

Abstract
Personalization of therapy to target specific molecular pathways has been placed in the forefront of cancer research. Initial reports from clinical trials designed to select patients for appropriate treatment on the basis of tumor characteristics not only have generated considerable excitement but also have identified several challenges. These challenges include the overcoming of regulatory and logistic difficulties, identification of the best selection biomarkers and diagnostic platforms that can be applied in the clinical setting, definition of relevant outcomes in small preselected patient populations, and the design of methods that facilitate rapid enrollment and interpretation of clinical trials by aggregating data across histologically diverse malignancies with common genetic alterations. Furthermore, because our knowledge of the functional consequences of many genetic alterations lags, investigators and sponsors struggle with choosing between ideal clinical trial designs and more practical ones. These challenges are amplified when more than one biomarker is used to select patients for a combination of targeted agents. This review summarizes the current status and challenges of clinical trials in the genomic era and proposes ways to address these challenges.
INTRODUCTION
Rapid advancement in next generation sequencing has revealed a complex landscape of genomic alterations, including mutations, amplifications, and deletions, that regulate molecular pathways common to a variety of tumor types.1-6 Some of these alterations are involved in tumor pathogenesis, cell growth, or survival and are therefore referred to as actionable. However, the majority of these genetic alterations are still of unclear significance. To better understand the clinical implications and actionability of these alterations, clinical trials have been initiated to select patients for novel targeted therapies on the basis of their genetic aberrations. Initial experience from these studies highlight considerable logistic and methodological difficulties and indicate that the benefit of genetically guided therapy may be restricted to a smaller patient subpopulation than originally predicted.7-11 This article briefly reviews the opportunities and challenges of clinical trials in the genomic era. It specifically focuses on the relevance to hematologic malignancies and recent insights from basket and umbrella trials as the prototype for genomic-based trials.
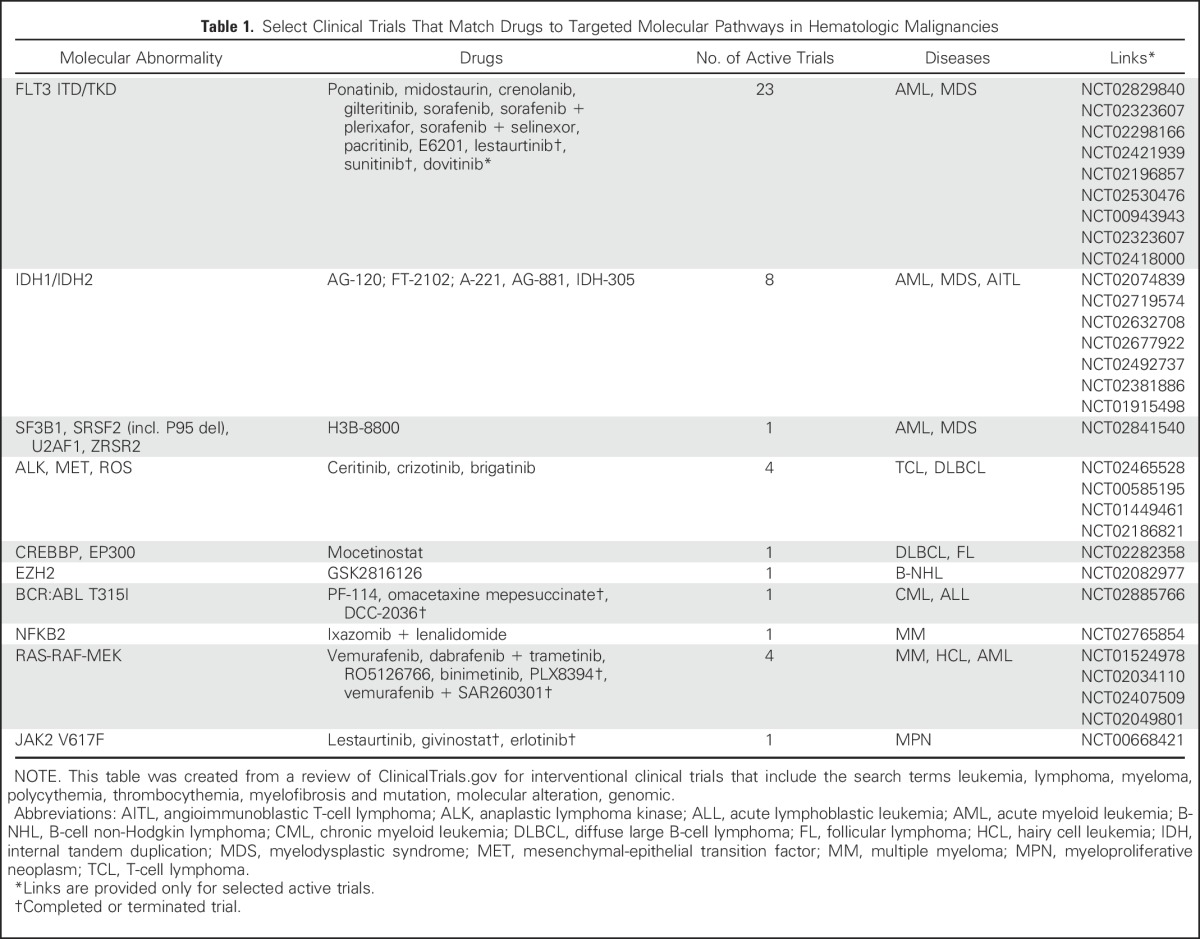
In a recent sequencing study of 3,696 tissue samples from patients with various hematologic malignancies, 95% had at least one known driver alteration, 82% of which were linked to a commercially available targeted therapy or one that is in clinical development.1 Hundreds of other alterations have been independently identified in acute myeloid leukemia (AML), chronic lymphocytic leukemia (CLL), multiple myeloma , and other hematologic malignancies.2-6,12-14 At the same time, a surge in novel therapies has occurred, with > 1,200 drugs currently in active development, 45% of which with a disclosed molecular target.15 When tested in unselected patient populations, the majority of these drugs fail to demonstrate a significant single-agent activity.16 In select diseases, some agents produce high response rates, such as those that target Bruton’s tyrosine kinase, phosphatidylinositol 3-kinase δ, and B-cell lymphoma 2 in patients with CLL, mantle cell lymphoma, Waldenstrom macroglobulinemia, and follicular lymphoma.17-19 However, when tested in unselected patients with other hematologic malignancies, these agents fail to achieve similar results.17,18 The resulting premise was that the matching of patients with targeted agents on the basis of the molecular abnormalities harbored by their malignancies would improve treatment outcomes (Table 1).
Table 1.
Select Clinical Trials That Match Drugs to Targeted Molecular Pathways in Hematologic Malignancies
PATIENT SELECTION ON THE BASIS OF GENETIC ALTERATIONS
Ideally, actionable genetic alterations should lead to new therapies that can be approved by regulatory agencies. For example, BRAFV600E predicts response to vemurafenib in melanoma, and KRAS mutations predict resistance to anti–epidermal growth factor receptor monoclonal antibodies in colorectal cancer.20,21 However, in many cases, identification of an association between a molecular insult and a targeted therapy may be obscure.
Often, the functional consequences are known only for some of the variants identified in a gene of interest (eg, TP53, CREBBP, EZH2). Consequently, investigators and sponsors are faced with the challenge of making decisions about which alterations to use for patient selection. Furthermore, although some new agents may preferentially target malignant cells that harbor specific genetic alterations, they may also demonstrate antiproliferative activity in tumors that do not carry such mutations. An example is the EZH2 inhibitor tazemetostat, which has demonstrated a clinical response in patients with lymphoma and solid tumors irrespective of the presence of its targeted tyrosine 641 mutation.23 Likewise, in myelofibrosis, responses to ruxolitinib, a Janus kinase 2 inhibitor, were seen in patients with and without the JAK2-V617F mutation.24 Finally, co-occurring alterations represent an additional challenge for prioritizing which mutation to target and for evaluating responses.25 DNMT3A mutations, for example, were identified in 30% of patients with AML but associated with a poorer prognosis only in patients with NPM1 mutations.26
To summarize, one of the main objectives of genomic-based trials is to generate insights into the relationship among mutations, malignancy, and response to novel drugs. How to differentiate driver from passenger mutations, what allele frequency should be considered significant, what copy number cutoffs should be used to define meaningful amplification, how to analyze mutations in tumor suppressor genes, how to handle the presence of subclones, and how to analyze variations in noncoding regions are all areas of active research, with special bioinformatics tools under development to predict and prioritize mutations by their actionability.22,27 Longitudinal molecular monitoring plays a subsequent role in the study of mechanisms of resistance and disease evolution. It is, therefore, of utmost importance that the data accumulated through genomic-driven trials will be shared across institutions to allow collaborative knowledge generation.28
BASKET CLINICAL TRIAL DESIGNS
Most of the currently identified genetic alterations are observed in small subsets of patients, which creates challenges for patient accrual and the application of standard designs characteristic of chemotherapy-based clinical trials. To mitigate these challenges, basket trials treat various cancers per their genomic characterization often by pooling together subjects irrespective of the histologic origin of their disease (Table 2).29 Preliminary reports from such studies highlight difficulties in providing appropriate treatment to match disease complexity and probability of response, choosing the right patients, defining relevant outcomes, overcoming regulatory and logistic hurdles, and aggregating results to generate and share new knowledge about actionable mutations and biomarkers of disease resistance and evolution.7-11 In fact, an interim analysis of the National Cancer Institute-Molecular Analysis for Therapy Choice (NCI-MATCH) trial demonstrated that after screening nearly 800 patients, only 9% were found to harbor a study-targeted actionable mutation.8 Other studies reported higher, but still limited, match rates.9,11
Table 2.
Platform Basket Trials (Multiple Histologies/Molecular Pathways) That Enroll Hematologic Malignancies
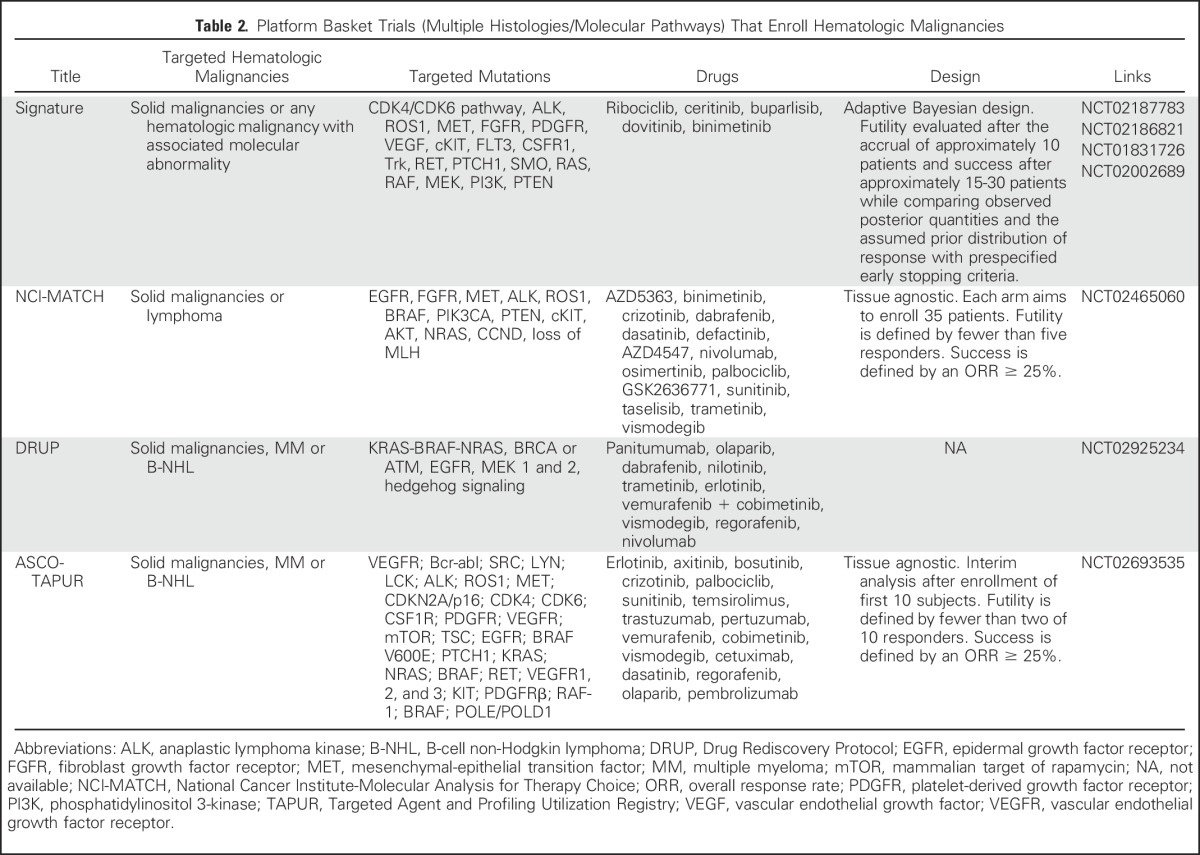
The baseline predicate of most basket trials is that molecular responses to targeted therapy can be evaluated, at least in part, independently from the tissue of origin and irrespective of the presence of additional molecular variants in tumors.29 Thus, patients with differing malignancies but similar molecular abnormalities can be aggregated to allow for faster enrollment. This assumption may only hold true for certain molecular pathways in select cancers and arguably has to be tested before aggregating data across baskets.29-31 Results from a recent basket study of nonmelanoma tumors with BRAFV600 mutations provide a prototypical example, with an 81% response rate for BRAF inhibitors in patients with advanced melanoma as opposed to 40% in hairy cell leukemia and non–small-cell lung carcinoma and only 5% in metastatic colorectal cancer.32,33 Further consideration should be given to the pooling of patients with the same histologic type of cancer but who are following different lengths of therapy because new mutations and new subclones often emerge during the course of therapy.34-36 Consequently, the utility of targeted therapy may be better demonstrated in trials that incorporate these drugs earlier in the disease course.37 Other possible avenues are the testing of therapies in patients with a complete response (CR) but a high risk of relapse or as a measure of a deepening response in patients with residual disease after conventional therapy.37 Notwithstanding, a recent trial that used such an approach to test enzastaurin (a protein kinase C inhibitor) in high-risk patients with diffuse large B-cell lymphoma (DLBCL) in their first CR failed to demonstrate a clinical benefit, even when the evaluation was limited to patients with PKC abnormalities.38 Finally, the limiting of studies to patients with very advanced disease or who are too frail to withstand any other treatment may further obscure the utility of targeted therapies. In this respect, several studies found that high postscreening attrition rates in up to 70% to 90% of patients in basket trials were partly attributed to a deteriorating clinical status and death.8,39
The SHIVA (Molecularly Targeted Therapy Based on Tumor Molecular Profiling Versus Conventional Therapy for Advanced Cancer) trial was the first prospective histology-agnostic randomized trial to compare genomically matched targeted therapy to physician choice. The study included 195 subjects with various metastatic solid malignancies and no standard therapeutic option in whom a molecular alteration existed. Objective response rates in both groups were 3% to 4%, and the median progression-free survival (PFS) was 2 months.9 One of the main criticisms of the SHIVA trial was that the therapy selected for some of the patients was inappropriate to the molecular target.40 However, similar preliminary results have been reported from the Novartis Signature trials, with a response rate of 2.4% in 469 patients who received tissue-agnostic, genetic-specific single-agent therapy.10
The findings from the SHIVA and Signature trials highlight that targeted monotherapy may not be enough to treat certain malignancies. In the context of hematologic malignancies, this seems particularly true for the more-aggressive cancers possibly because of higher replicative stress.41 For example, in Philadelphia-positive acute lymphoblastic leukemia, despite high response rates with monotherapy, intensive chemotherapy with tyrosine kinase inhibitors needs to be incorporated to achieve long-term survival.42 Furthermore, tumors characterized by a high number of synchronous mutations seem to be less susceptible to targeted therapy.43 To that effect, DLBCL contains, on average, up to 100 clonally represented gene alterations per case compared with 11 in CLL and 30 in multiple myeloma.14
An innovative approach to these challenges is seen in the design of the Beat AML trial, which is an umbrella trial (ie, multiple molecular pathways/drugs for a single histology) that will be recruiting elderly patients with de novo AML.44 Each arm will evaluate a different outcome measure determined by the clinical implication of the affected molecular pathway. Thus, in patients with NPM1 mutations in whom a high response rate is expected, the primary end point will be PFS, whereas in patients with IDH mutations, it will be the CR rate, with absence of molecular minimal residual disease as a secondary end point. For patients who harbor more than one abnormality, treatment priorities have been defined on the basis of the known severity of the alteration and the observed allelic burden. Finally, the protocol has inherent procedures for the introduction of novel combination therapies whereby safety is first evaluated through a phase Ib design with patients with relapsed/refractory disease before proceeding with accrual of patients with de novo disease to that arm.
ADAPTIVE TRIAL DESIGNS
Results from recent basket trials highlight the need to have protocol flexibility in place to eliminate ineffective drugs and to introduce new agents or patient cohorts as the trial evolves. With consideration given to the tremendous difficulties in recruiting patients on the basis of molecular alterations, study designs must identify the active baskets as early as possible (ie, enrollment of the minimum number of patients) while accumulating enough data to understand why the drug failed in other baskets and what the biology behind successes and failures is. In the context of large randomized trials, the dropping of ineffective treatment arms typically is performed with a planned interim analysis. However, in the nonrandomized setting of histology-agnostic basket trials, interim analyses are limited by small sample sizes, unbalanced data (because of variable accrual rates across histologies), and the potential heterogeneity in responses seen among patients with different malignancies. Furthermore, molecularly matched historical control estimates often are not available, which makes sound early decisions about efficacy or futility more challenging. Adaptive trial designs use multiple interim analyses of the data that accumulate as the trial progresses to adapt key features based on predefined rules. These may amend the required samples sizes, drug doses, and termination of futile arms (Fig 1). Although most current basket trial designs assume that data can be aggregated across histologies, we believe that similarities in efficacy must be demonstrated before such aggregation takes place. Novel designs for interim analyses can be used to assess such similarities across all or a subset of histologies and to determine whether data from several histologies can be combined to reach a more rapid conclusion. A recent simulation study demonstrated that such an approach improves the probability of identifying a drug that is truly effective in multiple histologies at a modest loss of power if the drug is only effective in a single histology.31 If similarities cannot be demonstrated, the efficacy of the drug can be evaluated separately for each histology by paying special consideration to statistical power and the false discovery rate.45
Fig 1.

Suggested design of a basket clinical trial (ie, single molecular pathway/drug combination in multiple histologies) for hematologic malignancies (shows a single basket). As a first step, histologic subtypes are evaluated separately to assess similarity in efficacy. Each subtype has its relevant response criteria. Subtypes with similar responses are aggregated to allow for faster enrollment. Subtypes with exceptional responses continue as standalone, and futile subtypes are terminated. The numbers and futility thresholds required in stage I and II depend on promising and nonpromising efficacy criteria on the basis of the patient population/molecular profile and the false-positive and false-negative error rates. Numbers are hypothetical and are used for illustration only. A response rate (RR) threshold of 50% was selected for efficacy to indicate that higher responses may be expected and required in the setting of molecularly matched therapy. CLL, chronic lymphocytic leukemia; DLBCL, diffuse large B-cell lymphoma; FL, follicular lymphoma; Ig, immunoglobulin; MM, multiple myeloma.
As an example of the implementation of these concepts, the Signature program is a tissue-agnostic basket trial of multiple agents. The trial implements a Bayesian adaptive design with a hierarchical model that allows the borrowing of information across histologies.30 Early interim analyses are used for evaluating futility (after the accrual of 10 patients) and success (after 15 to 30 patients) and compares observed posterior quantities and the assumed prior distribution of response with prespecified early stopping criteria.46 This approach enables the enrollment of subgroups of various sample sizes and can improve power when some commonality in clinical benefit is observed.47
DEFINITION OF APPROPRIATE OUTCOMES
The generation of a properly matched control group that would account for mutational characteristics, tissue of origin, stage of disease, and number of prior lines of therapy is a formidable task. In the absence of a properly matched control group, the most reliable clinical end points for assessing treatment effects are the rate and duration of responses,48 yet response may be short-lived and should not be regarded in and of itself as an unequivocal surrogate marker for a survival benefit.49 Such was the case with the use of idelalisib in mantle cell lymphoma, which resulted in an overall response rate of 70% but a median duration of 3 months.50
For malignancies that involve hematopoiesis, a correlation with survival might be clearer if response is confined to achievement of a stringent CR or the absence of minimal residual disease.51-53 In this regard, an additional goal of exploratory genomic clinical trials is to evaluate molecular biomarkers for attainment of response and their utility for follow-up.48 Conversely, targeted therapy may provide considerable survival benefit despite disease persistence. Patients with CLL treated with ibrutinib, for example, often display prolonged lymphocytosis composed of biologically inert leukemic cells, yet this does not indicate a poor outcome or impending relapse.54 For lymphomas, CR, as defined by a lack of fluorodeoxyglucose uptake on positron emission tomography and computed tomography imaging, may not be robust enough for evaluating responses to targeted therapies.55 An emerging solution is the use of deep sequencing to measure low levels of circulating tumor DNA, which have been demonstrated as good measures of response and relapse.56,57
Alternative end points may include the ratio PFStargeted:PFSprior line (ie, a comparison of PFS generated by the study drug with that of the preceding therapy) whereby a value > 1.3 has been considered clinically meaningful.7,58,59 In one such basket trial, Von Hoff et al58 reported that 27% of patients achieved this end point with a median PFStargeted:PFSprior line ratio of 2.9. Of note, the overall response rate was only 10%. Other trials have used freedom from progression at 16 weeks as the primary end point.8,10,60 However, if the PFS varies considerably among tumors of different origins, these end points may be of limited value.48
Special consideration should also be afforded to the evaluation of quality of life, safety, and tolerability. Molecularly targeted therapies are associated with unique off-target effects that may present at variable time points during what is often a very prolonged treatment course. The combination of several novel agents may be associated with further unexpected toxicities.61 For example, idelalisib has been associated with a considerable risk for colitis, which usually appears after several months of therapy, and when combined with lenalidomide and rituximab, results in unacceptable rates of hepatotoxicity.61,62 Therefore, adverse events reporting methods should capture unrecognized toxicities as well as delayed and chronic adverse events and gauge their ramifications on quality of life.63,64
Finally, many new targeted therapies are effective at controlling disease but require ongoing treatment, often for many years. Implementation of personalized therapy thus raises concerns about potentially prohibitive costs.65 Therefore, hand in hand with identifying efficient targeted therapies is a need to develop strategies for stopping therapy with retreatment at the time of progression or use of molecular biomarkers as a measure to avoid nonbeneficial treatment altogether.66,67
PRACTICAL CONSIDERATIONS: OVERCOMING REGULATORY AND LOGISTIC DIFFICULTIES
The need to evaluate a large number of therapies in a diverse patient population, often in several countries, introduces unique difficulties to clinical trial initiation. In a recent report from the Worldwide Innovative Networking Consortium in Personalized Cancer Medicine (WINTHER) trial, considerable delays in attaining Food and Drug Administration approval for the study led to > 3 years to activate the US sites. At international sites, the process was shorter but complicated by varying rules about testing novel therapies and the off-label use of approved drugs.7 Another challenge in this regard would be getting pharmaceutical companies to collaborate on the same trial, particularly for the evaluation of combination therapies.37
Long turnaround times for molecular sequencing and the requirement to perform all tests at Clinical Laboratory Improvement Amendments–certified laboratories have also emerged as prohibitive factors, especially where international collaborations are concerned.7 NCI-MATCH investigators reported backlogs in their central laboratory, which resulted in a 5-week delay on sequencing results.8 Similar timelines have been reported in studies that used local laboratories from the MD Anderson Cancer Center and Signature trials.7,39 Special attention should also be paid to technical factors, such as the appropriate acquisition of biopsy material, tissue purity, and cellularity. For example, in a study that evaluated everolimus treatment in DLBCL, responses were independent of the mammalian target of rapamycin/phosphatidylinositol 3-kinase pathway activation possibly as a result of degradation of biopsy material during long delays in tissue fixation.68 Of note, recent reports demonstrated a high success rate in tumor sequencing and highly concordant sequencing results across leading centers.8,9,69
Finally, one of the most important impediments to trial progress, as identified by the WINTHER investigators, has been medication acquisition.7 This is of particular concern in trials that involve community clinics and international sites.7,8,10
SUMMARY
Preliminary reports from major basket trials highlight key challenges in implementing precision oncology. These include difficulties with providing appropriate treatment to match disease complexity, overcoming regulatory and logistic hurdles, choosing the right patients, defining relevant outcomes, and aggregating results to identify actionable mutations and share new knowledge. Insights from these trials can be extended to any trials that use genomics for diagnosis, follow-up, or treatment allocation. Notwithstanding, these are exploratory platforms for screening multiple mutations and therapies to find those few target-drug combinations that could transform cancer care.
Footnotes
Supported by Memorial Sloan Kettering Cancer Center grant P30CA008748 and Memorial Sloan Kettering Specialized Programs of Research Excellence grant in lymphoma P50CA192997-01A1.
AUTHOR CONTRIBUTIONS
Conception and design: All authors
Collection and assembly of data: All authors
Data analysis and interpretation: All authors
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Clinical Trials in the Genomic Era
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/site/ifc.
Erel Joffe
No relationship to disclose
Alexia Iasonos
No relationship to disclose
Anas Younes
No relationship to disclose
REFERENCES
- 1.He J, Abdel-Wahab O, Nahas MK, et al. Integrated genomic DNA/RNA profiling of hematologic malignancies in the clinical setting. Blood. 2016;127:3004–3014. doi: 10.1182/blood-2015-08-664649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374:2209–2221. doi: 10.1056/NEJMoa1516192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. doi: 10.1182/blood-2013-08-518886. Papaemmanuil E, Gerstung M, Malcovati L, et al: Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 122:3616-3627, 2013; quiz 3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Quesada V, Conde L, Villamor N, et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat Genet. 2011;44:47–52. doi: 10.1038/ng.1032. [DOI] [PubMed] [Google Scholar]
- 5.Walker BA, Boyle EM, Wardell CP, et al. Mutational spectrum, copy number changes, and outcome: Results of a sequencing study of patients with newly diagnosed myeloma. J Clin Oncol. 2015;33:3911–3920. doi: 10.1200/JCO.2014.59.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Landau DA, Tausch E, Taylor-Weiner AN, et al. Mutations driving CLL and their evolution in progression and relapse. Nature. 2015;526:525–530. doi: 10.1038/nature15395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rodon J, Soria JC, Berger R, et al. Challenges in initiating and conducting personalized cancer therapy trials: Perspectives from WINTHER, a Worldwide Innovative Network (WIN) Consortium trial. Ann Oncol. 2015;26:1791–1798. doi: 10.1093/annonc/mdv191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. ECOG-ACRIN Cancer Research Group: NCI-MATCH/EAY131 Interim Analysis, 2016. http://ecog-acrin.org/nci-match-eay131/interim-analysis.
- 9.Le Tourneau C, Delord J-P, Gonçalves A, et al. Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): A multicentre, open-label, proof-of-concept, randomised, controlled phase 2 trial. Lancet Oncol. 2015;16:1324–1334. doi: 10.1016/S1470-2045(15)00188-6. [DOI] [PubMed] [Google Scholar]
- 10. Peguero JA, Knost JA, Bauer TM, et al: Successful implementation of a novel trial model: The Signature program. J Clin Oncol 33, 2015 (suppl; abstr 106) [Google Scholar]
- 11.Lopez-Chavez A, Thomas A, Rajan A, et al. Molecular profiling and targeted therapy for advanced thoracic malignancies: A biomarker-derived, multiarm, multihistology phase II basket trial. J Clin Oncol. 2015;33:1000–1007. doi: 10.1200/JCO.2014.58.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang J, Ding L, Holmfeldt L, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481:157–163. doi: 10.1038/nature10725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang J, Grubor V, Love CL, et al. Genetic heterogeneity of diffuse large B-cell lymphoma. Proc Natl Acad Sci U S A. 2013;110:1398–1403. doi: 10.1073/pnas.1205299110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vogelstein B, Papadopoulos N, Velculescu VE, et al: Cancer genome landscapes. Science 339:1546-1558, 2013. [DOI] [PMC free article] [PubMed]
- 15. Global Business Intelligence Research: Frontier Pharma: Hematological cancers – highly innovative pipeline continues trend towards targeted, patient-specific therapies, 2016. http://gbiresearch.com/report-store/market-reports/frontier-pharma/frontier-pharma-hematological-cancers-highly-innovative-pipeline-continues-trend-towards-targeted-patientspecific-therapies?utm_source=mediacenter&utm_medium=pr&utm_campaign=160621aa_gbi_h.
- 16.Arrowsmith J. Trial watch: Phase II failures: 2008-2010. Nat Rev Drug Discov. 2011;10:328–329. doi: 10.1038/nrd3439. [DOI] [PubMed] [Google Scholar]
- 17.Pon JR, Marra MA. Clinical impact of molecular features in diffuse large B-cell lymphoma and follicular lymphoma. Blood. 2016;127:181–186. doi: 10.1182/blood-2015-07-658401. [DOI] [PubMed] [Google Scholar]
- 18.Mehta-Shah N, Younes A. Novel targeted therapies in diffuse large B-cell lymphoma. Semin Hematol. 2015;52:126–137. doi: 10.1053/j.seminhematol.2015.01.007. [DOI] [PubMed] [Google Scholar]
- 19.Jain N, O’Brien S. Targeted therapies for CLL: Practical issues with the changing treatment paradigm. Blood Rev. 2016;30:233–244. doi: 10.1016/j.blre.2015.12.002. [DOI] [PubMed] [Google Scholar]
- 20.Linardou H, Dahabreh IJ, Kanaloupiti D, et al. Assessment of somatic k-RAS mutations as a mechanism associated with resistance to EGFR-targeted agents: A systematic review and meta-analysis of studies in advanced non-small-cell lung cancer and metastatic colorectal cancer. Lancet Oncol. 2008;9:962–972. doi: 10.1016/S1470-2045(08)70206-7. [DOI] [PubMed] [Google Scholar]
- 21.Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carr TH, McEwen R, Dougherty B, et al. Defining actionable mutations for oncology therapeutic development. Nat Rev Cancer. 2016;16:319–329. doi: 10.1038/nrc.2016.35. [DOI] [PubMed] [Google Scholar]
- 23. Morschhauser F, Salles G, McKay P, et al: Initial report from a phase 2 multi-center study of tazemetostat (EPZ-6438), an inhibitor of enhancer of Zeste-Homolog 2 (EZH2), in patients with relapsed or refractory B-cell non-Hodgkin lymphoma (NHL). Presented at ASH Meeting Lymphoma Biol, Colorado Springs, CO, June 18-21, 2016 (abstr 88525) [Google Scholar]
- 24.Harrison C, Kiladjian J-J, Al-Ali HK, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med. 2012;366:787–798. doi: 10.1056/NEJMoa1110556. [DOI] [PubMed] [Google Scholar]
- 25.Catenacci DVT. Expansion platform type II: Testing a treatment strategy. Lancet Oncol. 2015;16:1276–1278. doi: 10.1016/S1470-2045(15)00224-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gale RE, Lamb K, Allen C, et al. Simpson’s paradox and the impact of different DNMT3A mutations on outcome in younger adults with acute myeloid leukemia. J Clin Oncol. 2015;33:2072–2083. doi: 10.1200/JCO.2014.59.2022. [DOI] [PubMed] [Google Scholar]
- 27.Van Allen EM, Wagle N, Stojanov P, et al. Whole-exome sequencing and clinical interpretation of formalin-fixed, paraffin-embedded tumor samples to guide precision cancer medicine. Nat Med. 2014;20:682–688. doi: 10.1038/nm.3559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chakradhar S. Group mentality: Determining if targeted treatments really work for cancer. Nat Med. 2016;22:222–224. doi: 10.1038/nm0316-222. [DOI] [PubMed] [Google Scholar]
- 29.Simon R. Genomic alteration-driven clinical trial designs in oncology. Ann Intern Med. 2016;165:270–278. doi: 10.7326/M15-2413. [DOI] [PubMed] [Google Scholar]
- 30.Berry DA. The Brave New World of clinical cancer research: Adaptive biomarker-driven trials integrating clinical practice with clinical research. Mol Oncol. 2015;9:951–959. doi: 10.1016/j.molonc.2015.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cunanan K, Iasonos A, Shen R, et al. An efficient basket trial design. Meml Sloan-Kettering Cancer Center, Dept Epidemiol Biostat Work Pap Ser 31, 2016. [Google Scholar]
- 32. Dietrich S, Pircher A, Endris V, et al: BRAF inhibition in hairy cell leukemia with low-dose vemurafenib. Blood 127:2847-2855, 2016. [DOI] [PubMed]
- 33.Hyman DM, Puzanov I, Subbiah V, et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med. 2015;373:726–736. doi: 10.1056/NEJMoa1502309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Knight SJL, Yau C, Clifford R, et al. Quantification of subclonal distributions of recurrent genomic aberrations in paired pre-treatment and relapse samples from patients with B-cell chronic lymphocytic leukemia. Leukemia. 2012;26:1564–1575. doi: 10.1038/leu.2012.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ding L, Ley TJ, Larson DE, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481:506–510. doi: 10.1038/nature10738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Prasad V, Fojo T, Brada M. Precision oncology: Origins, optimism, and potential. Lancet Oncol. 2016;17:e81–e86. doi: 10.1016/S1470-2045(15)00620-8. [DOI] [PubMed] [Google Scholar]
- 37.Estey E, Levine RL, Löwenberg B. Current challenges in clinical development of “targeted therapies”: The case of acute myeloid leukemia. Blood. 2015;125:2461–2466. doi: 10.1182/blood-2015-01-561373. [DOI] [PubMed] [Google Scholar]
- 38.Crump M, Leppä S, Fayad L, et al. Randomized, double-blind, phase III trial of enzastaurin versus placebo in patients achieving remission after first-line therapy for high-risk diffuse large B-cell lymphoma. J Clin Oncol. 2016;34:2484–2492. doi: 10.1200/JCO.2015.65.7171. [DOI] [PubMed] [Google Scholar]
- 39.Meric-Bernstam F, Brusco L, Shaw K, et al. Feasibility of large-scale genomic testing to facilitate enrollment onto genomically matched clinical trials. J Clin Oncol. 2015;33:2753–2762. doi: 10.1200/JCO.2014.60.4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tsimberidou AM, Kurzrock R. Precision medicine: Lessons learned from the SHIVA trial. Lancet Oncol. 2015;16:e579–e580. doi: 10.1016/S1470-2045(15)00397-6. [DOI] [PubMed] [Google Scholar]
- 41.Barlow JH, Faryabi RB, Callén E, et al. Identification of early replicating fragile sites that contribute to genome instability. Cell. 2013;152:620–632. doi: 10.1016/j.cell.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Foà R, Vitale A, Vignetti M, et al. Dasatinib as first-line treatment for adult patients with Philadelphia chromosome-positive acute lymphoblastic leukemia. Blood. 2011;118:6521–6528. doi: 10.1182/blood-2011-05-351403. [DOI] [PubMed] [Google Scholar]
- 43.Wheler JJ, Janku F, Naing A, et al. Cancer therapy directed by comprehensive genomic profiling: A single center study. Cancer Res. 2016;76:3690–3701. doi: 10.1158/0008-5472.CAN-15-3043. [DOI] [PubMed] [Google Scholar]
- 44. Leukemia & Lymphoma Society: Beat AML master trial: Information for healthcare professionals. http://www.lls.org/beat-aml/beat-aml-for-healthcare-professionals.
- 45.Cunanan K, Gonen M, Shen R, et al. Basket trials in oncology: A trade-off between complexity and efficiency. J Clin Oncol. doi: 10.1200/JCO.2016.69.9751. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kang BP, Slosberg E, Snodgrass S, et al. The Signature program: Bringing the protocol to the patient. Clin Pharmacol Ther. 2015;98:124–126. doi: 10.1002/cpt.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Berry SM, Broglio KR, Groshen S, et al. Bayesian hierarchical modeling of patient subpopulations: Efficient designs of phase II oncology clinical trials. Clin Trials. 2013;10:720–734. doi: 10.1177/1740774513497539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Simon R, Roychowdhury S. Implementing personalized cancer genomics in clinical trials. Nat Rev Drug Discov. 2013;12:358–369. doi: 10.1038/nrd3979. [DOI] [PubMed] [Google Scholar]
- 49.Burnett AK, Hills RK, Hunter AE, et al. The addition of gemtuzumab ozogamicin to low-dose Ara-C improves remission rate but does not significantly prolong survival in older patients with acute myeloid leukaemia: Results from the LRF AML14 and NCRI AML16 pick-a-winner comparison. Leukemia. 2013;27:75–81. doi: 10.1038/leu.2012.229. [DOI] [PubMed] [Google Scholar]
- 50. Kahl BS, Spurgeon SE, Furman RR, et al: A phase 1 study of the PI3Kδ inhibitor idelalisib in patients with relapsed/refractory mantle cell lymphoma (MCL). Blood 123:3398-3405, 2014. [DOI] [PMC free article] [PubMed]
- 51.Thompson PA, Wierda WG. Eliminating minimal residual disease as a therapeutic end point: Working toward cure for patients with CLL. Blood. 2016;127:279–286. doi: 10.1182/blood-2015-08-634816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Paiva B, van Dongen JJM, Orfao A. New criteria for response assessment: Role of minimal residual disease in multiple myeloma. Blood. 2015;125:3059–3068. doi: 10.1182/blood-2014-11-568907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Goekbuget N, Kantarjian H, Brüggemann M, et al. An evaluation of molecular response in a phase 2 open-label, multicenter confirmatory study in patients (pts) with relapsed/refractory B-precursor acute lymphoblastic leukemia (r/r ALL) receiving treatment with the BiTE® antibody construct blinatumomab. Blood. 2014;124:3704. [Google Scholar]
- 54.Woyach JA, Smucker K, Smith LL, et al. Prolonged lymphocytosis during ibrutinib therapy is associated with distinct molecular characteristics and does not indicate a suboptimal response to therapy. Blood. 2014;123:1810–1817. doi: 10.1182/blood-2013-09-527853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cheson BD, Ansell S, Schwartz L, et al: Refinement of the Lugano classification response criteria for lymphoma in the era of immunomodulatory therapy. Blood 128:2489-2496, 2016. [DOI] [PubMed]
- 56.Kurtz DM, Green MR, Bratman S V, et al. Noninvasive monitoring of diffuse large B-cell lymphoma by immunoglobulin high-throughput sequencing. Blood. 2015;125:3679–3687. doi: 10.1182/blood-2015-03-635169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Assouline SE, Nielsen TH, Yu S, et al. Phase 2 study of panobinostat with or without rituximab in relapsed diffuse large B-cell lymphoma. Blood. 2016;128:185–194. doi: 10.1182/blood-2016-02-699520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Von Hoff DD, Stephenson JJ, Jr, Rosen P, et al. Pilot study using molecular profiling of patients’ tumors to find potential targets and select treatments for their refractory cancers. J Clin Oncol. 2010;28:4877–4883. doi: 10.1200/JCO.2009.26.5983. [DOI] [PubMed] [Google Scholar]
- 59.Tsimberidou A-M, Iskander NG, Hong DS, et al. Personalized medicine in a phase I clinical trials program: The MD Anderson Cancer Center initiative. Clin Cancer Res. 2012;18:6373–6383. doi: 10.1158/1078-0432.CCR-12-1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. ClinicalTrials.gov: TAPUR: Testing the Use of Food and Drug Administration (FDA) Approved Drugs That Target a Specific Abnormality in a Tumor Gene in People With Advanced Stage Cancer, 2016. https://clinicaltrials.gov/ct2/show/NCT02693535.
- 61.Cheah CY, Nastoupil LJ, Neelapu SS, et al. Lenalidomide, idelalisib, and rituximab are unacceptably toxic in patients with relapsed/refractory indolent lymphoma. Blood. 2015;125:3357–3359. doi: 10.1182/blood-2015-03-633156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Coutré SE, Barrientos JC, Brown JR, et al. Management of adverse events associated with idelalisib treatment: Expert panel opinion. Leuk Lymphoma. 2015;56:2779–2786. doi: 10.3109/10428194.2015.1022770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cabarrou B, Boher JM, Bogart E, et al. How to report toxicity associated with targeted therapies? Ann Oncol. 2016;27:1633–1638. doi: 10.1093/annonc/mdw218. [DOI] [PubMed] [Google Scholar]
- 64.Thanarajasingam G, Atherton PJ, Novotny PJ, et al. Longitudinal adverse event assessment in oncology clinical trials: The Toxicity over Time (ToxT) analysis of Alliance trials NCCTG N9741 and 979254. Lancet Oncol. 2016;17:663–670. doi: 10.1016/S1470-2045(16)00038-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Djalalov S, Beca J, Hoch JS, et al. Cost effectiveness of EML4-ALK fusion testing and first-line crizotinib treatment for patients with advanced ALK-positive non-small-cell lung cancer. J Clin Oncol. 2014;32:1012–1019. [Google Scholar]
- 66.Saußele S, Richter J, Hochhaus A, et al. The concept of treatment-free remission in chronic myeloid leukemia. Leukemia. 2016;30:1638–1647. doi: 10.1038/leu.2016.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cardoso F, van’t Veer LJ, Bogaerts J, et al. 70-gene signature as an aid to treatment decisions in early-stage breast cancer. N Engl J Med. 2016;375:717–729. doi: 10.1056/NEJMoa1602253. [DOI] [PubMed] [Google Scholar]
- 68.Barnes JA, Jacobsen E, Feng Y, et al. Everolimus in combination with rituximab induces complete responses in heavily pretreated diffuse large B-cell lymphoma. Haematologica. 2013;98:615–619. doi: 10.3324/haematol.2012.075184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Van Allen EM, Robinson D, Morrissey C, et al. A comparative assessment of clinical whole exome and transcriptome profiling across sequencing centers: Implications for precision cancer medicine. Oncotarget. 2016;7:52888–52899. doi: 10.18632/oncotarget.9184. [DOI] [PMC free article] [PubMed] [Google Scholar]