

Abstract
Klebsiella pneumoniae shows increasing emergence of multidrug-resistant lineages, including strains resistant to all available antimicrobial drugs. We conducted whole-genome sequencing of 178 highly drug-resistant isolates from a tertiary hospital in Lahore, Pakistan. Phylogenetic analyses to place these isolates into global context demonstrate the expansion of multiple independent lineages, including K. quasipneumoniae.
Keywords: Klebsiella, ESBL, extended-spectrum β-lactamase, antimicrobial resistance, neonatal infection, multidrug resistance, Pakistan, bacteria, Australia, United Kingdom, Klebsiella pneumonia, children
Klebsiella spp. are gram-negative bacteria that are widely distributed in the environment, and K. pneumoniae is a common cause of infection in humans (1). Increasingly, K. pneumoniae is reported as a cause of invasive bloodborne infections, particularly in healthcare settings and in immunocompromised patients (2). Of concern is that infection-associated K. pneumoniae is often multidrug resistant (MDR) and can harbor resistance determinants against most, if not all, commonly used antimicrobial drugs, posing a major threat to public health. The World Health Organization recently highlighted finding new treatments against MDR Enterobacteriaceae (including Klebsiella) as priority 1 (critical) (http://www.who.int/mediacentre/news/releases/2017/bacteria-antibiotics-needed/en/).
K. pneumoniae is a major pathogen in economically developed settings, and multiple outbreaks in different countries have been reported. Less is known about its prevalence in economically challenged areas, including lower and middle income countries (LMIC). Reports are now appearing about Klebsiella-associated infections in Nepal (3) and in Indonesia, Laos, and Vietnam (1). Klebsiella can spread rapidly in hospital environments, and the increasing prevalence of MDR strains has raised concern among major health organizations (4,5). Thus, high-resolution insight into the diversity of Klebsiella spp. isolated in LMICs will provide vital data for improving epidemiologic management of infections and for better understanding of the mechanisms of spread between LMICs and more developed countries.
The Study
Clinical samples were collected during a 22-month period (May 2010–February 2012) from The Children’s Hospital & The Institute of Child Health, Lahore (Lahore, Pakistan), the largest tertiary care hospital in the region (Figure 1, panel A). The hospital had a capacity of 650 beds during the study period but is under pressure to handle up to 2,000 inpatients at any given time. The primary catchment area is Lahore (population ≈10 million); the hospital also receives patients from the greater area of Punjab province (population ≈100 million) (Figure 1, panel A). The Ethical Committee of The Children’s Hospital & Institute of Child Health, Lahore, approved the study.
Figure 1.
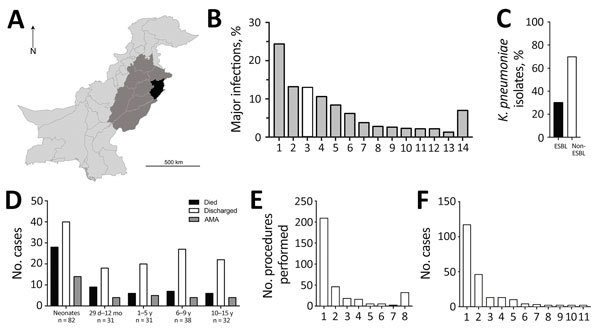
Statistical overview of bacterial isolates from clinical samples collected during May 2010–February 2012 from The Children’s Hospital & The Institute of Child Health, Lahore, Pakistan. A) Map of Pakistan highlighting the main catchment area of Lahore (black, population ≈10 million) and the wider area of Punjab (medium gray, population ≈100 million). B) A total of 5,475 samples collected from children resulted in laboratory-positive cultures; the 5 most frequently occurring bacterial species accounted for ≈70% of total bacterial infections, and Klebsiella pneumoniae (white bar) was the third most dominant (710 isolates). 1, Escherichia coli; 2, coagulase-negative Staphylococcus; 3, K. pneumoniae; 4, Pseudomonas aeruginosa; 5, K. oxytoca; 6, Staphylococcus aureus; 7, Acinetobacter spp.; 8, Enterococcus faecalis; 9, Citrobacter spp.; 10, Streptococcus pyogenes; 11, Burkhoderia cepacia; 12, Enterobacter clocae; 13, Salmonella enterica var. Typhi; 14, others (>100 species). C) The proportion of ESBL-producing K. pneumoniae (214 isolates) among all K. pneumoniae isolates demonstrated high prevalence of antimicrobial resistance. D) A total of 38.3% of ESBL-producing K. pneumoniae infections occurred in neonates (<29 d), an age group that also showed the highest fatality rate (34.1%). Patients who were removed from the hospital against medical advice (AMA) typically were critically ill and were taken home by the family to avoid dying in the hospital. E) The apparent hierarchy shown in panel E closely correlated with interventions given. IV line (97.7%), urinary catheter (27.5%), and ETT (8.4%) were the 3 most commonly administered procedures among sampled patients, although no temporal relationship between procedure and sample collection could be established. 1, IV line; 2, urinary catheter; 3, ETT; 4, PD catheter; 5, surgery; 6, NG tube; 7, CVP; 8, others. F) A total of 54.6% of ESBL-producing K. pneumoniae isolates were from patient blood samples, followed by urine (21.5%), CSF (6%), and ETT (6%). 1, Blood; 2, urine; 3, CSF; 4, ETT; 5, PD catheter; 6, tracheal secretions; 7, pus; 8, CVP tip; 9, ear swab; 10, pleural fluid; 11, wound swab. CSF, cerebrospinal fluid; CVP, central venous catheter tip; ESBL, extended-spectrum β-lactamase; ETT, endotracheal tube; IV, intravenous; NG, nasogastric; PD, peritoneal dialysis catheter. The regional map was derived from the Global Administrative Areas online resource (http://www.gadm.org/).
A total of 44,260 samples were collected in the course of routine sampling from children; 5,475 (12.4%) resulted in laboratory-positive cultures. Of these, 710 (13.0%) samples were positively identified as K. pneumoniae, the third most dominant isolate after Escherichia coli (1,336 [24.4%]) and coagulase-negative staphylococci (724 [13.2%]) (Figure 1, panel B). We screened all K. pneumoniae isolates for resistance to ceftazidime (30 μg disc, zone of inhibition <17 mm) or cefotaxime (30 μg disc, zone of inhibition <22 mm). We further tested K. pneumoniae isolates that were resistant to any of these indicator drugs using the Clinical and Laboratory Standards Institute combined-disc confirmatory test (6); extended-spectrum β-lactamase (ESBL) production was confirmed when the zone of inhibition by either cephalosporin drug increased by >5 mm in the presence of clavulanate. A total of 214 of K. pneumoniae isolates were ESBL-positive (Figure 1, panel C); most were isolated from children with bloodstream infections (Figure 1, panel D). The outcomes were severe, especially among neonatal patients (Figure 1, panel D); 56 died, 31 were taken home against medical advice, and 127 were discharged (Figure 1, panel D). Almost all patients infected with ESBL Klebsiella had received an intravenous line (209 [97.7%]) (Figure 1, panel E,F), and a high number received a urinary catheter (46 [21.5%]).
We performed whole-genome sequencing on 178 isolates (Technical Appendix Table 1). We prepared Illumina sequencing libraries (Illumina, San Diego, CA, USA) with a 450-bp insert size according to the manufacturer’s protocols and sequenced them on an Illumina HiSeq2000 with 100-bp–long paired-end reads before assembly using an open-source high-throughput assembly and improvement pipeline as described (7) (https://github.com/sanger-pathogens/) and annotated using prokka (8). Initial clustering using mash (9) enabled aligning of these isolates to published reference sequences (Technical Appendix Figure 1, panel A). The clustering indicated a strong structure for the isolates that fell within the species K. pneumoniae (Technical Appendix Figure 1, panel B). However, the analysis also revealed a large group of sequences most similar to K. quasipneumoniae; closer inspection focusing on this species showed strongest similarity to subspecies similipneumoniae (Technical Appendix Figure 1, panel C) (10). We combined several independent datasets: a large global collection (1); 2 hospital outbreaks obtained in a comparable time frame, 1 of which was based in Nepal in 2012 (3); and a hospital study from Spain that also focused on diversity within ESBL-producing strains (11) (Technical Appendix Table 2). We applied the pan-genome pipeline Roary version 3.7.0 (12) with a blastp (https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE=Proteins) percentage identity of 90% and a core definition of 99%, resulting in a core gene alignment comprising 1,793 genes for all studies (Figure 2) and 3,486 genes for the strains of this study (Technical Appendix Figure 2). We first extracted single-nucleotide polymorphisms using snp-sites version 2.3.2 (13), then calculated a maximum-likelihood tree using RAxML version 8.2.8 (14) with the general time-reversible model and 100 bootstrap repeats. The core gene phylogeny (Figure 2) shows a wide distribution of the isolates from Pakistan across different lineages rather than 1 clonal lineage. The diversity of our strain collection is further emphasized through the diversity of multilocus sequence types (STs). No single ST dominates (Figure 2 outer ring; Technical Appendix Figure 2); however, a large group of isolates belongs to ST15, which is known to be problematic. The presence of K. quasipneumoniae isolates agrees with an overall lower percentage of reads mapped against K. pneumoniae (Technical Appendix Table 1) and with recent descriptions of virulent K. quasipneumoniae strains (1,9,15). Assessing the metadata in phylogenetic context highlights the association of the K. quasipneumoniae lineage with patients in the neonatal ward, suggestive of its nosocomial residency (Technical Appendix Figure 2). However, other main lineages (e.g., ST15, ST48) show a dynamic spread across wards and age groups, indicating against >1 resident lineages but instead a frequent movement of K. pneumoniae through the hospital, general population, or both.
Figure 2.
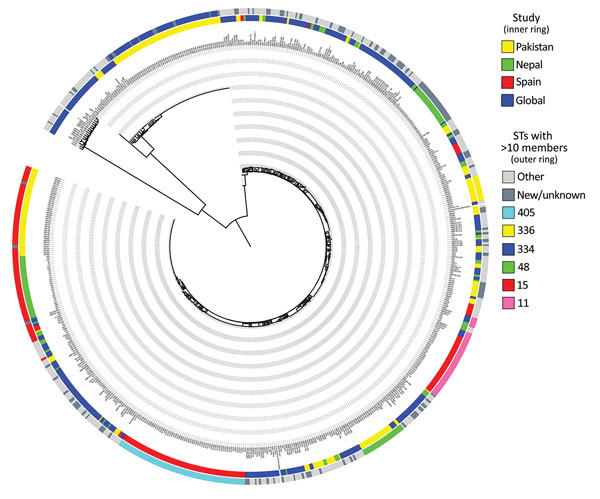
Phylogenetic analysis demonstrating the diversity of Klebsiella pneumoniae isolates from clinical samples collected during May 2010–February 2012 from The Children’s Hospital & The Institute of Child Health, Lahore, Pakistan, in a global context. The core gene tree based on the alignment derived from Roary (12) was calculated using RAxML (14) and shows the wide diversity of samples analyzed in this study (inner ring, yellow) in context with a large-scale global analysis (inner ring, blue [4]) and 2 hospital outbreaks, which show a more clonal pattern (inner ring: red, outbreak in Spain [11]; green, outbreak in Nepal [3]). The sequence types observed (outer ring) also reflect the diversity; most sequence types have <10 members even in this combined collection. STs, sequence types.
The high number of K. quasipneumoniae isolates, even if potentially restricted to most sequences derived from a lineage potentially resident in a specific ward, highlights the importance of a diverse set of sampling sites to be studied. It also highlights the need for continued monitoring of new emerging strains and that our knowledge of the diversity of potentially problematic lineages is far from exhaustive.
Conclusions
The Klebsiella isolates in this study represented the Klebsiella isolates routinely present in infections over a protracted period. Our findings highlight a consistent problem with ESBL-encoding strains belonging to a multitude of lineages. We observed sporadic single-isolate lineages, as well as smaller, related clusters of 5–10 strains per lineage, in addition to 2 larger clusters of strains. More studies are needed to better delineate the distinguishing features for successful spread and persistence of lineages such as the ST15 cluster. Also, the large spread of K. quasipneumoniae is unusual. Further intense monitoring of LMIC hospital environments is urgently needed to prevent the persistence of resident lineages with very high base-level drug resistance, which, through the inevitable acquisition of a few more genes, would lead to untreatable infections.
Details of sequenced Klebsiella pneumoniae strains from children, Pakistan; GenBank accession numbers of published K. pneumoniae strains included in this analysis; whole-genome clustering; patient metadata in phylogenetic context.
Acknowledgments
This work was supported by National Health and Medical Research Council program grants (0606788 to R.A.S. and T. L.; 1092262 to R.A.S., G.D., and T.L.); the Wellcome Trust (206194); and the Higher Education Commission of Pakistan and The Children’s Hospital & The Institute of Child Health, Lahore, Pakistan. H.E. was supported by a scholarship from Higher Education Commission Pakistan under the International Research Support Initiative Program.
Biography
Dr. Ejaz is a microbiologist who is working as Assistant Professor in Aljouf University, Aljouf, Saudi Arabia. He also worked at The Children’s Hospital Lahore, Pakistan, and University of Melbourne, Australia. His primary research interests include medical bacteriology.
Footnotes
Suggested citation for this article: Ejaz H, Wang N, Wilksch JJ, Page AJ, Cao H, Gujaran S, et al. Phylogenetic analysis of Klebsiella pneumoniae from hospitalized children, Pakistan. Emerg Infect Dis. 2017 Nov [date cited]. https://doi.org/10.3201/eid2311.170833
These authors contributed equally to this article.
Current affiliation: CAMS, Aljouf University, Aljouf, Saudi Arabia.
References
- 1.Holt KE, Wertheim H, Zadoks RN, Baker S, Whitehouse CA, Dance D, et al. Genomic analysis of diversity, population structure, virulence, and antimicrobial resistance in Klebsiella pneumoniae, an urgent threat to public health. Proc Natl Acad Sci U S A. 2015;112:E3574–81. 10.1073/pnas.1501049112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee CR, Lee JH, Park KS, Kim YB, Jeong BC, Lee SH. Global dissemination of carbapenemase-producing Klebsiella pneumoniae: epidemiology, genetic context, treatment options, and detection methods. Front Microbiol. 2016;7:895. 10.3389/fmicb.2016.00895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chung The H, Karkey A, Pham Thanh D, Boinett CJ, Cain AK, Ellington M, et al. A high-resolution genomic analysis of multidrug-resistant hospital outbreaks of Klebsiella pneumoniae. EMBO Mol Med. 2015;7:227–39. 10.15252/emmm.201404767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.World Health Organization. Antimicrobial resistance: global report on surveillance 2014. Geneva: The Organization; 2014. [Google Scholar]
- 5.Centers for Disease Control and Prevention. New carbapenem-resistant Enterobacteriaceae warrant additional action by healthcare providers [cited 2017 Jan 5]. https://emergency.cdc.gov/han/han00341.asp
- 6.Clinical and Laboratory Standards Institute. Performance standards for antimicrobial susceptibility testing; twentieth informational supplement. M100-S20. Wayne (PA): The Institute; 2010. [Google Scholar]
- 7.Page AJ, De Silva N, Hunt M, Quail MA, Parkhill J, Harris SR, et al. Robust high-throughput prokaryote de novo assembly and improvement pipeline for Illumina data. Microb Genom. 2016;2:e000083. 10.1099/mgen.0.000083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–9. 10.1093/bioinformatics/btu153 [DOI] [PubMed] [Google Scholar]
- 9.Ondov BD, Treangen TJ, Melsted P, Mallonee AB, Bergman NH, Koren S, et al. Mash: fast genome and metagenome distance estimation using MinHash. Genome Biol. 2016;17:132. 10.1186/s13059-016-0997-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brisse S, Passet V, Grimont PA. Description of Klebsiella quasipneumoniae sp. nov., isolated from human infections, with two subspecies, Klebsiella quasipneumoniae subsp. quasipneumoniae subsp. nov. and Klebsiella quasipneumoniae subsp. similipneumoniae subsp. nov., and demonstration that Klebsiella singaporensis is a junior heterotypic synonym of Klebsiella variicola. Int J Syst Evol Microbiol. 2014;64:3146–52. 10.1099/ijs.0.062737-0 [DOI] [PubMed] [Google Scholar]
- 11.Pérez-Vázquez M, Oteo J, García-Cobos S, Aracil B, Harris SR, Ortega A, et al. Phylogeny, resistome and mobile genetic elements of emergent OXA-48 and OXA-245 Klebsiella pneumoniae clones circulating in Spain. J Antimicrob Chemother. 2016;71:887–96. 10.1093/jac/dkv458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Page AJ, Cummins CA, Hunt M, Wong VK, Reuter S, Holden MT, et al. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics. 2015;31:3691–3. 10.1093/bioinformatics/btv421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Page AJ, Taylor B, Delaney AJ, Soares J, Seemann T, Keane JA, et al. SNP-sites: rapid efficient extraction of SNPs from multi-FASTA alignments. Microb Genom. 2016;2:e000056. 10.1099/mgen.0.000056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stamatakis A. Using RAxML to infer phylogenies. Curr Protoc Bioinformatics. 2015;51:6.14.1-14. [DOI] [PubMed]
- 15.Arena F, Henrici De Angelis L, Pieralli F, Di Pilato V, Giani T, Torricelli F, et al. Draft genome sequence of the first hypermucoviscous Klebsiella quasipneumoniae subsp. quasipneumoniae isolate from a bloodstream infection. Genome Announc. 2015;3:e00952–15. 10.1128/genomeA.00952-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Details of sequenced Klebsiella pneumoniae strains from children, Pakistan; GenBank accession numbers of published K. pneumoniae strains included in this analysis; whole-genome clustering; patient metadata in phylogenetic context.