

Abstract
Background
Salivary duct carcinomas (SDCa) are rare highly aggressive malignancies. Most patients die from distant metastatic disease within three years of diagnosis. There are limited therapeutic options for disseminated disease.
Results
11 cases showed androgen receptor expression and 6 cases showed HER2 amplification. 6 Somatic mutations with additional available targeted therapies were identified: EGFR (p.G721A: Gefitinib), PDGFRA (p.H845Y: Imatinib and Crenolanib), PIK3CA (p.H1047R: Everolimus), ERBB2 (p.V842I: Lapatinib), HRAS (p.Q61R: Selumetinib) and KIT (p.T670I: Sorafenib). Furthermore, alterations in PTEN, PIK3CA and HRAS that alter response to androgen deprivation therapy and HER2 inhibition were also seen.
Materials and Methods
Somatic mutation analysis was performed on DNA extracted from 15 archival cases of SDCa using the targeted Illumina TruSeq Amplicon Cancer Panel. Potential targetable genetic alterations were identified using extensive literature and international somatic mutation database (COSMIC, KEGG) search. Immunohistochemistry for androgen receptor and immunohistochemistry and fluorescent in situ hybridization for HER2 were also performed.
Conclusions
SDCa show multiple somatic mutations, some that are amenable to pharmacologic manipulation and others that confer resistance to treatments currently under investigation. These findings emphasize the need to develop testing and treatment strategies for SDCa.
Keywords: salivary duct carcinoma, somatic mutation analysis, targeted therapies, androgen receptor, HER2
INTRODUCTION
Salivary duct carcinoma (SDCa) is a highly aggressive primary salivary gland malignancy associated with poor prognosis accounting for 10–12% of salivary gland malignancies [1, 2]. Currently, surgical resection of the primary tumor, neck dissection, and adjuvant radiation with or without platinum-based chemotherapy forms the mainstay of treatment. Despite this aggressive multimodal approach, the majority of patients with SDCa die within three years of diagnosis due to distant metastatic disease [2, 3]. This highlights the need for effective systemic therapy that can produce a sustained response.
The wide availability of massive parallel sequencing technology has revealed the somatic mutational landscape of many malignancies [4–7]. The identification of driver mutations and downstream effector targets has allowed development of targeted therapies that have revolutionized the treatment of subsets of selected malignancies such as breast, melanoma and pulmonary adenocarcinoma, often with lower toxicity than conventional chemotherapy [5–8]. Identifying effective targeted therapies in rare aggressive malignancies, such as SDCa, is particularly challenging, as well-powered clinical trials are not feasible. The use of targeted therapies in SDCa remains experimental, with variable response rates [5, 9–10]. Alternatives to traditional randomized controlled trials are needed, including targeted gene panel analysis of retrospective archival cohorts, with consideration of agents targeted at specific mutations with proven benefit in more common cancers. This approach has the potential to advance treatment and improve patient outcomes in this rare but lethal tumor [11–14].
This study aims to identify and analyze clinically relevant somatic mutations in SDCa that are amenable to targeted therapy. Moreover, we aim to identify somatic mutations that may alter response to these therapies. The data generated by this study can be used as a starting point for preclinical and clinical studies investigating targeted systemic therapy options for SDCa.
RESULTS
Cohort characteristics
The final cohort included 15 cases of SDCa where DNA extracted from retrospective FFPE tissues passed quality control. The cohort included 14 males and 1 female with a median age of 58 years (range 41–74). The median follow-up was 21.5 months (range 4–78 months), during which time 8 (53.3%) patients died of SDCa. Regional metastases were seen in 10 patients and two patients developed distant metastases to the lung, bone and / or brain. All patients were treated with local resection and adjuvant radiotherapy. In addition, nine patients underwent a neck dissection and five patients received adjuvant platinum-based chemotherapy and one patient is currently receiving Trastuzumab (Table 1). There were 11 patients positive for AR expression and of these 6 patients also demonstrated HER2 amplification (Table 2). None of the tumors demonstrated estrogen or progesterone receptor expression.
Table 1. Clinical features, treatment and follow up.
| Case No. | Age (y) | Sex | Presentation | Treatment | Follow-up (months) |
|---|---|---|---|---|---|
| 1 | 63 | M | NK | Submandibular gland excision and RT | DOD (37) |
| 2 | 56 | M | Submandibular mass | Submandibular gland excision, SND (I), and RT | AWD (54) |
| 3 | 52 | M | NK | Parotidectomy and RT | DOD (38) |
| 4 | 72 | F | Parotid mass | Total parotid, SND, RT, and CRT (Doxorubicin and Cyclophosphamide) | NED (4) |
| 5 | 58 | M | Parotid mass | Total parotid, SND (II), and RT | NED (78) |
| 6 | 50 | M | Parotid mass, facial weakness | Radical parotid, SND (II, III, V), RT, and CRT (Carboplatin) | DOD (16) |
| 7 | 54 | M | Parotid mass | Total parotid, MRND, RT, and CRT (Cisplatin then Paclitaxel) | DOD (25) |
| 8 | 41 | M | Sublingual mass | Sublingual gland excision and RT | NED (52) |
| 9 | 63 | M | Parotid mass | Superficial parotid, SND (II-III), RT, CRT (Cisplatin) | DOD (13) |
| 10 | 64 | M | NK | Parotidectomy and RT | AWOD (18) |
| 11 | 58 | M | Parotid mass | Total Parotid, RT, and CRT (Cisplatin then Paclitaxel and Trastuzumab) | DOD (28) |
| 12 | 65 | M | Parotid mass | Superficial parotid and RT | NED (14) |
| 13 | 74 | M | Submandibular mass | Submandibular gland excision, SND (I), RT, and CRT (Cisplatin) | NK |
| 14 | 53 | M | Lung metastases | Radical parotid, RND, and RT | DOD (6) |
| 15 | 59 | M | Parotid mass | Superficial parotid, SND (II), and RT | DOD (9) |
Abbreviations: RND, radical neck dissection; RT, radiotherapy; MNRD, modified radical neck dissection; CRT, chemotherapy; SND, selective neck dissection; NK, not known; DOD, died of disease; AWD, alive with disease; AWOD, alive with other disease; NED, alive with no evidence of disease; DUC, died of unrelated cause.
Table 2. Clinico-pathological features of the salivary duct carcinoma cohort.
| Case no. | Tumor size | LVI | PNI | Margins | Stage | GATA3 | AR Expression | HER2 FISH |
|---|---|---|---|---|---|---|---|---|
| 1 | 48 mm | Not seen | Not seen | 0.6 mm | T3Nx | + | − | Non-amplified |
| 2 | 22 mm | Not seen | Not seen | 0.2 mm | T2N0 | + | + | Non-amplified |
| 3 | 25 mm | Present | Not seen | Involved | T2N2b | + | + | Amplified |
| 4 | 20 mm | Present | Not seen | Involved | T2N2b | + | + | Non-amplified |
| 5 | 42 mm | Present | Not seen | 1 mm | T3N2b | + | + | Non-amplified |
| 6 | 17 mm | Extensive | Extensive | Involved | T4aN2b | + | + | Amplified |
| 7 | 15 mm | Extensive | Present | Involved | T1N2b | + | + | Non-amplified |
| 8 | 40 mm | Present | Present | Involved | T2Nx | + | + | Amplified |
| 9 | 23 mm | Extensive | Present | Involved | T4aN2b | + | + | Amplified |
| 10 | 64 mm | Extensive | Present | Involved | T4aN2b | + | + | Amplified |
| 11 | 42 mm | Present | Present | 0.3 mm | T3N2b | + | + | Non-amplified |
| 12 | 50 mm | Not seen | Present | Involved | T3Nx | + | + | Amplified |
| 13 | 15 mm | Present | Present | 0.7 mm | T1N0 | + | − | Non-amplified |
| 14 | 40 mm | Extensive | Present | Involved | T2N2bM1 | + | − | Non-amplified |
| 15 | 35 mm | Extensive | Not seen | Involved | T2N2bM1 | + | − | Non-amplified |
Abbreviations: LVI, lymphovascular invasion; PNI, perineural invasion; N/A, not applicable; AR, androgen Receptor; GATA3, GATA binding protein 3; HER2. human epidermal growth factor receptor 2.
Somatic mutations in SDCa
The mean coverage of 15 cases was 6087 (± 1848) with 95.1% (± 2.6%) uniformity of coverage at 0.2X of mean. Significant heterogeneity was present in terms of the number of somatic mutations present per patient (Mean = 19, range 1–154) A total of 289 non-synonymous mutations were identified in the 48-gene panel in 14 patients; after exclusion of a single patient where normal tissue was not available for comparison. These included 250 (86.5%) missense mutations, 14 (4.8%) nonsense mutations, 13 (4.5%) small indel mutations and 11 (3.8%) splice site changes.
The most commonly mutated genes in the cohort were TP53 (n = 10), PIK3CA (N = 8), PTEN (N = 8), FBXW7 (n = 8), ATM (N = 7), GNAQ (N = 6), and HRAS (N = 4) (Figure 1), (Supplementary Table 1).
Figure 1. Frequency of somatic mutations found in the SDCa cohort.
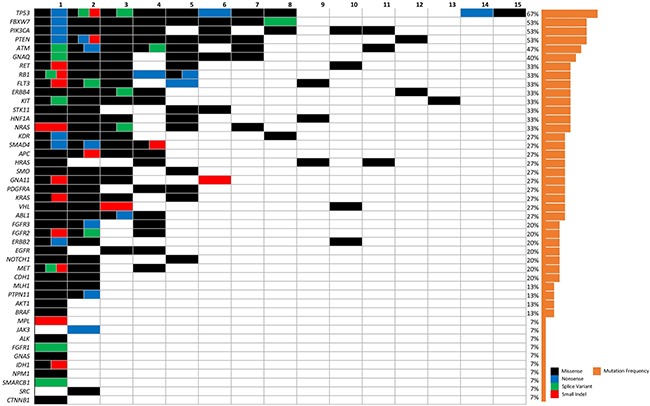
The columns represent individual patients and rows represent specific type of mutation and relative frequency. Color legend of the aberrations represented including missense (black), nonsense (blue), splice variant (green) and small indel (red). Cases with more than one aberration are represented by a split cell with multiple colors.
The four signaling pathways of carcinogenesis and progression affected by the somatic mutations identified in > 60% patients using this targeted panel were the phosphatidylinositol 3′-kinase (PI3K)-Akt signaling pathway (N = 11), mitogen-activated protein kinase signaling pathway (MAPK) (N = 11), p53 signaling pathway (N = 10) and Janus kinase/signal transducers and activators of transcription pathway (JAK/STAT) (N = 9) (Figure 2).
Figure 2. Schematic representation of androgen receptor pathway.
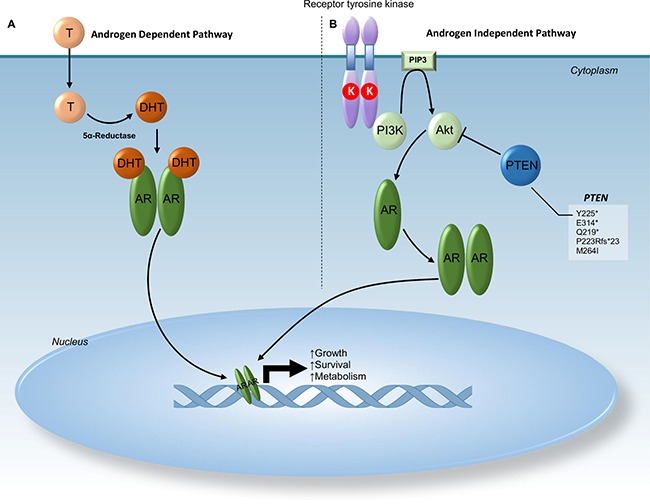
Section (A) demonstrates activation of androgen receptor (AR) by testosterone (T) under normal conditions. Signals from dihydrotestosterone cause dimerization of AR and relocation to the nucleus initiating metabolic activities and growth. Section (B) demonstrates activation of AR mediated by tyrosine kinase receptors. Dimerization of the tyrosine kinase receptor leads to activation of PI3K and phosphorylation of AKT. Phosphorylated AKT can then cause dimerization of AR leading to relocation of AR to the nucleus initiating metabolic activities and growth. Normally functional PTEN inhibits the AR dimerization initiated by phosphorylated AKT. Truncation and frameshift mutations in PTEN as seen in this cohort lead to the loss of this PTEN inhibition. Thus, somatic mutations in PTEN result in lack of sensitivity to androgen deprivation therapy. Abbreviations: AR - Androgen receptor; Akt - V-Akt Murine Thymoma Viral Oncogene Homolog; Chr - Chromosome; DHT - Dihydrotestosterone; K (red circle) - Kinase; PIP3 - Phosphatidylinositol (3,4,5)-Triphosphate; PTEN - Phosphatase and Tensin Homolog; T – Testosterone.
Potential therapeutic targets
Changes potentially amenable to targeted therapy were identified in 12 (80%) cases. Of these, 11 patients expressed androgen receptor by immunohistochemistry and 6 patients demonstrated HER2 amplification by FISH. Specific mutations such as PDGFRA H845Y, HRAS Q61R, PIK3CA H1047R, KIT T670I, EFGR G721A and ERBB2 V842I with potential targeted therapies were also observed in patients who had AR expression or HER2 amplification. Table 3 summarizes the genetic alterations with the potential targeted therapies. Two patients had only a TP53 mutation, currently not amenable to targeted therapy.
Table 3. Analysis of SDCa for somatic mutations with potential therapeutic targets and clinical trials.
| Case # | Gene Symbol | Gene Name | Mutation | COSMIC ID | Functional effect | Drug |
|---|---|---|---|---|---|---|
| 4 | PIK3CA | Phosphatidylinositol-4,5-Bisphosphate 3-Kinase, Catalytic Subunit Alpha | c.3140A>G, p.H1047R | COSM775 | Poor prognosis in breast cancer [15] Increased sensitivity to PI3K pathway inhibitors [16] |
Everolimus [16], BEZ-235 (Dactolisib) [17] |
| 11 | PIK3CA | Phosphatidylinositol-4,5-Bisphosphate 3-Kinase, Catalytic Subunit Alpha | c.1633G>A, p.E545K | COSM763 | Increased catalytic activity resulting in enhanced downstream signaling and oncogenic transformation in vitro[18] Associated with AKT1 activation in breast cancer [15] |
NVP-BYL719 (Alpelisib) [19] |
| 2 | PDGFRA | Platelet Derived Growth Factor Receptor Alpha Polypeptide | c.2533C>T, H845Y | COSM96893 | Increased autophosphorylation of PDGFRA [7] Responsiveness to Imatinib and Crenolanib in melanoma [7] |
Imatinib, Crenolanib [7] |
| 10 | ERBB2 | Erb-B2 Receptor Tyrosine Kinase | c.2524G>A, p.V842I | COSM14065 | Increases phosphorylation of signaling proteins [8] | Lapatinib, Neratinib, Trastuzumab[8] |
| 2 | KIT | KIT Proto-Oncogene Receptor Tyrosine Kinase | c.2009C>T, p.T670I | COSM12708 | Confers resistance to Imatinib [20] Gain of function, leads to constitutive phosphorylation of KIT [20] | Sorafenib [20] |
| 11 | HRAS | Harvey Rat Sarcoma Viral Oncogene Homolog | c.182A>G, p.Q61R | COSM499 | Sensitized towards MEK inhibitor treatment [21] | Selumetinib and MEK162 (Binimetinib) and Everolimus [21] |
| 4 | HRAS | Harvey Rat Sarcoma Viral Oncogene Homolog | c.37G>C, p.G13R | COSM486 | Unknown | Current Phase 1b clinical trials of MLN2480 and MLN0128 or Alisertib or Paclitaxel |
| 1 | EGFR | Epidermal Growth Factor Receptor | c.2162G>C, p.G721A | COSM28510 | Unknown | Gefitinib, Erlotinib [22] |
Specific genetic alterations were examined to assess potential suitability for targeted therapies and recorded when the functional effect and or targeted therapy had been defined in the literature.
*ongoing trial recruitment/experimental
Somatic mutations that may alter susceptibility to androgen deprivation or HER2 inhibition therapy
We also identified somatic alterations that may confer potential resistance to androgen deprivation or HER2 inhibition therapy (Table 4). PTEN loss as has been reported to reduce sensitivity to androgen deprivation and HER2 inhibition. This was seen in two patients with androgen receptor expression (Figures 2 and 3).
Table 4. Mutations conferring resistance to androgen deprivation therapy and Herceptin treatment.
| Case # | Gene Symbol | Gene Name | Mutation | COSMIC ID | Functional effect | Predicted Resistance |
|---|---|---|---|---|---|---|
| 1 | PTEN | Phosphatase and Tensin Homolog | c.675T>A, p.Y225* | COSM5291* | Truncated protein leading to predicted loss of function | Androgen Deprivation Therapy [23]; Trastuzumab [24] |
| 2 | c.940G>T, p.E314* | COSM5305 | ||||
| c.655C>T, p.Q219* | COSM5155 | |||||
| c.937_940delAAGG, p.P223Rfs*23 | - | Frameshift mutation resulting in a deletion/insertion | ||||
| 4 | c.792G>A, p.M264I | COSM5351742 | Increased phosphorylation of PTEN protein, may cause configuration into inactive form [25] | |||
| 4 | PIK3CA | Phosphatidylinositol-4,5-Bisphosphate 3-Kinase, Catalytic Subunit Alpha | c.3140A>G, p.H1047R | COSM775 | Poor prognosis in breast cancer [15]Increased sensitivity to PI3K pathway inhibitors [16] | Trastuzumab [26] |
| 11 | c.1633G>A, p.E545K | COSM763 | Increased catalytic activity resulting in enhanced downstream signaling and oncogenic transformation in vitro [18]Associated with AKT1 activation in breast cancer [15] | Trastuzumab [26] and Cisplatin [18] | ||
| 11 | HRAS | Harvey Rat Sarcoma Viral Oncogene Homolog | c.182A>G, p.Q61R | COSM499 | May cause MAPK pathway activation [27]Interfere with GTP hydrolysis leading to hyperactive RAS kinase activity [21] | Trastuzumab and Lapatinib [4, 28] |
| 1, 9 | c.142G>A, p.G48R | COSM5555612 | An activating mutation that may cause upregulation of MAPK signaling pathway [29] | |||
| 4 | c.37G>C, p.G13R | COSM486 | Unknown |
*COSM5291 denotes the mutation PTEN c.675T>G, which has the consequence of producing p.Y225* as the TAG codon is produced. The change c.675T>A produces the codon TAA which also results in a premature stop codon and truncation.
Figure 3. Schematic representation of HER2 signaling pathway.
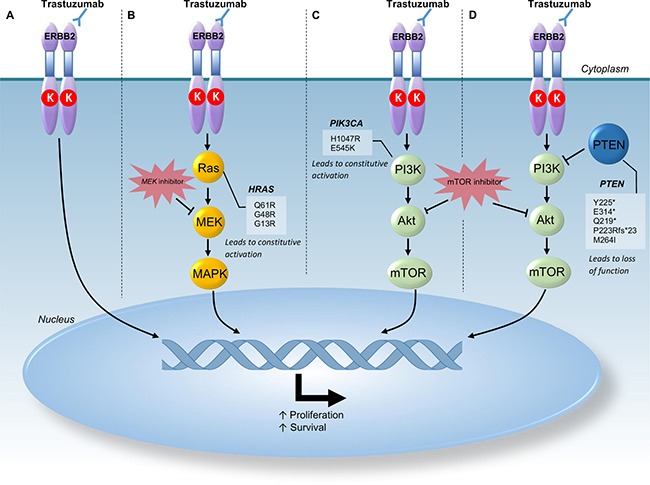
Activation of this pathway leads to proliferation and survival of cells. Section (A) demonstrates normal inhibition of HER2 signaling pathway by Trastuzumab in cancers without mutations that may confer resistance. Section (B) demonstrates mutations in HRAS which lead to constitutive activation of the Ras gene downstream of Trastuzumab inhibition thus leading to resistance to Trastuzumab. MEK inhibitors such as Selumetinib act downstream of these activating mutations and can overcome the effects of HRAS mutations. Section (C) demonstrates mutations in PIK3Ca which lead to constitutive activation of this pathway downstream of Trastuzumab inhibition thus, leading to resistance of Trastuzumab therapy. Section (D) demonstrates mutations in PTEN that lead to truncation or a frameshift mutation resulting in a loss of function. PTEN inhibits the activation of PI3K hence, the loss of function is thought to contribute to resistance to Trastuzumab therapy. In both section C and section D a potential therapy downstream of these mutations conferring resistance to Trastuzumab include mTOR inhibitors such as Everolimus. Abbreviations: Chr - Chromosome; MAPK - Mitogen-Activated Protein Kinase; mTOR - Mechanistic Target of Rapamycin; PI3K - Phosphatidylinositol-4,5-Bisphosphonate 3-Kinase.
Similarly, PIK3CA and HRAS mutations that are known to reduce sensitivity to HER2 inhibition were seen in one patient that demonstrated HER2 amplification. Figure 3 illustrates the role of PIK3CA and HRAS in downstream signaling of the HER2 signaling pathway.
DISCUSSION
Massive parallel sequencing technologies are changing the management of several common aggressive malignancies such as pulmonary adenocarcinoma and melanoma [5, 6]. Similar efforts would also benefit patients with rare malignancies such as SDCa who develop systemic metastases despite conventional therapies. The current study demonstrates that 80% of SDCa harbor alterations in pathways that may be susceptible to currently available systemic therapies, thus providing evidence towards implementation of precision medicine in these rare neoplasms. The study has also identified somatic alterations in PIK3CA and HRAS that alter sensitivity to the currently proposed therapies in SDCa such as androgen deprivation therapy and Trastuzumab. On the other hand, these alterations in PIK3CA and HRAS are susceptible to (mTOR) and mitogen extracellular signal-regulated kinases (MEK) inhibitors.
There are several reports of patients treated with androgen deprivation therapy, Trastuzumab and Venmurafinib for metastatic SDCa with partial to complete response [30–35]. However, these modalities have not been accepted as mainstream treatments for SDCa. Clinical trials for evaluating the effectiveness of new therapies are inherently difficult to undertake for rare malignancies [36] and the molecular pathways of carcinogenesis and progression amenable to pharmacologic modulation need to be explored. Dalin et al. have laid the groundwork by performing comprehensive whole exome sequencing of 16 examples of SDCa [37]. Our study demonstrates overlapping findings. Approximately 75% of patients in both studies demonstrate androgen receptor expression, 30% demonstrate HER2 amplification and nearly 70% patients harbor genetic alterations with published clinical or pre-clinical evidence supporting targeted therapies for AR, HER2 amplification, PDGFRA H845Y, HRAS Q61R, PIK3CA H1047R, KIT T670I and ERBB2 V842I [7, 8, 16, 20, 21, 32, 33, 37, 38].
Clinical trials with Trastuzumab have been attempted in SDCa previously [39]. However, testing for HER2 amplification or factors that may alter response to HER2 inhibition was not undertaken prior to treatment resulting in variable response amongst the patients [39]. The findings of our study and that of Dalin et al. suggest that only 30–40% of patients with SDCa demonstrate HER2 amplification [37]. The experience described by Limaye et al. would indicate that HER2 expression is essential for response with Trastuzumab and should be undertaken prior to initiation of treatment [34]. Furthermore, the current study has also identified mutations in PIK3CA, HRAS and PTEN that have been shown to confer resistance to HER2 inhibition highlighting the essential role of somatic mutation testing in selecting patients for this therapy [4, 37, 40].
Our study also includes a subgroup of patients (4/15) that lack both androgen expression and HER2 amplification. Two patients also harbored both HRAS mutations that may be amenable to MAPK/MEK inhibitors such as Selumetinib [21] and PIK3CA H1047R mutations susceptible to mTOR inhibitors such as Everolimus [16, 41]. The coexistence of PIK3CA and HRAS mutations as observed in the current cohort is well documented in SDCa [4, 37]. These patients may benefit from therapeutic approaches using MEK and mTOR inhibitors synergistically [21]. Other mutations observed in isolated cases included EGFR G721A, which has been shown to respond to Gefinitib or Erlotinib in non-small cell lung cancer [22] and PDGFRA H845Y which has been shown to respond to Imatinib [7]. However, the later changes have been described in relatively small number of patients till date and warrant further investigations, particularly in SDCa. While BRAF V600E mutations were not identified in the current cohort, BRAF inhibitors such as vemurafenib may also be useful in a select group of SDCa that demonstrate these alterations as described by Nardi et al. [35, 42]. These findings highlight the role of somatic mutation testing in the management of SDCa.
While targeted therapies are often better tolerated than conventional chemotherapeutics, they are not without toxicities. Trastuzumab causes rare but clinically important cardiomyopathy [43]. Agents targeting the androgen pathway often cause side-effects such as sexual dysfunction, hot flushes, mood swings and fatigue [44]. Decline in bone and cardiovascular health can be seen in the long term [45]. EGFR inhibitors, such as Erlotinib and Gefinitib, most frequently cause skin and gastrointestinal toxicity [46]. Rational therapeutic choices based on appropriate testing, such as somatic mutation analysis is crucial to improve patient outcomes and minimize unwarranted toxicity.
It is increasingly recognized that the genotype of the tumor alone is not a reliable predictor of response to targeted therapies and transcriptomic profiling may also be useful [36]. Our approach of using formalin fixed paraffin embedded tissues from a retrospective cohort and a targeted panel is not without limitations. However, fresh tissue, ideal for genomic studies, may not always be available for rare malignancies and is less suited to clinical practice. Formalin fixed tissues generally yield short fragmented, covalently modified and cross linked DNA, but do allow analysis in the clinical setting. The findings of our study demonstrate the feasibility of using FFPE material and targeted panel sequencing. This allows for wider availability and cost effective testing providing results within a clinically relevant time frame. Analysis of FFPE tissue from a retrospective cohort with known clinical outcomes from multiple institutions can assist with development of international multi-institutional studies. Rigorous quality checks as employed in this study ensure that the results provide high quality data. Furthermore, it is encouraging that the targeted panel utilized in this study provided similar clinically relevant information as compared to the more expensive and technically challenging comprehensive methods such as whole exome sequencing. Thus, our pragmatic and cost effective approach can be widely adopted and allows for collection and development of evidence for targeted therapies in rare malignancies.
In conclusion, the majority of SDCa demonstrate molecular changes that may be amenable to pharmacologic modulations that are well established and tolerated for other cancers such as androgen deprivation therapy in prostate cancer and Trastuzumab in breast cancer [23, 26]. Testing for these molecular alterations is essential for rational therapeutic decisions. Early testing can also identify alterations that may predict resistance to these drugs. The findings of our study and that of the others provide a strong biologic argument for exploring these treatment options with pragmatically designed clinical trials [4, 37].
MATERIALS AND METHODS
Patients
A total of 195 cases of primary salivary gland neoplasms identified in the database of Sydney Head and Neck Cancer Institute (2000–2015) and held in the archives of the department of Tissue Pathology and Diagnostic Oncology at Royal Prince Alfred Hospital were reviewed to identify 25 cases of SDCa. Of these, suitable quality DNA extracted from FFPE tissue was available for somatic mutation testing in 15 cases after rigorous quality control. SDCa was defined as an adenocarcinoma with apocrine morphology resembling high-grade breast ductal carcinoma in situ (DCIS) [47]. Less common patterns include micropapillary, mucinous or sarcomatoid morphology [48]. The tumors demonstrated immunostaining for GATA3 and lacked immunoreactivity with p63 and CK 5/6. All relevant clinicopathologic information including age, gender, tumor site, tumor size, lymphovascular, perineural invasion, neck node involvement, details of local and regional failure were obtained.
All histopathologic slides were reviewed and highly cellular areas of the tumor without necrosis, stroma, inflammatory infiltrate or haemorrhage were identified. The neoplastic cell content ranged from approximately 20–80% in these areas selected for further analysis.
Immunohistochemistry
Immunohistochemistry was performed on all cases. Androgen receptor (AR441; Dako; 1:250) staining was performed on the Leica Microsystems Bond-III automated staining platform. The Ventana BenchMark Ultra was utilized to stain HER2 (4B5; Ventana; prediluted), estrogen receptor (ER) (SP1; Ventana; prediluted), and progesterone receptor (PR) (1E2; Ventana; prediluted), as per standard protocol. The intensity of HER2 staining was graded using a widely accepted 4-point system, as defined in the literature for breast ductal carcinoma [49]. All immunohistochemistry slides were reviewed by 2 pathologists (RG and PL).
HER2 fluorescence in situ hybridization
Interphase fluorescence in-situ hybridization (FISH) for HER2 was performed on all cases using the Vysis PathVysion HER2 DNA Probe Kit (Abbott Molecular, Des Plaines, IL) as per the manufacturer's instructions, except that Invitrogen Pretreatment Solution (Life Technologies, Carlsbad, CA) was used at 98–102°C for 20 minutes. This enumeration FISH probe quantifies HER2/neu gene copy number using a SpectrumOrange labeled 190kb DNA fragment and quantifies the chromosome 17 copy number using a SpectrumGreen labeled alpha satellite DNA fragment. Signals were counted in at least 100 tumor nuclei using an epifluorescence microscope. HER2 FISH was interpreted as per the current American Society of Clinical Oncology Guidelines for breast cancer [50].
Somatic mutation analysis
Malignant tissue selected as described above was macrodissected from the paraffin blocks for deoxyribonucleic acid (DNA) extraction. 6–8 cores, each approximately 1mm in thickness, were taken from different areas with highest tumor cellularity. Case matched normal tissue from either the adjacent normal salivary gland parenchyma or benign neck lymph nodes was used as germline control for mutation filtering. DNA extraction was performed using truXTRAC® formalin fixed paraffin embedded (FFPE) DNA microTUBE kit (Covaris, Woburn, MA, USA) as per the manufacturer’s instructions. Fifteen samples that had sufficient DNA that passed the quality control (QC) checks using Illumina FFPE QC Kit were used for library preparation. The TruSeq Amplification Cancer Panel® (Illumina, San Diego, USA) was utilized to assess 48 clinically relevant genes: ABL1, ALK, AKT1, APC, ATM, BRAF, CDH1, CDKN2A, CSF1R, CTNNB1, EGFR, ERBB2, ERBB4, FBXW7, FGFR1, FGFR2, FGFR3, FLT3, GNA11, GNAQ, GNAS, HNF1A, HRAS, IDH1, JAK2, JAK3, KDR, KIT, KRAS, MET, MLH1, MPL, NOTCH1, NPM1, NRAS, PDGFRA, PIK3CA, PTEN, PTPN11, RB1, RET, SMAD4, SMARCB1, SMO, SRC, STK11, TP53, VHL. The reads were aligned to the human reference genome using Isis Smith-Waterman-Gotoh (v2.6), the variants were called using Illumina Somatic Variant Caller (v4.0) and annotated using Illumina variant studio (v2.3). Somatic variants were identified after deducting the normal/germline variants observed in the matched normal samples from those observed in the tumor samples. Variant positions with at least 500 X read depth and those variant alleles observed >5% were included in further analysis. In-silico analysis using PolyPhen 2, (Harvard, USA) and SIFT (J Craig Venter Institute, USA) was performed. Catalogue of Somatic Mutations in Cancer (COSMIC), Functional Analysis through Hidden Markov Models (FATHMM) for pathogenicity [51], Kyoto Encyclopedia of Genes and Genomes (KEGG) and GeneCards (Weizmann Institute of Science, Israel) databases were used for functional annotation of the identified variants and to understand their potential interactions in the relevant signal transduction pathways. The literature was extensively searched using Pubmed (English Language) for all changes identified as significant by COSMIC and FATHMM for their functional characterization in other malignancies, cell cultures or animal models. A literature search using PubMed for English language literature was performed to identify potential targetable gene variants with currently approved targeted therapies.
At an individual case level, specific genetic alterations with a described targeted therapy were sought by literature search and analyzed for potential suitability using experience from other cancers, drug availability and potential side effects.
SUPPLEMENTARY MATERIALS TABLE
Acknowledgments
Grant sponsor: Sydney Cancer Center Grant, Sydney Breast Cancer Foundation and the philanthropic support from Mr. David Paradice, the Tag Family Foundation, the O’Sullivan family, the Wald family, and ICAP.
Footnotes
CONFLICTS OF INTEREST
We do not have any conflicts of interest or financial disclosures to declare
REFERENCES
- 1.Guzzo M, Di Palma S, Grandi C, Molinari R. Salivary duct carcinoma: clinical characteristics and treatment strategies. Head Neck. 1997;19:126–33. doi: 10.1002/(sici)1097-0347(199703)19:2<126::aid-hed7>3.0.co;2-6. https://doi.org/10.1002/(SICI)1097-0347(199703)19:2<126::AID-HED7>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 2.Otsuka K, Imanishi Y, Tada Y, Kawakita D, Kano S, Tsukahara K, Shimizu A, Ozawa H, Okami K, Sakai A, Sato Y, Ueki Y, Sato Y, et al. Clinical Outcomes and Prognostic Factors for Salivary Duct Carcinoma: A Multi-Institutional Analysis of 141 Patients. Ann Surg Oncol. 2016;23:2038–45. doi: 10.1245/s10434-015-5082-2. https://doi.org/10.1245/s10434-015-5082-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Luk PP, Weston JD, Yu B, Selinger CI, Ekmejian R, Eviston TJ, Lum T, Gao K, Boyer M, O’Toole SA, Clark JR, Gupta R. Salivary duct carcinoma: clinicopathologic features, morphologic spectrum, and somatic mutations. Head Neck. 2016;38:E1838–47. doi: 10.1002/hed.24332. https://doi.org/10.1002/hed.24332. [DOI] [PubMed] [Google Scholar]
- 4.Chiosea SI, Williams L, Griffith CC, Thompson LD, Weinreb I, Bauman JE, Luvison A, Roy S, Seethala RR, Nikiforova MN. Molecular characterization of apocrine salivary duct carcinoma. Am J Surg Pathol. 2015;39:744–52. doi: 10.1097/PAS.0000000000000410. https://doi.org/10.1097/PAS.0000000000000410. [DOI] [PubMed] [Google Scholar]
- 5.Chan BA, Hughes BG. Targeted therapy for non-small cell lung cancer: current standards and the promise of the future. Transl Lung Cancer Res. 2015;4:36–54. doi: 10.3978/j.issn.2218-6751.2014.05.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, Hamid O, Schuchter L, Cebon J, Ibrahim N, Kudchadkar R, Burris HA, 3rd, Falchook G, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367:1694–703. doi: 10.1056/NEJMoa1210093. https://doi.org/10.1056/NEJMoa1210093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dai J, Kong Y, Si L, Chi Z, Cui C, Sheng X, Mao L, Li S, Lian B, Yang R, Liu S, Xu X, Guo J. Large-scale analysis of PDGFRA mutations in melanomas and evaluation of their sensitivity to tyrosine kinase inhibitors imatinib and crenolanib. Clin Cancer Res. 2013;19:6935–42. doi: 10.1158/1078-0432.CCR-13-1266. https://doi.org/10.1158/1078-0432.CCR-13-1266. [DOI] [PubMed] [Google Scholar]
- 8.Bose R, Kavuri SM, Searleman AC, Shen W, Shen D, Koboldt DC, Monsey J, Goel N, Aronson AB, Li S, Ma CX, Ding L, Mardis ER, Ellis MJ. Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer Discov. 2013;3:224–37. doi: 10.1158/2159-8290.CD-12-0349. https://doi.org/10.1158/2159-8290.CD-12-0349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alvarez RH, Valero V, Hortobagyi GN. Emerging targeted therapies for breast cancer. J Clin Oncol. 2010;28:3366–79. doi: 10.1200/JCO.2009.25.4011. https://doi.org/10.1200/JCO.2009.25.4011. [DOI] [PubMed] [Google Scholar]
- 10.El Zouhairi M, Charabaty A, Pishvaian MJ. Molecularly targeted therapy for metastatic colon cancer: proven treatments and promising new agents. Gastrointest Cancer Res. 2011;4:15–21. [PMC free article] [PubMed] [Google Scholar]
- 11.Bell SA, Tudur Smith C. A comparison of interventional clinical trials in rare versus non-rare diseases: an analysis of ClinicalTrials.gov. Orphanet J Rare Dis. 2014;9:170. doi: 10.1186/s13023-014-0170-0. https://doi.org/10.1186/s13023-014-0170-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Munoz J, Kurzrock R. Targeted therapy in rare cancers—adopting the orphans. Nat Rev Clin Oncol. 2012;9:631–42. doi: 10.1038/nrclinonc.2012.160. https://doi.org/10.1038/nrclinonc.2012.160. [DOI] [PubMed] [Google Scholar]
- 13.Billingham L, Malottki K, Steven N. Research methods to change clinical practice for patients with rare cancers. Lancet Oncol. 2016;17:e70–80. doi: 10.1016/S1470-2045(15)00396-4. https://doi.org/10.1016/S1470-2045(15)00396-4. [DOI] [PubMed] [Google Scholar]
- 14.Boyd N, Dancey JE, Gilks CB, Huntsman DG. Rare cancers: a sea of opportunity. Lancet Oncol. 2016;17:e52–61. doi: 10.1016/S1470-2045(15)00386-1. https://doi.org/10.1016/S1470-2045(15)00386-1. [DOI] [PubMed] [Google Scholar]
- 15.Mangone FR, Bobrovnitchaia IG, Salaorni S, Manuli E, Nagai MA. PIK3CA exon 20 mutations are associated with poor prognosis in breast cancer patients. Clinics (Sao Paulo) 2012;67:1285–90. doi: 10.6061/clinics/2012(11)11. https://doi.org/10.6061/clinics/2012(11)11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Janku F, Wheler JJ, Naing A, Falchook GS, Hong DS, Stepanek VM, Fu S, Piha-Paul SA, Lee JJ, Luthra R, Tsimberidou AM, Kurzrock R. PIK3CA mutation H1047R is associated with response to PI3K/AKT/mTOR signaling pathway inhibitors in early-phase clinical trials. Cancer Res. 2013;73:276–84. doi: 10.1158/0008-5472.CAN-12-1726. https://doi.org/10.1158/0008-5472.CAN-12-1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lui VW, Hedberg ML, Li H, Vangara BS, Pendleton K, Zeng Y, Lu Y, Zhang Q, Du Y, Gilbert BR, Freilino M, Sauerwein S, Peyser ND, et al. Frequent mutation of the PI3K pathway in head and neck cancer defines predictive biomarkers. Cancer Discov. 2013;3:761–69. doi: 10.1158/2159-8290.CD-13-0103. https://doi.org/10.1158/2159-8290.CD-13-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arjumand W, Merry CD, Wang C, Saba E, McIntyre JB, Fang S, Kornaga E, Ghatage P, Doll CM, Lees-Miller SP. Phosphatidyl inositol-3 kinase (PIK3CA) E545K mutation confers cisplatin resistance and a migratory phenotype in cervical cancer cells. Oncotarget. 2016;7:82424–82439. doi: 10.18632/oncotarget.10955. https://doi.org/10.18632/oncotarget.10955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang X, Ding J, Meng LH. PI3K isoform-selective inhibitors: next-generation targeted cancer therapies. Acta Pharmacol Sin. 2015;36:1170–76. doi: 10.1038/aps.2015.71. https://doi.org/10.1038/aps.2015.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guo T, Agaram NP, Wong GC, Hom G, D’Adamo D, Maki RG, Schwartz GK, Veach D, Clarkson BD, Singer S, DeMatteo RP, Besmer P, Antonescu CR. Sorafenib inhibits the imatinib-resistant KITT670I gatekeeper mutation in gastrointestinal stromal tumor. Clin Cancer Res. 2007;13:4874–81. doi: 10.1158/1078-0432.CCR-07-0484. https://doi.org/10.1158/1078-0432.CCR-07-0484. [DOI] [PubMed] [Google Scholar]
- 21.Kiessling MK, Curioni-Fontecedro A, Samaras P, Atrott K, Cosin-Roger J, Lang S, Scharl M, Rogler G. Mutant HRAS as novel target for MEK and mTOR inhibitors. Oncotarget. 2015;6:42183–96. doi: 10.18632/oncotarget.5619. https://doi.org/10.18632/oncotarget.5619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu JM, Han Y, Duan HQ, Gao EM, Zhang Y, Liu XQ, Zhang JS, Toschi L, Galetta D, Azzariti A, Paradiso A. EGFR mutations and HER2/3 protein expression and clinical outcome in Chinese advanced non-small cell lung cancer patients treated with gefitinib. J Cancer Res Clin Oncol. 2009;135:771–82. doi: 10.1007/s00432-008-0512-1. https://doi.org/10.1007/s00432-008-0512-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jia S, Gao X, Lee SH, Maira SM, Wu X, Stack EC, Signoretti S, Loda M, Zhao JJ, Roberts TM. Opposing effects of androgen deprivation and targeted therapy on prostate cancer prevention. Cancer Discov. 2013;3:44–51. doi: 10.1158/2159-8290.CD-12-0262. https://doi.org/10.1158/2159-8290.CD-12-0262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ettl T, Baader K, Stiegler C, Müller M, Agaimy A, Zenk J, Kühnel T, Gosau M, Zeitler K, Schwarz S, Brockhoff G. Loss of PTEN is associated with elevated EGFR and HER2 expression and worse prognosis in salivary gland cancer. Br J Cancer. 2012;106:719–26. doi: 10.1038/bjc.2011.605. https://doi.org/10.1038/bjc.2011.605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kechagioglou P, Papi RM, Provatopoulou X, Kalogera E, Papadimitriou E, Grigoropoulos P, Nonni A, Zografos G, Kyriakidis DA, Gounaris A. Tumor suppressor PTEN in breast cancer: heterozygosity, mutations and protein expression. Anticancer Res. 2014;34:1387–400. [PubMed] [Google Scholar]
- 26.Kataoka Y, Mukohara T, Shimada H, Saijo N, Hirai M, Minami H. Association between gain-of-function mutations in PIK3CA and resistance to HER2-targeted agents in HER2-amplified breast cancer cell lines. Ann Oncol. 2010;21:255–62. doi: 10.1093/annonc/mdp304. https://doi.org/10.1093/annonc/mdp304. [DOI] [PubMed] [Google Scholar]
- 27.Miller MB, Bi WL, Ramkissoon LA, Kang YJ, Abedalthagafi M, Knoff DS, Agarwalla PK, Wen PY, Reardon DA, Alexander BM, Laws ER, Jr, Dunn IF, Beroukhim R, et al. MAPK activation and HRAS mutation identified in pituitary spindle cell oncocytoma. Oncotarget. 2016;7:37054–63. doi: 10.18632/oncotarget.9244. https://doi.org/10.18632/oncotarget.9244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zoppoli G, Moran E, Soncini D, Cea M, Garuti A, Rocco I, Cirmena G, Grillo V, Bagnasco L, Icardi G, Ansaldi F, Parodi S, Patrone F, et al. Ras-induced resistance to lapatinib is overcome by MEK inhibition. Curr Cancer Drug Targets. 2010;10:168–75. doi: 10.2174/156800910791054211. https://doi.org/10.2174/156800910791054211. [DOI] [PubMed] [Google Scholar]
- 29.Šolman M, Ligabue A, Blaževitš O, Jaiswal A, Zhou Y, Liang H, Lectez B, Kopra K, Guzmán C, Härmä H, Hancock JF, Aittokallio T, Abankwa D. Specific cancer-associated mutations in the switch III region of Ras increase tumorigenicity by nanocluster augmentation. eLife. 2015;4:e08905. doi: 10.7554/eLife.08905. https://doi.org/10.7554/eLife.08905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yamamoto N, Minami S, Fujii M. Clinicopathologic study of salivary duct carcinoma and the efficacy of androgen deprivation therapy. Am J Otolaryngol. 2014;35:731–35. doi: 10.1016/j.amjoto.2014.07.007. https://doi.org/10.1016/j.amjoto.2014.07.007. [DOI] [PubMed] [Google Scholar]
- 31.Soper MS, Iganej S, Thompson LD. Definitive treatment of androgen receptor-positive salivary duct carcinoma with androgen deprivation therapy and external beam radiotherapy. Head Neck. 2014;36:E4–7. doi: 10.1002/hed.23383. https://doi.org/10.1002/hed.23383. [DOI] [PubMed] [Google Scholar]
- 32.Kaidar-Person O, Billan S, Kuten A. Targeted therapy with trastuzumab for advanced salivary ductal carcinoma: case report and literature review. Med Oncol. 2012;29:704–06. doi: 10.1007/s12032-011-9884-1. https://doi.org/10.1007/s12032-011-9884-1. [DOI] [PubMed] [Google Scholar]
- 33.Krishnamurthy J, Krishnamurty DM, Baker JJ, Zhen W, Lydiatt D, Ganti AK. Salivary duct carcinoma responding to trastuzumab-based therapy: case report and review of the literature. Head Neck. 2013;35:E372–75. doi: 10.1002/hed.23307. https://doi.org/10.1002/hed.23307. [DOI] [PubMed] [Google Scholar]
- 34.Limaye SA, Posner MR, Krane JF, Fonfria M, Lorch JH, Dillon DA, Shreenivas AV, Tishler RB, Haddad RI. Trastuzumab for the treatment of salivary duct carcinoma. Oncologist. 2013;18:294–300. doi: 10.1634/theoncologist.2012-0369. https://doi.org/10.1634/theoncologist.2012-0369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hyman DM, Puzanov I, Subbiah V, Faris JE, Chau I, Blay JY, Wolf J, Raje NS, Diamond EL, Hollebecque A, Gervais R, Elez-Fernandez ME, Italiano A, et al. Vemurafenib in Multiple Nonmelanoma Cancers with BRAF V600 Mutations. N Engl J Med. 2015;373:726–36. doi: 10.1056/NEJMoa1502309. https://doi.org/10.1056/NEJMoa1502309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Voest EE, Bernards R. DNA-Guided Precision Medicine for Cancer: A Case of Irrational Exuberance? Cancer Discov. 2016;6:130–32. doi: 10.1158/2159-8290.CD-15-1321. https://doi.org/10.1158/2159-8290.CD-15-1321. [DOI] [PubMed] [Google Scholar]
- 37.Dalin MG, Desrichard A, Katabi N, Makarov V, Walsh LA, Lee KW, Wang Q, Armenia J, West L, Dogan S, Wang L, Ramaswami D, Ho AL, et al. Comprehensive Molecular Characterization of Salivary Duct Carcinoma Reveals Actionable Targets and Similarity to Apocrine Breast Cancer. Clin Cancer Res. 2016;22:4623–33. doi: 10.1158/1078-0432.CCR-16-0637. https://doi.org/10.1158/1078-0432.CCR-16-0637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nashed M, Casasola RJ. Biological therapy of salivary duct carcinoma. J Laryngol Otol. 2009;123:250–52. doi: 10.1017/S0022215108002314. https://doi.org/10.1017/S0022215108002314. [DOI] [PubMed] [Google Scholar]
- 39.Haddad R, Colevas AD, Krane JF, Cooper D, Glisson B, Amrein PC, Weeks L, Costello R, Posner M. Herceptin in patients with advanced or metastatic salivary gland carcinomas. A phase II study. Oral Oncol. 2003;39:724–27. doi: 10.1016/s1368-8375(03)00097-6. https://doi.org/10.1016/S1368-8375(03)00097-6. [DOI] [PubMed] [Google Scholar]
- 40.Dillon LM, Miller TW. Therapeutic targeting of cancers with loss of PTEN function. Curr Drug Targets. 2014;15:65–79. doi: 10.2174/1389450114666140106100909. https://doi.org/10.2174/1389450114666140106100909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mazumdar T, Byers LA, Ng PK, Mills GB, Peng S, Diao L, Fan YH, Stemke-Hale K, Heymach JV, Myers JN, Glisson BS, Johnson FM. A comprehensive evaluation of biomarkers predictive of response to PI3K inhibitors and of resistance mechanisms in head and neck squamous cell carcinoma. Mol Cancer Ther. 2014;13:2738–50. doi: 10.1158/1535-7163.MCT-13-1090. https://doi.org/10.1158/1535-7163.MCT-13-1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nardi V, Sadow PM, Juric D, Zhao D, Cosper AK, Bergethon K, Scialabba VL, Batten JM, Borger DR, Iafrate AJ, Heist RS, Lawrence DP, Flaherty KT, et al. Detection of novel actionable genetic changes in salivary duct carcinoma helps direct patient treatment. Clin Cancer Res. 2013;19:480–90. doi: 10.1158/1078-0432.CCR-12-1842. https://doi.org/10.1158/1078-0432.CCR-12-1842. [DOI] [PubMed] [Google Scholar]
- 43.Guglin M, Cutro R, Mishkin JD. Trastuzumab-induced cardiomyopathy. J Card Fail. 2008;14:437–44. doi: 10.1016/j.cardfail.2008.02.002. https://doi.org/10.1016/j.cardfail.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 44.Kumar RJ, Barqawi A, Crawford ED. Adverse events associated with hormonal therapy for prostate cancer. Rev Urol. 2005;7(Suppl 5):S37–43. [PMC free article] [PubMed] [Google Scholar]
- 45.Cheung AS, Pattison D, Bretherton I, Hoermann R, Lim Joon D, Ho E, Jenkins T, Hamilton EJ, Bate K, Chan I, Zajac JD, Grossmann M. Cardiovascular risk and bone loss in men undergoing androgen deprivation therapy for non-metastatic prostate cancer: implementation of standardized management guidelines. Andrology. 2013;1:583–89. doi: 10.1111/j.2047-2927.2013.00093.x. https://doi.org/10.1111/j.2047-2927.2013.00093.x. [DOI] [PubMed] [Google Scholar]
- 46.Hirsh V. Managing treatment-related adverse events associated with egfr tyrosine kinase inhibitors in advanced non-small-cell lung cancer. Curr Oncol. 2011;18:126–38. doi: 10.3747/co.v18i3.877. https://doi.org/10.3747/co.v18i3.877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barnes L, Eveson JW, Reichart P, Sidransky D. World Health Organization classification of tumours: pathology and genetics of head and neck tumours. IARC Press: Lyon. 2005:430. [Google Scholar]
- 48.Simpson RH. Salivary duct carcinoma: new developments—morphological variants including pure in situ high grade lesions; proposed molecular classification. Head Neck Pathol. 2013;7(Suppl 1):S48–58. doi: 10.1007/s12105-013-0456-x. https://doi.org/10.1007/s12105-013-0456-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wolff AC, Hammond ME, Schwartz JN, Hagerty KL, Allred DC, Cote RJ, Dowsett M, Fitzgibbons PL, Hanna WM, Langer A, McShane LM, Paik S, Pegram MD, et al. American Society of Clinical Oncology, and College of American Pathologists American Society of Clinical Oncology/College of American Pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. J Clin Oncol. 2007;25:118–45. doi: 10.1200/JCO.2006.09.2775. https://doi.org/10.1200/JCO.2006.09.2775. [DOI] [PubMed] [Google Scholar]
- 50.Wolff AC, Hammond ME, Hicks DG, Dowsett M, McShane LM, Allison KH, Allred DC, Bartlett JM, Bilous M, Fitzgibbons P, Hanna W, Jenkins RB, Mangu PB, et al. American Society of Clinical Oncology, and College of American Pathologists Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. J Clin Oncol. 2013;31:3997–4013. doi: 10.1200/JCO.2013.50.9984. https://doi.org/10.1200/JCO.2013.50.9984. [DOI] [PubMed] [Google Scholar]
- 51.Shihab HA, Gough J, Cooper DN, Day IN, Gaunt TR. Predicting the functional consequences of cancer-associated amino acid substitutions. Bioinformatics. 2013;29:1504–10. doi: 10.1093/bioinformatics/btt182. https://doi.org/10.1093/bioinformatics/btt182. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.