

Abstract
The genomic CDKN2A/B locus, encoding p16INK4a among others, is linked to an increased risk for cardiovascular disease and type 2 diabetes. Obesity is a risk factor for both cardiovascular disease and type 2 diabetes. p16INK4a is a cell cycle regulator and tumour suppressor. Whether it plays a role in adipose tissue formation is unknown. p16INK4a knock-down in 3T3/L1 preadipocytes or p16INK4a deficiency in mouse embryonic fibroblasts enhanced adipogenesis, suggesting a role for p16INK4a in adipose tissue formation. p16INK4a-deficient mice developed more epicardial adipose tissue in response to the adipogenic peroxisome proliferator activated receptor gamma agonist rosiglitazone. Additionally, adipose tissue around the aorta from p16INK4a-deficient mice displayed enhanced rosiglitazone-induced gene expression of adipogenic markers and stem cell antigen, a marker of bone marrow-derived precursor cells. Mice transplanted with p16INK4a-deficient bone marrow had more epicardial adipose tissue compared to controls when fed a high-fat diet. In humans, p16INK4a gene expression was enriched in epicardial adipose tissue compared to other adipose tissue depots. Moreover, epicardial adipose tissue from obese humans displayed increased expression of stem cell antigen compared to lean controls, supporting a bone marrow origin of epicardial adipose tissue. These results show that p16INK4a modulates epicardial adipose tissue development, providing a potential mechanistic link between the genetic association of the CDKN2A/B locus and cardiovascular disease risk.
Keywords: p16INK4a, CDKN2A, adipogenesis, perivascular adipose tissue, bone marrow
Introduction
Genome-wide association studies show a robust association of a region near the tumour suppressor locus CDKN2A/B with cardiovascular disease (CVD) and type 2 diabetes (T2D).1,2 CDKN2A codes for p16INK4a and its alternative reading frame transcript variant p14ARF (p19ARF in mice), whereas CDKN2B codes for p15INK4b. p16INK4a and p15INK4b are cyclin-dependent kinase (CDK) inhibitors, controlling cell cycle progression, whereas p14ARF modulates the cell cycle by initiating p53-dependent cell cycle arrest.2 We previously showed that p16INK4a affects the inflammatory phenotype of murine3 and human4 macrophages.
Obesity, hallmarked by excessive adipose tissue (AT) expansion, is a risk factor for CVD and T2D. The human body contains different AT depots, located beneath the skin (subcutaneous AT (scAT)) and in the abdominal cavity surrounding the internal organs (visceral AT (vAT)). In addition, AT is found in different locations surrounding blood vessels, named perivascular AT (pvAT), thought to play a role in CVD.5 A specific type of pvAT called epicardial AT (ecAT) surrounds the coronary arteries of the heart. ecAT is fat located between the myocardium and visceral pericardium, that is, onto the myocardium. More distally, pericardial adipose tissue (pcAT) is the fat depot outside the visceral pericardium, that is, within the pericardial sac. Although pvAT is thought to develop from its own precursors, it shares many similarities between white and brown AT (bAT).6 Besides structural support, pvAT is being increasingly appreciated as an active modulator of vascular tone and function. Given the close role of pvAT with obesity, it may be an important link between the metabolic syndrome and atherosclerosis.6
During adipogenesis, cell cycle regulation contributes to adipocyte formation.7 During adipogenesis, precursor cells go through 1 or 2 rounds of cells division, a process called mitotic clonal expansion. However, the potential role of cell cycle mediators does not entirely depend on this clonal mechanism. It has been shown, for example, that CDK4, whose function is controlled by p16INK4a, directly interacts and activates the master adipogenic regulator peroxisome proliferator activated receptor gamma (PPARγ) besides its role in the cell cycle.8 Currently, it is unknown whether CDKN2A/B gene products play a role in adipogenesis. We therefore investigated whether p16INK4a is involved in adipogenesis and AT formation.
Experimental procedures
Cell culture
3T3-L1 cells were cultured in growth medium containing Dulbecco’s modified Eagle’s medium (Invitrogen, Paris, France) and 10% foetal calf serum (GE healthcare, Lille, France). For differentiation, fully confluent cell monolayers were incubated for 48 h in medium containing 10% HyClone Cosmic Calf Serum (GE Healthcare, Lille, France), methylisobutylxanthine (0.5 mM), insulin (5 μM) and dexamethasone (1 μM). Hereafter, adipocytes were cultured in post-differentiation medium (Dulbecco’s modified Eagle’s medium, 10% HyClone Cosmic Calf Serum, 5 μM insulin). Small interfering RNA (siRNA) experiments were performed overnight in pre-confluent 3T3/L1 cells using siRNA for CDKN2A [043107-00-005; Thermo Scientific (ON-TARGET plus SMART pool siRNA)], control [D-001810-10-20; Thermo Scientific (ON-TARGET plus non-targeting pool siRNA), Villebon-sur-Yvette, France] or PPARγ [L-040712-00-0005, Thermo Scientific (ON-TARGET plus SMART pool siRNA)] using INTERFERin reagent (Polyplus-transfection) according to manufacturer’s instructions. Transfection efficiency was measured by quantitative polymerase chain reaction (qPCR). Treatments with the CDK4 inhibitor (PD0332991) were done using 1 µM of PD0332991 in non-differentiated 3T3-L1, dimethyl sulfoxide (DMSO) as vehicle.
Mouse embryonic fibroblasts (MEFs) were derived from 13.5-day-old wild-type and p16−/−embryos and plated in six-well plates (300,000 cells/well). Adipocyte differentiation was initiated after 2 days of confluence. MEFs were differentiated with AmnioMAX-C100 medium (Invitrogen, Paris, France), 7.5% AmnioMAX-C100 supplement (Invitrogen, Paris, France), 7.5% foetal calf serum, 0.5 mM 3-isobutyl-1-methylxanthine, 1 μM dexamethasone, 5 μM insulin. From days 3 to 8, cells were incubated with medium supplemented with 5 μM insulin and 1 μM rosiglitazone. At different time points, cells were lysed and homogenized for RNA isolation, or fixed in 4% paraformaldehyde and stained with Oil Red O. All experiments were performed at least in triplicate.
Flow cytometry
3T3-L1 (1 × 106 to 1 × 107) were trypsinized, centrifuged at 850g for 5 min and resuspended in 0.5 mL of 4°C phosphate-buffered saline (PBS), fixed in cold 70% ethanol for 30 min, and washed twice in PBS by centrifugation at 850g for 5 min. Fixed cells were treated with 5 µg/mL of DNase-free RNase A (Sigma, Lesquin, France) and incubated in 20 µg/mL of propidium iodide (PI, sigma, Lesquin, France) for 15 min at room temperature. DNA content frequency was acquired using a FACS Canto II flow cytometer equipped with the FACS Diva software (BD Biosciences, Franklin Lakes, NJ, USA). Cell doublets were excluded from the analysis using doublet discrimination gating method: FSC-A/SSC-A gate followed by a PI-A/PI-W gate. Cell cycle distribution was analysed using FACSDiva6.0 (Tree Star, Inc., Ashland, OR, USA).
Mice
p16INK4a-deficient (p16−/−) mice on a C67BL/6J background were kindly provided by P. Krimpenfort (Netherlands Cancer Institute, Amsterdam). Female mice were treated with rosiglitazone (15 mg/kg) for 5 weeks. For bone marrow transplantation, 8-week-old female ldlr−/− mice were lethally irradiated (8 Gy) and tail vein injected the next day with 107 bone marrow cells isolated from 8-week-old p16−/− or littermate control (p16+/+) donor mice. Mice received autoclaved acidified water (pH = 2) supplemented with neomycin (100 mg/L) (Cat.N1142, Sigma-Aldrich, St. Louis, MO, USA) and polymyxin B sulphate (60,000 U/L) (Cat.21850029, Invitrogen, Paris, France) 1 week before and 4 weeks after transplantation. Mice were studied 6 weeks post-transplantation allowing complete repopulation by the donor bone marrow. To ensure that donor bone marrow efficiently replaced the resident blood cell population, DNA was extracted from whole blood with an Illustra blood kit (GE Health-care, Lille, France). PCR was performed with the forward 5′-GCA-GTG-TTG-CAG-TTT-GAA-CCC-3′ and reverse 5′-TGT-GGC-AAC-TGA-TTC-AGT-TTG-3′ primers, yielding products of different lengths depending on the genotype, separated on a 1.5% agarose gel and quantified with the Gel Doc XR system (Bio-Rad, Marnes-la-Coquette, France). Over 95% of host blood cells were from donor origin. All protocols were conducted with the approval of the ethical review boards of the Pasteur Institute, Lille, France (CEEA 13/2010) and Maastricht University, Maastricht, The Netherlands (DEC 2007-117).
RNA extraction and analysis
Total RNA was obtained from tissues using the guanidiniumthiocyanate (GSCN)/phenol/chloroform extraction method and from cells using EXTRACT-ALL (Thermo Fisher Scientific, Villebon-sur-Yvette, France), DNase-I treated and complementary DNA (cDNA) was generated using cDNA reverse transcription kit (Applied Biosystems, Villebon-sur-Yvette, France). Real-time qPCR detection was conducted on a Stratagene Mx3005P (Agilent technologies) using Brilliant II SYBR Green reagent (Agilent Technologies, Les Ulis, France) and specific primers. Messenger RNA (mRNA) levels were normalized to cyclophilin mRNA and fold induction was calculated using the delta CT method. Murine primers: PPARγ forward CCGTGATGGAAGACCACTCG, PPARγ reverse AGGCCTGTTGTAGAGCTGGGTC, C/EBPalpha forward TAGGTTTCTGGGCTTTGTGG, C/EBPalpha reverse CACACTGCCATTGCACAAG, adiponectin forward GTGATGGCAGAGATGGCACT, adiponectin reverse GCTTCTCCAGGCTCTCCTTT, perilipin forward TGTACAGGGTGCCAGCAA, perilipin reverse GGGATCTTTTCCTCCAGGTG, cyclophilin forward GCATACGGGTCCTGGCATCTTGTCC, cyclophilin reverse ATGGTGATCTTCTTGCTGGTCTTGC, stem cell antigen (SCA)-1 forward GGCCCTACTGTGTGCAGAA, SCA-1 reverse TTACTTTCCTTGTTTGAGAATCCA. Human primers: p16INKA forward GAGCAGCATGGAGCCTTC, p16INKA reverse GGCCTCCGACCGTAACTATT, SCA-2 forward AAGATCTTCTTGCCAGTGC, SCA-2 reverse CAGACACAGTCACGCAGTAG.
pvAT measurements
Mice were euthanized by cervical dislocation and the heart of each animal was rinsed with PBS and fixed with 4% phosphate-buffered paraformaldehyde (pH = 7.4). Serial 10-µm-thick sections were cut and lipid deposition was quantitatively analysed by Oil red-O staining.
Human adipose tissue collection
ScAT and vAT were obtained as previously described4 (Table 1). The study was approved by the ethics committee of the CHRU of Lille, France, under the ABOS and OMENTOB frameworks. None of the patients had clinical symptoms of systemic inflammation or malignancies. Human ecAT and pcAT was obtained from patients undergoing cardiac surgery for coronary artery by-pass graft at the Lille University Regional Hospital (Table 2). The study was approved by the local Patient Protection Committee (Lille, France) and patients gave informed consent. ecAT is defined as the fat located between the myocardium and visceral pericardium, that is, onto the myocardium. pcAT is defined as the fat depot outside the visceral pericardium, that is, within the pericardial sac.
Table 1.
Clinical and biologic characteristics of lean versus obese patients undergoing bariatric surgery.
| Lean |
Obese |
p value | |
|---|---|---|---|
| n = 10 | n = 13 | ||
| Age | 47.5 ± 13.13 | 44.08 ± 7.09 | 0.4308* |
| Gender (m/w) | 5/5 | 0/13 | 0.0075** |
| BMI (kg/m2) | 24.62 ± 1.8 | 52.04 ± 7.45 | <0.0001* |
| Hypertensive (yes/no) | 6/4 | 5/8 | 0.4136** |
| Fasting plasma glucose (g/L) | 0.82 ± 0.18 | 0.92 ± 0.07 | 0.0652* |
| Fasting plasma insulin (mU/L) | 5.54 ± 2.77 | 9.72 ± 3.55 | 0.0080* |
| HOMA-IR | 0.81 ± 0.42 | 1.44 ± 0.51 | 0.0068* |
| Fasting plasma TG (g/L) | 1.25 ± 0.67 | 0.89 ± 0.3 | 0.0926* |
| Fasting plasma cholesterol (g/L) | 2.03 ± 0.49 | 2.02 ± 0.31 | 0.9172* |
| Fasting plasma LDL-C (g/L) | 1.21 ± 0.42 | 1.32 ± 0.26 | 0.4535* |
| Fasting plasma HDL-C (g/L) | 0.57 ± 0.15 | 0.52 ± 0.08 | 0.2842* |
BMI: body mass index; HDL-C: high-density lipoprotein cholesterol; LDL-C: low-density lipoprotein cholesterol; TG: triglyceride; HOMO-IR: homeostatic model assessment of insulin resistance.
Data are presented as mean ± standard deviation. Statistical comparison was done by *Student’s t-test or **Fisher’s exact test.
Table 2.
Clinical and biologic characteristics of lean versus obese patients undergoing cardiac surgery for coronary artery by-pass graft.
| Lean |
Obese |
p value | |
|---|---|---|---|
| n = 14 | n = 11 | ||
| Age | 68.3 ± 8.3 | 63.55 ± 6.023 | 0.1252* |
| Gender (m/w) | 9/5 | 10/1 | 0.1804** |
| BMI (kg/m2) | 23.9 ± 2.94 | 32.38 ± 1.79 | <0.0001* |
| Hypertensive (yes/no) | 3/11 | 8/3 | 0.0172** |
| Fasting plasma glucose (g/L) | 1.02 ± 0.22 | 1 ± 0.13 | 0.9562*** |
| Fasting plasma TG (g/L) | 0.99 ± 0.46 | 1.63 ± 0.31 | 0.0015*** |
| Fasting plasma cholesterol (g/L) | 2.04 ± 0.39 | 1.64 ± 0.15 | 0.0043* |
| Fasting plasma LDL-C (g/L) | 1.34 ± 0.4 | 0.91 ± 0.12 | 0.0015*** |
| Fasting plasma HDL-C (g/L) | 0.5 ± 0.18 | 0.4 ± 0.074 | 0.0906* |
| ALAT (UI/L) | 21.64 ± 9.5 | 46.27 ± 50.7 | 0.0283*** |
| ASAT (UI/L) | 35.57 ± 26.92 | 40.82 ± 27.18 | 0.7841*** |
| Creatinine (mg/L) | 11.24 ± 4.19 | 11.8 ± 4.96 | 0.8245*** |
| CRP (mg/L) | 3.26 ± 0.85 | 3.68 ± 0.95 | 0.2506* |
BMI: body mass index; HDL-C: high-density lipoprotein cholesterol; LDL-C: low-density lipoprotein cholesterol; TG: triglyceride; CRP: C-reactive protein; ASAT: aspartate-aminotransferase; ALAT: alanine-aminotransferase.
Data are presented as mean ± standard deviation. Statistical comparison was done by *Student’s t-test, **Fisher’s exact test or ***Mann–Whitney test.
Statistics
Data are expressed as mean ± standard error of mean (SEM). Results were analysed by unpaired two-tailed Student’s t-test, one-way analysis of variance (ANOVA) or two-way ANOVA as appropriate using GraphPad Prism software (La Jolla, CA, USA). p < 0.05 was considered statistically significant.
Results
p16INK4a knock-down (Supplemental Figure 1) in 3T3-L1 cells using siRNA targeting CDKN2A increased cell division during clonal expansion (Figure 1(a)) and concomitantly enhanced adipogenesis, illustrated by enhanced lipid accumulation (Figure 1(b)) and increased expression of adipogenic genes such as PPARγ, c/ebpα, adiponectin and perilipin (Figure 1(c)). Inhibiting CDK4 activity with the pharmacological inhibitor PD0332991 or simultaneously knocking down PPARγ with siRNA blunted the effects of p16INK4a knock-down (Figure 1(b) and (c)). Given the key role of PPARγ in adipogenesis,7 we tested whether p16INK4a deficiency affects adipogenic differentiation of MEFs. Indeed, primary p16−/− MEFs displayed increased adipogenesis upon PPARγ activation compared to MEFs from p16+/+ littermates, evidenced by enhanced expression of PPARγ (Figure 2(a)), c/ebpα (Figure 2(b)), adiponectin (Figure 2(c)) and perilipin (Figure 2(d)) at several time points of adipogenic differentiation. These data demonstrate that p16INK4a deficiency increases adipogenesis via a CDK4-PPARγ-dependent pathway.
Figure 1.

p16INK4a knock-down enhances clonal expansion and adipogenesis 3T3/L1 cells: (a) 3T3/L1 preadipocytes were either treated with control siRNA (white bars) or siRNA targeting CDKN2A (black bars). Cells were stained with propidium iodide and cell cycle status was analysed by flow cytometry before differentiation (start) and 24 h or 48 h after starting differentiation, (b, c) 3T3/L1 preadipocytes were either treated with control siRNA (white bars) or siRNA targeting CDKN2A (black bars), the CDK4 inhibitor PD0332991 (white bars with black dots), siRNA targeting CDKN2A combined with PD0332991 (black bars with white dots), siRNA targeting PPARγ (striped bars), or siRNA targeting CDKN2A combined with siRNA targeting PPARγ (black bars with white stripes), (b) Oil Red O staining of neutral lipids. Scale bar is 100 µm, (c) gene expression levels of PPARγ, C/EBPα, adiponectin and perilipin were measured by qPCR before (preadip.) and after differentiation into mature adipocytes (adip.).
Data are represented as mean and SEM. Differences were assessed with one-way ANOVA using Bonferroni post-test. ***p < 0.001, **p < 0.01, *p < 0.05 ns = not significant.
Figure 2.
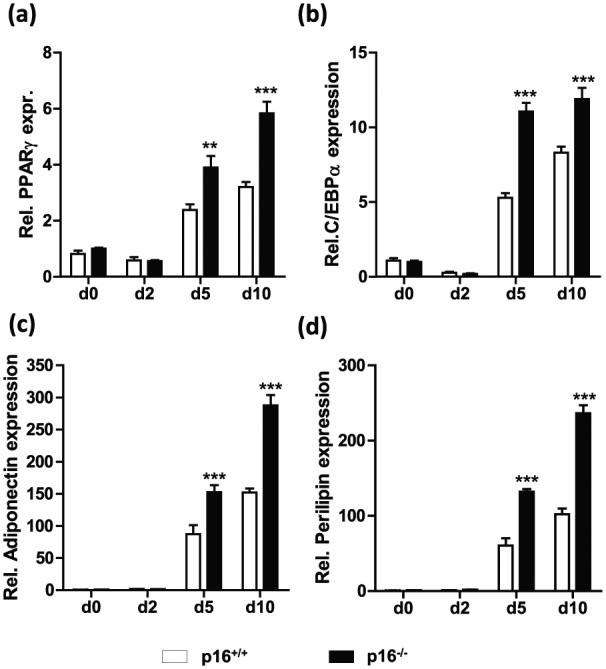
p16INK4a-deficient MEFs display enhanced adipogenesis: mRNA expression of PPARγ (a), C/EBPα (b), adiponectin (c), and perilipin (d) was measured in p16+/+ (white bars) and p16−/− (black bars) MEFs by qPCR before differentiation (d0), and after indicated time points (d = days) of adipogenic differentiation.
Differences were assessed with two-way ANOVA using Bonferroni post-test. ***p < 0.001, **p < 0.01 and *p < 0.05, ns = not significant.
To assess a role of p16INK4a in AT formation in vivo, we analysed AT depots from p16+/+ and p16−/− mice. Neither scAT, gonadal AT [ovarian AT (oAT) or ecAT] nor bAT weight was different between the genotypes in either male or female mice (Supplemental Figure 2A and B). Moreover, the weight of scAT and ecAT from p16+/+ and p16−/− mice did not change upon feeding a high-fat diet (data not shown), suggesting that p16 deficiency has no major impact on adipogenesis in vivo under these conditions. Since our results show that the effect of p16INK4a deficiency on adipogenesis in vitro depends on PPARγ, we assessed the effect of rosiglitazone on AT depots from p16+/+ and p16−/− mice. Rosiglitazone treatment induced bAT formation equally in p16+/+ and p16−/− mice, while oAT or scAT were unaffected (Supplemental Figure 2C). Since recent studies show that AT surrounding the murine heart is induced by PPARγ activation,9 perivascular AT was analysed. Rosiglitazone induced the appearance of adipocytes at the aortic valve area, that is, aortic pvAT which anatomically corresponds to human ecAT located between the myocardium and visceral pericardium (Figure 3(a) and (b)). Interestingly, pvAT surface was significantly larger in p16−/− mice than in wild-type littermates (Figure 3(a) and (b)). Since these murine AT depots were too small to obtain enough mRNA for reliable qPCR analysis, mRNA was measured in thoracic aortas with their surrounding pvAT. Rosiglitazone-treated p16−/− mice expressed higher levels of adipogenic genes such as PPARγ (Figure 3(c)) and perilipin (Figure 3(d)), in pvAT from thoracic aortas compared to littermate controls. Expression of the macrophage marker F4/80 was unaltered (Figure 3(e)), indicating that p16INK4a deficiency does not influence pvAT macrophage content. Mesenchymal stem cell precursors especially reside in the perivascular region.10 Moreover, rosiglitazone has been shown to contribute to murine peripheral adipogenesis by promoting recruitment of SCA-1-positive bone marrow-derived progenitor cells,11 prompting us to measure SCA-1 expression. Interestingly, while SCA-1 expression was unaffected by rosiglitazone treatment in other ATs (Supplemental Figure 3A–C), SCA-1 mRNA was induced by rosiglitazone in pvAT from thoracic aortas and this induction was markedly enhanced in p16−/− mice (Figure 3(f)). Therefore, we next investigated the contribution of bone marrow p16INK4a deficiency on pvAT development. The mice were fed a western-type diet as a physiologically relevant stimulus of pvAT development. Bone marrow-restricted p16INK4a deficiency resulted in enhanced formation of aortic pvAT (Figure 4(a) and (b)). Mice transplanted with p16INK4a-deficient bone marrow displayed slightly lower body weight gain compared with controls. However, this difference did not reach statistical significance (Supplemental Figure 4A). In addition, oAT was lower in mice transplanted with p16INK4a-deficient bone marrow (Supplemental Figure 4B), arguing that increased adipogenesis is restricted to the aortic region in these animals. Thus, our results show that p16INK4a deficiency in bone marrow cells enhances western diet-induced pvAT formation in mice.
Figure 3.

p16INK4a deficiency enhances rosiglitazone-induced pvAT formation in vivo: p16+/+ (n = 10) and p16−/− (n = 9) littermates were treated with rosiglitazone (15 mg/kg) for 5 weeks or vehicle and cardiac AT was stained for neutral lipids with Oil Red O (a). Scale bare represents 500 µm. Stained surface area was quantified at different locations from the valve area (b). Aortic perivascular AT gene expression of PPARγ (c), perilipin (d), F4/80 (e) and SCA-1 (f) was measured with qPCR, expressed as fold change relative to untreated controls (set at 1).
Data are represented as mean and SEM. Differences were assessed with either Student’s t-test or two-way ANOVA using Bonferroni post-test where appropriate. **p < 0.01 and *p < 0.05, ns = not significant.
Figure 4.

Bone marrow p16INK4a deficiency enhances diet-induced pvAT formation: Lethally irradiated ldlr−/− mice were transplanted with either p16+/+ (n = 10) or p16−/− (n = 11) bone marrow. After reconstitution, animals were fed a western diet and AT formation in the hearts was quantified by measuring Oil Red O surface area (a, b) in hearts from p16+/+ transplanted (BMTp16+/+) or p16−/− transplanted (BMTp16−/−) animals. Scale bars represent 500 µm (top panel) or 100 µm (lower panel).
Data are represented as mean and SEM. Differences were assessed with Student’s t-test. **p < 0.01.
Finally, we investigated gene expression of p16INK4a in scAT, vAT, pcAT and ecAT isolated from human subjects. Compared to the other AT depots, p16INK4a expression is especially high in ecAT (Figure 5(a)), explaining why ecAT is more sensitive to modulation by p16INK4a than other ATs. Although SCA-1 has no human ortholog, the neighbouring SCA-2 gene is conserved in humans and may have similar functions.12 Interestingly, SCA-2 expression is higher in ecAT from obese patients compared to lean controls (Figure 5(b)), while its expression is not influenced by obesity in other AT depots (Figure 5(c) to (e)).
Figure 5.

p16INK4a and SCA expression in human adipose tissue depots: mRNA expression analysis of p16INK4A in human scAT (n = 11), vAT (n = 12), pcAT (n = 18) and ecAT (n = 25) (a) and of the human stem cell marker SCA-2 in ecAT (b) and pcAT (e) (n = 25, lean n = 14, obese n = 11) and in vAT (c) and scAT (d) (n = 23, lean n = 10, obese n = 13), from lean (white bars) or obese (black bars) individuals.
Data are represented as mean and SEM. Differences were assessed with either student’s t-test, one-way or two-way ANOVA using Bonferroni post-test where appropriate. ***p < 0.001, **p < 0.01 and *p < 0.05, ns = not significant.
Discussion
Although the link between CDKN2A and CVD and T2D was discovered in 2007,1,2 it remained unclear via which pathways this locus influences CVD. Our results show that p16INK4a plays a role in mitotic clonal expansion-mediated adipogenesis and in the development of murine pvAT close to the heart.
We show for the first time a functional role for p16INK4a in adipocyte differentiation, likely due to an enhancement of mitotic clonal expansion, crucial for adipogenesis of murine cells.13 Although the employed siRNA targeting CDKN2A knocks down both p16INK4a and p19ARF, our data show that the effects of CDKN2A knock-down depend on CDK4 activity, which is controlled by p16INK4a but not p19ARF.14 Moreover, similar results were obtained using primary MEFs from mice specifically lacking p16INK4a. CDK4 was shown to promote adipogenesis through PPARγ activation.8 In line, our data show that the effects of CDKN2A knock-down depend on PPARγ. In addition, CDK4 was recently shown to potentiate insulin signalling in adipocytes, thereby inhibiting lipolysis and enhancing lipogenesis.15 Therefore, we cannot exclude that metabolic changes are at least partially involved in the enhanced adipogenesis after p16INK4a knock-down.
In vivo, p16INK4a deficiency did not affect rosiglitazone-induced adipogenesis in bAT, vAT or scAT. Interestingly, p16INK4a deficiency increased the formation of pvAT surrounding the aortic root. In addition, by performing bone marrow transplantation, we found increased pvAT formation in mice transplanted with p16INK4a-deficient bone marrow fed a high-fat high-cholesterol diet. We did not observe increased body weight or oAT, suggesting that the effects of p16INK4a deficiency are specific for pvAT formation. The origin of adipocyte precursor cells currently remains subject of debate and conflicting data exist whether or not adipocyte progenitors derive from hematopoietic cells.16 In support of a bone marrow origin of at least some adipocytes, is the fact that adipogenesis in white AT depots occurs in close proximity to the vasculature without sharing a common progenitor with vascular cells.16 To our knowledge, the potential hematopoietic origin of pvAT has not yet been investigated. Although our study was not designed to univocally identify a bone marrow origin of pvAT, our results suggest that increased adipogenesis caused by p16INK4a deficiency in bone marrow cells contributes to aortic pvAT formation.
In human ecAT, p16INK4a mRNA is more abundant than in other AT depots. This result suggests that ecAT may be, more than other AT depots, sensitive to p16INK4a function or regulation. Interestingly, SCA-2 gene expression was higher in ecAT isolated from obese compared to lean individuals. In line, murine aortic pvAT from p16−/− mice displayed higher SCA-1 mRNA levels than wild-type controls. Murine SCA-1 is a surface molecule associated with stem cells thought to function as a co-signalling molecule with roles in haematopoiesis, bone formation and muscle homeostasis.12 SCA-1 does not have a clear human ortholog since the region encoding this and five additional related genes has been deleted through evolution. However, the region flanking this deleted 500 kb are conserved in humans and it is possible that at least some of the important roles of SCA-1 in murine development are taken over by one or a number of human-related proteins,12 such as SCA-2. However, it remains to be determined whether SCA-2 shares the same role in bone marrow stem cells as SCA-1 in mice.
The development of fat covering the human heart and aorta is thought to contribute to CVD. These fat depots, which are in direct contact with the epicardium and the arteries, are a source of free fatty acids, adipokines, and inflammatory cytokines.5 ecAT is thus believed to directly interact with cardiomyocytes and the coronary vasculature through paracrine signalling. Recent epidemiological studies have linked ecAT volume with CVD, such as atrial fibrillation and coronary atherosclerosis.5
In conclusion, our results shed light on the potential role of p16INK4a in pvAT development. Given the known association of pvAT with CVD, our data provide a new mechanism to explain one of the most profound GWAS associations for CVD risk.
Footnotes
Author contributions: K.W., Y.D., S.A.H., J.V., X.M., A.C., M.T., B.D., E.B., C.D., E.V, R.P. performed experiments. K.W., Y.D., S.A.H., C.G.S., F.P., D.M., B.S., R.P. interpreted the results. K.W., R.P. and B.S. wrote the article. K.W., F.P., D.M., B.S. and R.P. designed experiments. K.W. and Y.D. contributed equally to this article.
Declaration of conflicting interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding: This work was supported by grants from European Genomic Institute for Diabetes (E.G.I.D., ANR-10-LABX-46), the Fondation pour la Recherche Médicale (DCV20070409276, B.S.), an EFSD/GlaxoSmithKline Research Grant (B.S. and K.W.), the Cost Action (BM0602), and the Conseil régional Nord Pas-de-Calais and FEDER. B.S. is a member of the Institut Universitaire de France. K.W. was supported by the Netherlands Organization for Scientific Research (NWO-Veni Grant # 91612056), a European FP7 Marie Curie grant (FP7-PEOPLE-2012-CIG grant # 322070), a European Atherosclerosis Society grant and the Dutch Heart Foundation (NHS-2013T143). Y.D. was supported by INSERM (n° 00540009).
References
- 1. McPherson R, Pertsemlidis A, Kavaslar N, et al. A common allele on chromosome 9 associated with coronary heart disease. Science 2007; 316: 1488–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hannou SA, Wouters K, Paumelle R, et al. Functional genomics of the CDKN2A/B locus in cardiovascular and metabolic disease: what have we learned from GWASs? Trends Endocrinol Metab 2015; 26: 176–184. [DOI] [PubMed] [Google Scholar]
- 3. Cudejko C, Wouters K, Fuentes L, et al. p16INK4a deficiency promotes IL-4-induced polarization and inhibits proinflammatory signaling in macrophages. Blood 2011; 118: 2556–2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fuentes L, Wouters K, Hannou SA, et al. Downregulation of the tumour suppressor p16INK4A contributes to the polarisation of human macrophages toward an adipose tissue macrophage (ATM)-like phenotype. Diabetologia 2011; 54: 3150–3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hassan M, Latif N, Yacoub M. Adipose tissue: friend or foe? Nat Rev Cardiol 2012; 9: 689–702. [DOI] [PubMed] [Google Scholar]
- 6. Brown NK, Zhou Z, Zhang J, et al. Perivascular adipose tissue in vascular function and disease: a review of current research and animal models. Arterioscler Thromb Vasc Biol 2014; 34: 1621–1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rosen ED, MacDougald OA. Adipocyte differentiation from the inside out. Nat Rev Mol Cell Biol 2006; 7: 885–896. [DOI] [PubMed] [Google Scholar]
- 8. Abella A, Dubus P, Malumbres M, et al. Cdk4 promotes adipogenesis through PPARgamma activation. Cell Metab 2005; 2: 239–249. [DOI] [PubMed] [Google Scholar]
- 9. Yamaguchi Y, Cavallero S, Patterson M, et al. Adipo-genesis and epicardial adipose tissue: a novel fate of the epicardium induced by mesenchymal transformation and PPARgamma activation. Proc Natl Acad Sci U S A 2015; 112: 2070–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Crisan M, Yap S, Casteilla L, et al. A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell 2008; 3: 301–313. [DOI] [PubMed] [Google Scholar]
- 11. Crossno JT, Majka SM, Grazia T, et al. Rosiglitazone promotes development of a novel adipocyte population from bone marrow-derived circulating progenitor cells. J Clin Invest 2006; 116: 3220–3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Holmes C, Stanford WL. Concise review: stem cell antigen-1: expression, function, and enigma. Stem Cells 2007; 25: 1339–1347. [DOI] [PubMed] [Google Scholar]
- 13. Tang QQ, Otto TC, Lane MD. Mitotic clonal expansion: a synchronous process required for adipogenesis. Proc Natl Acad Sci U S A 2003; 100: 44–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gil J, Peters G. Regulation of the INK4b-ARF-INK4a tumour suppressor locus: all for one or one for all. Nat Rev Mol Cell Biol 2006; 7: 667–677. [DOI] [PubMed] [Google Scholar]
- 15. Lagarrigue S, Lopez-Mejia IC, Denechaud PD, et al. CDK4 is an essential insulin effector in adipocytes. J Clin Invest 2016; 126: 335–348. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 16. Berry R, Jeffery E, Rodeheffer MS. Weighing in on adipocyte precursors. Cell Metab 2014; 19: 8–20. [DOI] [PMC free article] [PubMed] [Google Scholar]