

Abstract
As predictive markers for anti-EGFR therapy, KRAS and BRAF mutations are routinely detected in primary and metastatic colorectal cancer (CRC) cells, but seldom in circulating tumor cells (CTCs). Detecting mutations in CTCs could help explain mutational differences between tumor cells at local sites and distant metastases, thereby improving treatment outcomes. Here, we conducted a systematic review and meta-analysis to compare KRAS and BRAF mutations in paired CTCs and primary tumors from CRC patients, to detect any possible discordance. A total of 244 CRC patients from nine studies were included. Our subgroup meta-analysis demonstrated that the total odds ratio for mutations in CTCs was only 55% of that in primary tumors in the stage IV subgroup. We also found low heterogeneity among studies and differences in mutations between CTCs and primary tumors in the stage IV subgroup (I2 = 0%, P = 0.01). We observed a higher frequency of KRAS mutations in CTCs than in primary tumors at early stages (I + II), a similar frequency in stage III, and a lower frequency in stage IV. There were also differences among the Epcam-targeted CTC enrichment, PCR-based mutation profiling, and ≥ 3 CTCs enriched (I2 = 0%, P = 0.03) subgroups. These finding indicate mutational discordance between CTCs and primary CRCs, particularly in the stage IV and KRAS subgroups. We suggest large-sample studies stratified by clinical stage and KRAS subtype are urgently warranted to accurately evaluate mutational variations in CTCs compared to primary and metastatic CRC cells.
Keywords: colorectal cancer, KRAS mutation, BRAF mutation, circulating tumor cells, clinical stage
INTRODUCTION
Colorectal cancer (CRC) is the third leading cause of cancer-related death worldwide [1]. The endothelial growth factor receptor (EGFR) relative pathway regulates the expression of genes involved in the proliferation, angiogenesis, and metastasis of CRC cells [2, 3]. Mutations in the Kirsten rat sarcoma (KRAS) viral oncogene have been reported to be present in 30–40% of CRC patients and to correlate with clinical resistance to anti-EGFR drugs in metastatic CRCs [4–6]. Indeed, KRAS mutations are a predictive biomarker for anti-EGFR resistance in metastatic CRC (mCRC) [7, 8]. Furthermore, KRAS mutations in primary tumors correlate with poor prognosis and short term survival [9, 10]. Therefore, only CRC patients with wild type KRAS usually receive anti-EGFR therapy [11, 12]. BRAF (proto-oncogeneB-Raf), also part of the EGFR pathway, is also mutated in 10% of mCRC patients [8, 13, 14]. In addition, retrospective analyses have shown that wild-type BRAF is necessary for successful response to anti-EGFR therapies in metastatic CRCs (mCRCs) [13, 15]. KRAS and BRAF mutations together serve as predictive markers for anti-EGFR therapies [16, 17].
Recently, studies exploring the correlation between KRAS mutational status in primary and metastatic tumors of CRC patients revealed intra-tumor heterogeneity or mutation discordance after anti-EGFR treatment failure in patients with wild type KRAS [18–21]. Furthermore, other studies suggested that resistance to anti-EGFR therapy possibly stemmed from selection of preexisting minor sub-clones harboring mutations [5, 11]. The mutational status of metastatic tumors does not always correspond with those in primary lesions. Circulating tumor cells (CTCs) can carry mutant variants from local tumor sites to distant metastases [22]. Recent improvements in DNA-sequencing technology have allowed high resolution by exploring sub-clone heterogeneity between primary and metastatic tumors [23, 24], but recovering sufficient tumor cells is still challenging to exceed the available sequencing analytical platforms [25]. Genotyping of CTCs might improve the monitoring of response to targeted EGFR therapies by identifying genomic profiles and predicting disease metastasis prior to clinical progression [26–29]. High concordance in mutations has been observed between CTCs and primary tissues in mCRC patients and the presumption that discordance exists in both wild type and mutation sub-populations has been challenged [30–34]. It is also possible that mutated sub-clones in primary tumors shed as CTCs into peripheral blood were not present in the tumor tissues used for genomic analyses [35–38]. On the other hand, some studies have detected mutations in CTCs at the sub-clone level by deep sequencing of primary tumor tissues [29]. However, based on genotype and phenotype profiling, accurate detection of the mutational status of CTCs is still challenging [39]. Here, we tested for KRAS and BRAF mutations in CTCs and paired primary CRCs. We also investigated genetic heterogeneity between CTCs and primary tumors.
MATERIALS AND METHODS
The protocol of our system review was published in PROSPERO: CRD42016042107
Search strategy
The identification of potentially relevant studies was performed through a comprehensive and systematic search in PubMed, EMBASE, Web of Science Databases, and Google Scholar by using the following keywords “colorectal cancer”, “circulating tumor cells” and “mutation”. Searching details: “neoplastic cells, circulating” [MeSH Terms] OR (“neoplastic” [All Fields] AND “cells” [All Fields] AND “circulating” [All Fields]) OR “circulating neoplastic cells” [All Fields] OR (“circulating” [All Fields] AND “tumor” [All Fields] AND “cell” [All Fields]) OR “circulating tumor cell” [All Fields] AND (“colorectal neoplasms” [MeSH Terms] OR (“colorectal” [All Fields] AND “neoplasms” [All Fields]) OR “colorectal neoplasms” [All Fields] OR (“colorectal” [All Fields] AND “cancer” [All Fields]) OR “colorectal cancer” [All Fields]) AND “mutation” [MeSH Terms] OR “mutation” [All Fields]. The latest search was updated on September 30, 2016. Bibliographies of eligible studies, review articles, the reference lists of each selected study, and other relevant publications were also reviewed to identify all potentially relevant studies.
Inclusion and exclusion criteria
To qualify as relevant, a study had to fulfill the following criteria: (1) circulating tumor cells were enriched and isolated from CRC patients; (2) KRAS and BRAF mutations had to be present in CTCs and primary tumors; (3) the correlations of KRAS mutation in CTCs and paired primary tissues were assessed; (4) the correlations of KRAS mutation in CTCs and paired primary tissues based on treatment outcome (progression or stable) were assessed; (5) the correlations of BRAF mutation in CTCs and paired primary tissues were assessed; (6) to have been published as a full paper in English up until September 30, 2016. Studies were excluded from our analysis if any of the following conditions occurred: (1) not analyzing KRAS or BRAF mutation in CTCs; (2) CTCs-related analyses without KRAS or BRAF mutation; (3) the samples for KRAS or BRAF mutation analyses were not from CRC patients; (4) small sample analysis (less than four cases); (5) non-human sample analysis.
Data extraction and quality assessment
Two investigators independently screened the studies and extracted the data from included studies by using standard data abstraction forms. For each study, relevant data were compiled as follows: name of first author, year of publication, total number of patients included, patients’ gender, tumor location (colon/rectum), clinical stage, time of blood sample draws, number of patients with CTCs detected, cutoff number of CTCs, enrichment method and antibody staining of CTCs, number of mutations in CTCs, tissue samples, and in both CTCs and tissue samples combined, subtype mutation of CTCs, methods for mutation detection, tumor status (stable vs progression), and NOS score. The mutational status of paired CTCs and tumor tissue samples from each patient along with clinical data were evaluated. Then the data from a total of nine qualifying studies were used for further analysis. The quality of each study was independently assessed by two investigators using the Newcastle–Ottawa Scale (NOS) [40].
Statistical methods
Data were presented as odds ratio (OR) with its 95% confidence interval(CI) to show the agreement of KRAS and BRAF mutation in paired CTCs and primary tumor samples while risk ratios (RR) at 95% CI were presented to show the agreement of KRAS mutation in paired CTCs and tissue samples based on tumors status (stable or progression). The individual OR and RR were combined into pooled ORs and RRs, and initial analyses were performed with a fixed effect model assuming homogeneity in the individual studies. Heterogeneity was evaluated by Q-test and I2 test. When statistically significant heterogeneity among the studies was found by Q-test (P < 0.05) or I2 > 50%, subgroups were classified by gene subtype, stage, and CTC enrichment, with isolation approach stratification or random effect models being applied for further meta-analysis. Otherwise, the fixed effects model was adopted. Sensitivity analyses were conducted to identify whether results of the meta-analysis were affected by exclusion of any individual study and to testify the reliability of the conclusions. All P values were 2-sided and all analyses were performed using Review Manager 5.3.
RESULTS
Overview of included studies and quality assessment
From 317 studies retrieved, nine studies that focus on comparing CTC-related mutations with paired tumor tissue of CRCs were included for systematic review (See Figure 1, Table 1). Table 1 summarizes details as name of first author (year of publication), total number of patients included, gender, tumor location, clinical stage, time of blood sample draws, number of patients with CTCs, cutoff number of CTCs, enrichment method and antibody staining of CTCs, number of mutation in CTCs, tissue samples, and in both CTCs and tissue samples combined, subtype mutation of CTCs, methods for mutation detection, tumor status, and NOS score. According to the NOS quality assessment, all the selected studies have high quality with a median score of 8.11 stars and were thus subject to further meta-analyses. Among the total 315 CRC patients, 181 (57.46%) were males and 134 (42.54%) females, with 228 (72.38%) colon carcinomas and 87 (27.62%) rectal carcinomas. Among 315 CRC patients, KRAS and BRAF mutations were detected in CTCs and primary tumors from 244 CRC patients. KRAS codon12 and codon13 mutations were detected in 28.27% (69/244) and 5.73% (14/244) of the cases, separately, while BRAF mutations were detected in 5.11% (7/137) of the cases. According to the UICC Classification of Colorectal Cancer, eight cases (3.28%) were classified as stage I, 24 cases (9.83%) as stage II, 47 cases (19.26 %) as stage III, and stage IV had the majority population with 165 cases (67.62%). The concordance of KRAS mutation in CTCs with paired primary tissues of CRCs was compared in all nine studies, but only four studies were evaluated for BRAF mutation of CTCs, three studies were analyzed for KRAS mutation of CTCs with tumor status, and one study assessed mutations among primary tumors, CTCs, and metastatic lesions [30–38].
Figure 1. Diagram for retrieval of studies.
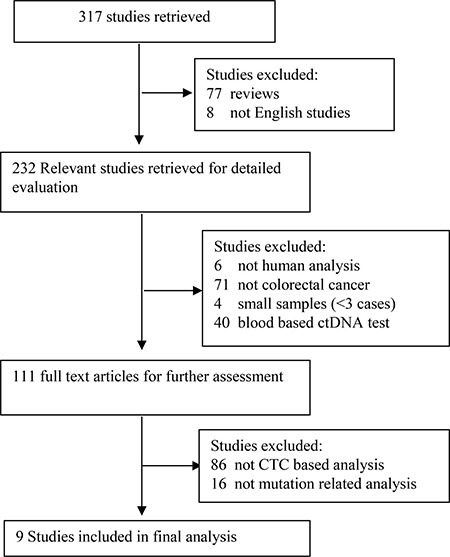
Table 1. Characteristics of studies involved in mutation analysis of CTCs.
| First author (year of publication) |
1 Buim, M.E (2015) | 2 Fabbri, F (2013) | 3 Harb, W (2013) | 4 Raimondi, C (2014) | 5 Kalikaki, A (2014) | 6 Lyberopoulou, A (2015) | 7 Mostert, B (2013) | 8 Steinert G (2014) | 9 Mohamed Suhaimi, N.A (2015) | Total |
|---|---|---|---|---|---|---|---|---|---|---|
| Total number of patients included | 26 | 40 | 15 | 40 | 31 | 52 | 43 | 28 | 40 | 315 |
| Patients’ gender (male/female) |
18/8 | 24/16 | 11/4 | 15/25 | 22/9 | 33/19 | 28/15 | 11/17 | 19/21 | 181/134 |
| Tumor location (colon/rectal) |
16 /10 | 29/11 | 12/3 | 28/12 | 31/0 | 41/11 | 26/17 | 23/5 | 22/18 | 228/87 |
| Number of patients with CTCs detected | 21 | 16 | 14 | 30 | 23 | 52 | 26 | 22 | 40 | 244 |
| Clinical stage | IV 21 | IV 16 | II 4 III 2 IV 8 |
IV 30 | IV 23 | II 6 III 24 IV 22 |
IV 26 | III 6 IV 16 |
I 8 II 14 III 15 IV 3 |
I 8 II 24 III 47 IV 165 |
| Time of blood sample draws | After chemotherapy and monoclonal therapy | Before treatment | After operation | After chemotherapy and monoclonal therapy | After chemotherapy and monoclonal therapy | Before treatment | Before tumor resection | Before or in the operation | After operation | |
| Cutoff number of CTCs | 2 cells | 5 cell | 5 cells | 1 cell | 1 cell | 2 cells | 3 cells | 2 cells | 1 cell | |
| Enrichment and antibody staining of CTCs | Isolation by size of epithelial tumors ( ISET) CD45- cells |
Density gradient centrifugation CK +/Hoechst +/ CD45- cells |
IsoFlux System EpCAM+ cell |
CellSearch EPCAM+/CK+/ DAPI +/CD45- cells |
CellSearch EPCAM+/CK+/ DAPI +/CD45- cells |
Density gradient centrifugation EpCAM+/ Vimentin+/CK+/CD45- cells |
CellSearch TM EPCAM+ |
CellSearch System EPCAM+/CK+ cells |
Size-based filtration unit CK+/DAPI+/ CD45- cell |
|
| Number of mutation in CTCs detected | KRAS: codons12 7/21 |
KRAS: codons12 3/16 |
KRAS: codons12 1/14 codons13 5/14 |
KRAS: codons12 6/30 |
KRAS: codons12 6/23 codons13 1/23 |
KRAS: codons12 29/52 codons13 0/52 BRAF: 4/52 |
KRAS: codons12 4/26 codons13 1/26 BRAF: 1/26 |
KRAS: codons12 4/22 codons13 2/22 BRAF: 1/19 |
KRAS: codons12 9/40 codons13 5/40 BRAF: 1/40 |
KRAS: codons12 69/244 codons13 14/244 BRAF: 7/137 |
| Number of mutation in tissue samples | KRAS: codons12 6/21 codons13 3/21 |
KRAS: codons12 6/16 codons13 3/16 |
KRAS: codons12 1/14 codons13 1/14 |
KRAS: codons12 13/30 |
KRAS: codons12 7/23 codons13 1/23 |
KRAS: codons12 26/52 codons13 0/52 BRAF: 6/52 |
KRAS: codons12 8/26 codons13 1/26 BRAF: 1/26 |
KRAS: codons12 8/22 codons13 1/22 BRAF: 3/19 |
KRAS: codons12 9/40 codons13 2/40 BRAF: 5/ 40 |
KRAS: codons12 84/244 codons13 12/244 BRAF: 15/137 |
| Number of mutations in both CTC and tissue samples | KRAS: 5 cases |
KRAS: 2 cases |
KRAS: 1 case |
KRAS: 2 cases |
KRAS: 5 cases |
KRAS: 26 cases BRAF: 4 cases |
KRAS: 4 cases BRAF: 1 case |
KRAS: 5 cases BRAF: 1 case |
KRAS: 9 cases BRAF: 1 case |
KRAS: 59 cases BRAF: 7 case |
| Subtype mutation of CTCs | KRAS codons12 codons13 | KRAS codons12 codons13 | KRAS codons12 codons13 | KRAS codons12 codons13 | KRAS codons12 codons13 | KRAS codons12 codons13 BRAF (V600E) |
KRAS codons12 codons13 BRAF (V600E) |
KRAS codons12 codons13 BRAF (V600E) |
KRAS codons12 codons13 BRAF (V600E) |
|
| Methods for mutative detection of CTCs | Pyrosequencing | Pyrosequencing | castPCR | RT-PCR | PNA-mediated PCR | RFLP assay and ASPCR | nested ASB PCR | aCGH PCR | HRM assay and ASPCR | |
| Tumor status (progression/stable) | 5/16 | NA | 7/6 | NA | 6/17 | NA | NA | NA | NA | 18/39 |
| NOS score | 8 | 8 | 8 | 8 | 9 | 8 | 8 | 8 | 8 | 8.11 (mean) |
NA: not available; castPCR: Competitive Allele-Specific TaqMan PCR; PNA: peptide nucleic acid; RFLP: restriction fragment length polymorphism; nested ASB PCR: nested Allele-Specific Blocker PCR; aCGH PCR: array comparative genomic hybridization PCR; HRM: High resolution melt; ASPCR: Allele-specific PCR; NOS score: Newcastle–Ottawa Scale score.
Correlation of KRAS mutation in paired CTCs and primary tumors
We summarized the data from all nine studies based on KRAS mutation with stage and codon subgroup stratification (See Table 2). From the results calculated by McNemar Test and Kappa value, significant discordance (McNemar value < 0.001 for codon12+13 and codon12, 0.289 for codon13) and poor agreement (Kappa value 0.377 for codon12+13, 0.397 for codon12 and 0.476 for codon13) of mutations in paired CTCs and primary CRC tumors were observed in the stage IV subgroup. Conversely, high concordance and better agreement between those two study populations (McNemar value 0.687 for codon12+13 and 0.500 for codon12, Kappa value 0.744 for codon12+13 and 0.913 for codon12) were observed in the stage III subgroup.
Table 2. Relationship of KRAS mutation in paired CTCs with tissue samples of CRC patients based on stage III and IV stratification.
| Stage IV | Stage III | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CTCs (n) | McNemar-Test | Kappa | P value | CTCs (n) | McNemar-Test | Kappa | P value | |||||||
| Mutation Codon 12+13 | Wild type | Total | Mutation Codon 12+13 | Wild type | Total | |||||||||
| Tissue sample (n) |
Mutation Codon 12+13 | 35 | 39 | 74 | < 0.001* | 0.377 | < 0.001* | Mutation Codon 12+13 | 19 | 2 | 21 | 0.687 | 0.744 | < 0.001* |
| Wild type | 10 | 81 | 91 | Wild type | 4 | 22 | 26 | |||||||
| Total | 45 | 120 | 165 | Total | 23 | 24 | 47 | |||||||
| Mutation Codon12 |
Wild type | Total | McNemar-Test | Kappa | P value | Mutation Codon12 | Wild type | Total | McNemar-Test | Kappa | P value | |||
| Tissue sample (n) |
Mutation Codon12 |
30 | 34 | 64 | < 0.001* | 0.397 | < 0.001* | Mutation Codon12 | 19 | 2 | 21 | 0.500 | 0.913 | < 0.001* |
| Wild type | 10 | 91 | 101 | Wild type | 0 | 26 | 26 | |||||||
| Total | 40 | 125 | 165 | Total | 19 | 28 | 47 | |||||||
| Mutation Codon13 |
Wild type | Total | McNemar-Test | Kappa | P value | Mutation Codon13 |
Wild type | Total | McNemar-Test | Kappa | P value | |||
| Tissue sample (n) |
Mutation Codon13 |
4 | 6 | 10 | 0.289 | 0.476 | < 0.001* | Mutation Codon13 | 0 | 0 | 0 | NA | NA | NA |
| Wild type | 2 | 153 | 155 | Wild type | 4 | 43 | 47 | |||||||
| Total | 6 | 159 | 165 | Total | 4 | 43 | 47 | |||||||
*:indicate P <0.05, NA: not available.
| Stage IV | Stage III | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CTCs (n) | McNemar-Test | Kappa | P value | CTCs (n) | McNemar-Test | Kappa | P value | |||||||
| Mutation Codon 12+13 | Wild type | Total | Mutation Codon 12+13 | Wild type | Total | |||||||||
| Tissue sample (n) |
Mutation Codon 12+13 | 33 | 41 | 74 | < 0.001* | 0.321 | < 0.001* | Mutation Codon 12+13 | 17 | 4 | 21 | 1.000 | 0.614 | < 0.001* |
| Wild type | 13 | 77 | 90 | Wild type | 5 | 21 | 26 | |||||||
| Total | 46 | 118 | 164 | Total | 22 | 25 | 47 | |||||||
| Mutation Codon12 |
Wild type | Total | McNemar-Test | Kappa | P value | Mutation Codon12 | Wild type | Total | McNemar-Test | Kappa | P value | |||
| Tissue sample (n) |
Mutation Codon12 |
29 | 36 | 65 | 0.001* | 0.335 | < 0.001* | Mutation Codon12 | 20 | 2 | 22 | 0.500 | 0.914 | < 0.001* |
| Wild type | 13 | 86 | 99 | Wild type | 0 | 25 | 25 | |||||||
| Total | 42 | 122 | 164 | Total | 20 | 27 | 47 | |||||||
| Mutation Codon13 |
Wild type | Total | McNemar-Test | Kappa | P value | Mutation Codon13 |
Wild type | Total | McNemar-Test | Kappa | P value | |||
| Tissue sample (n) |
Mutation Codon13 |
5 | 27 | 32 | < 0.001* | 0.147 | 0.025* | Mutation Codon13 | 4 | 0 | 4 | 0.125 | 0.624 | < 0.001* |
| Wild type | 6 | 126 | 132 | Wild type | 4 | 39 | 43 | |||||||
| Total | 11 | 153 | 164 | Total | 8 | 39 | 47 | |||||||
Pooled data analysis of KRAS mutation in paired CTCs and primary tumors (stage I–IV)
With reference to stage I-IV CRC patients, the odds ratio (OR) of paired CTCs and tumor tissues was 0.87 (95% CI; 0.60, 1.26) for KRAS codon12+13 mutation, 0.77 (95% CI; 0.51, 1.14) for KRAS codon12 mutation, and 1.32 (95% CI; 0.53, 3.29) for KRAS codon13 mutation (see Figure 2). No heterogeneity or discordance between studies was observed for three KRAS sub-type mutations in CRC patients (I2 = 27%, test for overall effect P = 0.45 for codon12+13, I2 = 0%, P = 0.19 for codon12 and I2 = 0%, P = 0.55 for codon13).
Figure 2. Analyses of KRAS codon12+13, codon12 and codon13 mutation in paired CTCs and primary tumors (stage I-IV).
Pooled data analysis of KRAS sub-type mutation in paired CTCs and primary tumors (stage IV)
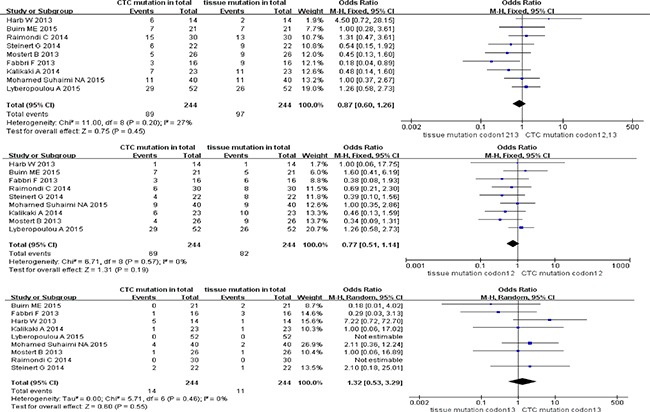
With reference to KRAS sub-type mutation in stage IV CRC patients, high homogeneity (all I2 = 0%) between studies and pooled ORs was observed with 0.55 (95% CI; 0.35, 0.88) for KRAS codon12+13 mutation, 0.69 (95% CI; 0.43, 1.11) for KRAS codon12 mutation, and 0.59 (95% CI; 0.21, 1.64) for KRAS codon13 mutation. Furthermore, only the KRAS codon12+13 mutation sub-type group presented discordance of mutation ratio in paired CTCs and primary tumors (P = 0.01 for codon12+13 mutation, P = 0.12 for codon12 mutation, and P = 0.31 for codon13 mutation) (Figure 3).
Figure 3. Pooled data analysis of KRAS codon12+13, codon12, codon13 mutation in paired CTCs and primary tumors (stage IV).
Pooled data analysis of KRAS sub-type mutation in paired CTCs and primary tumors (stage III)
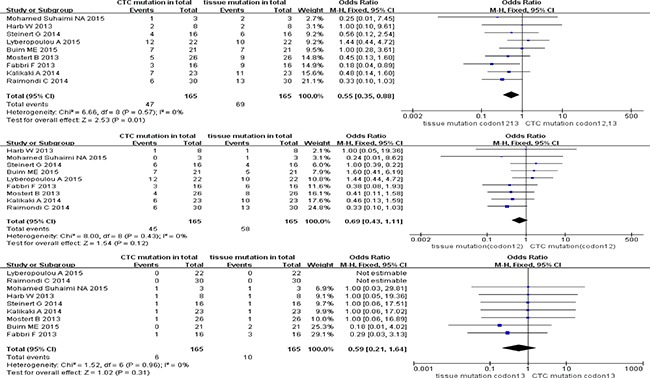
The pooled ORs of KRAS sub-type mutation of paired CTCs and primary tumors in stage III were 1.19 (95% CI; 0.53, 2.69) for KRAS codon12+13 mutation, 0.91 (95% CI; 0.38, 2.16) for KRAS codon12 mutation, and 6.83 (95% CI; 0.91, 51.00) for KRAS codon13 mutation. All three KRAS sub-type groups showed high homogeneity (I2 = 0) of studies and concordance of mutation ratio in paired CTCs and primary tumor cohorts (test for overall effect P range from 0.06 to 0.82) (Figure 4).
Figure 4. Pooled data analysis of KRAS codon12+13, codon12, codon13 mutation in paired CTCs and primary tumors (stage III).
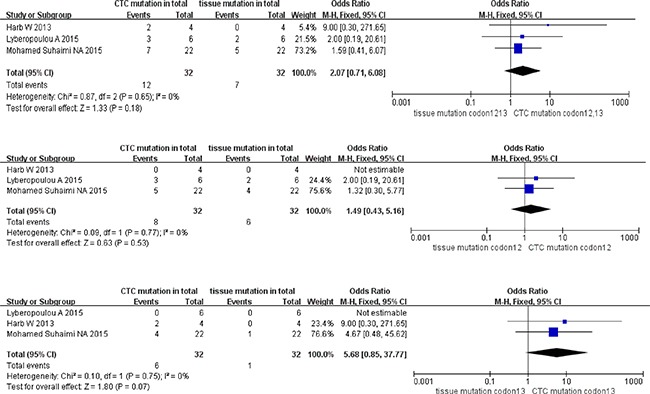
Pooled data analysis of KRAS sub-type mutation in paired CTCs and primary tumors (stage I + II)
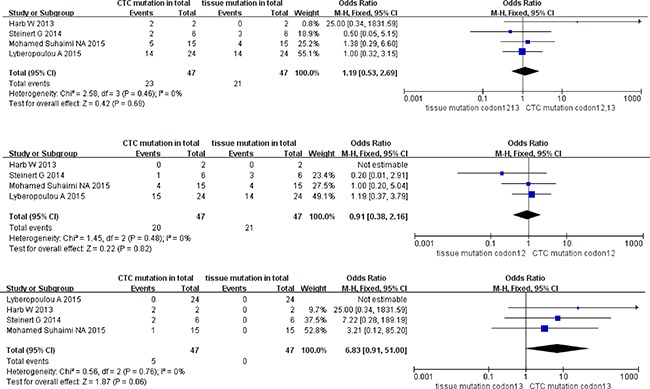
The total ORs of KRAS sub-type mutation in paired CTCs and primary tumors at stage I+II were 2.07 (95% CI; 0.71, 6.08) for KRAS codon12+13 mutation, 1.49 (95% CI; 0.43, 5.16) for KRAS codon12 mutation, and 5.68 (95% CI; 0.85, 37.77) for KRAS codon13 mutation. However, no statistic divergence was observed in paired CTCs and primary tumors despite high homogeneity in all three KRAS sub-type groups (all I2 = 0% with P = 0.18 for codon12+13 mutation, P = 0.53 for codon12 mutation and P = 0.07 for codon13 mutation) (Figure 5).
Figure 5. Pooled data analysis of KRAS codon12+13, codon12, codon13 mutation in paired CTCs and primary tumors (stage I–II).
Pooled data analysis of subgroup stratified as enrichment methods of CTC, methods for detecting CTC mutations, and cutoff number of CTCs (stage IV)
We classified the qualifying studies based on enrichment approach of CTCs into a subset of six studies with Epcam-targeted and three without Epcam-targeted CTC enrichment. We stratified such subsets for further analysis. As a result, ORs in paired CTCs and primary tumors were 0.58 (95% CI: 0.34, 0.99) and 0.47 (95% CI: 0.19, 1.19) for the subsets with and without Epcam-targeted enrichment, respectively. We observed low heterogeneity and significant discordance in studies using the Epcam-targeted enrichment approach (I2 = 0%, P = 0.05) (Figure 6A). In parallel, we divided the methods for detecting mutations in CTCs into PCR-based (OR 0.57, 95% CI: 0.34, 0.96) and pyrosequencing-based (OR 0.45, 95% CI: 0.08, 2.43) subgroups. We found low heterogeneity in studies and divergence of KRAS mutation ratio in the PCR-based subgroup (I2 = 0%, P = 0.04) (Figure 6B). Finally, the cutoff number of CTCs was used to classify the studies as 1–2 CTCs (OR 0.64, 95% CI: 0.37, 1.09) and ≥ 3 CTCs (OR 0.38, 95% CI: 0.16, 0.92) subgroups. We found low heterogeneity in studies with significant difference of OR in the ≥ 3 CTCs subgroup (I2 = 0%, P = 0.03) (Figure 6C).
Figure 6A. Subgroup analysis based on enrichment method of CTC in paired CTCs and primary tumors (stage IV).
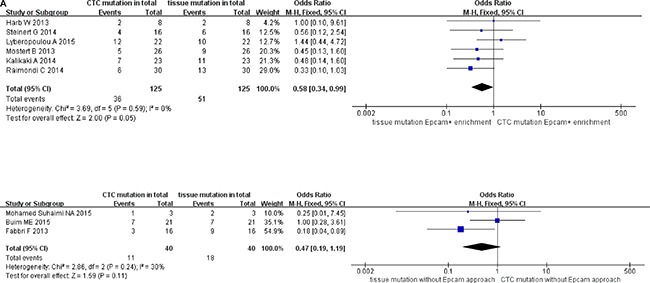
Figure 6B. Subgroup analysis based on methods for detecting CTC mutations in paired CTCs and primary tumors (stage IV).
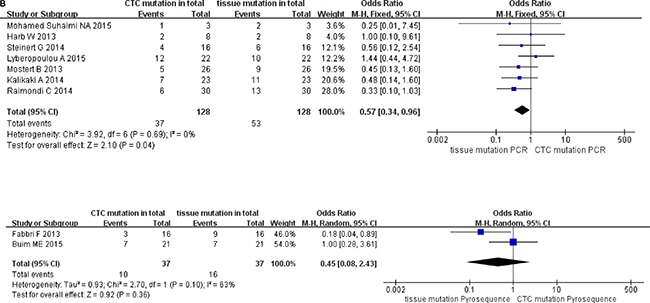
Figure 6C. Subgroup analysis based on cutoff number of CTCs in paired CTCs and primary tumors (stage IV).
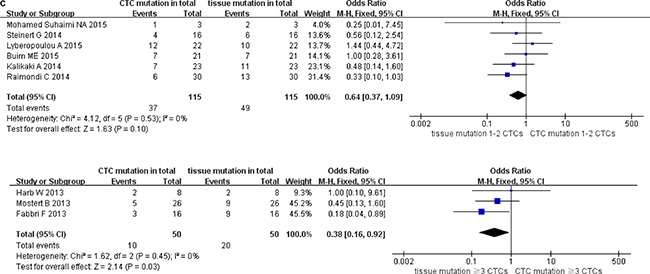
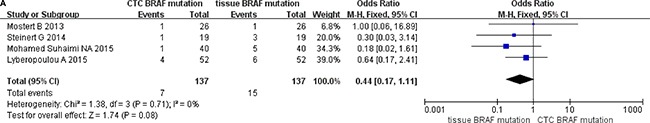
Data analysis of BRAF mutation in paired CTCs with primary tumors
A total of 137 cases from four studies were analyzed based on detection of BRAF mutations in paired CTCs with primary tumors of CRC patients. The pooled OR was 0.44 (95% CI: 0.14, 1.11); however, we found no difference in paired subgroup populations with high homogeneity between studies (I2 = 0%, P = 0.08) (Figure 7A).
Figure 7A. Data analysis of BRAF mutation in paired CTCs and primary tumors.
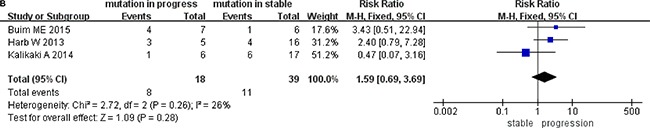
Data analysis of tumor status in CTCs with KRAS mutation
By comparing the differences between the tumor progression and the stable cohorts in CTCs with KRAS mutation, the risk ratio associated with different tumor status was 1.59 (95% CI; 0.69, 3.69). We saw no difference between studies, and measured low heterogeneity (I2 = 26%, P = 0.28) (Figure 7B).
Figure 7B. Data analysis of tumor status in CTCs with KRAS mutation.
DISCUSSION
Our meta-analysis compared the discordance of both KRAS and BFRF mutation in paired CTCs with primary tumors from CRC patients. Variations in genotype and phenotype between primary tumors and metastasis are effective prognosis biomarkers [15, 41, 42]. However, because of lacking specific tumor biomarkers and the limitations of molecular profiling technology for single cell analysis, CTC-related mutations in CRC are still under active investigation. Even though numerous studies correlated CTC counts with prognosis, we found here through our meta-analysis of the literature that few mutation-centric CRC studies focus on CTCs. From nine studies included, only one study compared KRAS and BRAF mutations in primary tumors, and CTCs and metastatic lesions. However, the data of mutations from metastatic lesions were not included in our meta-analysis because no other similar studies were available to carry out statistics [30]. Additional studies comparing the mutational status of CTCs combined with primary tumors and metastatic lesions are required in future CRC-related analysis.
Our findings here revealed discrepancies between KRAS mutations in CTCs and paired primary tumors by pooled meta-analysis. Even though high concordance between primary tumors and clinical metastases has been previously demonstrated, discordant sub-populations of genetic mutation intra-tumor or during the metastatic process are commonly reported [10, 14, 21, 43]. We grouped individual data drawn from each qualifying study into stage IV, stage III, and stage I + II subgroups. We observed high discordance of mutation in CTCs with paired primary tumors in the stage IV and III subgroups, demonstrating that CTCs do not always show genotype agreement with primary lesions, especially in metastatic patients [14, 18–20]. However, the limited number of samples from only three studies in the early stage subgroup, as well as few CTCs enriched in peripheral blood, decreased the confidence of our analysis for the stage I + II subgroup. Therefore, a larger cohort will be necessary to explore the molecular properties of CTCs and their relationship with primary tumors in CRC patients at early stages.
Recently, clinical and experimental findings demonstrated that in human CRCs, KRAS codon12 mutations were much more frequent in metastatic than in non-metastatic tumors, and were more aggressive than KRAS codon13 mutation [44–46]. Our findings revealed a higher frequency for codon12 mutations than for codon13 mutations in both CTCs (69 cases:14 cases) and primary tumors (84 cases:12 cases), in agreement with previous findings [16, 44–46]. Additionally, we found more KRAS mutations in CTCs than in primary tumors at early stage (stage I + II), the same in stage III, but fewer in stage IV. This higher frequency of KRAS mutations that we observed in CTCs may correlate with their aggressiveness and resistance to anti-tumor therapy. Our analysis demonstrated more KRAS mutations in CTCs than in tumor tissues at an early stage, but lower KRAS mutations in advanced stages of CRC. Furthermore, the decrease of KRAS codon12 mutations in CTCs at stage I–II vs stage IV suggested that codon12 mutations may be a more sensitive biomarker for dynamically tracking mutation variation in CTCs.
According to recent reports, Epcam- CTC can be more aggressive than Epcam+ CTC, but the EMT process renders Epcam- CTCs undetectable, which decreases the clinical application of molecular profiling for CTC enrichment based on Epcam [47–49]. To decrease selection bias, we performed subgroup analysis based on Epcam expression in stage IV patients. The six Epcam-targeted subgroups showed disagreement between CTCs and paired tumor tissues in stage IV patients, suggesting that target Epcam enrichment might be more suitable for identifying aggressive sources of CTCs with mutations than profiles based on size and density gradient. A similar result was found through gene sequencing and RT-PCR. PCR-based techniques for detecting genetic alterations have been proposed to provide advantages such as high sensitivity and specificity, albeit at a high cost [50–53]. Low-level mutations sometimes cannot be detected by conventional Sanger sequencing when they are masked by a high background level of wild-type [50]. The ≥ 3 CTCs subgroup showed significant discordance of CTCs with paired tumor tissues, suggesting that increased numbers of CTCs may account for increased mutations. Based on our analyses, Epcam-targeted CTC enrichment, PCR-based mutation profiling, and ≥ 3 CTCs enrichment may improve comparisons in mutation variation between CTCs and paired tumor tissues [36, 48–50, 53]. In addition to KRAS mutations, BRAF mutations are reported only in 8%–15% of CRC cells and are associated with poor response to anti-EGFR therapy [8, 54]. We also analyzed discordance of BRAF V600E mutation between CTCs and paired primary CRC tumors but detected no differences. Due to the low proportion of BRAF mutation in primary tumors and CTCs, BRAF mutations were not very significant to our meta-analysis. Considering the small number of studies included in our analysis, further studies with larger cohorts are warranted.
We observed a high concordance of KRAS status was observed in CTCs and primary tumors, contrary to discrepancies reported in previous studies [30, 36, 37]. We also analyzed clinical stage, mutation sub-type, methods of CTC enrichment, mutation profiling, and cutoff number of CTCs to decrease bias between studies. However, certain limitations must be considered when interpreting our results. First, only nine studies were included in our meta-analysis, with few samples per study further limiting subgroup analyses. Second, each study had insufficient samples regarding various characteristics such as stage, mutation sub-type, cutoff number of CTCs, CTC enrichment methods, and mutation profiling, which yielded few samples in each subgroup. However, homogeneity was observed in stage IV subgroups and other KRAS sub-types, suggesting the necessity of subgroup partition based on stage and codon type in future studies. Third, publication bias might affect our results even though no significant publication bias was observed in our meta-analysis. Despite these limitations, our meta-analysis evaluated the effects of KRAS and BRAF mutation in CTCs of CRC patients by using all qualifications with intensive subgroup analyses. Therefore, large sample studies of CRCs focusing on more detailed characteristics are urgently needed to investigate genotype profiling not only regarding mutational variations, but also tumor progression and prognosis. In conclusion, our analyses have provided insight into the molecular profiling and genetic detection of mutational variations in paired CTCs and primary tumors. We found mutation discordance between CTCs and primary CRCs, particularly in the stage IV and KRAS subgroups. Large sample studies stratified by clinical stage and KRAS sub-type are warranted to accurately evaluate mutational variations in CTCs compare to primary and metastatic CRC cells.
ACKNOWLEDGMENTS AND FUNDING
This work was funded by grants from the Medicine and Health Science Research Fund of Zhejiang Province (no. 2008B021) and the Science Foundation for Fostering Talents of Zhejiang Cancer Hospital (no. 2012YC008).
Footnotes
CONFLICTS OF INTEREST
None to declare.
REFERENCES
- 1.Siegel R, Desantis C, Jemal A. Colorectal cancer statistics, 2014. CA Cancer J Clin. 2014;64:104–17. doi: 10.3322/caac.21220. [DOI] [PubMed] [Google Scholar]
- 2.Phipps AI, Buchanan DD, Makar KW, Win AK, Baron JA, Lindor NM, Potter JD, Newcomb PA. KRAS-mutation status in relation to colorectal cancer survival: the joint impact of correlated tumour markers. Br J Cancer. 2013;108:1757–64. doi: 10.1038/bjc.2013.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Karapetis CS, Khambata-Ford S, Jonker DJ, O’Callaghan CJ, Tu D, Tebbutt NC, Simes RJ, Chalchal H, Shapiro JD, Robitaille S, Price TJ, Shepherd L, Au HJ, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. 2008;359:1757–65. doi: 10.1056/NEJMoa0804385. [DOI] [PubMed] [Google Scholar]
- 4.Jancík S, Drábek J, Radzioch D, Hajdúch M. Clinical relevance of KRAS in human cancers. J Biomed Biotechnol. 2010;2010:150960. doi: 10.1155/2010/150960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, Valtorta E, Schiavo R, Buscarino M, Siravegna G, Bencardino K, Cercek A, Chen CT, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012;486:532–36. doi: 10.1038/nature11156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Siena S, Sartore-Bianchi A, Di Nicolantonio F, Balfour J, Bardelli A. Biomarkers predicting clinical outcome of epidermal growth factor receptor-targeted therapy in metastatic colorectal cancer. J Natl Cancer Inst. 2009;101:1308–24. doi: 10.1093/jnci/djp280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Normanno N, Tejpar S, Morgillo F, De Luca A, Van Cutsem E, Ciardiello F. Implications for KRAS status and EGFR-targeted therapies in metastatic CRC. Nat Rev Clin Oncol. 2009;6:519–27. doi: 10.1038/nrclinonc.2009.111. [DOI] [PubMed] [Google Scholar]
- 8.De Roock W, De Vriendt V, Normanno N, Ciardiello F, Tejpar S. KRAS, BRAF, PIK3CA, and PTEN mutations: implications for targeted therapies in metastatic colorectal cancer. Lancet Oncol. 2011;12:594–603. doi: 10.1016/S1470-2045(10)70209-6. [DOI] [PubMed] [Google Scholar]
- 9.Tie J, Lipton L, Desai J, Gibbs P, Jorissen RN, Christie M, Drummond KJ, Thomson BN, Usatoff V, Evans PM, Pick AW, Knight S, Carne PW, et al. KRAS mutation is associated with lung metastasis in patients with curatively resected colorectal cancer. Clin Cancer Res. 2011;17:1122–30. doi: 10.1158/1078-0432.CCR-10-1720. [DOI] [PubMed] [Google Scholar]
- 10.Artale S, Sartore-Bianchi A, Veronese SM, Gambi V, Sarnataro CS, Gambacorta M, Lauricella C, Siena S. Mutations of KRAS and BRAF in primary and matched metastatic sites of colorectal cancer. J Clin Oncol. 2008;26:4217–19. doi: 10.1200/JCO.2008.18.7286. [DOI] [PubMed] [Google Scholar]
- 11.Diaz LA, Jr, Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, Allen B, Bozic I, Reiter JG, Nowak MA, Kinzler KW, Oliner KS, Vogelstein B. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature. 2012;486:537–40. doi: 10.1038/nature11219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schmoll HJ, Van Cutsem E, Stein A, Valentini V, Glimelius B, Haustermans K, Nordlinger B, van de Velde CJ, Balmana J, Regula J, Nagtegaal ID, Beets-Tan RG, Arnold D, et al. ESMO Consensus Guidelines for management of patients with colon and rectal cancer. a personalized approach to clinical decision making. Ann Oncol. 2012;23:2479–516. doi: 10.1093/annonc/mds236. [DOI] [PubMed] [Google Scholar]
- 13.Laurent-Puig P, Cayre A, Manceau G, Buc E, Bachet JB, Lecomte T, Rougier P, Lievre A, Landi B, Boige V, Ducreux M, Ychou M, Bibeau F, et al. Analysis of PTEN, BRAF, and EGFR status in determining benefit from cetuximab therapy in wild-type KRAS metastatic colon cancer. J Clin Oncol. 2009;27:5924–30. doi: 10.1200/JCO.2008.21.6796. [DOI] [PubMed] [Google Scholar]
- 14.Mao C, et al. Concordant analysis of KRAS, BRAF, PIK3CA mutations, and PTEN expression between primary colorectal cancer and matched metastases. Sci Rep-Uk; 2015:5. doi: 10.1038/srep08065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Roock W, Claes B, Bernasconi D, De Schutter J, Biesmans B, Fountzilas G, Kalogeras KT, Kotoula V, Papamichael D, Laurent-Puig P, Penault-Llorca F, Rougier P, Vincenzi B, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 2010;11:753–62. doi: 10.1016/S1470-2045(10)70130-3. [DOI] [PubMed] [Google Scholar]
- 16.Bihl MP, Hoeller S, Andreozzi MC, Foerster A, Rufle A, Tornillo L, Terracciano L. KRAS mutation testing in colorectal cancer: comparison of the results obtained using 3 different methods for the analysis of codons G12 and G13. Diagn Mol Pathol. 2012;21:14–23. doi: 10.1097/PDM.0b013e31822b831a. [DOI] [PubMed] [Google Scholar]
- 17.Haraldsdottir S, Bekaii-Saab T. Integrating anti-EGFR therapies in metastatic colorectal cancer. J Gastrointest Oncol. 2013;4:285–98. doi: 10.3978/j.issn.2078-6891.2013.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Swanton C. Intratumor heterogeneity: evolution through space and time. Cancer Res. 2012;72:4875–82. doi: 10.1158/0008-5472.CAN-12-2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fisher R, Pusztai L, Swanton C. Cancer heterogeneity: implications for targeted therapeutics. Br J Cancer. 2013;108:479–85. doi: 10.1038/bjc.2012.581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blanco-Calvo M, Á Concha, Figueroa A, Garrido F, Valladares-Ayerbes M. Colorectal Cancer Classification and Cell Heterogeneity: A Systems Oncology Approach. Int J Mol Sci. 2015;16:13610–32. doi: 10.3390/ijms160613610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Knijn N, Mekenkamp LJ, Klomp M, Vink-Börger ME, Tol J, Teerenstra S, Meijer JW, Tebar M, Riemersma S, van Krieken JH, Punt CJ, Nagtegaal ID. KRAS mutation analysis: a comparison between primary tumours and matched liver metastases in 305 colorectal cancer patients. Br J Cancer. 2011;104:1020–26. doi: 10.1038/bjc.2011.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maheswaran S, Haber DA. Circulating tumor cells: a window into cancer biology and metastasis. Curr Opin Genet Dev. 2010;20:96–99. doi: 10.1016/j.gde.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shah SP, Morin RD, Khattra J, Prentice L, Pugh T, Burleigh A, Delaney A, Gelmon K, Guliany R, Senz J, Steidl C, Holt RA, Jones S, et al. Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature. 2009;461:809–13. doi: 10.1038/nature08489. [DOI] [PubMed] [Google Scholar]
- 24.Mardis ER. Cancer genomics identifies determinants of tumor biology. Genome Biol. 2010;11:211. doi: 10.1186/gb-2010-11-5-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kosmidou V, Oikonomou E, Vlassi M, Avlonitis S, Katseli A, Tsipras I, Mourtzoukou D, Kontogeorgos G, Zografos G, Pintzas A. Tumor heterogeneity revealed by KRAS, BRAF, and PIK3CA pyrosequencing: KRAS and PIK3CA intratumor mutation profile differences and their therapeutic implications. Hum Mutat. 2014;35:329–40. doi: 10.1002/humu.22496. [DOI] [PubMed] [Google Scholar]
- 26.Schneck H, Blassl C, Meier-Stiegen F, Neves RP, Janni W, Fehm T, Neubauer H. Analysing the mutational status of PIK3CA in circulating tumor cells from metastatic breast cancer patients. Mol Oncol. 2013;7:976–86. doi: 10.1016/j.molonc.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Di Fiore F, Charbonnier F, Lefebure B, Laurent M, Le Pessot F, Michel P, Frebourg T. Clinical interest of KRAS mutation detection in blood for anti-EGFR therapies in metastatic colorectal cancer. Br J Cancer. 2008;99:551–52. doi: 10.1038/sj.bjc.6604451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gasch C, Bauernhofer T, Pichler M, Langer-Freitag S, Reeh M, Seifert AM, Mauermann O, Izbicki JR, Pantel K, Riethdorf S. Heterogeneity of epidermal growth factor receptor status and mutations of KRAS/PIK3CA in circulating tumor cells of patients with colorectal cancer. Clin Chem. 2013;59:252–60. doi: 10.1373/clinchem.2012.188557. [DOI] [PubMed] [Google Scholar]
- 29.Heitzer E, Auer M, Gasch C, Pichler M, Ulz P, Hoffmann EM, Lax S, Waldispuehl-Geigl J, Mauermann O, Lackner C, Höfler G, Eisner F, Sill H, et al. Complex tumor genomes inferred from single circulating tumor cells by array-CGH and next-generation sequencing. Cancer Res. 2013;73:2965–75. doi: 10.1158/0008-5472.CAN-12-4140. [DOI] [PubMed] [Google Scholar]
- 30.Mostert B, Jiang Y, Sieuwerts AM, Wang H, Bolt-de Vries J, Biermann K, Kraan J, Lalmahomed Z, van Galen A, de Weerd V, van der Spoel P, Ramírez-Moreno R, Verhoef C, et al. KRAS and BRAF mutation status in circulating colorectal tumor cells and their correlation with primary and metastatic tumor tissue. Int J Cancer. 2013;133:130–41. doi: 10.1002/ijc.27987. [DOI] [PubMed] [Google Scholar]
- 31.Fabbri F, Carloni S, Zoli W, Ulivi P, Gallerani G, Fici P, Chiadini E, Passardi A, Frassineti GL, Ragazzini A, Amadori D. Detection and recovery of circulating colon cancer cells using a dielectrophoresis-based device: KRAS mutation status in pure CTCs. Cancer Lett. 2013;335:225–31. doi: 10.1016/j.canlet.2013.02.015. [DOI] [PubMed] [Google Scholar]
- 32.Harb W, Fan A, Tran T, Danila DC, Keys D, Schwartz M, Ionescu-Zanetti C. Mutational Analysis of Circulating Tumor Cells Using a Novel Microfluidic Collection Device and qPCR Assay. Transl Oncol. 2013;6:528–38. doi: 10.1593/tlo.13367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Steinert G, Schölch S, Niemietz T, Iwata N, García SA, Behrens B, Voigt A, Kloor M, Benner A, Bork U, Rahbari NN, Büchler MW, Stoecklein NH, et al. Immune escape and survival mechanisms in circulating tumor cells of colorectal cancer. Cancer Res. 2014;74:1694–704. doi: 10.1158/0008-5472.CAN-13-1885. [DOI] [PubMed] [Google Scholar]
- 34.Mohamed Suhaimi NA, Foong YM, Lee DY, Phyo WM, Cima I, Lee EX, Goh WL, Lim WY, Chia KS, Kong SL, Gong M, Lim B, Hillmer AM, et al. Non-invasive sensitive detection of KRAS and BRAF mutation in circulating tumor cells of colorectal cancer patients. Mol Oncol. 2015;9:850–60. doi: 10.1016/j.molonc.2014.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Buim ME, Fanelli MF, Souza VS, Romero J, Abdallah EA, Mello CA, Alves V, Ocea LM, Mingues NB, Barbosa PN, Tyng CJ, Chojniak R, Chinen LT. Detection of KRAS mutations in circulating tumor cells from patients with metastatic colorectal cancer. Cancer Biol Ther. 2015;16:1289–95. doi: 10.1080/15384047.2015.1070991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Raimondi C, Nicolazzo C, Gradilone A, Giannini G, De Falco E, Chimenti I, Varriale E, Hauch S, Plappert L, Cortesi E, Gazzaniga P. Circulating tumor cells: exploring intratumor heterogeneity of colorectal cancer. Cancer Biol Ther. 2014;15:496–503. doi: 10.4161/cbt.28020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kalikaki A, Politaki H, Souglakos J, Apostolaki S, Papadimitraki E, Georgoulia N, Tzardi M, Mavroudis D, Georgoulias V, Voutsina A. KRAS genotypic changes of circulating tumor cells during treatment of patients with metastatic colorectal cancer. PLoS One. 2014;9:e104902. doi: 10.1371/journal.pone.0104902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lyberopoulou A, Aravantinos G, Efstathopoulos EP, Nikiteas N, Bouziotis P, Isaakidou A, Papalois A, Marinos E, Gazouli M. Mutational analysis of circulating tumor cells from colorectal cancer patients and correlation with primary tumor tissue. PLoS One. 2015;10:e0123902. doi: 10.1371/journal.pone.0123902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wicha MS, Hayes DF. Circulating tumor cells: not all detected cells are bad and not all bad cells are detected. J Clin Oncol. 2011;29:1508–11. doi: 10.1200/JCO.2010.34.0026. [DOI] [PubMed] [Google Scholar]
- 40.Deeks JJ, Dinnes J, D’Amico R, Sowden AJ, Sakarovitch C, Song F, Petticrew M, Altman DG, International Stroke Trial Collaborative Group. European Carotid Surgery Trial Collaborative Group Evaluating non-randomised intervention studies. Health Technol Assess. 2003;7:iii–x. 1–173. doi: 10.3310/hta7270. [DOI] [PubMed] [Google Scholar]
- 41.Baldus SE, Schaefer KL, Engers R, Hartleb D, Stoecklein NH, Gabbert HE. Prevalence and heterogeneity of KRAS, BRAF, and PIK3CA mutations in primary colorectal adenocarcinomas and their corresponding metastases. Clin Cancer Res. 2010;16:790–99. doi: 10.1158/1078-0432.CCR-09-2446. [DOI] [PubMed] [Google Scholar]
- 42.Santini D, Loupakis F, Vincenzi B, Floriani I, Stasi I, Canestrari E, Rulli E, Maltese PE, Andreoni F, Masi G, Graziano F, Baldi GG, Salvatore L, et al. High concordance of KRAS status between primary colorectal tumors and related metastatic sites: implications for clinical practice. Oncologist. 2008;13:1270–75. doi: 10.1634/theoncologist.2008-0181. [DOI] [PubMed] [Google Scholar]
- 43.Oltedal S, Aasprong OG, Møller JH, Kørner H, Gilje B, Tjensvoll K, Birkemeyer EM, Heikkilä R, Smaaland R, Nordgård O. Heterogeneous distribution of K-ras mutations in primary colon carcinomas: implications for EGFR-directed therapy. Int J Colorectal Dis. 2011;26:1271–77. doi: 10.1007/s00384-011-1233-5. [DOI] [PubMed] [Google Scholar]
- 44.Guerrero S, Casanova I, Farré L, Mazo A, Capellà G, Mangues R. K-ras codon 12 mutation induces higher level of resistance to apoptosis and predisposition to anchorage-independent growth than codon 13 mutation or proto-oncogene overexpression. Cancer Res. 2000;60:6750–56. [PubMed] [Google Scholar]
- 45.Vizan P, Boros LG, Figueras A, Capella G, Mangues R, Bassilian S, Lim S, Lee WN, Cascante M. K-ras codon-specific mutations produce distinctive metabolic phenotypes in human fibroblasts. Cancer Res. 2005;65:5512–15. doi: 10.1158/0008-5472.CAN-05-0074. [DOI] [PubMed] [Google Scholar]
- 46.Modest DP, Reinacher-Schick A, Stintzing S, Giessen C, Tannapfel A, Laubender RP, Brodowicz T, Knittelfelder R, Vrbanec D, Schmiegel W, Heinemann V, Zielinski CC. Cetuximab-based or bevacizumab-based first-line treatment in patients with KRAS p.G13D-mutated metastatic colorectal cancer: a pooled analysis. Anticancer Drugs. 2012;23:666–73. doi: 10.1097/CAD.0b013e328352ff1d. [DOI] [PubMed] [Google Scholar]
- 47.Gorges TM, Tinhofer I, Drosch M, Röse L, Zollner TM, Krahn T, von Ahsen O. Circulating tumour cells escape from EpCAM-based detection due to epithelial-to-mesenchymal transition. BMC Cancer. 2012;12:178. doi: 10.1186/1471-2407-12-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gires O, Stoecklein NH. Dynamic EpCAM expression on circulating and disseminating tumor cells: causes and consequences. Cell Mol Life Sci. 2014;71:4393–402. doi: 10.1007/s00018-014-1693-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Grover PK, Cummins AG, Price TJ, Roberts-Thomson IC, Hardingham JE. Circulating tumour cells: the evolving concept and the inadequacy of their enrichment by EpCAM-based methodology for basic and clinical cancer research. Ann Oncol. 2014;25:1506–16. doi: 10.1093/annonc/mdu018. [DOI] [PubMed] [Google Scholar]
- 50.Tsiatis AC, Norris-Kirby A, Rich RG, Hafez MJ, Gocke CD, Eshleman JR, Murphy KM. Comparison of Sanger sequencing, pyrosequencing, and melting curve analysis for the detection of KRAS mutations: diagnostic and clinical implications. J Mol Diagn. 2010;12:425–32. doi: 10.2353/jmoldx.2010.090188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zuo Z, Chen SS, Chandra PK, Galbincea JM, Soape M, Doan S, Barkoh BA, Koeppen H, Medeiros LJ, Luthra R. Application of COLD-PCR for improved detection of KRAS mutations in clinical samples. Mod Pathol. 2009;22:1023–31. doi: 10.1038/modpathol.2009.59. [DOI] [PubMed] [Google Scholar]
- 52.Peeters M, Oliner KS, Parker A, Siena S, Van Cutsem E, Huang J, Humblet Y, Van Laethem JL, André T, Wiezorek J, Reese D, Patterson SD. Massively parallel tumor multigene sequencing to evaluate response to panitumumab in a randomized phase III study of metastatic colorectal cancer. Clin Cancer Res. 2013;19:1902–12. doi: 10.1158/1078-0432.CCR-12-1913. [DOI] [PubMed] [Google Scholar]
- 53.Simi L, Pratesi N, Vignoli M, Sestini R, Cianchi F, Valanzano R, Nobili S, Mini E, Pazzagli M, Orlando C. High-resolution melting analysis for rapid detection of KRAS, BRAF, and PIK3CA gene mutations in colorectal cancer. Am J Clin Pathol. 2008;130:247–53. doi: 10.1309/LWDY1AXHXUULNVHQ. [DOI] [PubMed] [Google Scholar]
- 54.Mao C, Liao RY, Qiu LX, Wang XW, Ding H, Chen Q. BRAF V600E mutation and resistance to anti-EGFR monoclonal antibodies in patients with metastatic colorectal cancer: a meta-analysis. Mol Biol Rep. 2011;38:2219–23. doi: 10.1007/s11033-010-0351-4. [DOI] [PubMed] [Google Scholar]