

Abstract
Purpose
Adoptive transfer of genetically modified T cells is being explored as a treatment for patients with metastatic cancer. Most current strategies use genes that encode major histocompatibility complex (MHC) class I–restricted T-cell receptors (TCRs) or chimeric antigen receptors to genetically modify CD8+ T cells or bulk T cells for treatment. Here, we evaluated the safety and efficacy of an adoptive CD4+ T-cell therapy using an MHC class II–restricted, HLA-DPB1*0401–restricted TCR that recognized the cancer germline antigen, MAGE-A3 (melanoma-associated antigen-A3).
Patients and Methods
Patients received a lymphodepleting preparative regimen, followed by adoptive transfer of purified CD4+ T cells, retrovirally transduced with MAGE-A3 TCR plus systemic high-dose IL-2. A cell dose escalation was conducted, starting at 107 total cells and escalating at half-log increments to approximately 1011 cells. Nine patients were treated at the highest dose level (0.78 to 1.23 × 1011 cells).
Results
Seventeen patients were treated. During the cell dose-escalation phase, an objective complete response was observed in a patient with metastatic cervical cancer who received 2.7 × 109 cells (ongoing at ≥ 29 months). Among nine patients who were treated at the highest dose level, objective partial responses were observed in a patient with esophageal cancer (duration, 4 months), a patient with urothelial cancer (ongoing at ≥ 19 months), and a patient with osteosarcoma (duration, 4 months). Most patients experienced transient fevers and the expected hematologic toxicities from lymphodepletion pretreatment. Two patients experienced transient grade 3 and 4 transaminase elevations. There were no treatment-related deaths.
Conclusion
These results demonstrate the safety and efficacy of administering autologous CD4+ T cells that are genetically engineered to express an MHC class II–restricted antitumor TCR that targets MAGE-A3. This clinical trial extends the reach of TCR gene therapy for patients with metastatic cancer.
INTRODUCTION
Adoptive cell transfer (ACT) is a personalized cancer immunotherapy that involves the administration of a patient’s own autologous immune cells.1 Transferred T cells can be genetically modified with a T-cell receptor (TCR) or a chimeric antigen receptor (CAR) to redirect them to attack the tumor. Administration of CAR-modified T cells that target B-cell lineage differentiation antigen CD19 can lead to objective responses in patients with B-cell cancers2-11; however, thus far, it has been challenging to extend CAR T-cell therapy to patients with solid tumors. In large part, this has been because solid cancers generally lack suitable cell-surface targets that only express on tumor cells but not on normal cells. Recognition of normal tissues by CAR T cells can potentially trigger unacceptable toxicities.12
In contrast to CARs, TCRs are capable of recognizing antigens that are derived from intracellular proteins. Most current TCR therapies use major histocompatibility complex (MHC) class I–restricted TCRs to genetically modify CD8+ T cells or bulk T cells for patient treatment; however, some evidence has suggested that CD4+ T cells alone could induce tumor regressions. In mice, established B16 melanoma could be eradicated by tumor-specific CD4+ T cells, whose activities could be further enhanced by either cytotoxic T-cell lymphocyte-4 blockade, OX40 stimulation, or Th17 polarization.13-15 In humans, a durable clinical response was observed in a patient with metastatic melanoma who was treated with an autologous HLA-DP4–restricted NY-ESO-1–specific CD4+ T-cell clone, as well as in a patient with metastatic cholangiocarcinoma who was treated with mutated ERBB2IP-reactive CD4+ T cells that were grown from tumor-infiltrating lymphocytes.16,17 These clinical studies indicate that transferring CD4+ T cells can induce long-term tumor regression in humans.
Cancer germline (CG) antigens, a class of tumor-associated antigens, show limited expression in normal adult tissues, except for germline-derived tissues. Of importance, germ cells lack expression of MHC molecules and are therefore protected from T cell–mediated immune surveillance. Conversely, CG antigens can show high levels of expression in a variety of cancer types.18,19 Among these antigens, MAGE-A3 (melanoma-associated antigen-A3) is the most frequently expressed CG antigen in a variety of cancer types and has been targeted by cancer immunotherapies, including ACT therapies.20-31 In a previous preclinical study, an MHC class II–restricted, HLA-DPB1*0401–restricted TCR that recognized MAGE-A3/A6 was isolated from the peripheral blood of a patient who received a MAGE-A3 peptide vaccine.32 The human constant regions of TCRα/β chains were replaced by mouse constant regions to enhance TCR pairing and reactivity.33 This TCR was demonstrated to recognize MAGE-A3 and its closest family member, MAGE-A6, which has 95.9% homology with MAGE-A3. Expression of MAGE-A3 and MAGE-A6 was not observed in any normal tissues, except testes.34 A clinical trial was thus designed and conducted to test whether ACT that used genetically modified CD4+ T cells targeting MAGE-A3 could induce tumor regression in patients with a variety of metastatic solid cancers. Previous animal studies indicated that IL-2 administration could significantly enhance T cell–mediated antitumor activity. As a result, our previous ACT clinical trials included high-dose IL-2 therapy; therefore, this clinical trial was designed to incorporate high-dose IL-2 therapy in patients after cell infusion.35-38
PATIENTS AND METHODS
Study Design
This clinical trial was designed to determine the maximum safe dose of the administration of autologous CD4+ cells that were retrovirally transduced with an HLA-DPB*0401–restricted MAGE-A3 TCR and whether this approach could result in clinical tumor regression in patients with metastatic cancer.
Before therapy, peripheral blood lymphocytes (PBLs) were isolated from patients by leukapheresis and separated by centrifugation on a lymphocyte separation medium cushion. CD8+ lymphocytes were labeled with clinical-grade CD8 antibody–coated magnetic particles, then depleted by using a CliniMACS clinical-scale cell separation apparatus (Miltenyi Biotec, Bergisch Gladbach, Germany). CD8+ T cell–depleted PBLs were stimulated by OKT3 antibody (50 ng/mL) and transduced with a γ-retroviral vector that encodes the HLA-DPB*0401–restricted MAGE-A3 TCR, as previously described.32 National Institutes of Health Guidelines for Research Involving Recombinant or Synthetic Nucleic Acid Molecules were followed. In the final cell products that were used for patient treatment, the median percentage of CD4+ T cells was 99% (range, 94% to 100%), and the median percentage of transduced CD4+ T cells was 90% (range, 77% to 92%).
Patients were treated with a nonmyeloablative chemotherapy preparative regimen (cyclophosphamide 60 mg/kg per day for 2 days and fludarabine 25 mg/m2 per day for 5 days), followed by a single intravenous infusion of autologous TCR-transduced CD4+ T cells. A cell dose escalation was conducted, treating one patient in each cohort, starting at 107 total cells and escalating at half-log increments. Nine patients were treated at the highest dose level (approximately 1011 cells). After cell infusion, patients received high-dose IL-2 intravenously at 720,000 IU/kg every 8 hours to physiologic tolerance.
Patients
Patients 18 to 70 years of age with a pathologically confirmed diagnosis of metastatic or locally advanced refractory/recurrent cancer were eligible for this clinical trial. All patients had progressive disease after at least one standard first-line therapy. Patients were required to have an Eastern Cooperative Oncology Group performance status of 0 or 1. In addition, to be eligible, patients must be HLA-DPB1*0401 positive and with tumors that contained > 50% MAGE-A–positive tumor cells. Immunohistochemistry of tumor specimens was performed and scored by the Laboratory of Pathology at the National Cancer Institute using an anti–MAGE-A antibody (6C1; Santa Cruz Biotechnologies, Santa Cruz, CA).34 Responses of patients to therapy were assessed by using Response Evaluation Criteria in Solid Tumors (RECIST version 1.0) guidelines and evaluated starting 1 month after cell administration and proceeding at regular intervals thereafter.
Immunologic Assay
Serum samples were obtained daily from patients during hospitalization. To determine the cytokine concentrations in serum samples, enzyme-linked immunosorbent assay for interferon gamma (IFN-γ), IL-6, IL-10, and TNF were performed according to manufacturer instructions (Thermo Fisher Scientific Life Sciences, Waltham, MA; and R&D Systems, Bio-Techne Corp, Minneapolis, MN). Before therapy and approximately 1 month after therapy, PBLs were isolated from patients by leukapheresis for flow cytometric analyses. Patients 7, 13, 14, and 17 did not undergo leukapheresis after therapy, rather, patients’ peripheral blood was collected into Vacutainer Cell Preparation Tubes (CPTs) (BD Bioscience, San Jose, CA), then tubes were spun down to isolate patients’ lymphocytes according to manufacturer instruction. PBL samples from patient 4 were unavailable. Anti-FOXP3 (236A/E7) and mouse anti-TCRβ (H57-597) antibodies were obtained from Thermo Fisher Scientific Life Sciences. Fixation/permeabilization and staining were performed according to manufacturer instructions (Thermo Fisher Scientific Life Sciences). Flow cytometric analyses were performed by using FACSCanto (BD Bioscience) and analyzed with FlowJo software (FlowJo, Ashland, OR).
Statistical Analysis
Mann-Whitney U test was used to test for correlations between T-cell persistence and cell doses (Prism; GraphPad Software, La Jolla, CA). Reported P values were two-tailed, and P < .05 were considered statistically significant.
RESULTS
Patient Characteristics
Seventeen patients with metastatic cancer were treated with TCR-transduced CD4+ T cells in this protocol (Table 1). One additional patient with metastatic bladder cancer did not receive any T cells or IL-2 as a result of toxicities after two doses of cyclophosphamide and two doses of fludarabine. All patients had measurable distant metastatic disease and had been previously treated with at least one standard first-line therapy. Because no unexpected dose-limiting toxicities—grade 3 and 4—were observed during the cell dose-escalation phase (dose cohort 1 to 8), the last nine patients (patient 9 to 17) were treated at the highest dose level (dose cohort 9). The median cell dose for dose cohort 9 was 1.00 × 1011 T cells (range, 0.6 to 1.23 × 1011 T cells). Because of the patient’s poor pulmonary function, patient 15 received a split dose of T cells with 3 × 1010 cells on day 0, then 3 × 1010 cells the next day. After cell infusion, patients also received high-dose IL-2, with the exception of three patients who had poor pulmonary function. Most patients were discharged after their whole blood cell counts were back to normal range. Because most patients were not local to the National Institutes of Health, they were hospitalized for the entire chemotherapy. Most patients were hospitalized for 13 days after cell infusion (range, 10 to 40 days).
Table 1.
Patient Characteristics, Therapies, and Responses

Clinical Responses
Eight patients were treated during the cell dose-escalation phase (low dose). Patient 6 with cervical cancer had been treated with radiation therapy and six cycles of cisplatin for primary cervical cancer and lymph node metastases; however, the patient later developed disease progression in the supraclavicular lymph nodes, which were positron emission tomography positive and biopsy proven to be recurrent cancer. The patient received 2.7 × 109 TCR-transduced CD4+ T cells and subsequently experienced an objective complete response in metastatic supraclavicular lymph nodes that is ongoing at 29 months (Fig 1A). Her remaining lymph node was positron emission tomography negative and less than 1 cm.
Fig 1.

Clinical responses of patients 6, 11, and 16. (A) Computed tomography (CT) scans of the neck of patient 6 with metastatic cervical cancer before (left) and 29 months after (right) T-cell therapy. (B) Magnetic resonance imaging scans of patient 11 with metastatic urothelial cancer before (left) and 18 months after (right) T-cell therapy. (C) CT scans of the chest of patient 16 with metastatic osteosarcoma before (left) and approximately 4 months after (right) T-cell therapy. Arrows indicate the locations of metastatic lesions before and after therapy.
Among nine patients who received the highest dose level of TCR-transduced CD4+ T cells (high dose), three patients experienced objective responses. Patient 9 with esophageal cancer was previously treated with radiation, FOLFOX (folinic acid-fluororuracil-oxaliplatin), and capecitabine. The patient experienced an objective partial response in mediastinal and paraesophageal lymph nodes, but experienced disease progression at 4 months with a new parasternal lesion (Appendix Fig A1, online only). Patient 11 with urothelial cancer had a primary tumor that involved his left ureter with metastases to the liver and intra-abdominal lymph nodes. The patient experienced disease progression through surgery and chemotherapy before T-cell therapy. ACT treatment resulted in an objective partial response that is ongoing for 19 months, with small residual disease in the periaortic lymph node and liver (Fig 1B). Lastly, Patient 16 with osteosarcoma was heavily pretreated, including surgery, chemotherapy, L-MTP-PE (liposomal muramyl tripeptide phosphatidyl ethanolamine), sorafenib, and radiation therapy. The patient experienced an objective partial response of metastatic lung lesions that lasted 4 months, at which time the growth of nontarget pulmonary lesions was observed (Fig 1C).
Adverse Events
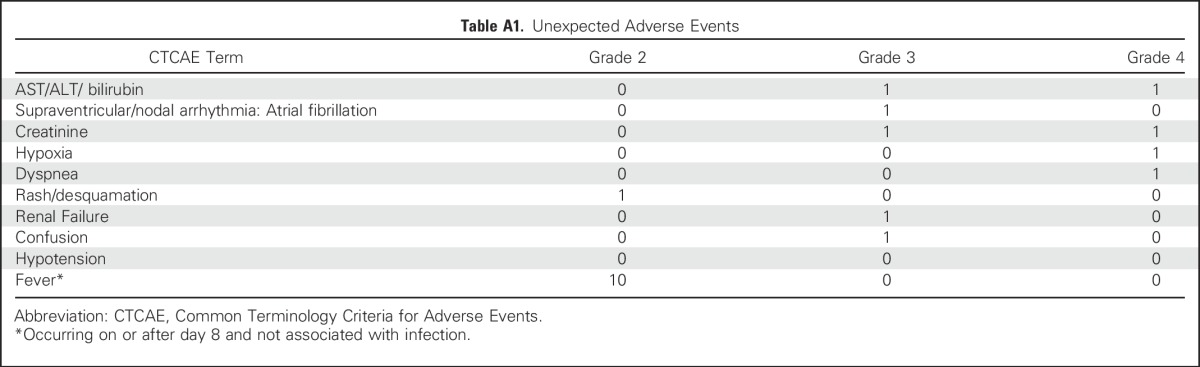
All patients experienced the expected transient grade 3 and 4 adverse events that resulted from nonmyeloablative chemotherapy preparative regimen and high-dose IL-2. Of note, 10 of 17 treated patients experienced prolonged high fever (> 39.0°C to 40.0°C, grade 2) after cell infusion (Appendix Table A1, online only). A 65-year-old patient with metastatic breast cancer received the nonmyeloablatve chemotherapy preparative regimen, 9 × 1010 cells, and one dose of IL-2. After therapy, the patient experienced grade 4 toxicities, including elevated ALT, AST, and creatinine. The patient subsequently developed respiratory failure and was hospitalized for more than 1 month before recovering. One additional patient experienced transient grade 3 toxicities in elevated ALT, AST, and creatinine that lasted 2 days.
Elevated Serum Cytokine Levels After Cell Infusion
Patients’ serum samples were collected during hospitalization, and serum cytokine concentrations of IL-6, IL-10, IFN-γ, and TNF were determined by using enzyme-linked immunosorbent assay. Elevated serum cytokines levels were detected 1 to 2 days after cell infusion (Appendix Fig A2, online only). Maximum cytokine levels are summarized in Fig 2. High levels of IL-6 were detected in all patients (range, 491 pg/mL to 9,686 pg/mL), and elevated IL-10 levels (> 100 pg/mL) were detected in 12 of 17 patients. In addition, elevated IFN-γ and TNF levels (> 100 pg/mL) were detected in nine patients and three patients, respectively. Of note, high levels of IFN-γ were detected in three patients (range, 4,898 pg/mL to 12,911 pg/mL); however, the levels of serum cytokines did not seem to correlate with clinical responses or symptoms, such as fevers.
Fig 2.

Elevated serum cytokine levels after adoptive transfer of MAGE-A3 (melanoma-associated antigen-A3) T-cell receptor–transduced CD4+ T cells. Serum samples were collected daily during hospitalization. Maximum serum cytokine levels and the corresponding time point after cell infusion are shown. Day 0 is the cell infusion day. Patients with objective responses are labeled in yellow. Low dose: 0.001 to 3 × 1010 total cells. High dose: Approximately 1011 total cells. n.d., not detectable. **Patients 5 and 8 had elevated interferon gamma (IFN-γ) levels before cell infusion (approximately 500 pg/mL and 4,000 pg/mL, respectively), and IFN-γ levels remained at similar levels after cell infusion.
Persistence of Genetically Modified T Cells in Peripheral Blood
Patients’ PBLs were collected approximately 1 month after cell infusion, and the frequency of TCR-transduced T cells was detected by antibody against the mouse TCRβ constant region. As shown in Figure 3, both the percentage and total number of TCR-transduced T cells were significantly higher in peripheral blood of patients who received the high cell dose compared with patients who received the low cell dose (P = .0002 and .0007, respectively; Figs 3B and 3C). In this small study, no relationship was observed between cell persistence and clinical response. Of note, both patient 9 and patient 16 experienced short-term clinical responses for 4 months. Infused genetically modified T cells still persisted well in the peripheral blood at the time of progression (223 cells/μL [20.1%] and 73 cells/μL [14.4%], respectively). These results suggest that tumors in patient 9 and patient 16 might have developed resistance to this T-cell therapy, such as loss of antigens or dysfunction of components in the antigen process and presentation pathways.
Fig 3.

After cell infusion, T-cell receptor (TCR)–transduced T cells persisted better in the high-dose group compared with the low-dose group. (A) Peripheral blood lymphocyte (PBL) samples before (top) and approximately 1 month after (bottom) T-cell therapy were stained with anti-mouse TCRβ constant region antibody (mTCRβ) to detect MAGE-A3 (melanoma-associated antigen-A3) TCR-transduced T cells. A representative result is shown. (B) Percentage of CD4+mTCRβ+ cells in lymphocyte populations at approximately 1 month in the low-dose and high-dose patients. (C) Cell numbers of CD4+mTCRβ+ cells in patients’ peripheral blood. The post-treatment PBL sample from patient 4 was not available. Statistical significance was determined by Mann-Whitney U test. Patients who experienced objective responses are labeled with gray circles. CR, complete response; PR, partial response.
DISCUSSION
Immune checkpoint blockade therapies can induce objective responses in a subset of patients with melanoma, renal cell carcinoma, non–small-cell lung cancer, and urothelial cancer, as well as tumors with mismatch-repair deficiency39-49; however, to date, these immunotherapies have had little impact in patients with other types of solid cancer. One alternative immunotherapy approach is the adoptive transfer of CAR or TCR-modified T cells; however, previous T-cell therapies targeting MAGE-A3 have failed. A clinical trial that used an HLA-A*01–restricted, affinity-enhanced TCR targeting MAGE-A3 led to lethal toxicity because of the recognition of a muscle protein, TTN (titin), in the heart by the altered TCR.50,51 In a different trial, ACT of T cells that were genetically engineered to express an HLA-A*0201–restricted TCR targeting MAGE-A3/9 recognized an HLA-A*0201–restricted MAGE-A12 epitope expressed in brain and led to lethal neurologic toxicity.34
In prior studies, cell transfer using cells that were engineered to express HLA-A*0201–restricted NY-ESO-1 TCR mediated a 55% objective response rate for patients with melanoma and a 61% objective response rate for patients with synovial sarcoma.52,53 In a recent study, the protein expression of MAGE-A and NY-ESO-1 in 3,668 tumor specimens was assessed by immunohistochemistry. The frequency of MAGE-A–positive tumor specimens was significantly higher than NY-ESO-1 in several common epithelial carcinomas, including cutaneous squamous cell carcinomas (SCCs; 52.8% v 2.8%), esophageal SCC (50% v 0%), head and neck SCC (41.1% v 0.65%), bladder urothelial cancer (40.4% v 8.3%), and cervical/anal SCC (37.5% v 0%).31 In addition, HLA-DPB1*0401 is present in 57% of the Caucasian population compared with a 47% incidence of HLA-A*0201.54 Thus, MAGE-A3 TCR immunotherapy described here could potentially be applicable for additional patients with solid cancers.
Our study provides direct evidence that objective tumor regressions can be mediated by MAGE-A3–specific CD4+ T cells in a variety of cancer types; however, additional studies are needed to understand the detailed mechanisms by which human CD4+ T cells mediate tumor regressions. Previous studies in the B16 melanoma mouse model suggested that tumor regressions were dependent on IFN-γ secreted by CD4+ T cells, as well as MHC class II molecules expressed on B16 melanoma after in vivo IFN-γ stimulation.13,15 Tumor regressions in mice were not dependent on endogenous CD8+ T cells, B cells, NK cells, or NK T cells.15,55 Thus, MAGE-A3–specific CD4+ T cells could possibly recognize tumor cells directly in vivo and kill tumor cells via IFN-γ or other pathways. Alternatively, MAGE-A3–specific CD4+ T cells might recognize MAGE-A3 cross-presented by antigen-presenting cells and secrete therapeutic cytokines or promote CD8+ T cell–mediated tumor regression by epitope spreading.56
The MAGE-A3 TCR used in this study was originally isolated from a T-cell clone with a regulatory T-cell (Treg) phenotype. In addition to being a naturally occurring and thymically selected TCR, this TCR was selected for clinical development because it had a higher affinity compared with another TCR that was isolated from an effector T-cell clone.32 This raised a potential possibility that genetically modified CD4+ T cells might convert to Tregs after adoptive transfer of CD4+ T cells into patients, and that Tregs might subsequently inhibit T-cell activity in vivo. Furthermore, previous studies in mice have suggested that high-affinity TCRs promoted the differentiation of both thymus-derived and peripherally derived Tregs.57 Despite these concerns, we were unable to detect any significant mTCR+FOXP3+ T cells in any patient’s peripheral blood 1 month after cell infusion, which suggested that transduced T cells did not convert to Tregs (Appendix Fig A3, online only). Data presented here suggested that other factors, such as the context of the tissue microenvironment, might play important roles in the conversion of Tregs.
In conclusion, this study demonstrated the safety of the adoptive transfer of genetically engineered CD4+ T cells targeting MAGE-A3 and demonstrated evidence of efficacy. Future clinical trials are needed to study the efficacy of this therapy in different types of cancer. Additional modifications may help to improve the efficacy of this therapy, such as manufacturing less-differentiated or Th17-polarized T cells by modifying the cell production process, and combining T-cell therapy with immune checkpoint blockade to prevent T-cell exhaustion.11,13,58,59 Combining multiple targets, such as neoantigen targets, at the same time may help overcome the challenge of tumor heterogeneity.60,61 Finally, clinical studies have demonstrated that tumors can also escape T cell–based immunotherapies by the loss of critical processing and presentation components of MHC class I, such as the deletion of β2-microglobulin. The ability to transfer MHC class II–restricted tumor-reactive T cells may be an effective way to prevent this demonstrated mechanism of tumor escape.62
ACKNOWLEDGMENT
We thank the National Cancer Institute Surgery Branch clinical team for the outstanding patient care.
Appendix
Fig A1.

Computed tomography scans of patient 9 with metastatic esophageal cancer before and 4 months after T-cell therapy.
Fig A2.

Serum cytokine levels before and after cell infusion. Patients 5 and 8 had elevated interferon gamma (IFN-γ) levels before cell infusion (approximately 500 pg/mL and 4,000 pg/mL, respectively), and the IFN-γ levels remained at similar levels after cell infusion (data not shown). CR, complete response; PR, partial response.
Fig A3.

MAGE-A3 (melanoma-associated antigen-A3) T-cell receptor (TCR)–transduced CD4+ T cells did not convert to regulatory T cells after cell infusion. (A) Intracellular staining for FOXP3 antibody was performed for peripheral blood lymphocyte samples before (top) and approximately 1 month after (bottom) T-cell therapy. (B) Percentage of CD4+mTCRβ+FOXP3+ cells in lymphocyte populations before (top) and 1 month after (bottom) cell infusion. (C) Percentage of CD4+mTCRβ−FOXP3+ cells in lymphocyte populations before (top) and 1 month after (bottom) cell infusion. Statistical significance was determined by Mann-Whitney U test.
Table A1.
Unexpected Adverse Events
Footnotes
Supported by the National Cancer Institute Intramural Research Program.
Presented in part at the American Association for Cancer Research Annual Meeting, New Orleans, LA, April 16-20, 2016, and the Society for Immunotherapy of Cancer Annual Meeting, National Harbor, MD, November 4-8, 2015.
Clinical trial information: NCT02111850.
AUTHOR CONTRIBUTIONS
Conception and design: Yong-Chen Lu, Xin Yao, Paul F. Robbins, Steven A. Feldman, Pierre van der Bruggen, James C. Yang, Steven A. Rosenberg
Provision of study materials or patients: Steven A. Feldman, Pierre van der Bruggen
Collection and assembly of data: Yong-Chen Lu, Linda L. Parker, Tangying Lu, Zhili Zheng, Mary Ann Toomey, Donald E. White, Yong F. Li, Christopher A. Klebanoff, Stephanie L. Goff, Richard M. Sherry, Udai S. Kammula, James C. Yang
Data analysis and interpretation: Yong-Chen Lu, Linda L. Parker, Zhili Zheng, Mary Ann Toomey, Yong F. Li, Steven A. Feldman, Christopher A. Klebanoff, Stephanie L. Goff, Richard M. Sherry, Udai S. Kammula, James C. Yang, Steven A. Rosenberg
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Treatment of Patients With Metastatic Cancer Using a Major Histocompatibility Complex Class II–Restricted T-Cell Receptor Targeting the Cancer Germline Antigen MAGE-A3
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/site/ifc.
Yong-Chen Lu
Patents, Royalties, Other Intellectual Property: Patents pending, held by the National Cancer Institute (Inst)
Linda L. Parker
No relationship to disclose
Tangying Lu
No relationship to disclose
Zhili Zheng
No relationship to disclose
Mary Ann Toomey
No relationship to disclose
Donald E. White
No relationship to disclose
Xin Yao
Employment: Cellular Biomedicine Group
Stock or Other Ownership: Cellular Biomedicine Group
Yong F. Li
No relationship to disclose
Paul F. Robbins
Patents, Royalties, Other Intellectual Property: Kite Pharma (Inst)
Steven A. Feldman
Patents, Royalties, Other Intellectual Property: Patents: Isolation of MAGE-A3 specific TCRs (commercial license from National Institutes of Health Office of Technology Transfer (OTT)); manufacture of CAR T cells (commercial license from National Institutes of Health OTT by KITE Pharma)
Pierre van der Bruggen
Research Funding: ITeos Therapeutics (Inst)
Patents, Royalties, Other Intellectual Property: Ludwig Cancer Research
Christopher A. Klebanoff
Stock or Other Ownership: Kite Pharma
Consulting or Advisory Role: Cell Design Laboratories, Bristol-Myers Squibb, RXi Pharmaceuticals
Stephanie L. Goff
No relationship to disclose
Richard M. Sherry
No relationship to disclose
Udai S. Kammula
No relationship to disclose
James C. Yang
Research Funding: Kite Pharma (Inst)
Patents, Royalties, Other Intellectual Property: Patents held by the National Cancer Institute (Inst)
Steven A. Rosenberg
Patents, Royalties, Other Intellectual Property: The National Cancer Institute has cooperative research and development agreements with Kite Pharma and Lion Biotechnologies (Inst)
REFERENCES
- 1.Rosenberg SA, Restifo NP: Adoptive cell transfer as personalized immunotherapy for human cancer. Science 348:62-68, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kochenderfer JN, Wilson WH, Janik JE, et al. : Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood 116:4099-4102, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Porter DL, Levine BL, Kalos M, et al. : Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med 365:725-733, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kalos M, Levine BL, Porter DL, et al. : T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med 3:95ra73, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kochenderfer JN, Dudley ME, Feldman SA, et al. : B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood 119:2709-2720, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brentjens RJ, Davila ML, Riviere I, et al. : CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med 5:177ra38, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maude SL, Frey N, Shaw PA, et al. : Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 371:1507-1517, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davila ML, Riviere I, Wang X, et al. : Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med 6:224ra25, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kochenderfer JN, Dudley ME, Kassim SH, et al. : Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol 33:540-549, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee DW, Kochenderfer JN, Stetler-Stevenson M, et al. : T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: A phase 1 dose-escalation trial. Lancet 385:517-528, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Turtle CJ, Hanafi LA, Berger C, et al. : Immunotherapy of non-Hodgkin’s lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Sci Transl Med 8:355ra116, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klebanoff CA, Rosenberg SA, Restifo NP: Prospects for gene-engineered T cell immunotherapy for solid cancers. Nat Med 22:26-36, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Muranski P, Boni A, Antony PA, et al. : Tumor-specific Th17-polarized cells eradicate large established melanoma. Blood 112:362-373, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hirschhorn-Cymerman D, Budhu S, Kitano S, et al. : Induction of tumoricidal function in CD4+ T cells is associated with concomitant memory and terminally differentiated phenotype. J Exp Med 209:2113-2126, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Quezada SA, Simpson TR, Peggs KS, et al. : Tumor-reactive CD4+ T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J Exp Med 207:637-650, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hunder NN, Wallen H, Cao J, et al. : Treatment of metastatic melanoma with autologous CD4+ T cells against NY-ESO-1. N Engl J Med 358:2698-2703, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tran E, Turcotte S, Gros A, et al. : Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 344:641-645, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Simpson AJ, Caballero OL, Jungbluth A, et al. : Cancer/testis antigens, gametogenesis and cancer. Nat Rev Cancer 5:615-625, 2005 [DOI] [PubMed] [Google Scholar]
- 19.Coulie PG, Van den Eynde BJ, van der Bruggen P, et al. : Tumour antigens recognized by T lymphocytes: At the core of cancer immunotherapy. Nat Rev Cancer 14:135-146, 2014 [DOI] [PubMed] [Google Scholar]
- 20.Rosenberg SA: Cell transfer immunotherapy for metastatic solid cancer—What clinicians need to know. Nat Rev Clin Oncol 8:577-585, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roeder C, Schuler-Thurner B, Berchtold S, et al. : MAGE-A3 is a frequent tumor antigen of metastasized melanoma. Arch Dermatol Res 296:314-319, 2005 [DOI] [PubMed] [Google Scholar]
- 22.Tajima K, Obata Y, Tamaki H, et al. : Expression of cancer/testis (CT) antigens in lung cancer. Lung Cancer 42:23-33, 2003 [DOI] [PubMed] [Google Scholar]
- 23.Filho PA, López-Albaitero A, Xi L, et al. : Quantitative expression and immunogenicity of MAGE-3 and -6 in upper aerodigestive tract cancer. Int J Cancer 125:1912-1920, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim J, Reber HA, Hines OJ, et al. : The clinical significance of MAGEA3 expression in pancreatic cancer. Int J Cancer 118:2269-2275, 2006 [DOI] [PubMed] [Google Scholar]
- 25.Luo G, Huang S, Xie X, et al. : Expression of cancer-testis genes in human hepatocellular carcinomas. Cancer Immun 2:11, 2002 [PubMed] [Google Scholar]
- 26.Han MH, Eom HS, Park WS, et al. : Detection of circulating lymphoma cells in patients with non-Hodgkin lymphoma using MAGE-A3 gene expression in peripheral blood. Leuk Res 34:1127-1131, 2010 [DOI] [PubMed] [Google Scholar]
- 27.Jungbluth AA, Ely S, DiLiberto M, et al. : The cancer-testis antigens CT7 (MAGE-C1) and MAGE-A3/6 are commonly expressed in multiple myeloma and correlate with plasma-cell proliferation. Blood 106:167-174, 2005 [DOI] [PubMed] [Google Scholar]
- 28.Gure AO, Chua R, Williamson B, et al. : Cancer-testis genes are coordinately expressed and are markers of poor outcome in non–small-cell lung cancer. Clin Cancer Res 11:8055-8062, 2005 [DOI] [PubMed] [Google Scholar]
- 29.Dhodapkar MV, Osman K, Teruya-Feldstein J, et al. : Expression of cancer/testis (CT) antigens MAGE-A1, MAGE-A3, MAGE-A4, CT-7, and NY-ESO-1 in malignant gammopathies is heterogeneous and correlates with site, stage and risk status of disease. Cancer Immun 3:9, 2003 [PubMed] [Google Scholar]
- 30.Park TS, Groh EM, Patel K, et al. : Expression of MAGE-A and NY-ESO-1 in primary and metastatic cancers. J Immunother 39:1-7, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kerkar SP, Wang ZF, Lasota J, et al. : MAGE-A is more highly expressed than NY-ESO-1 in a systematic immunohistochemical analysis of 3668 cases. J Immunother 39:181-187, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yao X, Lu YC, Parker LL, et al. : Isolation and characterization of an HLA-DPB1*04: 01-restricted MAGE-A3 T-cell receptor for cancer immunotherapy. J Immunother 39:191-201, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cohen CJ, Zhao Y, Zheng Z, et al. : Enhanced antitumor activity of murine-human hybrid T-cell receptor (TCR) in human lymphocytes is associated with improved pairing and TCR/CD3 stability. Cancer Res 66:8878-8886, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morgan RA, Chinnasamy N, Abate-Daga D, et al. : Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother 36:133-151, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rosenberg SA, Yang JC, White DE, et al. : Durability of complete responses in patients with metastatic cancer treated with high-dose interleukin-2: Identification of the antigens mediating response. Ann Surg 228:307-319, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mulé JJ, Shu S, Schwarz SL, et al. : Adoptive immunotherapy of established pulmonary metastases with LAK cells and recombinant interleukin-2. Science 225:1487-1489, 1984 [DOI] [PubMed] [Google Scholar]
- 37.Dudley ME, Wunderlich JR, Robbins PF, et al. : Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science 298:850-854, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morgan RA, Dudley ME, Wunderlich JR, et al. : Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 314:126-129, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nguyen LT, Ohashi PS: Clinical blockade of PD1 and LAG3—Potential mechanisms of action. Nat Rev Immunol 15:45-56, 2015 [DOI] [PubMed] [Google Scholar]
- 40.Hodi FS, O’Day SJ, McDermott DF, et al. : Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 363:711-723, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Topalian SL, Hodi FS, Brahmer JR, et al. : Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 366:2443-2454, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hamid O, Robert C, Daud A, et al. : Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med 369:134-144, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Le DT, Uram JN, Wang H, et al. : PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med 372:2509-2520, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Garon EB, Rizvi NA, Hui R, et al. : Pembrolizumab for the treatment of non–small-cell lung cancer. N Engl J Med 372:2018-2028, 2015 [DOI] [PubMed] [Google Scholar]
- 45.Brahmer J, Reckamp KL, Baas P, et al. : Nivolumab versus docetaxel in advanced squamous-cell non–small-cell lung cancer. N Engl J Med 373:123-135, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reck M, Rodríguez-Abreu D, Robinson AG, et al. : Pembrolizumab versus chemotherapy for PD-L1-positive non–small-cell lung cancer. N Engl J Med 375:1823-1833, 2016 [DOI] [PubMed] [Google Scholar]
- 47.Sharma P, Retz M, Siefker-Radtke A, et al. : Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): A multicentre, single-arm, phase 2 trial. Lancet Oncol 18:312-322, 2017 [DOI] [PubMed] [Google Scholar]
- 48.Plimack ER, Bellmunt J, Gupta S, et al. : Safety and activity of pembrolizumab in patients with locally advanced or metastatic urothelial cancer (KEYNOTE-012): A non-randomised, open-label, phase 1b study. Lancet Oncol 18:212-220, 2017 [DOI] [PubMed] [Google Scholar]
- 49.Balar AV, Galsky MD, Rosenberg JE, et al. : Atezolizumab as first-line treatment in cisplatin-ineligible patients with locally advanced and metastatic urothelial carcinoma: A single-arm, multicentre, phase 2 trial. Lancet 389:67-76, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Linette GP, Stadtmauer EA, Maus MV, et al. : Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood 122:863-871, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cameron BJ, Gerry AB, Dukes J, et al. : Identification of a Titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci Transl Med 5:197ra103, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Robbins PF, Morgan RA, Feldman SA, et al. : Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol 29:917-924, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Robbins PF, Kassim SH, Tran TL, et al. : A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: Long-term follow-up and correlates with response. Clin Cancer Res 21:1019-1027, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.González-Galarza FF, Takeshita LY, Santos EJ, et al. : Allele frequency net 2015 update: New features for HLA epitopes, KIR and disease and HLA adverse drug reaction associations. Nucleic Acids Res 43:D784-D788, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xie Y, Akpinarli A, Maris C, et al. : Naive tumor-specific CD4+ T cells differentiated in vivo eradicate established melanoma. J Exp Med 207:651-667, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Carreno BM, Magrini V, Becker-Hapak M, et al. : Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 348:803-808, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li MO, Rudensky AY: T cell receptor signalling in the control of regulatory T cell differentiation and function. Nat Rev Immunol 16:220-233, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Abate-Daga D, Hanada K, Davis JL, et al. : Expression profiling of TCR-engineered T cells demonstrates overexpression of multiple inhibitory receptors in persisting lymphocytes. Blood 122:1399-1410, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Irving M, Vuillefroy de Silly R, Scholten K, et al. : Engineering chimeric antigen receptor T-cells for racing in solid tumors: Don’t forget the fuel. Front Immunol 8:267, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.DuPage M, Mazumdar C, Schmidt LM, et al. : Expression of tumour-specific antigens underlies cancer immunoediting. Nature 482:405-409, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tran E, Robbins PF, Rosenberg SA: ‘Final common pathway’ of human cancer immunotherapy: Targeting random somatic mutations. Nat Immunol 18:255-262, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zaretsky JM, Garcia-Diaz A, Shin DS, et al. : Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med 375:819-829, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]