Vidovic D, Shulman GI, Spiegelman BM, Griffin PR. Antidiabetic actions of a non-agonist PPARgamma ligand blocking Cdk5-mediated phosphorylation. Nature. 2011;477:477–481. doi: 10.1038/nature10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.LeBrasseur NK, Kelly M, Tsao TS, Farmer SR, Saha AK, Ruderman NB, Tomas E. Thiazolidinediones can rapidly activate AMP-activated protein kinase in mammalian tissues. Am J Physiol Endocrinol Metab. 2006;291:E175–181. doi: 10.1152/ajpendo.00453.2005. [DOI] [PubMed] [Google Scholar]
- 12.Colca JR, McDonald WG, Cavey GS, Cole SL, Holewa DD, Brightwell-Conrad AS, Wolfe CL, Wheeler JS, Coulter KR, Kilkuskie PM, Gracheva E, Korshunova Y, Trusgnich M, Karr R, Wiley SE, Divakaruni AS, Murphy AN, Vigueira PA, Finck BN, Kletzien RF. Identification of a mitochondrial target of thiazolidinedione insulin sensitizers (mTOT)--relationship to newly identified mitochondrial pyruvate carrier proteins. PLoS One. 2013;8:e61551. doi: 10.1371/journal.pone.0061551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Divakaruni AS, Wiley SE, Rogers GW, Andreyev AY, Petrosyan S, Loviscach M, Wall EA, Yadava N, Heuck AP, Ferrick DA, Henry RR, McDonald WG, Colca JR, Simon MI, Ciaraldi TP, Murphy AN. Thiazolidinediones are acute, specific inhibitors of the mitochondrial pyruvate carrier. Proc Natl Acad Sci U S A. 2013;110:5422–5427. doi: 10.1073/pnas.1303360110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Colca JR, McDonald WG, Waldon DJ, Leone JW, Lull JM, Bannow CA, Lund ET, Mathews WR. Identification of a novel mitochondrial protein (“mitoNEET”) cross-linked specifically by a thiazolidinedione photoprobe. Am J Physiol Endocrinol Metab. 2004;286:E252–260. doi: 10.1152/ajpendo.00424.2003. [DOI] [PubMed] [Google Scholar]
- 15.Leesnitzer LM, Parks DJ, Bledsoe RK, Cobb JE, Collins JL, Consler TG, Davis RG, Hull-Ryde EA, Lenhard JM, Patel L, Plunket KD, Shenk JL, Stimmel JB, Therapontos C, Willson TM, Blanchard SG. Functional consequences of cysteine modification in the ligand binding sites of peroxisome proliferator activated receptors by GW9662. Biochemistry. 2002;41:6640–6650. doi: 10.1021/bi0159581. [DOI] [PubMed] [Google Scholar]
- 16.Lee G, Elwood F, McNally J, Weiszmann J, Lindstrom M, Amaral K, Nakamura M, Miao S, Cao P, Learned RM, Chen JL, Li Y. T0070907, a selective ligand for peroxisome proliferator-activated receptor gamma, functions as an antagonist of biochemical and cellular activities. J Biol Chem. 2002;277:19649–19657. doi: 10.1074/jbc.M200743200. [DOI] [PubMed] [Google Scholar]
- 17.Itoh T, Fairall L, Amin K, Inaba Y, Szanto A, Balint BL, Nagy L, Yamamoto K, Schwabe JW. Structural basis for the activation of PPARgamma by oxidized fatty acids. Nat Struct Mol Biol. 2008 doi: 10.1038/nsmb.1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hughes TS, Giri PK, de Vera IM, Marciano DP, Kuruvilla DS, Shin Y, Blayo AL, Kamenecka TM, Burris TP, Griffin PR, Kojetin DJ. An alternate binding site for PPARγ ligands. Nat Commun. 2014;5:3571. doi: 10.1038/ncomms4571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hughes TS, Shang J, Brust R, de Vera IM, Fuhrmann J, Ruiz C, Cameron MD, Kamenecka TM, Kojetin DJ. Probing the Complex Binding Modes of the PPARgamma Partial Agonist 2-Chloro-N-(3-chloro-4-((5-chlorobenzo[d]thiazol-2-yl)thio)phenyl)-4-(trifluorome thyl)benzenesulfonamide (T2384) to Orthosteric and Allosteric Sites with NMR Spectroscopy. J Med Chem. 2016;59:10335–10341. doi: 10.1021/acs.jmedchem.6b01340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Y, Wang Z, Furukawa N, Escaron P, Weiszmann J, Lee G, Lindstrom M, Liu J, Liu X, Xu H, Plotnikova O, Prasad V, Walker N, Learned RM, Chen JL. T2384, a novel antidiabetic agent with unique peroxisome proliferator-activated receptor gamma binding properties. J Biol Chem. 2008;283:9168–9176. doi: 10.1074/jbc.M800104200. [DOI] [PubMed] [Google Scholar]
- 21.Bae H, Jang JY, Choi CS-S, Lee J-J, Kim H, Jo A, Lee K-J, Choi JC, Suh SW, Park SB. Mechanistic elucidation guided by covalent inhibitors for the development of anti-diabetic PPARγ ligands. Chem Sci advance article. 2016 doi: 10.1039/C6SC01279E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arnold LA, Estebanez-Perpina E, Togashi M, Jouravel N, Shelat A, McReynolds AC, Mar E, Nguyen P, Baxter JD, Fletterick RJ, Webb P, Guy RK. Discovery of small molecule inhibitors of the interaction of the thyroid hormone receptor with transcriptional coregulators. J Biol Chem. 2005;280:43048–43055. doi: 10.1074/jbc.M506693200. [DOI] [PubMed] [Google Scholar]
- 23.Wang Y, Chirgadze NY, Briggs SL, Khan S, Jensen EV, Burris TP. A second binding site for hydroxytamoxifen within the coactivator-binding groove of estrogen receptor β. Proc Natl Acad Sci U S A. 2006;103:9908–9911. doi: 10.1073/pnas.0510596103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Estébanez-Perpiñá E, Arnold AA, Nguyen P, Rodrigues ED, Mar E, Bateman R, Pallai P, Shokat KM, Baxter JD, Guy RK, Webb P, Fletterick RJ. A surface on the androgen receptor that allosterically regulates coactivator binding. Proc Natl Acad Sci U S A. 2007;104:16074–16079. doi: 10.1073/pnas.0708036104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Estébanez-Perpiñá E, Arnold LA, Jouravel N, Togashi M, Blethrow J, Mar E, Nguyen P, Phillips KJ, Baxter JD, Webb P, Guy RK, Fletterick RJ. Structural insight into the mode of action of a direct inhibitor of coregulator binding to the thyroid hormone receptor. Mol Endocrinol. 2007;21:2919–2928. doi: 10.1210/me.2007-0174. [DOI] [PubMed] [Google Scholar]
- 26.Moore TW, Mayne CG, Katzenellenbogen JA. Minireview: Not picking pockets: nuclear receptor alternate-site modulators (NRAMs) Mol Endocrinol. 2010;24:683–695. doi: 10.1210/me.2009-0362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Buzon V, Carbo LR, Estruch SB, Fletterick RJ, Estebanez-Perpina E. A conserved surface on the ligand binding domain of nuclear receptors for allosteric control. Mol Cell Endocrinol. 2012;348:394–402. doi: 10.1016/j.mce.2011.08.012. [DOI] [PubMed] [Google Scholar]
- 28.Caboni L, Lloyd DG. Beyond the ligand-binding pocket: targeting alternate sites in nuclear receptors. Med Res Rev. 2013;33:1081–1118. doi: 10.1002/med.21275. [DOI] [PubMed] [Google Scholar]
- 29.Scheepstra M, Leysen S, van Almen GC, Miller JR, Piesvaux J, Kutilek V, van Eenennaam H, Zhang H, Barr K, Nagpal S, Soisson SM, Kornienko M, Wiley K, Elsen N, Sharma S, Correll CC, Trotter BW, van der Stelt M, Oubrie A, Ottmann C, Parthasarathy G, Brunsveld L. Identification of an allosteric binding site for RORgammat inhibition. Nat Commun. 2015;6:8833. doi: 10.1038/ncomms9833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tice CM, Zheng YJ. Non-canonical modulators of nuclear receptors. Bioorg Med Chem Lett. 2016;26:4157–4164. doi: 10.1016/j.bmcl.2016.07.067. [DOI] [PubMed] [Google Scholar]
- 31.Bernardes A, Souza PC, Muniz JR, Ricci CG, Ayers SD, Parekh NM, Godoy AS, Trivella DB, Reinach P, Webb P, Skaf MS, Polikarpov I. Molecular mechanism of peroxisome proliferator-activated receptor alpha activation by WY14643: a new mode of ligand recognition and receptor stabilization. J Mol Biol. 2013;425:2878–2893. doi: 10.1016/j.jmb.2013.05.010. [DOI] [PubMed] [Google Scholar]
- 32.Bruning JB, Chalmers MJ, Prasad S, Busby SA, Kamenecka TM, He Y, Nettles KW, Griffin PR. Partial agonists activate PPARgamma using a helix 12 independent mechanism. Structure. 2007;15:1258–1271. doi: 10.1016/j.str.2007.07.014. [DOI] [PubMed] [Google Scholar]
- 33.Hughes TS, Chalmers MJ, Novick S, Kuruvilla DS, Chang MR, Kamenecka TM, Rance M, Johnson BA, Burris TP, Griffin PR, Kojetin DJ. Ligand and receptor dynamics contribute to the mechanism of graded PPARγ agonism. Structure. 2012;20:139–150. doi: 10.1016/j.str.2011.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marciano DP, Kuruvilla DS, Boregowda SV, Asteian A, Hughes TS, Garcia-Ordonez R, Corzo CA, Khan TM, Novick SJ, Park H, Kojetin DJ, Phinney DG, Bruning JB, Kamenecka TM, Griffin PR. Pharmacological repression of PPARgamma promotes osteogenesis. Nat Commun. 2015;6:7443. doi: 10.1038/ncomms8443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Violin JD, Crombie AL, Soergel DG, Lark MW. Biased ligands at G-protein-coupled receptors: promise and progress. Trends Pharmacol Sci. 2014;35:308–316. doi: 10.1016/j.tips.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 36.Christopoulos A, Changeux JP, Catterall WA, Fabbro D, Burris TP, Cidlowski JA, Olsen RW, Peters JA, Neubig RR, Pin JP, Sexton PM, Kenakin TP, Ehlert FJ, Spedding M, Langmead CJ. International Union of Basic and Clinical Pharmacology. XC multisite pharmacology: recommendations for the nomenclature of receptor allosterism and allosteric ligands. Pharmacol Rev. 2014;66:918–947. doi: 10.1124/pr.114.008862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shiau AK, Barstad D, Loria PM, Cheng L, Kushner PJ, Agard DA, Greene GL. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell. 1998;95:927–937. doi: 10.1016/s0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- 38.Ohtera A, Miyamae Y, Yoshida K, Maejima K, Akita T, Kakizuka A, Irie K, Masuda S, Kambe T, Nagao M. Identification of a New Type of Covalent PPARgamma Agonist using a Ligand-Linking Strategy. ACS Chem Biol. 2015;10:2794–2804. doi: 10.1021/acschembio.5b00628. [DOI] [PubMed] [Google Scholar]
- 39.Powell E, Kuhn P, Xu W. Nuclear Receptor Cofactors in PPARgamma-Mediated Adipogenesis and Adipocyte Energy Metabolism. PPAR Res. 2007;2007:53843. doi: 10.1155/2007/53843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Koppen A, Kalkhoven E. Brown vs white adipocytes: the PPARgamma coregulator story. FEBS Lett. 2010;584:3250–3259. doi: 10.1016/j.febslet.2010.06.035. [DOI] [PubMed] [Google Scholar]
- 41.Viswakarma N, Jia Y, Bai L, Vluggens A, Borensztajn J, Xu J, Reddy JK. Coactivators in PPAR-Regulated Gene Expression. PPAR Res. 2010;2010 doi: 10.1155/2010/250126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Acton JJ, 3rd, Black RM, Jones AB, Moller DE, Colwell L, Doebber TW, Macnaul KL, Berger J, Wood HB. Benzoyl 2-methyl indoles as selective PPARgamma modulators. Bioorg Med Chem Lett. 2005;15:357–362. doi: 10.1016/j.bmcl.2004.10.068. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.