

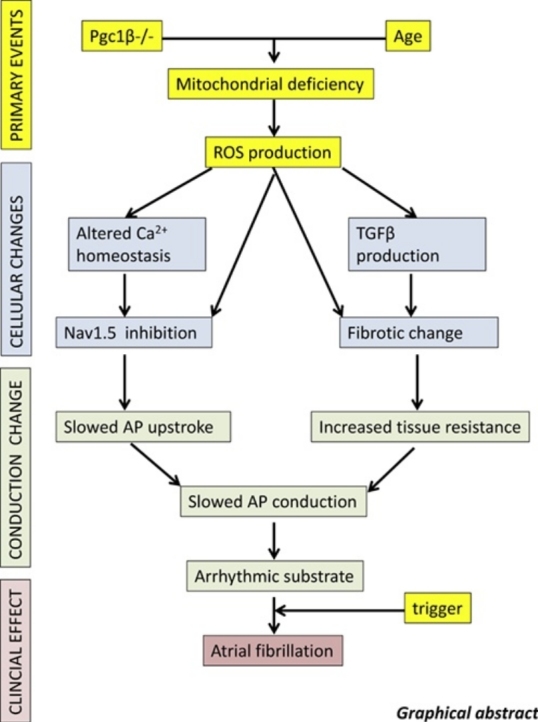
Graphical abstract
Keywords: Peroxisome proliferator activated receptor-γ coactivator-1 (PGC-1), Atrial, Action potential, Wavelength, Cardiac conduction, Cardiac arrhythmias, Atrial fibrillation
Highlights
-
•
Ageing and several chronic conditions are associated with mitochondrial dysfunction.
-
•
We investigated atrial arrhythmia in energetically deficient Pgc-1β-/- murine hearts.
-
•
Pgc-1β-/- hearts showed increased age-dependent arrhythmic and fibrotic changes.
-
•
These were attributed to slowed action potential depolarization and conduction.
-
•
The latter offer mechanisms for the atrial arrhythmia observed in these conditions.
Abstract
Introduction
Ageing and several age-related chronic conditions including obesity, insulin resistance and hypertension are associated with mitochondrial dysfunction and represent independent risk factors for atrial fibrillation (AF).
Materials and methods
Atrial arrhythmogenesis was investigated in Langendorff-perfused young (3–4 month) and aged (>12 month), wild type (WT) and peroxisome proliferator activated receptor-γ coactivator-1β deficient (Pgc-1β−/−) murine hearts modeling age-dependent chronic mitochondrial dysfunction during regular pacing and programmed electrical stimulation (PES).
Results and discussion
The Pgc-1β−/− genotype was associated with a pro-arrhythmic phenotype progressing with age. Young and aged Pgc-1β−/− hearts showed compromised maximum action potential (AP) depolarization rates, (dV/dt)max, prolonged AP latencies reflecting slowed action potential (AP) conduction, similar effective refractory periods and baseline action potential durations (APD90) but shortened APD90 in APs in response to extrasystolic stimuli at short stimulation intervals. Electrical properties of APs triggering arrhythmia were similar in WT and Pgc-1β−/− hearts. Pgc-1β−/− hearts showed accelerated age-dependent fibrotic change relative to WT, with young Pgc-1β−/− hearts displaying similar fibrotic change as aged WT, and aged Pgc-1β−/− hearts the greatest fibrotic change. Mitochondrial deficits thus result in an arrhythmic substrate, through slowed AP conduction and altered repolarisation characteristics, arising from alterations in electrophysiological properties and accelerated structural change.
1. Introduction
Atrial fibrillation (AF) affects 1–3% of the population in the developed world (DeWilde et al., 2006, Friberg and Bergfeldt, 2013, Majeed et al., 2001) and is associated with significant morbidity and mortality, including five-fold increases in risks of stroke (Wolf et al., 1991) and significant increases in risks of all-cause mortality (Benjamin et al., 1998, Chamberlain et al., 2015, Friberg et al., 2007). Recent studies forecast a substantial increase in the incidence and prevalence of AF in the coming decades, with potentially 9 million cases in the United States (Miyasaka et al., 2006) and 18 million in Europe by 2060 (Krijthe et al., 2013), underpinning its recognition as a global epidemic. Its prevalence appears to a great extent to be a function of age, affecting 0.1% of individuals under the age of 55, reaching 20% of those above the age of 80 (Go et al., 2001, Zoni-Berisso et al., 2014). The aetiological significance of this age-related incidence or indeed the mechanisms underlying the initiation and perpetuation of AF remain incompletely explained. It is however clear that AF is a dynamic process, which at its inception is characterised by fleeting episodes of the abnormal rhythm triggered by focal ectopic activity in the pulmonary vein sleeves (Haïssaguerre et al., 1998). With time, these episodes become more protracted and eventually permanent through progressive electrical and structural remodeling ultimately producing a tissue substrate itself conducive to arrhythmia maintenance. Treatment in these latter stages is far less efficacious (Cappato et al., 2010), highlighting the need to target therapies to the upstream processes.
There is now growing appreciation for roles of metabolic, and in particular mitochondrial, dysfunction in the pathogenesis of AF. Mitochondrial dysfunction is a recognised feature of ageing (Sun et al., 2016) as well as a number of the constituents of the metabolic syndrome including obesity (Bournat and Brown, 2010), insulin resistance (Patti and Corvera, 2010) and hypertension (Dikalov and Ungvari, 2013), all recognised risk factors for AF (Menezes et al., 2013). Abnormal mitochondrial structure and function have been reported in animal models of AF (Ausma et al., 1997, Morillo et al., 1995). Moreover, analysis of cardiomyocytes from human patients with AF demonstrate increased DNA damage (Lin et al., 2003, Tsuboi et al., 2001), structural abnormalities (Bukowska et al., 2008) and evidence of impaired function (Ad et al., 2005, Lin et al., 2003). Whether the observed mitochondrial abnormalities are a cause or consequence of AF, and the mechanisms through which these changes occur remain unclear.
Disruptions in normal mitochondrial activity are known to be pro-arrhythmic, through reduced provision of ATP and/or aberrant production of reactive oxygen species (ROS), and therefore are a potential upstream mediator of arrhythmogenesis (Faivre and Findlay, 1990, Fosset et al., 1988, Manning et al., 1984). Much of this work has however been in the context of acute, profound mitochondrial impairment during ischaemia-reperfusion and focussed on mechanisms of ventricular arrhythmias. Interestingly, Chen & colleagues recently demonstrated increased ectopic activity, burst firing and shortening of the action potential duration (APD) in pulmonary veins and left atria of rabbit hearts subjected to ischaemia-reperfusion (Lin et al., 2012). Moreover, sustained arrhythmias are generally thought to occur through maladaptive changes in the electrophysiological properties of a tissue, promoting the formation of re-entrant circuits. Such would arise through slowed conduction of the depolarising wavefront and/or shortening of the effective refractory period (ERP). Indeed, time-dependent alterations in both atrial conduction (Gaspo et al., 1997a, Zheng et al., 2016) and repolarisation (Bosch et al., 1999, Daoud et al., 1996, Gaspo et al., 1997b) properties have been reported in animal models and human AF. Reductions in the inward sodium currents (INa), a major determinant of conduction velocity (King et al., 2013a), have been reported secondary to excess mitochondrial ROS production (Liu et al., 2010). Gap junction activity is also known to be similarly sensitive to mitochondrial function (Li et al., 2016, Sovari et al., 2013) and may also contribute to conduction slowing. Mitochondrial impairment and cardiac oxidative stress in general are also recognised to reduce action potential durations and ERPs (Chen et al., 2007, Kurokawa et al., 2011, Lesnefsky et al., 1991), both of which favour re-entry and arrhythmia.
The electrophysiological sequelae of chronic mitochondrial dysfunction have however been challenging to study, confounded by the early and often terminal development of contractile dysfunction. However, such evaluation now appears feasible in murine models lacking members of the peroxisome proliferator activated receptor-γ coactivator-1 (PGC-1) family of transcriptional coactivators. The PGC-1 family include PGC-1α and PGC-1β, which though found with a reasonable degree of ubiquity, are preferentially expressed in tissues with high oxidative capacity such as the heart, brain and skeletal muscle (Riehle and Abel, 2012) and act as key regulators of mitochondrial mass and function (Finck and Kelly, 2006, Lin et al., 2005). In cardiac cells, the PGC-1 coactivators interact with nuclear respiratory factor-1, estrogen related receptor-α and peroxisome proliferator-activated receptor-α, leading to increased mitochondrial biogenesis (Huss et al., 2004, Vega et al., 2000). They also act to upregulate expression of nuclear and/or mitochondrial encoded mitochondrial proteins involved in the tricarboxylic acid cycle, fatty acid β–oxidation and components of the oxidative phosphorylation complex (Arany et al., 2005). PGC-1 protein expression itself is increased by upstream signals such as those arising from cold exposure and aerobic exercise, thereby serving as a link between cellular energy stores and external stimuli ultimately coordinating mitochondrial activity with cellular energy demands (Sonoda et al., 2007). Interestingly, their expression levels are found to be reduced in obesity, insulin resistance, type II diabetes mellitus and ageing, correlating with the mitochondrial dysfunction that is seen in these conditions and implicating it in the their pathogenesis (Dillon et al., 2012, Leone and Kelly, 2011, Mootha et al., 2003).
Murine hearts lacking either Pgc-1α or Pgc-1β do not develop cardiac failure under conditions of baseline activity. Homozygous deficiency of Pgc-1α is associated with a mild baseline cardiac phenotype with no overt contractile dysfunction, but cardiac failure does develop following transverse aortic banding (Arany et al., 2005). Similarly, genetic ablation of Pgc-1β does not appear to be detrimental to cardiac function at baseline, but adrenergic challenge elicits blunted rate responses potentially reflecting underlying electrophysiological abnormalities (Lelliott et al., 2006). Indeed, Langendorff-perfused Pgc-1β−/− murine hearts displayed features consistent with increased vulnerability to ventricular arrhythmia, with greater episodes of action potential alternans, a recognised harbinger for arrhythmia, and more frequent episodes of ventricular tachycardia during programmed electrical stimulation (Gurung et al., 2011). Isolated cardiomyocytes in the latter study showed altered patterns of ion channel expression, spontaneous diastolic Ca2+ transients, and pro-arrhythmic after-depolarisation events. The electrophysiological alterations and any associated change in arrhythmic propensity in the atria of these hearts have hitherto not been investigated.
The present study investigates the atrial electrophysiological properties secondary to chronic mitochondrial insufficiency in murine hearts lacking Pgc-1β. The phenotypic effects of such a mitochondrial deficit are likely to be cumulative, evolving with advancing age. Young and aged WT and genetically modified hearts were therefore studied at both the whole heart and cellular level, assessing arrhythmic tendency and correlating this with electrophysiological parameters and structural changes.
2. Materials & methods
2.1. Experimental animals
This research has been regulated under the Animals (Scientific Procedures) Act 1986 Amendment Regulations 2012 following ethical review by the University of Cambridge Animal Welfare and Ethical Review Body (AWERB). Age-matched wild type (WT) and Pgc-1β−/− mice with an inbred C57/B6 genetic background were used for the study. Pgc-1β−/− mice were generated using a triple LoxP targeting vector as previously described (Lelliott et al., 2006). Mice aged between 3 and 4 months were classified as young, and aged mice were defined as those older than 12 months. Mice were housed in plastic cages within a temperature-controlled room maintained at 21 ± 1 °C and subjected to 12 h dark/light cycles. Sterile rodent chow (RM3 Maintenance Diet, SDS, Witham, Essex, UK) and water were available ad libitum.
2.2. Experimental solutions
All buffering media utilised in the study were based on Krebs-Henseleit (KH) solution, containing NaCl (119 mM), NaHCO3 (25 mM), KCl (4 mM), MgCl2 (1 mM), KH2PO4 (1.2 mM), CaCl2 (1.8 mM), glucose (10 mM) and sodium pyruvate (1.8 mM), pH adjusted to 7.4 and bubbled with 95% O2/5% CO2 (British Oxygen Company, Manchester, UK). All chemical reagents were purchased from Sigma-Aldrich (Dorset, Poole, UK) except where otherwise indicated. Hearts were electromechanically uncoupled using blebbistatin (20 μM, Selleckchem, Houston, USA) to minimize motion artifact during the microelectrode studies, permitting stable impalement of the cardiomyocyte.
2.3. Whole heart Langendorff preparation
Electrocardiograph and microelectrode studies were performed using a horizontal Langendorff perfusion system adapted for the murine heart incorporated into a Faraday cage, together with a light microscope (objective × 5, eyepiece × 5, W. Watson and Sons Limited, London, UK), custom built head stage and a warmed bath superfused with the buffering media. All equipment was electrically insulated. The stimulating and recording electrodes were positioned at appropriate positions on the right and left atrium respectively using two precision micromanipulators (Prior Scientific Instruments, Cambridge, UK).
Mice were anticoagulated with heparin sodium 200 IU (Sigma-Aldrich, Poole, UK) prior to sacrifice, administered into the intra-peritoneal space with a 27G hypodermic needle. Following an interval of 10 min, mice were killed by cervical dislocation (Schedule 1: UK Animals (Scientific Procedures) Act (1986)), a sternotomy and cardiectomy rapidly performed and the excised heart placed in ice-cold bicarbonate-buffered KH solution. The proximal segment of the aorta was identified and cannulated with a modified 21G hypodermic needle, and secured in place with an aneurysm clip (Harvard Apparatus, Kent, UK) and a 5-0 braided silk suture. The cannulated heart was mounted on to the Langendorff apparatus and retrogradely perfused with KH solution at a constant flow rate of 2.05 ml min−1 by a peristaltic pump (MINIPULS3, Gilson, Luton, UK) passing first through 200 μm and 5 μm Millipore filters (Millipore, Watford, UK) and maintained at 37 °C by a water jacket and circulator (model C-85A, Techne, Cambridge, UK). Upon perfusion, hearts were selected for experimentation if they demonstrated sustained intrinsic activity with a basic cycle length (BCL) <200 ms and 1:1 atrioventricular conduction (AV) for 10 min. Preparations meeting these criteria were subsequently perfused with 150 ml KH solution containing 20 μM blebbistatin and then normal KH solution throughout the remainder of the study.
2.4. Volume conductor electrocardiographic recordings
Whole heart volume conductor electrocardiographic (ECG) recordings were taken concurrently with intracellular recordings to distinguish between isolated cellular and generalised atrial phenomena. Two unipolar ECG leads were immersed into the warmed bath flanking the right and left atria respectively. Signals were amplified using a model NL104A amplifier (NeuroLog; Digitimer, Hertfordshire, UK), filtered at low and high cut-off frequencies of 5 and 500 Hz (model NL125/126 filter) and digitized using a model 1401 interface (Cambridge Electronic Design) for analysis with Spike II software (Cambridge Electronic Design).
2.5. Whole heart intracellular microelectrode recordings
Glass micropipettes were drawn from 1.2 mm outer diameter and 0.69 mm internal diameter borosilicate glass (Harvard Apparatus, Cambridge, UK) using a homebuilt microelectrode puller, and cut above the shoulders to an appropriate length. The microelectrodes were backfilled with 3 M KCl immediately before use, with tip resistances ranging between 15 and 25 MΩ. The filled microelectrode was mounted on to a right-angled microelectrode holder connected to a high-input impedance direct-current microelectrode amplifier system (University of Cambridge, Cambridge, UK). Intracellular voltage was measured relative to that of the Ag/AgCl reference electrode. AP recordings were used for analysis if obtained from an impalement associated with the abrupt appearance of a resting membrane potential (RMP) between −65 mV and −90 mV, stable and normal AP morphology and an AP amplitude >75 mV.
Hearts were placed in the anatomical position within the bath with the left atrium (LA) reflected back and fixed in position using three A1 insect pins. A bipolar platinum-coated stimulating electrode (NuMed, New York, USA) was positioned against the epicardial surface of the right atrium, pacing the heart using square-wave stimuli of 2 ms duration using a constant voltage stimulator (model DS2A-Mk.II, Digitimer, Welwyn Garden City, Herts., UK) controlled by Spike II software (Cambridge Electrical Design, Cambridge, UK) and delivering a voltage that was twice the diastolic excitation threshold plus 0.5 mV. The initial experiments examined the hearts under conditions of regular pacing at a basic cycle length (BCL) of 125 ms (8 Hz). Hearts were then studied using a programmed electrical stimulation (PES) protocol comprising drive trains of eight regularly paced (S1) beats at a BCL of 125 ms, followed by an isolated premature extra stimulus (S2) every ninth beat. The S2 stimulus was imposed at progressively shortening S1-S2 coupling intervals, initially being 89 ms and reducing by 1 ms every subsequent cycle to a final coupling interval of 5 ms. The protocol was terminated upon establishment of the atrial effective refractory period (ERP), defined as the first S1-S2 coupling interval at which the S2 stimulus failed to successfully elicit an AP, or sustained arrhythmia was observed.
2.6. Quantification of AP parameters and arrhythmic incidence
The electrophysiological parameters were calculated from each AP individually and averaged across the protocol to give an overall mean for each heart. The AP amplitude was measured from the baseline to the peak voltage excursion and the AP duration was measured as 90% recovery to baseline (APD90) (Fig. 1). AP latencies were measured as the time elapsed between the pacing stimulus and peak of the AP. Maximum rates of AP depolarization (dV/dt)max were calculated from the differentiated intracellular AP waveform. The incidence of abnormal atrial rhythms were determined from the regular pacing and PES protocols, correlating cellular phenomena with tissue-level activity. Isolated non-triggered APs were classified as ectopic beats, two successive non-triggered beats termed a couplet and atrial tachycardia (AT) was defined as an episode consisting of ≥3 consecutive non-triggered beats.
Fig. 1.
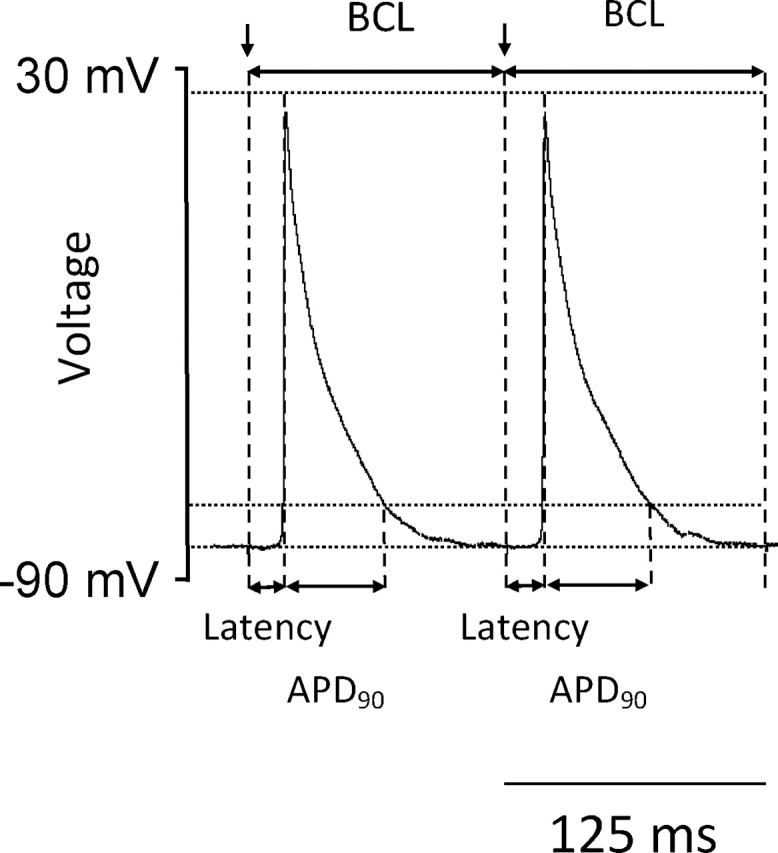
Basic measures of atrial action potential (AP) propagation, activation and recovery. AP amplitude was measured from the baseline to the peak voltage excursion. AP duration was measured as 90% recovery to baseline (APD90), and AP latencies were measured as the time elapsed between the pacing stimulus and peak of the AP. Maximum rates of depolarization (dV/dt)max were calculated from the first differential of the intracellular AP waveform.
2.7. Quantification of cardiac fibrosis
The quantification of cardiac fibrosis was performed as previously described (Jeevaratnam et al., 2011). Briefly, the excised heart was first flushed with KH solution and then perfused for five minutes with 4% buffered paraformaldehyde before being immersed in the paraformaldehyde overnight. Following the fixation process, longitudinal cardiac sections were cut and subjected to routine tissue processing and paraffin embedding. Serial sections of 7 μm thickness were then taken and stained with picrosirius red for fibrotic change. All sections were subsequently viewed, magnified and digitally acquired using the Nano Zoomer 2.0 Digital Pathology system (Hamamatsu, Hertfordshire, UK). A custom made 17 cm × 30 cm morphometric grid was superimposed on each magnified photomicrograph and each successive 1 cm × 1 cm square, corresponding to 0.2 mm × 0.2 mm area of tissue, was scored first for the presence or absence of cardiac tissue, and in turn for presence of fibrosis. The degree of fibrosis was quantified as the proportion of squares occupied by cardiac tissue showing evidence of fibrotic change. The analysis was performed independently by two investigators blinded to the animal genotype and age, and an inter-class correlation coefficient analysis (ICC) was performed to assess the consistency of their results, which can be interpreted as follows: 0–0.2 indicates poor agreement; 0.3–0.4 indicates fair agreement; 0.5–0.6 indicates moderate agreement; 0.7–0.8 indicates strong agreement; and >0.8 indicates almost perfect agreement.
2.8. Statistical analysis
Data from AP recordings were analysed using a custom written programme in the python programming language and all statistical analysis performed in the R programming language (R Core Team, 2015). Discrete incidences of abnormal rhythms were separated according to their type and the pacing protocol, either regular pacing or PES, in which they were observed. As often multiple episodes of AT were seen during a single protocol, the propensity to arrhythmia was expressed as protocols with one or more episodes of AT expressed relative to the total number studied, and compared using the Fisher Exact Test. Parameters describing electrophysiological properties measured during regular pacing were compared using a two-way analysis of variance (ANOVA) testing for significant effects of genotype, ageing, and an interaction between the two. Where the F-ratio yielded a significant result, post-hoc Tukey honestly significant testing was performed. Similar electrophysiological measurements from PES protocols were compared in two separate ways. Firstly, differences spanning the duration of the protocol were compared using ANOVA analysis of area under the curve (AUC) values for each group. To further evaluate the temporal nature of any differences between groups, mean protocol start and protocol end values for each experimental group were compared in the same manner as data from the regular pacing protocol. Where the data from PES protocols was normalised, the corresponding data from regular pacing was used as reference values. Data are expressed as mean ± standard error of the mean (SEM), and in all cases a p < 0.05 was taken to be significant, with application of Bonferroni correction where appropriate.
3. Results
The experiments evaluated the influence of ageing and mitochondrial dysfunction, through homozygous deficiency in Pgc-1β, upon atrial arrhythmic tendency and the associated electrophysiological alterations. Simultaneous ECG and intracellular microelectrode readings were recorded from Langendorff perfused WT and Pgc-1β−/− hearts and the presence or otherwise of an arrhythmic substrate was then correlated with structural changes at the organ level.
3.1. Pgc-1β−/− hearts develop an age-related arrhythmic phenotype
Volume conductor ECGs and intracellular action potential (AP) recordings were first obtained from Langendorff perfused hearts during regular pacing at a BCL of 125 ms (8 Hz) mimicking murine resting heart rates, thus enabling quantification of occurrence of spontaneous arrhythmia and electrophysiological characterisation under conditions of baseline activity. Fig. 2(a(i)) demonstrates a typical ECG recording from a young WT heart during regular 8 Hz pacing and Fig. 2(a(ii)) is the simultaneous intracellular AP from a left atrial (LA) cardiomyocyte. The intracellular recordings confirmed normally polarised resting membrane potentials (RMPs) statistically indistinguishable between groups (young WT: −76.62 ± 1.37 mV, n = 26; aged WT: −76.72 ± 1.47 mV, n = 27, young Pgc-1β−/−: -75.82 ± 0.68 mV, n = 34; aged Pgc-1β−/−: −77.43 ± 1.49 mV, n = 25). Similarly, AP amplitudes confirmed positive AP overshoots through all experimental groups, consistent with intracellular recordings from viable atrial cardiomyocytes in situ, though AP amplitudes were marginally lower in aged Pgc-1β−/− hearts (young WT: 96.76 ± 1.14 mV, n = 26; aged WT: 96.92 ± 1.55 mV, n = 27, young Pgc-1β−/−: 93.19 ± 1.12 mV, n = 34; aged Pgc-1β−/−: 91.21 ± 1.63 mV, n = 25, p < 0.05). No spontaneous arrhythmias were observed from hearts in any experimental group during the regular pacing protocols.
Fig. 2.

Typical recordings from Langendorff-perfused WT hearts during regular pacing and programmed electrical stimulation. Electrocardiograph (ECG) (i) and left atrial intracellular action potential (AP) recordings (ii) during (a) regular 8 Hz pacing and (b) a protocol imposing programmed electrical stimulation with a refractory outcome. The timings of stimulus delivery are given by the dashed bar above the AP recordings, and corresponding stimulation artefacts can be seen on the ECG and AP traces, preceding the respective complexes. In panel (B), arrows indicate the imposition of S2 extrastimuli. The first two S2 stimuli trigger APs, whereas the third S2 stimulus fails to elicit a response, thus representing a refractory outcome.
Hearts were then subjected to a programmed electrical stimulation (PES) protocol consisting of repeated cycles of nine beats, of which the first eight (S1) beats were separated by a regular interval of 125 ms and the ninth was a premature extra stimulus (S2) at an initial S1-S2 coupling interval of 89 ms that was decremented by 1 ms with each successive cycle. This permitted evaluation of the arrhythmic tendency of hearts in response to provocation with an imposed premature S2 beat, the alterations in electrophysiological parameters with varying coupling intervals, and differences in atrial effective refractory periods (ERPs) between groups. Fig. 2(b) shows typical (i) ECG and (ii) AP recordings during PES pacing from a young WT heart with a refractory as opposed to an arrhythmic outcome. Several abnormal rhythms were observed during PES pacing as exemplified in Fig. 3. These were triggered by the S2 premature stimulus and included isolated ectopic beats (Fig. 3(a)), paired beats termed a couplet (Fig. 3(b)) and episodes of atrial tachycardia (AT) defined as three or more consecutive non-stimulated beats (Fig. 3(c)).
Fig. 3.

Examples of intracellular records of abnormal rhythms elicited by S2 premature stimuli during PES pacing, including (a) isolated ectopic beats, (b) paired beats forming couplets and (c) episodes of atrial tachycardia (AT) defined as three or more consecutive non-stimulated beats. The timings of stimulus delivery are given by the dashed bar above the AP recordings and arrows indicate the onset of the abnormal rhythm.
Table 1 summarises the number of episodes of the different abnormal rhythms observed during PES, stratified by experimental group. Incidences of atrial tachycardia (AT) were significantly greater in aged Pgc-1β−/− hearts compared to any other group with respect to the overall proportion affected (p < 0.05, Fisher Exact Test) and the number of arrhythmic events per individual heart, suggesting an arrhythmic phenotype associated with mitochondrial dysfunction that progresses with age. AT was more frequently observed in the aged Pgc-1β−/− group compared to either of the WT groups, with no difference in incidence observed between the young and aged WT groups. Although a similar proportion of young and aged WT hearts were arrhythmic, arrhythmic event rates were higher in the latter further reinforcing an effect of age upon arrhythmic risk. Thus ANOVA of mean AT events per heart demonstrated a significant effect of genotype (F = 7.13, p < 0.01) with Pgc-1β−/− hearts having higher event rates and age (F = 7.26, p < 0.01), but no interactive effect (F = 2.37, p > 0.05). Post hoc Tukey tests demonstrated significant differences in rates of AT between aged Pgc-1β−/− hearts and young WT hearts (p < 0.01), aged WT hearts (p < 0.05) and young Pgc-1β−/− hearts (p < 0.05).
Table 1.
Summary of arrhythmic events during programmed electrical stimulation.
| Experimental group | No. that developed AT (n/total) | Ectopic beats (mean ± SEM) | Couplets (mean ± SEM) | AT (mean ± SEM) | Critical coupling interval (mean ± SEM) |
|---|---|---|---|---|---|
| Young Wild Type | 5/27 | 1.48 ± 0.50 | 0.41 ± 0.26 | 0.26** ± 0.11 | 28.71 # ± 3.46 |
| Aged Wild Type | 4/27 | 0.79 ± 0.53 | 1.17 ± 0.62 | 0.48† ± 0.36 | 32.71 ± 4.21 |
| Young Pgc-1β−/− | 11/34 | 1.22 ± 0.47 | 0.42 ± 0.21 | 0.86‡ ± 0.35 | 35.70 ± 1.35 |
| Aged Pgc-1β−/− | 12/25* | 1.64 ± 0.66 | 1.28 ± 0.55 | 2.64**,†,‡ ± 0.70 | 39.39 # ± 1.12 |
Symbols denote significant difference based on post hoc analysis, performed if the F value from two-way ANOVA was significant. Single, double and triple symbols denote p < 0.05, p < 0.01 and p < 0.001 respectively.
This increased propensity to AT is further highlighted on analysis of the critical coupling intervals, given by the S1-S2 coupling interval at which arrhythmia was triggered as shown in Fig. 4. Episodes of AT in WT hearts (Fig. 4(a) and (b)) were triggered predominantly at latter parts of the PES protocol corresponding to shorter S1-S2 coupling intervals. In contrast Pgc-1β−/− hearts developed arrhythmias at earlier stages of the protocol and over a wider range of coupling intervals (Fig. 4(c) and (d)), with aged Pgc-1β−/− hearts particularly appearing vulnerable throughout the duration of the protocol. The mean critical coupling interval was longer in Pgc-1β−/− hearts (F = 8.35, p < 0.01) and aged hearts (F = 3.93, p < 0.05), though no interactive effect was observed (F = 0.004, p > 0.05). Post hoc analysis demonstrated significant differences between aged Pgc-1β−/− and young WT hearts (p < 0.05) and a trend to significance between aged Pgc-1β−/− and aged WT hearts (p < 0.10).
Fig. 4.

Stratification of the occurrence of AT episodes by critical coupling intervals in young ((a), (c)) and old ((b), (d)), WT ((a), (b)) and Pgc-1β−/− hearts ((c), (d)). Pgc-1β−/− hearts display vulnerability to arrhythmia earlier, and through wider range of S1-S2 coupling intervals.
3.2. Action potential parameters during regular pacing
These differing arrhythmic profiles were next compared with electrophysiological parameters corresponding to AP initiation, propagation and recovery during regular pacing at 8 Hz. AP initiation was measured first through maximum rates of AP depolarisation (dV/dt)max, derived from left atrial intracellular microelectrode recordings. (dV/dt)max measurements quantify the depolarisation of the cardiomyocyte membrane capacitance by regenerative inward Na+ current. Reductions in (dV/dt)max are known to correlate with compromised conduction velocity of an AP wavefront, potentially providing a substrate permissive to AP re-entry and arrhythmia. Pgc-1β−/− hearts showed significantly lower values of (dV/dt)max compared to WT (F = 18.41, p < 0.001) but there was no effect of age (F = 0.17, p > 0.05) or interaction between age and genotype (F = 0.001, p > 0.05). On post hoc Tukey testing, each Pgc-1β−/− group, whether young or aged, showed significantly lower (dV/dt)max values than either of the WT groups (Table 2).
Table 2.
Action potential properties in WT and Pgc-1β-/- hearts during regular 8 Hz pacing.
| Experimental group | (dV/dt)max (V s−1) | AP latency (ms) | APD90 duration (ms) | Effective refractory period (ms) | Wavelength |
|---|---|---|---|---|---|
| Young Wild Type | 171.06*, † ± 6.60 | 17.69***, †††, ‡‡ ± 0.23 | 24.87 ± 1.18 | 26.62 ± 1.27 | 4.21*, † ± 0.25 |
| Aged Wild Type | 171.73‡‡, # ± 5.15 | 23.93*** ± 1.24 | 25.51 ± 1.59 | 27.33 ± 1.77 | 4.34‡‡, ## ± 0.29 |
| Young Pgc-1β-/- | 141.28*, ‡‡ ± 7.19 | 24.61††† ± 0.65 | 23.97 ± 0.93 | 29.82 ± 0.54 | 3.28*, ‡‡ ± 0.16 |
| Aged Pgc-1β-/- | 142.39†, # ± 8.08 | 23.48‡‡ ± 1.66 | 23.28 ± 2.16 | 28.33 ± 1.33 | 3.15†, ## ± 0.25 |
All values are given as mean (±SEM).
Symbols denote significant difference based on post hoc analysis, performed if the F value from two-way ANOVA was significant. Single, double and triple symbols denote p < 0.05, p < 0.01 and p < 0.001 respectively.
AP conduction through respective cardiac chambers is determined not only by the properties of the inward Na+ current, reflected in the cellular (dV/dt)max values, but also the total membrane capacitance and resistance (King et al., 2013a). AP propagation was therefore further assessed through comparisons of AP latency times, measured as the time intervening between stimulus delivery at the right atrial pacing site and the peak AP voltage measured at the left atrial (LA) recording site. In all experiments, the stimulating electrode was consistently positioned at the posterior aspect of the RA and recordings were made from the central region of the LA, minimising variability in distances between the respective electrodes. ANOVA of AP latency times demonstrated significant effects of genotype (F = 9.91, p < 0.01), age (F = 5.32, p < 0.05) and an interaction of the two (F = 12.47, p < 0.001). As detailed in Table 2, these differences were driven by young WT hearts, which had significantly shorter AP latency times than aged WT hearts (p < 0.001), young Pgc-1β−/− hearts (p < 0.001), and aged Pgc-1β−/− hearts (p < 0.01), There was no significant difference between the aged WT hearts and either of the Pgc-1β−/− groups.
Pro-arrhythmic tissue substrate has also been associated with altered repolarisation associated with action potential shortening or prolongation (Killeen et al., 2007, Sabir et al., 2007a, Sabir et al., 2007b). Electrophysiological parameters describing repolarisation are given in Table 2. No differences in APD at 90% repolarisation (APD90) were found during regular 8 Hz pacing either through effects of genotype (F = 1.07, p > 0.05), age (F = 0.001, p > 0.05) or an interaction of the two (F = 0.20, p > 0.05). Repolarisation properties were further assessed through measurement of the effective refractory period (ERP) obtained from the PES protocol, defined as the longest S1-S2 coupling interval at which the S2 stimulus failed to trigger an AP. The ERP generally correlates with the APD, and in keeping with this no differences between groups were observed when compared according to genotype (F = 3.36, p > 0.05) or age (F = 0.04, p > 0.05).
3.3. Action potential parameters following premature extrasystolic stimuli
The trigger-substrate model of arrhythmogenesis involves initiation of the abnormal rhythm through an arrhythmogenic trigger, such as an extrasystole, occurring within a pro-arrhythmic substrate capable of sustaining the arrhythmia (Antzelevich et al., 1999, Kalin et al., 2010). The PES protocol consisted of pulse trains of S1 beats 125 ms apart, punctuated every ninth beat by a premature S2 stimulus, mimicking such triggering extrasystoles. The external application of these premature beats thus controlled for incidence of ectopic stimuli between groups and so assessed for the presence of such an arrhythmogenic substrate.
Fig. 5(a(i)) plots mean (dV/dt)max values for APs triggered by S2 stimuli across the range of coupling intervals explored during the PES protocol. All experimental groups displayed the expected progressively reduced (dV/dt)max values with shortening of the S1-S2 coupling interval. In common with findings obtained during regular pacing, an analysis of the areas beneath the data curve (AUC) demonstrated that the overall rates of depolarisation were significantly higher in WT than Pgc-1β−/− hearts (F = 6.41, p < 0.05) (Table 3); there were no effects of age (F = 0.84, p > 0.05) or interacting effects of genotype and age (F = 0.27, p > 0.05). The difference between WT and Pgc-1β−/− hearts was most pronounced at the beginning of the protocol (Fig. 5(a(ii))) (ANOVA − genotype: F = 13.19, p < 0.001; age: F = 0.15, p > 0.05; interaction: F = 0.002, p > 0.05) and was of a similar magnitude as had been observed during 8 Hz pacing. On post hoc analysis, each WT group showed significantly higher (dV/dt)max values compared against either of the Pgc-1β−/− groups. In contrast, no difference in (dV/dt)max values was observed at the shortest coupling intervals at the end of the protocol, whether tested for effects of genotype (F = 0.09, p > 0.05), age (F = 0.18, p > 0.05) or interaction between these factors (F = 0.31, p > 0.05).
Fig. 5.

Plots of mean ± SEM (a) (dV/dt)max, (b) AP latency and (c) time to 90% AP recovery (APD90) and (d) AP wavelength for APs obtained in response to S2 stimuli (i) through the range of coupling intervals explored, reducing from 89 ms to 30 ms. Panel (ii) for each provides a comparison of these values at the beginning and termination of the pacing protocol, corresponding to a refractory outcome or the onset of sustained arrhythmia. The symbols denote significant differences between each pair, obtained from post hoc Tukey testing, which was conducted if the ANOVA indicated a significant outcome. Single, double and triple symbols denote p < 0.05, p < 0.01 and p < 0.001 respectively.
Table 3.
Area under the curve analysis for S2 triggered APs during programmed electrical stimulation.
| Experimental group | (dV/dt)max (V × 10−3) | AP latency (ms2) | APD90 (ms2) | Wavelength (ms) |
|---|---|---|---|---|
| Young Wild Type | 6.68 ± 0.32 | 1.06††, ‡ ± 0.04 | 1.06 ± 0.05 | 0.085 ± 0.005 |
| Aged Wild Type | 6.11 ± 0.35 | 1.21 ± 0.05 | 0.93 ± 0.06 | 0.082 ± 0.007 |
| Young Pgc-1β-/- | 5.44 ± 0.37 | 1.39†† ± 0.08 | 0.95 ± 0.05 | 0.066 ± 0.005 |
| Aged Pgc-1β-/- | 5.30 ± 0.64 | 1.38‡ ± 0.01 | 0.82 ± 0.06 | 0.065 ± 0.005 |
All values are given as mean (±SEM).
Symbols denote significant difference based on post hoc analysis, performed if the F value from two-way ANOVA was significant. Single, double and triple symbols denote p < 0.05, p < 0.01 and p < 0.001 respectively.
Similar plots for AP latencies are given in Fig. 5(b(i)) and 5(b(ii)). In keeping with findings during regular 8 Hz pacing and the observed differences in (dV/dt)max values, AP latency times were significantly prolonged in Pgc-1β−/− hearts when surveyed through the entirety of the protocol on AUC analyses (F = 12.98, p < 0.001), but there were no independent effects of age (F = 1.11, p > 0.05) or compound effects of age and genotype (F = 1.36, p > 0.05). Young WT hearts had significantly shorter AP latency times compared to young Pgc-1β−/− hearts (p < 0.01) and aged Pgc-1β−/− hearts (p < 0.05) on post hoc testing. ANOVA of AP latencies at the longest S1-S2 intervals demonstrated significant effects of genotype (F = 19.23, p < 0.001), age (F = 4.79, p < 0.05) and interacting effects of genotype and age (F = 6.12, p < 0.05). Here the AP latency times for young WT hearts were significantly shorter than all other groups including aged WT (p < 0.01), young Pgc-1β−/− (p < 0.001) and aged Pgc-1β−/− hearts (p < 0.001). AP latencies progressively lengthened in all groups as the S1-S2 interval shortened but to varying degrees. Thus, at the shortest S1-S2 intervals, a significant difference between WT and Pgc-1β−/− hearts persisted (F = 10.15, p < 0.001); however significant effects of age (F = 1.19, p > 0.05) or interaction (F = 2.79, p > 0.05) were no longer evident. AP latency times remained significantly shorter in young WT hearts when compared with young Pgc-1β−/− (p < 0.01) and aged Pgc-1β−/− hearts (p < 0.05); however the lengthening of AP latency in aged WT was less pronounced than that of the Pgc-1β−/− hearts and thus the difference with the young WT hearts was no longer significant.
The adaptation of AP duration, given by APD90 values, through progressively shortening S1–S2 coupling intervals is shown in Fig. 5(c(i)). Overall APD90 times did not differ between experimental groups (ANOVA – genotype: F = 2.95, p > 0.05; age: F = 1.71, p > 0.05; interaction: F = 0.002, p > 0.05), reflecting the findings during regular 8 Hz pacing. Accordingly, APD90 times at the beginning of the protocol, corresponding to the longest S1-S2 intervals, also did not differ between groups (ANOVA – genotype: F = 1.63, p > 0.05; age: F = 0.02, p > 0.05; interaction: F = 1.30, p > 0.05). APD90 times in all groups displayed the expected shortening as the S1-S2 interval decreased; however a small but significant difference in APD90 between WT and Pgc-1β−/− hearts was seen at the shortest coupling intervals (F = 6.60, p < 0.05), where Pgc-1β−/− hearts had shorter APD90. No differences were noted based upon age (F = 0.02, p > 0.05), or interacting effects of genotype and age (F = 1.52, p > 0.05). There were no significant differences between groups on post hoc Tukey testing.
Reductions in the AP wavelength have been suggested to correlate with increased arrhythmic risk, indicating the presence of substrate favorable to AP re-entry (Davidenko et al., 1995, Krogh-Madsen et al., 2012, Pandit and Jalife, 2013, Weiss et al., 2005). It has previously been calculated from terms relating to AP conduction and AP duration (Matthews et al., 2013), and similar analyses were conducted in the present study. Pgc-1β−/− hearts had significantly shorter wavelength values at resting hearts as measured during 8 Hz pacing (F = 20.62, p < 0.001), however there were no effects of ageing (F = 0.01, p > 0.05) or interacting effects of the two variables (F = 0.32, p > 0.05) (Table 2). AP wavelength profiles for beats triggered by S2 extrastimuli during PES pacing are shown in Fig. 5(d). Wavelengths were shorter throughout the protocol in Pgc-1β−/− hearts (F = 9.19, p < 0.01), with no independent effect of age (F = 0.03, p > 0.05) or interacting effects of genotype and age (F = 0.01, p > 0.05). The differences between WT and Pgc-1β−/− hearts noted during regular 8 Hz pacing was similarly evident at the longest S1-S2 intervals (F = 18.93, p < 0.01) with no other significances noted (ageing: F = 0.01, p > 0.05; genotype – age interaction: F = 1.20, p > 0.05) (Fig. 5(d(ii))). Post hoc Tukey tests revealed significant differences between young WT and aged Pgc-1β−/− hearts (p < 0.05), aged WT and young Pgc-1β−/− hearts (p < 0.01), and aged WT and aged Pgc-1β−/− hearts (p < 0.01). Wavelength values were reduced in all groups as the S1-S2 coupling interval shortened, correlating with the increased vulnerability to arrhythmias seen in all hearts. However, AP wavelengths for young Pgc-1β−/− hearts more closely converged to those of both WT groups, whereas wavelengths remained shorter in aged Pgc-1β−/− hearts. Thus though Pgc-1β−/− hearts continued to display significantly shorter wavelengths at the shortest S1-S2 intervals (ANOVA – genotype: F = 5.00, p < 0.05; age: F = 0.51, p > 0.05, interaction: F = 2.40, p > 0.05), no significant differences were noted between any groups on post hoc testing.
3.4. Relative changes in action potential parameters following premature extrasystolic stimuli
The energetic dysfunction associated with mitochondrial impairment would be expected to particularly compromise cardiac activity in the stressed state. Indeed Pgc-1α deficient hearts show normal contractile function at baseline but develop pronounced cardiac failure in response to aortic banding. Pgc-1β−/− hearts are known to develop chronotropic incompetence in response to adrenergic challenge despite normal resting heart rates. To further characterise the cardiac phenotype in response to increasing metabolic demand in the form of shortening pacing intervals, electrophysiological parameters during PES pacing were normalised internally to their corresponding values measured during regular 8 Hz pacing. The normalised profiles for the relevant parameters are shown in Fig. 6(a–c). Normalised (dV/dt)max (Fig. 6a) and normalised APD90 values (Fig. 6c) displayed similar reductions with shortening S1-S2 intervals, and there were no significant differences in AUC values for either parameter (Table 4). Despite differing absolute AP latency times, normalised AP latency profiles for young and aged WT hearts were similar (Fig. 6(b)). In contrast Pgc-1β−/− hearts displayed greater increments in normalised latency with shortening S1-S2 intervals, with aged Pgc-1β−/− hearts appearing most compromised. ANOVA analysis of AUC values for normalised AP latency showed no independent effects of genotype (F = 0.49, p > 0.05) or age (F = 0.05, p > 0.05), but a significant interacting effect of the two (F = 4.31, p < 0.05) with aged Pgc-1β−/− hearts having the highest AUC values. No significant differences were seen on individual comparisons with post hoc Tukey testing.
Fig. 6.

Plots of mean ± SEM (a) (dV/dt)max, (b) AP latency, (c) time to 90% AP recovery (APD90) in APs obtained in response to S2 stimuli, normalized to their corresponding values obtained during regular 8 Hz pacing through progressively shortening S1-S2 coupling intervals.
Table 4.
Area under the curve analysis for S2 triggered APs during programmed electrical stimulation, normalised to corresponding values from regular pacing.
| Experimental group | (dV/dt)max (V × 10−3) | AP latency (ms2) | APD90 (ms2) |
|---|---|---|---|
| Young Wild Type | 0.045 ± 0.001 | 0.039 ± 0.02 | 0.025 ± 0.001 |
| Aged Wild Type | 0.041 ± 0.002 | 0.035 ± 0.002 | 0.023 ± 0.002 |
| Young Pgc-1β-/- | 0.042 ± 0.002 | 0.037 ± 0.002 | 0.023 ± 0.001 |
| Aged Pgc-1β-/- | 0.041 ± 0.003 | 0.043 ± 0.003 | 0.023 ± 0.002 |
All values are given as mean (±SEM).
Symbols denote significant difference based on post hoc analysis, performed if the F value from two-way ANOVA was significant. Single, double and triple symbols denote p < 0.05, p < 0.01 and p < 0.001 respectively.
3.5. Contrasting impacts of (dV/dt)max upon AP latency in WT and Pgc-1β−/− hearts
Findings from the regular 8 Hz and PES pacing protocols suggested an arrhythmic substrate in Pgc-1β−/− hearts through compromised conduction parameters, with few alterations in repolarisation characteristics. Here, Pgc-1β−/− hearts displayed deficits in (dV/dt)max that were independent of age, and a corresponding altered conduction through the myocardium, reflected by prolonged AP latency times. However, the latter, differed from (dV/dt)max, in appearing to be influenced by age to some degree. This prompted further exploration of the relationship between these conduction parameters. Mean AP latency times from extrasystolic S2 beats recorded during the PES protocols are plotted against their corresponding mean (dV/dt)max values for each experimental group in Fig. 7. Reductions in (dV/dt)max with shortening S1-S2 coupling intervals is associated with increasing AP latency times for all groups, suggesting that much of the increase in AP latency observed with progressively shortening coupling intervals is attributable to concurrent reductions in (dV/dt)max. However differing associations between these parameters were seen in WT and Pgc-1β−/− hearts. As shown in Fig. 7(a), prolongation of AP latency independent of (dV/dt)max occurs in WT hearts with age, such that for any given (dV/dt)max value, the AP latency time is longer in aged WT hearts compared to young WT hearts. In contrast, young and aged Pgc-1β−/− hearts displayed a more homogeneous association between (dV/dt)max and AP latency, with values in line with those of aged WT hearts. Thus young Pgc-1β−/− hearts develop electrophysiological features resembling those of normal ageing, which may explain their increased propensity to arrhythmia.
Fig. 7.

Dependences of AP latency times upon (dV/dt)max through the programmed electrical stimulation protocol compared in (a) young and old WT and (b) young and aged Pgc-1β−/− hearts.
3.6. Compromised conduction triggering arrhythmia in all hearts
We next explored the bearing of the observed electrophysiological parameters upon the initiation of arrhythmic events. The mean values of the relevant electrophysiological parameters from the first S2 AP that triggered an episode of AT in a given heart are given in Table 5. Pgc-1β−/− hearts showed significantly higher values of (dV/dt)max for triggering S2 APs compared to WT (F = 4.55, p < 0.05), but there were no effects of age (F = 0.00, p > 0.05) or interacting effects of age and genotype (F = 0.28, p > 0.05). No significant differences were found upon individual comparisons during post hoc analysis. Interestingly, ANOVA analysis of AP latencies for the same S2 trigger APs revealed no differences with respect to genotype (F = 0.001, p > 0.05), age (F = 0.15, p > 0.05) or interaction of the two (F = 3.46, p > 0.05). No significant differences were seen in APD90 between trigger S2 APs of the different experimental groups (ANOVA – genotype: F = 1.02, p > 0.05; age: F = 0.37, p > 0.05; interaction: F = 0.39, p > 0.05). Similarly AP wavelengths for trigger S2 APs were also indistinguishable between groups (ANOVA – genotype: F = 1.96, p > 0.05; age: F = 0.07, p > 0.05; interaction: F = 1.04, p > 0.05). Thus AT was initiated in WT and Pgc-1β−/− hearts through premature beats that were indistinguishable in terms of measures of conduction across the tissue. The differing profiles of conduction between the groups, and the earlier development of compromise in Pgc-1β−/− hearts may explain their increased vulnerability to arrhythmia.
Table 5.
AP parameters for S2 triggered APs that initiated the first episode of atrial tachycardia during programmed electrical stimulation.
| Experimental group | (dV/dt)max (V s−1) | AP latency (ms) | APD90 (ms) | Wavelength |
|---|---|---|---|---|
| Wild Type Young | 100.10 ± 5.68 | 27.42 ± 1.52 | 15.96 ± 0.50 | 1.58 ± 0.10 |
| Wild Type Aged | 112.53 ± 10.63 | 33.90 ± 3.39 | 17.60 ± 3.59 | 1.98 ± 0.44 |
| Pgc-1β-/- Young | 142.03 ± 11.77 | 32.86 ± 1.77 | 15.17 ± 0.61 | 2.12 ± 0.16 |
| Pgc-1β-/- Aged | 137.46 ± 14.65 | 28.57 ± 2.56 | 15.43 ± 0.85 | 2.04 ± 0.18 |
All values are given as mean (±SEM).
Symbols denote significant difference based on post hoc analysis, performed if the F value from two-way ANOVA was significant. Single, double and triple symbols denote p < 0.05, p < 0.01 and p < 0.001 respectively.
3.7. Increased fibrotic change with Pgc-1β ablation
The influence of ageing and genotype upon the relationship between rates of depolarisation and latency prompted histological assessment for fibrotic change. Fibrosis is known to impede AP conduction through the myocardium through decoupling of adjacent myocytes, resulting in disrupted gap junction functioning and consequent increases in resistivity. In addition, fibroblast fusion with myocytes increases membrane capacitance. Histological assessment was conducted blindly by two investigators independently and the ICC, a measure of consistency between their findings, was 0.88 suggesting a high degree of consistency in the results.
Fig. 8(a) represents typical histological sections from young and aged WT and Pgc-1β−/− hearts, the quantification of fibrotic change is shown in Fig. 8(b). Genotype (F = 33.02, p < 0.001) and age (F = 4.75, p < 0.05) independently increases levels of fibrosis in the atria, but there was no evidence of interaction between the two. These findings complement the changes in latency noted in the earlier electrophysiological studies. The fibrotic change witnessed in WT aged hearts compared to WT young hearts explains the increased latency seen in this group. Young Pgc-1β−/− hearts show similar levels of fibrosis to aged WT hearts further supporting the suggestion of premature ageing in this group.
Fig. 8.

(a) Representative images of histological samples used for morphological assessment of fibrotic change. (b) The degree of fibrotic change was assessed as the proportion of morphometric squares covering tissue that demonstrated evidence of fibrosis as detected by picrosirius red staining. The numbers of hearts examined: young WT (n = 6), aged WT (n = 6), young Pgc-1β−/− (n = 6), aged Pgc-1β−/− (n = 6). Symbols denotes pairs of points showing significant differences from post hoc Tukey testing, where single, double and triple symbols denote p < 0.05, p < 0.01 and p < 0.001 respectively. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article).
4. Discussion
Atrial fibrillation is characterised by an irregular, often rapid atrial rhythm that may be asymptomatic in the short term, but nevertheless carries substantial risks of long term morbidity and mortality. Ageing represents the major risk factor for AF itself: AF affects <0.1% in those under the age of 50, 4% in individuals aged 60–69 and up to 20% in those aged above 85 (Go et al., 2001, Krijthe et al., 2013, Zoni-Berisso et al., 2014). A decline in mitochondrial function is correspondingly observed in ageing, and has been postulated to account for age-related decrements in organ function with associated susceptibility to disease (Biala et al., 2015, Lane et al., 2015), including predisposition to AF (Ad et al., 2005, Montaigne et al., 2013, Tsuboi et al., 2001). The present study therefore investigated the electrophysiological alterations associated with ageing and mitochondrial dysfunction in murine atria with homozygous deficiency of the transcriptional co-activator Pgc-1β. We report an age-related increase in arrhythmic incidence that was exacerbated by mitochondrial dysfunction. This propensity to arrhythmia developed predominantly through deficits in parameters pertaining to AP conduction at the cellular and tissue level.
The heart is a highly oxidative organ and served by a rich network of mitochondria, which account for up to 30% of myocardial volume and produce approximately 95% of its cellular ATP (Schaper et al., 1985). Understandably mitochondrial dysfunction is associated with altered cardiac electrical properties, giving rise to AP heterogeneities that provide a substrate for arrhythmia, and has been the subject of much attention in the context of ventricular arrhythmogenesis (Brown and O’Rourke, 2010, Yang et al., 2014). Mitochondrial abnormalities have also been reported in studies of AF but their role in its pathogenesis have been relatively unexplored. Evidence of altered mitochondrial structure was found in dog (Morillo et al., 1995) and goat (Ausma et al., 1997) models of AF. Here, AF was induced by rapid atrial pacing with the noted mitochondrial defects appearing as a possible consequence of the pacing protocol and/or AF itself. Similarly, evidence of mitochondrial abnormalities have been reported in tissue samples obtained during cardiac surgery from AF patients (Emelyanova et al., 2016, Lin et al., 2003, Slagsvold et al., 2014, Tsuboi et al., 2001). In these studies the selected cohorts had established AF and it remains difficult to distinguish whether the observed mitochondrial lesions were caused by or resulted from AF, or indeed were a confound of ageing or other age-related conditions associated with metabolic compromise. However baseline mitochondrial deficits were found to predict development of post-operative AF following cardiac surgery in patients with no prior history of AF (Ad et al., 2005, Montaigne et al., 2013), suggesting a more direct role in its pathogenesis. Furthermore, Marks and colleagues recently reported that diastolic Ca2+ leak, through progressive oxidation of ryanodine receptors, was associated with age-dependent development of AF in a murine model (Xie et al., 2015). Reductions in mitochondrial ROS production attenuated these diastolic Ca2+ transients and prevented AF.
Electrophysiological alterations secondary to chronic mitochondrial impairment have not been well characterised to date. The PGC-1 family of transcriptional coactivators, which includes PGC-1α and PGC-1β, serve as key modulators of cellular metabolic activity, particularly in oxidative tissues such as the heart and brain (Riehle and Abel, 2012). Their present study combines the hitherto separate use of murine cardiac models in metabolic and electrophysiological studies. First, these demonstrated that Pgc-1α and Pgc-1β murine models do replicate the clinically reported energetic changes. Both Pgc-1α-/- (Arany et al., 2005, Huss et al., 2004, Vega et al., 2000) and Pgc-1β-/- mice (Lelliott et al., 2006) showed blunted expression of oxidative phosphorylation genes and reduced mitochondrial enzymatic activities, capacities for mitochondrial ATP synthesis and cytosolic ATP levels. Conversely, overexpression in specific Pgc-1 family members increased expression of nuclear and mitochondrial genes, mitochondrial density and their oxidative capacity (Lehman et al., 2000, Russell et al., 2004).
Secondly, murine models have previously successfully replicated and clarified human phenotypes in arrhythmogenic conditions involving monogenic ion channel disorders. Murine hearts have similar overall anatomies and capacities to generate polymorphic arrhythmia (Martin et al., 2011, Matthews et al., 2013, Matthews et al., 2012, Matthews et al., 2010, Vaidya et al., 1999) involving formation of drifting rotors generating scroll waves even in their small volumes of tissue, as in human hearts (Gray et al., 1995). Mouse cardiac, particular ventricular AP waveforms show shorter, triangulated recovery waveforms distinct from those in humans, reflecting differing contributions from the various K+ current subtypes. Nevertheless, APs of mouse atria and ventricles resemble those of human hearts in their rapid depolarisation phases driven by inward INa (Guo et al., 1999) and their resulting transmural AP conduction velocities (Higuchi and Nakaya, 1984, Liu et al., 2004). Monogenic murine cardiac models successfully demonstrated clinically observed atrial and ventricular arrhythmic phenotypes and their mechanisms following loss of Scn5a function replicating Brugada Syndrome and following challenge by class I, Na+ channel blocking drugs, both situations compromising AP conduction (Dautova et al., 2010, Guzadhur et al., 2013, Martin et al., 2012).
Thirdly, deficiencies in either or both Pgc-1α and Pgc-1β do induce cardiac phenotypes. Mice deficient in both genes develop a lethal low cardiac output state and conduction system disease (Lai et al., 2008). Finally, Pgc-1β deficiency is not associated with cardiac dysfunction at baseline (Lelliott et al., 2006), but is associated with increased susceptibility to ventricular arrhythmias (Gurung et al., 2011). This makes it an appropriate model to investigate electrophysiological alterations secondary to mitochondrial dysfunction in murine atria. The modified Langendorff preparation utilised here permitted simultaneous volume conductor ECG and intracellular microelectrode recordings during regular pacing and programmed electrical stimulation applying premature extra stimuli, enabling assessment of AP activation and recovery properties
Hearts were first paced at a frequency of 8 Hz, reflecting murine resting heart rates and therefore providing steady state electrophysiological characterisation. No arrhythmias were observed in any group during regular pacing. This is consistent with previous reports that Pgc-1β ablation is not associated with a pronounced cardiac phenotype under conditions of baseline activity. In contrast arrhythmias were seen in all experimental groups during programmed electrical stimulation. Incidences of arrhythmia increased with age in both WT and Pgc-1β−/− hearts, in keeping with the cumulative risk of both atrial and ventricular arrhythmias with age seen in the clinical setting (Deo and Albert, 2012, Zoni-Berisso et al., 2014). Young WT hearts displayed the fewest episodes of AT of all groups, and the incidence was higher in aged WT hearts. The mitochondrial theory of ageing posits progressive deterioration in mitochondrial function, through accumulation of mutations in mitochondrial DNA and impaired autophagy, underpinning the ageing process and may contribute to this increased vulnerability to arrhythmia. Accordingly Pgc-1β−/− hearts, possessing a pronounced mitochondrial defect, had even higher incidences of AT. Here aged Pgc-1β−/− hearts displayed the greatest propensity to arrhythmia of all groups, in terms of the proportion of hearts that were arrhythmic and the overall number of episodes of arrhythmia.
The electrophysiological alterations underlying the greater propensity to arrhythmia in Pgc-1β−/−− hearts were examined with intracellular AP recordings from the left atrium, which suggested that these occurred primarily through abnormalities in AP conduction. At the cellular level, young and aged Pgc-1β−/− hearts had significantly reduced (dV/dt)max values compared to WT hearts during regular pacing. There was no difference in (dV/dt)max based upon age in either group. A similar pattern was also observed in APs triggered by S2 stimuli during the PES protocol. As would be expected, (dV/dt)max values progressively reduced with shortening of the coupling interval in all groups. At the longest coupling intervals (dV/dt)max values differed between WT and Pgc-1β−/− hearts to similar extents as during 8 Hz pacing, whereas they converged to become indistinguishable at the shortest coupling intervals. Thus Pgc-1β−/− hearts demonstrated compromise at modest levels of stress represented by the longer coupling intervals, and correlated with their increased susceptibility to arrhythmia through greater parts of the protocol.
Reduced atrial conduction velocities have been reported as an early feature in patients with AF (Zheng et al., 2016) and potentially play a significant role in providing a substrate for its maintenance in the long term (Miyamoto et al., 2009, Park et al., 2009). Values of (dV/dt)max are known to correlate with peak Na+ currents (INa) (Hondeghem and Katzung, 1977) and conduction velocity in skeletal and cardiac cells (Fraser et al., 2011, Usher-Smith et al., 2006). Thus, INa dominates over Ca2+ current, ICa, as the dominant inward, depolarising, current in atrial cardiomyocytes. It accounts for significantly greater maximum conductances (7.8 nS/pF vs. 0.12 nS/pF (Van Wagoner et al., 1999)) and maximum currents (∼85 pA/pF (Burashnikov et al., 2007, Sossalla et al., 2010) vs. ∼6 to 9 pA/pF (Carnes et al., 2007, Courtemanche et al., 1998)), at voltages corresponding to the most rapidly rising phase of the action potential (–30 mV vs +10 mV (Van Wagoner et al., 1999)). Time-dependent reductions in INa and consequent reductions in atrial conduction velocity have previously been implicated in the pathogenesis in a canine model of AF (Gaspo et al., 1997a). Furthermore SCN5A gene variants, which encodes the cardiac sodium channel responsible for the inward Na+ current, are associated with increased risk of developing AF (Darbar et al., 2008, Olson et al., 2005).
Mitochondrial dysfunction can alter INa through a number of potential mechanisms. Firstly reductions in INa in cardiomyocytes were observed in response to metabolic stress (Liu et al., 2009) and could be recovered with application of the mitochondrial ROS scavenger mitoTEMPO (Liu et al., 2010). Secondly, fluctuations in cytosolic [Ca2+] could also potential modify sodium channel properties through binding in its C-terminal region, either directly at an EF hand motif (Wingo et al., 2004) and indirectly through an IQ domain sensitive to calmodulin/calmodulin kinase II (Mori et al., 2000). Elevated intracellular [Ca2+] caused reductions in INa density and (dV/dt)max in cardiomyocytes in vitro (Casini et al., 2009), and in whole hearts following diastolic Ca2+ leaks, through application of caffeine (Zhang et al., 2009), known to increase diastolic Ca2+ release, or mutations associated with diastolic Ca2+ release (Glukhov et al., 2013, King et al., 2013b, Li et al., 2014). Abnormal diastolic Ca2+ transients have been recorded in cardiomyocytes in Pgc-1β−/− hearts (Gurung et al., 2011).
The conduction of an AP wavefront through tissue is influenced by the membrane capacitance and its resistance, in addition to (dV/dt)max (Jeevaratnam et al., 2011, King et al., 2013a). Conduction was therefore further assessed through evaluation of AP latency times. These were significantly prolonged in Pgc-1β−/− hearts compared to WT during regular pacing, with young WT hearts having significantly shorter AP latency durations than any other experimental group including aged WT hearts. During PES pacing, AP latency times increased with shortening of the S1-S2 coupling interval in all groups, but with differing magnitudes. Conduction slowing was most pronounced in Pgc-1β−/− hearts, particularly aged Pgc-1β−/− hearts, at the shorter coupling intervals correlating with their greater vulnerability to arrhythmia during the PES protocols. The differing comparisons of (dV/dt)max and AP latency between groups were further explored by evaluating the dependence of AP latency upon (dV/dt)max within groups. In all cases AP latency lengthened with reductions in (dV/dt)max, in keeping with the known relationship between (dV/dt)max, INa and conduction velocity (Hunter et al., 1975). However young and aged WT hearts displayed distinct relationships between AP latency and (dV/dt)max, with age-related delays in latency observed at any given (dV/dt)max value. In contrast this correlation was indistinguishable between young and aged Pgc-1β−/− hearts, where both resembled the conduction slowing seen with ageing in WT hearts.
Myocardial fibrosis is associated with increased tissue capacitance and resistance, contributing to conduction slowing independent of the influence of (dV/dt)max and may explain the conduction properties described in the present study. Fibrotic change is thought to be a key element of the remodeling seen in AF (Frustaci et al., 1997, Kostin et al., 2002). Progressive fibrosis is a common feature of cardiac ageing in animal (Eghbali et al., 1989, Jeevaratnam et al., 2012, Lin et al., 2008, Orlandi et al., 2004) and human (Gazoti Debessa et al., 2001) studies. Age-dependent fibrosis was similarly recorded in the present study, in both WT and Pgc-1β−/− hearts, and mitochondrial dysfunction through Pgc-1β ablation was associated with a further additive effect on the degree of fibrosis. Aged Pgc-1β−/− hearts therefore displayed the greatest degree of fibrosis whereas young WT hearts had the least and AP latency times correlated with the observed degrees of fibrosis in the respective groups.
The fibrosis-mediated reductions in cardiac conduction reported here could potentially occur through increased coupling of fibroblasts to cardiomyocytes through Cx43 and/or Cx45, thereby increasing membrane capacitance (Camelliti et al., 2004, Chilton et al., 2007, Van Veen et al., 2005). More direct disruption of gap junctions has also been reported consequently increasing tissue resistance, further slowing conduction (Xie et al., 2009). Interestingly, mice lacking the mitochondrial sirtuin SIRT3 display augmented mitochondrial ROS production and enhanced cardiac fibrosis (Hafner et al., 2010). Upregulated antioxidant capacity through a mitochondrial specific overexpression of catalase protected against features of cardiac ageing including myocardial fibrosis (Dai et al., 2009). Furthermore, transforming growth factor-β1 (TGF-β1) is thought to have a significant role in age-related myocardial fibrosis (Brooks and Conrad, 2000, Rosenkranz et al., 2002). Mice over expressing TGF-β1 develop pronounced atrial fibrosis, have reduced atrial conduction velocities and greater inducibility to atrial tachyarrhythmias including AF (Verheule et al., 2004). Serum levels of TGF-β1 are increased in individuals with AF compared to control (Lin et al., 2015). TGF-β1 activity is also enhanced by oxidative stress (Barcellos-Hoff and Dix, 1996, Sullivan et al., 2008).
A pro-arrhythmic substrate can also develop through altered repolarisation properties including reductions in the APD or shortening of the atrial ERP. Reductions in APD have been documented in AF and were also seen in the present study, consistent with the previously reported effect of mitochondrial dysfunction upon AP repolarisation properties (Brown and O’Rourke, 2010). However these were witnessed in aged Pgc-1β−/− hearts and most pronounced at shorter S1–S2 coupling intervals in the PES protocol and would favour re-entry and arrhtymogenesis. These parameters pertaining to AP recovery can be combined with those of AP activation to give the AP wavelength, defined as the distance travelled by the depolarising wave over one refractory period (Allessie et al., 1977). Shortening of the AP wavelength favours re-entry whereas its lengthening is thought to be protective (Davidenko et al., 1995, Pandit and Jalife, 2013, Spector, 2013, Weiss et al., 2005, Zaitsev et al., 2000) AP wavelength was shorter in Pgc-1β−/− hearts than WT heart, both at circumstances mimicking resting heart rates and PES pacing. With reductions in the S1-S2 coupling interval, the shorter AP wavelengths in aged Pgc-1β−/−hearts persisted, whereas those for young Pgc-1β−/− hearts and WT hearts overlapped, correlating with the differing arrhythmic susceptibilities observed in the present study.
Finally, the influence of these electrophysiological parameters upon arrhythmia induction was examined in the first S2 AP that provoked an episode of AT in each heart. Interestingly (dV/dt)max values were higher for S2 beats triggering AT in Pgc-1β−/− hearts than WT hearts; however no difference was seen in AP latency times. Thus the higher (dV/dt)max values of Pgc-1β−/− hearts appear to offset the increased fibrotic changes in these hearts, which therefore achieve a similar overall conduction velocity. Similarly, no difference in APD90 or AP wavelength were seen in S2 beats triggering arrhythmias in WT and Pgc-1β−/− hearts. This suggested critical electrophysiological thresholds common to all groups, below which the susceptibility to arrhythmias is increased. The differing parameters measured in the current experiments indicate these circumstances arise earlier through ageing and mitochondrial dysfunction, thus widening the range of S1-S2 intervals at which the Pgc-1β−/− hearts were at risk of arrhythmia compared to WT hearts. Accordingly, the critical coupling intervals were longer in Pgc-1β−/− hearts compared to WT hearts, and in particular in aged Pgc-1β−/− hearts.
5. Conclusion
Together, the present experiments associate an atrial arrhythmic phenotype secondary to a chronic mitochondrial deficit, modeled by in murine Pgc-1β−/− hearts. This is attributable to maladaptive alterations in action potential conduction arising from electrophysiological changes at the cellular level and structural, fibrotic, changes at the tissue level.
Conflicts of interest
None declared.
Acknowledgements
We acknowledge financial support from the Medical Research Council (MR/M001288/1), the Wellcome Trust (105727/Z/14/Z), British Heart Foundation (PG/14/79/31102 and PG/15/12/31280), Sudden Arrhythmic Death Syndrome (SADS) UK, The McVeigh Benefaction and the Fundamental Research Grant Scheme, Ministry of Education, Malaysia (FRGS/2/2014/SKK01/PERDANA/02/1).
References
- Ad N., Schneider A., Khaliulin I., Borman J.B., Schwalb H. Impaired mitochondrial response to simulated ischemic injury as a predictor of the development of atrial fibrillation after cardiac surgery: in vitro study in human myocardium. J. Thorac. Cardiovasc. Surg. 2005;129:41–45. doi: 10.1016/j.jtcvs.2004.03.058. [DOI] [PubMed] [Google Scholar]
- Allessie M.A., Bonke F.I., Schopman F.J. Circus movement in rabbit atrial muscle as a mechanism of tachycardia: III. The leading circle concept: a new model of circus movement in cardiac tissue without the involvement of an anatomical obstacle. Circ. Res. 1977;41:9–18. doi: 10.1161/01.res.41.1.9. [DOI] [PubMed] [Google Scholar]
- Arany Z., He H., Lin J., Hoyer K., Handschin C., Toka O., Ahmad F., Matsui T., Chin S., Wu P.-H., Rybkin I.I., Shelton J.M., Manieri M., Cinti S., Schoen F.J., Bassel-Duby R., Rosenzweig A., Ingwall J.S., Spiegelman B.M. Transcriptional coactivator PGC-1 alpha controls the energy state and contractile function of cardiac muscle. Cell Metab. 2005;1:259–271. doi: 10.1016/j.cmet.2005.03.002. [DOI] [PubMed] [Google Scholar]
- Ausma J., Wijffels M., Thoné F., Wouters L., Allessie M., Borgers M. Structural changes of atrial myocardium due to sustained atrial fibrillation in the goat. Circulation. 1997;96:3157–3163. doi: 10.1161/01.cir.96.9.3157. [DOI] [PubMed] [Google Scholar]
- Barcellos-Hoff M.H., Dix T.A. Redox-mediated activation of latent transforming growth factor-beta 1. Mol. Endocrinol. 1996;10:1077–1083. doi: 10.1210/mend.10.9.8885242. [DOI] [PubMed] [Google Scholar]
- Benjamin E.J., Wolf P.A., D’Agostino R.B., Silbershatz H., Kannel W.B., Levy D. Impact of atrial fibrillation on the risk of death: the Framingham Heart Study. Circulation. 1998;98:946–952. doi: 10.1161/01.cir.98.10.946. [DOI] [PubMed] [Google Scholar]
- Biala A.K., Dhingra R., Kirshenbaum L.A. Mitochondrial dynamics: orchestrating the journey to advanced age. J. Mol. Cell. Cardiol. 2015;83:37–43. doi: 10.1016/j.yjmcc.2015.04.015. [DOI] [PubMed] [Google Scholar]
- Bosch R.F., Zeng X., Grammer J.B., Popovic K., Mewis C., Kühlkamp V. Ionic mechanisms of electrical remodeling in human atrial fibrillation. Cardiovasc. Res. 1999;44:121–131. doi: 10.1016/s0008-6363(99)00178-9. [DOI] [PubMed] [Google Scholar]
- Bournat J.C., Brown C.W. Mitochondrial dysfunction in obesity. Curr. Opin. Endocrinol. Diabetes Obes. 2010;17:446–452. doi: 10.1097/MED.0b013e32833c3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks W.W., Conrad C.H. Myocardial fibrosis in transforming growth factor β1Heterozygous mice. J. Mol. Cell. Cardiol. 2000;32:187–195. doi: 10.1006/jmcc.1999.1065. [DOI] [PubMed] [Google Scholar]
- Brown D.A., O’Rourke B. Cardiac mitochondria and arrhythmias. Cardiovasc. Res. 2010;88:241–249. doi: 10.1093/cvr/cvq231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukowska A., Schild L., Keilhoff G., Hirte D., Neumann M., Gardemann A., Neumann K.H., Röhl F.-W., Huth C., Goette A., Lendeckel U. Mitochondrial dysfunction and redox signaling in atrial tachyarrhythmia. Exp. Biol. Med. 2008;233:558–574. doi: 10.3181/0706-RM-155. [DOI] [PubMed] [Google Scholar]
- Burashnikov A., Di Diego J.M., Zygmunt A.C., Belardinelli L., Antzelevitch C. Atrium-selective sodium channel block as a strategy for suppression of atrial fibrillation: differences in sodium channel inactivation between atria and ventricles and the role of ranolazine. Circulation. 2007;116:1449–1457. doi: 10.1161/CIRCULATIONAHA.107.704890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camelliti P., Devlin G.P., Matthews K.G., Kohl P., Green C.R. Spatially and temporally distinct expression of fibroblast connexins after sheep ventricular infarction. Cardiovasc. Res. 2004;62:415–425. doi: 10.1016/j.cardiores.2004.01.027. [DOI] [PubMed] [Google Scholar]
- Cappato R., Calkins H., Chen S.-A., Davies W., Iesaka Y., Kalman J., Kim Y.-H., Klein G., Natale A., Packer D., Skanes A., Ambrogi F., Biganzoli E. Updated worldwide survey on the methods, efficacy, and safety of catheter ablation for human atrial fibrillation. Circ. Arrhythm. Electrophysiol. 2010;3:32–38. doi: 10.1161/CIRCEP.109.859116. [DOI] [PubMed] [Google Scholar]
- Carnes C.A., Janssen P.M.L., Ruehr M.L., Nakayama H., Nakayama T., Haase H., Bauer J.A., Chung M.K., Fearon I.M., Gillinov A.M., Hamlin R.L., Van Wagoner D.R. Atrial glutathione content, calcium current, and contractility. J. Biol. Chem. 2007;282:28063–28073. doi: 10.1074/jbc.M704893200. [DOI] [PubMed] [Google Scholar]
- Casini S., Verkerk A.O., van Borren M.M.G.J., van Ginneken A.C.G., Veldkamp M.W., de Bakker J.M.T., Tan H.L. Intracellular calcium modulation of voltage-gated sodium channels in ventricular myocytes. Cardiovasc. Res. 2009;81:72–81. doi: 10.1093/cvr/cvn274. [DOI] [PubMed] [Google Scholar]
- Chamberlain A.M., Gersh B.J., Alonso A., Chen L.Y., Berardi C., Manemann S.M., Killian J.M., Weston S.A., Roger V.L. Decade-long trends in atrial fibrillation incidence and survival: a community study. Am. J. Med. 2015;128:260–267. doi: 10.1016/j.amjmed.2014.10.030. (e1.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F., De Diego C., Xie L.-H., Yang J.-H., Klitzner T.S., Weiss J.N. Effects of metabolic inhibition on conduction, Ca transients, and arrhythmia vulnerability in embryonic mouse hearts. AJP Heart Circ. Physiol. 2007;293:H2472–H2478. doi: 10.1152/ajpheart.00359.2007. [DOI] [PubMed] [Google Scholar]
- Chilton L., Giles W.R., Smith G.L. Evidence of intercellular coupling between co-cultured adult rabbit ventricular myocytes and myofibroblasts. J. Physiol. 2007;583:225–236. doi: 10.1113/jphysiol.2007.135038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtemanche M., Ramirez R.J., Nattel S. Ionic mechanisms underlying human atrial action potential properties: insights from a mathematical model. Am. J. Physiol. 1998;275:H301–H321. doi: 10.1152/ajpheart.1998.275.1.H301. [DOI] [PubMed] [Google Scholar]
- Dai D.-F., Santana L.F., Vermulst M., Tomazela D.M., Emond M.J., MacCoss M.J., Gollahon K., Martin G.M., Loeb L.A., Ladiges W.C., Rabinovitch P.S. Overexpression of catalase targeted to mitochondria attenuates murine cardiac aging. Circulation. 2009;119:2789–2797. doi: 10.1161/CIRCULATIONAHA.108.822403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daoud E.G., Bogun F., Goyal R., Harvey M., Man K.C., Strickberger S.A., Morady F. Effect of atrial fibrillation on atrial refractoriness in humans. Circulation. 1996;94:1600–1606. doi: 10.1161/01.cir.94.7.1600. [DOI] [PubMed] [Google Scholar]
- Darbar D., Kannankeril P.J., Donahue B.S., Kucera G., Stubblefield T., Haines J.L., George A.L., Roden D.M. Cardiac sodium channel (SCN5A) variants associated with atrial fibrillation. Circulation. 2008;117:1927–1935. doi: 10.1161/CIRCULATIONAHA.107.757955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dautova Y., Zhang Y., Grace A.A., Huang C.L.-H. Atrial arrhythmogenic properties in wild-type and Scn5a+/− murine hearts. Exp. Physiol. 2010;95:994–1007. doi: 10.1113/expphysiol.2010.053868. [DOI] [PubMed] [Google Scholar]
- Davidenko J.M., Salomonsz R., Pertsov A.M., Baxter W.T., Jalife J. Effects of pacing on stationary reentrant activity. Theoretical and experimental study. Circ. Res. 1995;77:1166–1179. doi: 10.1161/01.res.77.6.1166. [DOI] [PubMed] [Google Scholar]
- DeWilde S., Carey I., Emmas C., Richards N., Cook D. Trends in the prevalence of diagnosed atrial fibrillation, its treatment with anticoagulation and predictors of such treatment in UK primary care. Heart. 2006;92:1064–1070. doi: 10.1136/hrt.2005.069492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deo R., Albert C.M. Epidemiology and genetics of sudden cardiac death. Circulation. 2012;125:620–637. doi: 10.1161/CIRCULATIONAHA.111.023838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikalov S.I., Ungvari Z. Role of mitochondrial oxidative stress in hypertension. Am. J. Physiol. Heart Circ. Physiol. 2013;305:H1417–H14127. doi: 10.1152/ajpheart.00089.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon L.M., Rebelo A.P., Moraes C.T. The role of PGC-1 coactivators in aging skeletal muscle and heart. IUBMB Life. 2012;64:231–241. doi: 10.1002/iub.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eghbali M., Eghbali M., Robinson T.F., Seifter S., Blumenfeld O.O. Collagen accumulation in heart ventricles as a function of growth and aging. Cardiovasc. Res. 1989;23:723–729. doi: 10.1093/cvr/23.8.723. [DOI] [PubMed] [Google Scholar]
- Emelyanova L., Ashary Z., Cosic M., Negmadjanov U., Ross G., Rizvi F., Olet S., Kress D., Sra J., Tajik A.J., Holmuhamedov E.L., Shi Y., Jahangir A. Selective downregulation of mitochondrial electron transport chain activity and increased oxidative stress in human atrial fibrillation. Am. J. Physiol. – Hear. Circ. Physiol. 2016;311:H54–H63. doi: 10.1152/ajpheart.00699.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faivre J.F., Findlay I. Action potential duration and activation of ATP-sensitive potassium current in isolated guinea-pig ventricular myocytes. Biochim. Biophys. Acta. 1990;1029:167–172. doi: 10.1016/0005-2736(90)90450-3. [DOI] [PubMed] [Google Scholar]
- Finck B.N., Kelly D.P. PGC-1 coactivators: inducible regulators of energy metabolism in health and disease. J. Clin. Invest. 2006;116:615–622. doi: 10.1172/JCI27794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fosset M., De Weille J.R., Green R.D., Schmid-Antomarchi H., Lazdunski M. Antidiabetic sulfonylureas control action potential properties in heart cells via high affinity receptors that are linked to ATP-dependent K+ channels. J. Biol. Chem. 1988;263:7933–7936. [PubMed] [Google Scholar]
- Fraser J.A., Huang C.L.-H., Pedersen T.H. Relationships between resting conductances, excitability, and t-system ionic homeostasis in skeletal muscle. J. Gen. Physiol. 2011;138:95–116. doi: 10.1085/jgp.201110617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friberg L., Bergfeldt L. Atrial fibrillation prevalence revisited. J. Intern. Med. 2013;274:461–468. doi: 10.1111/joim.12114. [DOI] [PubMed] [Google Scholar]
- Friberg L., Hammar N., Pettersson H., Rosenqvist M. Increased mortality in paroxysmal atrial fibrillation: report from the Stockholm Cohort-Study of Atrial Fibrillation (SCAF) Eur. Heart J. 2007;28:2346–2353. doi: 10.1093/eurheartj/ehm308. [DOI] [PubMed] [Google Scholar]
- Frustaci A., Chimenti C., Bellocci F., Morgante E., Russo M.A., Maseri A. Histological substrate of atrial biopsies in patients with lone atrial fibrillation. Circulation. 1997;96:1180–1184. doi: 10.1161/01.cir.96.4.1180. [DOI] [PubMed] [Google Scholar]
- Gaspo R., Bosch R.F., Bou-Abboud E., Nattel S. Tachycardia-induced changes in Na+ current in a chronic dog model of atrial fibrillation. Circ. Res. 1997;81:1045–1052. doi: 10.1161/01.res.81.6.1045. [DOI] [PubMed] [Google Scholar]
- Gaspo R., Bosch R.F., Talajic M., Nattel S. Functional mechanisms underlying tachycardia-induced sustained atrial fibrillation in a chronic dog model. Circulation. 1997;96:4027–4035. doi: 10.1161/01.cir.96.11.4027. [DOI] [PubMed] [Google Scholar]
- Gazoti Debessa C.R., Mesiano Maifrino L.B., Rodrigues de Souza R. Age related changes of the collagen network of the human heart. Mech. Ageing Dev. 2001;122:1049–1058. doi: 10.1016/s0047-6374(01)00238-x. [DOI] [PubMed] [Google Scholar]
- Glukhov A.V., Kalyanasundaram A., Lou Q., Hage L.T., Hansen B.J., Belevych A.E., Mohler P.J., Knollmann B.C., Periasamy M., Györke S., Fedorov V.V. Calsequestrin 2 deletion causes sinoatrial node dysfunction and atrial arrhythmias associated with altered sarcoplasmic reticulum calcium cycling and degenerative fibrosis within the mouse atrial pacemaker complex. Eur. Heart J. 2013:1–13. doi: 10.1093/eurheartj/eht452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go A.S., Hylek E.M., Phillips K.A., Chang Y., Henault L.E., Selby J.V., Singer D.E. Prevalence of diagnosed atrial fibrillation in adults: national implications for rhythm management and stroke prevention: the AnTicoagulation and Risk Factors in Atrial Fibrillation (ATRIA) Study. JAMA. 2001;285:2370–2375. doi: 10.1001/jama.285.18.2370. [DOI] [PubMed] [Google Scholar]
- Gray R.A., Jalife J., Panfilov A., Baxter W.T., Cabo C., Davidenko J.M., Pertsov A.M. Nonstationary vortexlike reentrant activity as a mechanism of polymorphic ventricular tachycardia in the isolated rabbit heart. Circulation. 1995;91:2454–2469. doi: 10.1161/01.cir.91.9.2454. [DOI] [PubMed] [Google Scholar]
- Guo W., Xu H., London B., Nerbonne J.M. Molecular basis of transient outward K+ current diversity in mouse ventricular myocytes. J. Physiol. 1999;521(Pt. 3):587–599. doi: 10.1111/j.1469-7793.1999.00587.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurung I.S., Medina-Gomez G., Kis A., Baker M., Velagapudi V., Neogi S.G., Campbell M., Rodriguez-Cuenca S., Lelliott C., McFarlane I., Oresic M., Grace A.A., Vidal-Puig A., Huang C.L.-H. Deletion of the metabolic transcriptional coactivator PGC1β induces cardiac arrhythmia. Cardiovasc. Res. 2011;92:29–38. doi: 10.1093/cvr/cvr155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzadhur L., Jeevaratnam K., Matthews G., Grace A., Huang C.L.-H. Electrophysiological mechanisms underlying the initiation of atrial arrhythmia in genetically modified murine hearts. Trends Comp. Biochem. Physiol. 2013;17:59–80. [Google Scholar]
- Haïssaguerre M., Jaïs P., Shah D.C., Takahashi A., Hocini M., Quiniou G., Garrigue S., Le Mouroux A., Le Métayer P., Clémenty J. Spontaneous initiation of atrial fibrillation by ectopic beats originating in the pulmonary veins. N. Engl. J. Med. 1998;339:659–666. doi: 10.1056/NEJM199809033391003. [DOI] [PubMed] [Google Scholar]
- Hafner A.V., Dai J., Gomes A.P., Xiao C.-Y., Palmeira C.M., Rosenzweig A., Sinclair D.A. Regulation of the mPTP by SIRT3-mediated deacetylation of CypD at lysine 166 suppresses age-related cardiac hypertrophy. Aging (Albany. NY) 2010;2:914–923. doi: 10.18632/aging.100252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi T., Nakaya Y. T wave polarity related to the repolarization process of epicardial and endocardial ventricular surfaces. Am. Heart J. 1984;108:290–295. doi: 10.1016/0002-8703(84)90614-8. [DOI] [PubMed] [Google Scholar]
- Hondeghem L.M., Katzung B.G. Time- and voltage-dependent interactions of antiarrhythmic drugs with cardiac sodium channels. Biochim. Biophys. Acta. 1977;472:373–398. doi: 10.1016/0304-4157(77)90003-x. [DOI] [PubMed] [Google Scholar]
- Hunter P.J., McNaughton P.A., Noble D. Analytical models of propagation in excitable cells. Prog. Biophys. Mol. Biol. 1975;30:99–144. doi: 10.1016/0079-6107(76)90007-9. [DOI] [PubMed] [Google Scholar]
- Huss J.M., Torra I.P., Staels B., Giguère V., Kelly D.P. Estrogen-related receptor alpha directs peroxisome proliferator-activated receptor alpha signaling in the transcriptional control of energy metabolism in cardiac and skeletal muscle. Mol. Cell. Biol. 2004;24:9079–9091. doi: 10.1128/MCB.24.20.9079-9091.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeevaratnam K., Tee Poh, Zhang S., Rewbury Y., Guzadhur R., Duehmke L., Grace R., Lei A.A., Huang M., Huang C.L.-H. Delayed conduction and its implications in murine Scn5a+/− hearts: independent and interacting effects of genotype, age, and sex. Pflugers Arch. Eur. J. Physiol. 2011;461:29–44. doi: 10.1007/s00424-010-0906-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeevaratnam K., Rewbury R., Zhang Y., Guzadhur L., Grace A.A., Lei M., Huang C.L.-H. Frequency distribution analysis of activation times and regional fibrosis in murine Scn5a+/− hearts: the effects of ageing and sex. Mech. Ageing Dev. 2012;133:591–599. doi: 10.1016/j.mad.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Killeen M.J., Gurung I.S., Thomas G., Stokoe K.S., Grace A.A., Huang C.L.-H. Separation of early afterdepolarizations from arrhythmogenic substrate in the isolated perfused hypokalaemic murine heart through modifiers of calcium homeostasis. Acta Physiol. (Oxf) 2007;191:43–58. doi: 10.1111/j.1748-1716.2007.01715.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King J., Huang C.L.-H., Fraser J.A. Determinants of myocardial conduction velocity: implications for arrhythmogenesis. Front. Physiol. 2013;4:154. doi: 10.3389/fphys.2013.00154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King J., Zhang Y., Lei M., Grace A.A., Huang C.L.-H., Fraser J.A. Atrial arrhythmia, triggering events and conduction abnormalities in isolated murine RyR2-P2328S hearts. Acta Physiol. 2013;207:308–323. doi: 10.1111/apha.12006. [DOI] [PubMed] [Google Scholar]
- Kostin S., Klein G., Szalay Z., Hein S., Bauer E.P., Schaper J. Structural correlate of atrial fibrillation in human patients. Cardiovasc. Res. 2002;54:361–379. doi: 10.1016/s0008-6363(02)00273-0. [DOI] [PubMed] [Google Scholar]
- Krijthe B.P., Kunst A., Benjamin E.J., Lip G.Y.H., Franco O.H., Hofman A., Witteman J.C.M., Stricker B.H., Heeringa J. Projections on the number of individuals with atrial fibrillation in the European Union, from 2000 to 2060. Eur. Heart J. 2013;34:2746–2751. doi: 10.1093/eurheartj/eht280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh-Madsen T., Abbott G.W., Christini D.J. Effects of electrical and structural remodeling on atrial fibrillation maintenance: a simulation study. PLoS Comput. Biol. 2012;8:e1002390. doi: 10.1371/journal.pcbi.1002390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurokawa S., Niwano S., Niwano H., Ishikawa S., Kishihara J., Aoyama Y., Kosukegawa T., Masaki Y., Izumi T. Progression of ventricular remodeling and arrhythmia in the primary hyperoxidative state of glutathione-depleted rats. Circ. J. 2011;75:1386–1393. doi: 10.1253/circj.cj-10-1089. [DOI] [PubMed] [Google Scholar]
- Lai L., Leone T.C., Zechner C., Schaeffer P.J., Kelly S.M., Flanagan D.P., Medeiros D.M., Kovacs A., Kelly D.P. Transcriptional coactivators PGC-1alpha and PGC-lbeta control overlapping programs required for perinatal maturation of the heart. Genes Dev. 2008;22:1948–1961. doi: 10.1101/gad.1661708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane R.K., Hilsabeck T., Rea S.L. The role of mitochondrial dysfunction in age-related diseases. Biochim. Biophys. Acta – Bioenergy. 2015;1847:1387–1400. doi: 10.1016/j.bbabio.2015.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehman J.J., Barger P.M., Kovacs A., Saffitz J.E., Medeiros D.M., Kelly D.P. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J. Clin. Invest. 2000;106:847–856. doi: 10.1172/JCI10268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lelliott C.J., Medina-Gomez G., Petrovic N., Kis A., Feldmann H.M., Bjursell M., Parker N., Curtis K., Campbell M., Hu P., Zhang D., Litwin S.E., Zaha V.G., Fountain K.T., Boudina S., Jimenez-Linan M., Blount M., Lopez M., Meirhaeghe A., Bohlooly-Y M., Storlien L., Strömstedt M., Snaith M., Oresic M., Abel E.D., Cannon B., Vidal-Puig A. Ablation of PGC-1beta results in defective mitochondrial activity, thermogenesis, hepatic function, and cardiac performance. PLoS Biol. 2006:4. doi: 10.1371/journal.pbio.0040369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leone T.C., Kelly D.P. Transcriptional control of cardiac fuel metabolism and mitochondrial function. Cold Spring Harb. Symp. Quant. Biol. 2011;76:175–182. doi: 10.1101/sqb.2011.76.011965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesnefsky E.J., Williams G.R., Rubinstein J.D., Hogue T.S., Horwitz L.D., Reiter M.J. Hydrogen peroxide decreases effective refractory period in the isolated heart. Free Radic. Biol. Med. 1991;11:529–535. doi: 10.1016/0891-5849(91)90133-n. [DOI] [PubMed] [Google Scholar]
- Li N., Chiang D.Y., Wang S., Wang Q., Sun L., Voigt N., Respress J.L., Ather S., Skapura D.G., Jordan V.K., Horrigan F.T., Schmitz W., Müller F.U., Valderrabano M., Nattel S., Dobrev D., Wehrens X.H.T. Ryanodine receptor-mediated calcium leak drives progressive development of an atrial fibrillation substrate in a transgenic mouse model. Circulation. 2014;129:1276–1285. doi: 10.1161/CIRCULATIONAHA.113.006611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Jin D., Hata M., Takai S., Yamanishi K., Shen W., El-Darawish Y., Yamanishi H., Okamura H. Dysfunction of mitochondria and deformed gap junctions in the heart of IL-18-deficient mice. Am. J. Physiol. Heart Circ. Physiol. 2016;311:H313–H325. doi: 10.1152/ajpheart.00927.2015. [DOI] [PubMed] [Google Scholar]
- Lin P.H., Lee S.H., Su C.P., Wei Y.H. Oxidative damage to mitochondrial DNA in atrial muscle of patients with atrial fibrillation. Free Radic. Biol. Med. 2003;35:1310–1318. doi: 10.1016/j.freeradbiomed.2003.07.002. [DOI] [PubMed] [Google Scholar]
- Lin J., Handschin C., Spiegelman B.M. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005;1:361–370. doi: 10.1016/j.cmet.2005.05.004. [DOI] [PubMed] [Google Scholar]
- Lin J., Lopez E.F., Jin Y., Van Remmen H., Bauch T., Han H.-C., Lindsey M.L. Age-related cardiac muscle sarcopenia: combining experimental and mathematical modeling to identify mechanisms. Exp. Gerontol. 2008;43:296–306. doi: 10.1016/j.exger.2007.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y.-K., Lai M.-S., Chen Y.-C., Cheng C.-C., Huang J.-H., Chen S.-A., Chen Y.-J., Lin C.-I. Hypoxia and reoxygenation modulate the arrhythmogenic activity of the pulmonary vein and atrium. Clin. Sci. (Lond). 2012;122:121–132. doi: 10.1042/CS20110178. [DOI] [PubMed] [Google Scholar]
- Lin X., Wu N., Shi Y., Wang S., Tan K., Shen Y., Dai H., Zhong J. Association between transforming growth factor β1 and atrial fibrillation in essential hypertensive patients. Clin. Exp. Hypertens. 2015;37:82–87. doi: 10.3109/10641963.2014.913600. [DOI] [PubMed] [Google Scholar]
- Liu G., Iden J.B., Kovithavongs K., Gulamhusein R., Duff H.J., Kavanagh K.M. In vivo temporal and spatial distribution of depolarization and repolarization and the illusive murine T wave. J. Physiol. 2004;555:267–279. doi: 10.1113/jphysiol.2003.054064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M., Sanyal S., Gao G., Gurung I.S., Zhu X., Gaconnet G., Kerchner L.J., Shang L.L., Huang C.L.-H., Grace A., London B., Dudley S.C. Cardiac Na+ current regulation by pyridine nucleotides. Circ. Res. 2009;105:737–745. doi: 10.1161/CIRCRESAHA.109.197277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M., Liu H., Dudley S.C. Reactive oxygen species originating from mitochondria regulate the cardiac sodium channel. Circ. Res. 2010;107:967–974. doi: 10.1161/CIRCRESAHA.110.220673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majeed A., Moser K., Carroll K. Trends in the prevalence and management of atrial fibrillation in general practice in England and Wales, 1994–1998: analysis of data from the general practice research database. Heart. 2001;86:284–288. doi: 10.1136/heart.86.3.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning A.S., Coltart D.J., Hearse D.J. Ischemia and reperfusion-induced arrhythmias in the rat: effects of xanthine oxidase inhibition with allopurinol. Circ. Res. 1984;55:545–548. doi: 10.1161/01.res.55.4.545. [DOI] [PubMed] [Google Scholar]
- Martin C.A., Guzadhur L., Grace A.A., Lei M., Huang C.L.-H. Mapping of reentrant spontaneous polymorphic ventricular tachycardia in a Scn5a+/− mouse model. Am. J. Physiol. Heart Circ. Physiol. 2011;300:H1853–H1862. doi: 10.1152/ajpheart.00034.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin C.A., Matthews G.D.K., Huang C.L.-H. Sudden cardiac death and inherited channelopathy: the basic electrophysiology of the myocyte and myocardium in ion channel disease. Heart. 2012;98:536–543. doi: 10.1136/heartjnl-2011-300953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews G.D.K., Martin C.A., Grace A.A., Zhang Y., Huang C.L.-H. Regional variations in action potential alternans in isolated murine Scn5a (+/−) hearts during dynamic pacing. Acta Physiol. (Oxf) 2010;200:129–146. doi: 10.1111/j.1748-1716.2010.02138.x. [DOI] [PubMed] [Google Scholar]
- Matthews G., Guzadhur L., Grace A., Huang C.-H. Nonlinearity between action potential alternans and restitution, which both predict ventricular arrhythmic properties in Scn5a+/− and wild-type murine hearts. J. Appl. Physiol. 2012;112:1847–1863. doi: 10.1152/japplphysiol.00039.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews G., Guzadhur L., Sabir I., Grace A., Huang C.-H. Action potential wavelength restitution predicts alternans and arrhythmia in murine Scn5a+/− hearts. J. Physiol. 2013 doi: 10.1113/jphysiol.2013.254938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menezes A.R., Lavie C.J., DiNicolantonio J.J., O’Keefe J., Morin D.P., Khatib S., Milani R.V. Atrial fibrillation in the 21 st century: a current understanding of risk factors and primary prevention strategies. Mayo Clin. Proc. 2013;88:394–409. doi: 10.1016/j.mayocp.2013.01.022. [DOI] [PubMed] [Google Scholar]
- Miyamoto K., Tsuchiya T., Narita S., Yamaguchi T., Nagamoto Y., Ando -i S., Hayashida K., Tanioka Y., Takahashi N. Bipolar electrogram amplitudes in the left atrium are related to local conduction velocity in patients with atrial fibrillation. Europace. 2009;11:1597–1605. doi: 10.1093/europace/eup352. [DOI] [PubMed] [Google Scholar]
- Miyasaka Y., Barnes M.E., Gersh B.J., Cha S.S., Bailey K.R., Abhayaratna W.P., Seward J.B., Tsang T.S.M. Secular trends in incidence of atrial fibrillation in Olmsted County, Minnesota, 1980–2000, and implications on the projections for future prevalence. Circulation. 2006;114:119–125. doi: 10.1161/CIRCULATIONAHA.105.595140. [DOI] [PubMed] [Google Scholar]
- Montaigne D., Marechal X., Lefebvre P., Modine T., Fayad G., Dehondt H., Hurt C., Coisne A., Koussa M., Remy-Jouet I., Zerimech F., Boulanger E., Lacroix D., Staels B., Neviere R. Mitochondrial dysfunction as an arrhythmogenic substrate. J. Am. Coll. Cardiol. 2013;62:1466–1473. doi: 10.1016/j.jacc.2013.03.061. [DOI] [PubMed] [Google Scholar]
- Mootha V.K., Lindgren C.M., Eriksson K.-F., Subramanian A., Sihag S., Lehar J., Puigserver P., Carlsson E., Ridderstråle M., Laurila E., Houstis N., Daly M.J., Patterson N., Mesirov J.P., Golub T.R., Tamayo P., Spiegelman B., Lander E.S., Hirschhorn J.N., Altshuler D., Groop L.C. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- Mori M., Konno T., Ozawa T., Murata M., Imoto K., Nagayama K. Novel interaction of the voltage-dependent sodium channel (VDSC) with calmodulin: does VDSC acquire calmodulin-mediated Ca2+-sensitivity? Biochemistry. 2000;39:1316–1323. doi: 10.1021/bi9912600. [DOI] [PubMed] [Google Scholar]
- Morillo C.A., Klein G.J., Jones D.L., Guiraudon C.M. Chronic rapid atrial pacing. Structural functional, and electrophysiological characteristics of a new model of sustained atrial fibrillation. Circulation. 1995;91:1588–1595. doi: 10.1161/01.cir.91.5.1588. [DOI] [PubMed] [Google Scholar]
- Olson T.M., Michels V.V., Ballew J.D., Reyna S.P., Karst M.L., Herron K.J., Horton S.C., Rodeheffer R.J., Anderson J.L. Sodium channel mutations and susceptibility to heart failure and atrial fibrillation. JAMA. 2005;293:447–454. doi: 10.1001/jama.293.4.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlandi A., Francesconi A., Marcellini M., Ferlosio A., Spagnoli L. Role of ageing and coronary atherosclerosis in the development of cardiac fibrosis in the rabbit. Cardiovasc. Res. 2004;64:544–552. doi: 10.1016/j.cardiores.2004.07.024. [DOI] [PubMed] [Google Scholar]
- Pandit S.V., Jalife J. Rotors and the dynamics of cardiac fibrillation. Circ. Res. 2013;112:849–862. doi: 10.1161/CIRCRESAHA.111.300158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J., Pak H., Kim S., Jang J., Choi J., Lim H., Hwang C., Kim Y. Electrophysiologic characteristics of complex fractionated atrial electrograms in patients with atrial fibrillation. J. Cardiovasc. Electrophysiol. 2009;20:266–272. doi: 10.1111/j.1540-8167.2008.01321.x. [DOI] [PubMed] [Google Scholar]
- Patti M.-E., Corvera S. The role of mitochondria in the pathogenesis of type 2 diabetes. Endocr. Rev. 2010;31:364–395. doi: 10.1210/er.2009-0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. URL http://www.R-project.org/.
- Riehle C., Abel E.D. PGC-1 proteins and heart failure. Trends Cardiovasc. Med. 2012;22:98–105. doi: 10.1016/j.tcm.2012.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenkranz S., Flesch M., Amann K., Haeuseler C., Kilter H., Seeland U., Schlüter K.-D., Böhm M. Alterations of β-adrenergic signaling and cardiac hypertrophy in transgenic mice overexpressing TGF-β 1. Am. J. Physiol. – Heart Circ. Physiol. 2002;283:H1253–H1262. doi: 10.1152/ajpheart.00578.2001. [DOI] [PubMed] [Google Scholar]
- Russell L.K., Mansfield C.M., Lehman J.J., Kovacs A., Courtois M., Saffitz J.E., Medeiros D.M., Valencik M.L., McDonald J.A., Kelly D.P. Cardiac-specific induction of the transcriptional coactivator peroxisome proliferator-activated receptor gamma coactivator-1alpha promotes mitochondrial biogenesis and reversible cardiomyopathy in a developmental stage-dependent manner. Circ. Res. 2004;94:525–533. doi: 10.1161/01.RES.0000117088.36577.EB. [DOI] [PubMed] [Google Scholar]
- Sabir I.N., Fraser J.A., Killeen M.J., Grace A.A., Huang C.L.-H. The contribution of refractoriness to arrhythmic substrate in hypokalemic Langendorff-perfused murine hearts. Pflugers Arch. 2007;454:209–222. doi: 10.1007/s00424-007-0217-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabir I.N., Killeen M.J., Goddard C.A., Thomas G., Gray S., Grace A.A., Huang C.L.-H. Transient alterations in transmural repolarization gradients and arrhythmogenicity in hypokalaemic Langendorff-perfused murine hearts. J. Physiol. 2007;581:277–289. doi: 10.1113/jphysiol.2007.128637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaper J., Meiser E., Stämmler G. Ultrastructural morphometric analysis of myocardium from dogs, rats, hamsters, mice, and from human hearts. Circ. Res. 1985;56:377–391. doi: 10.1161/01.res.56.3.377. [DOI] [PubMed] [Google Scholar]
- Slagsvold K.H., Johnsen A.B., Rognmo O., Høydal M., Wisløff U., Wahba A. Comparison of left versus right atrial myocardium in patients with sinus rhythm or atrial fibrillation – an assessment of mitochondrial function and microRNA expression. Physiol. Rep. 2014;2 doi: 10.14814/phy2.12124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonoda J., Mehl I.R., Chong L.-W., Nofsinger R.R., Evans R.M. PGC-1beta controls mitochondrial metabolism to modulate circadian activity, adaptive thermogenesis, and hepatic steatosis. Proc. Natl. Acad. Sci. U. S. A. 2007;104:5223–5228. doi: 10.1073/pnas.0611623104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sossalla S., Kallmeyer B., Wagner S., Mazur M., Maurer U., Toischer K., Schmitto J.D., Seipelt R., Schöndube F.A., Hasenfuss G., Belardinelli L., Maier L.S. Altered Na+ currents in atrial fibrillation. effects of ranolazine on arrhythmias and contractility in human atrial myocardium. J. Am. Coll. Cardiol. 2010;55:2330–2342. doi: 10.1016/j.jacc.2009.12.055. [DOI] [PubMed] [Google Scholar]
- Sovari A.A., Rutledge C.A., Jeong E.-M., Dolmatova E., Arasu D., Liu H., Vahdani N., Gu L., Zandieh S., Xiao L., Bonini M.G., Duffy H.S., Dudley S.C. Mitochondria oxidative stress, connexin-43 remodeling, and sudden arrhythmic death. Circ. Arrhythm. Electrophysiol. 2013;6:623–631. doi: 10.1161/CIRCEP.112.976787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spector P. Principles of cardiac electric propagation and their implications for re-entrant arrhythmias. Circ. Arrhythm. Electrophysiol. 2013;6:655–661. doi: 10.1161/CIRCEP.113.000311. [DOI] [PubMed] [Google Scholar]
- Sullivan D.E., Ferris M., Pociask D., Brody A.R. The latent form of TGFβ 1 is induced by TNFα through an ERK specific pathway and is activated by asbestos-derived reactive oxygen species in vitro and in vivo. J. Immunotoxicol. 2008;5:145–149. doi: 10.1080/15476910802085822. [DOI] [PubMed] [Google Scholar]
- Sun N., Youle R.J., Finkel T. The mitochondrial basis of aging. Mol. Cell. 2016;61:654–666. doi: 10.1016/j.molcel.2016.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuboi M., Hisatome I., Morisaki T., Tanaka M., Tomikura Y., Takeda S., Shimoyama M., Ohtahara A., Ogino K., Igawa O., Shigemasa C., Ohgi S., Nanba E. Mitochondrial DNA deletion associated with the reduction of adenine nucleotides in human atrium and atrial fibrillation. Eur. J. Clin. Invest. 2001;31:489–496. doi: 10.1046/j.1365-2362.2001.00844.x. [DOI] [PubMed] [Google Scholar]
- Usher-Smith J.A., Xu W., Fraser J.A., Huang C.L.-H. Alterations in calcium homeostasis reduce membrane excitability in amphibian skeletal muscle. Pflugers Arch. 2006;453:211–221. doi: 10.1007/s00424-006-0132-z. [DOI] [PubMed] [Google Scholar]
- Vaidya D., Morley G.E., Samie F.H., Jalife J. Reentry and fibrillation in the mouse heart. A challenge to the critical mass hypothesis. Circ. Res. 1999;85:174–181. doi: 10.1161/01.res.85.2.174. [DOI] [PubMed] [Google Scholar]
- Van Veen T.A.B., Stein M., Royer A., Le Quang K., Charpentier F., Colledge W.H., Huang C.L.-H., Wilders R., Grace A.A., Escande D., de Bakker J.M.T., van Rijen H.V.M. Impaired impulse propagation in Scn5a-knockout mice: combined contribution of excitability, connexin expression, and tissue architecture in relation to aging. Circulation. 2005;112:1927–1935. doi: 10.1161/CIRCULATIONAHA.105.539072. [DOI] [PubMed] [Google Scholar]
- Van Wagoner D.R., Pond A.L., Lamorgese M., Rossie S.S., McCarthy P.M., Nerbonne J.M. Atrial L-type Ca2+ currents and human atrial fibrillation. Circ. Res. 1999;85:428–436. doi: 10.1161/01.res.85.5.428. [DOI] [PubMed] [Google Scholar]
- Vega R.B., Huss J.M., Kelly D.P. The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor alpha in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol. Cell. Biol. 2000;20:1868–1876. doi: 10.1128/mcb.20.5.1868-1876.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verheule S., Sato T., Iv T.E., Engle S.K., Otten D., Rubart-Von Der Lohe M., Nakajima H.O., Nakajima H., Field L.J., Olgin J.E. Increased vulnerability to atrial fibrillation in transgenic mice with selective atrial fibrosis caused by overexpression of TGF-beta1. Circ. Res. 2004;94:1458–1465. doi: 10.1161/01.RES.0000129579.59664.9d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss J.N., Qu Z., Chen P.S., Lin S.F., Karagueuzian H.S., Hayashi H., Garfinkel A., Karma A. The dynamics of cardiac fibrillation. Circulation. 2005;112:1232–1240. doi: 10.1161/CIRCULATIONAHA.104.529545. [DOI] [PubMed] [Google Scholar]
- Wingo T.L., Shah V.N., Anderson M.E., Lybrand T.P., Chazin W.J., Balser J.R. An EF-hand in the sodium channel couples intracellular calcium to cardiac excitability. Nat. Struct. Mol. Biol. 2004;11:219–225. doi: 10.1038/nsmb737. [DOI] [PubMed] [Google Scholar]
- Wolf P.A., Abbott R.D., Kannel W.B. Atrial fibrillation as an independent risk factor for stroke: the Framingham Study. Stroke. 1991;22:983–988. doi: 10.1161/01.str.22.8.983. [DOI] [PubMed] [Google Scholar]
- Xie Y., Garfinkel A., Camelliti P., Kohl P., Weiss J.N., Qu Z. Effects of fibroblast-myocyte coupling on cardiac conduction and vulnerability to reentry: a computational study. Heart Rhythm. 2009;6:1641–1649. doi: 10.1016/j.hrthm.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie W., Santulli G., Reiken S.R., Yuan Q., Osborne B.W., Chen B.-X., Marks A.R. Mitochondrial oxidative stress promotes atrial fibrillation. Sci. Rep. 2015;5:11427. doi: 10.1038/srep11427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K.-C., Bonini M.G., Dudley S.C. Mitochondria and arrhythmias. Free Radic. Biol. Med. 2014;71:351–361. doi: 10.1016/j.freeradbiomed.2014.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaitsev A.V., Berenfeld O., Mironov S.F., Jalife J., Pertsov A.M. Distribution of excitation frequencies on the epicardial and endocardial surfaces of fibrillating ventricular wall of the sheep heart. Circ. Res. 2000;86:408–417. doi: 10.1161/01.res.86.4.408. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Schwiening C., Killeen M.J., Zhang Y., Ma A., Lei M., Grace A.A., Huang C.L.-H. Pharmacological changes in cellular Ca2+ homeostasis parallel initiation of atrial arrhythmogenesis in murine Langendorff-perfused hearts. Clin. Exp. Pharmacol. Physiol. 2009;36:969–980. doi: 10.1111/j.1440-1681.2009.05170.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y., Xia Y., Carlson J., Kongstad O., Yuan S. Atrial average conduction velocity in patients with and without paroxysmal atrial fibrillation. Clin. Physiol. Funct. Imaging. 2016;12342 doi: 10.1111/cpf.12342. [DOI] [PubMed] [Google Scholar]
- Zoni-Berisso M., Lercari F., Carazza T., Domenicucci S. Epidemiology of atrial fibrillation: European perspective. Clin. Epidemiol. 2014;6:213–220. doi: 10.2147/CLEP.S47385. [DOI] [PMC free article] [PubMed] [Google Scholar]