

Abstract
The role of Ras signaling during tooth development is poorly understood. Ras proteins—which are activated by many upstream pathways, including receptor tyrosine kinase cascades—signal through multiple effectors, such as the mitogen-activated protein kinase (MAPK) and PI3K pathways. Here, we utilized the mouse incisor as a model to study how the MAPK and PI3K pathways regulate dental epithelial stem cells and amelogenesis. The rodent incisor—which grows continuously throughout the life of the animal due to the presence of epithelial and mesenchymal stem cells—provides a model for the study of ectodermal organ renewal and regeneration. Utilizing models of Ras dysregulation as well as inhibitors of the MAPK and PI3K pathways, we found that MAPK and PI3K regulate dental epithelial stem cell activity, transit-amplifying cell proliferation, and enamel formation in the mouse incisor.
Keywords: MAPK, PI3K, cervical loop, cell differentiation, ameloblast, enamel
Introduction
The role of Ras signaling and its effectors during tooth development is not well understood. Ras signaling is activated by multiple upstream pathways, including receptor tyrosine kinase (RTK) signaling cascades (Fig. 1A). When ligands such as fibroblast growth factors (FGFs) bind RTKs, the small, monomeric GTPase Ras proteins (encoded by HRAS, NRAS, and KRAS) are recruited to the membrane and converted to active Ras-GTP. Activated Ras proteins then signal through multiple effector pathways, including mitogen-activated protein kinase (MAPK)/Raf/MEK/ERK and PI3K/AKT, to regulate cell proliferation and differentiation.
Figure 1.

Phosphorylation of ERK, MEK, and AKT in the mouse laCL. (A) Schematic of the receptor tyrosine kinase (RTK)/Ras signaling pathway. Fibroblast growth factors bind to RTKs and activate Ras, which signals through Raf/MEK/ERK and PI3K/AKT. Spry proteins antagonize Ras signaling. (B) The mouse hemimandible contains 3 molars and 1 incisor, with the laCL and liCL present at the proximal end of the incisor. (B′) Magnified schematic of the boxed region in panel B shows the location of the laCL, liCL, pre-am, am, and od. (B′′) Magnified view of boxed region in panel B′ shows the different compartments composing the laCL: DESCs reside in the SR and OEE of the proximal laCL and give rise to the IEE and T-A cells. T-A cells generate pre-am, which differentiate into enamel-producing am. (C) Immunofluorescence with an antibody against pERK in the mouse incisor showed a biphasic expression profile with intense expression in T-A (white arrowhead), low or no expression in pre-am (*), and moderate expression in am (blue arrowhead) regions. (C′) Magnified boxed region shows scattered, low-intensity pERK staining in the OEE and SR (yellow and orange arrowheads, respectively). (D) pMEK was detected in the OEE, SR, and T-A. (D′) Magnified boxed region shows polarized membrane staining in the OEE (yellow arrowhead) and weak, evenly distributed membrane staining in the SR (orange arrowhead). In the T-A, pMEK staining was evenly distributed along the membrane (white arrowhead), or there was punctate nuclear staining (green arrowhead). (E) pAKT was detected in the OEE, SR, and T-A. (E′) Magnified boxed region shows weak, diffuse cytoplasmic staining and bright, punctate staining in the OEE and proximal SR (yellow and orange arrowheads). Cells in the T-A showed a similar pattern of diffuse and punctate cytoplasmic staining, plus nuclear pAKT staining (green arrowhead). Scale bars = 100 μm. am, ameloblasts; DESC, dental epithelial stem cell; ERK, extracellular signal-regulated kinases; IEE, inner enamel epithelium; laCL, labial cervical loop; liCL, lingual cervical loop; od, odontoblasts; OEE, outer enamel epithelium; pre-am, preameloblast; Spry, Sprouty; SR, stellate reticulum; T-A, transit amplifying.
To better understand downstream MAPK and PI3K signaling in dental epithelial stem cell (DESC) regulation and ameloblast differentiation, we utilized the mouse incisor as a model. The mouse incisor (Fig. 1B–B′′) is a remarkable tooth that is continuously renewed throughout the life of the animal due to the presence of epithelial and mesenchymal stem cell populations (Lapthanasupkul et al. 2012; Zhao et al. 2014), and here, we focus on the epithelial component of the incisor. The continuous renewal of the incisor epithelium is fueled by DESCs in the labial cervical loop (laCL) at the proximal end of the incisor (Harada et al. 1999; Parsa et al. 2010; Seidel et al. 2010; Juuri et al. 2012; Biehs et al. 2013). DESCs are housed in the proximal part of the laCL, although whether they reside in the outer enamel epithelium (OEE) or stellate reticulum (SR) remains an open question. The DESCs generate the inner enamel epithelium (IEE), where the transit-amplifying (T-A) cells undergo several rounds of division and move distally along the labial aspect of the incisor. T-A cells differentiate into ameloblasts that produce enamel on the labial surface of the incisor.
Previously, a role for upstream effectors and regulators of Ras signaling was identified in development and maintenance of the laCL in the mouse. Fgf10 is expressed in the mesenchyme surrounding the apical portion of the cervical loop and underlying the IEE, and Fgf10 deletion results in a morphologically abnormal cervical loop and hypoplastic incisor (Harada et al. 2002). Furthermore, combined inactivation of Fgf3 and Fgf10 in Fgf3−/−;Fgf10+/− animals results in thin or absent incisor enamel and incomplete formation of the laCL (Wang et al. 2007). Hyperactivating RTK signaling by combined deletion of Spry2 and Spry4 (Spry2+/−; Spry4−/− mice)—which encode proteins that, in part, negatively regulate Ras signaling (de Maximy et al. 1999; Minowada et al. 1999)—results in generation of ameloblasts that form enamel on the lingual aspect of the incisor (Klein et al. 2008; Boran et al. 2009).
Further downstream, a mouse model carrying an HrasG12V gain-of-function mutation (Hras GOF; Chen et al. 2009) has thin, hypomineralized enamel and hyperproliferative and disorganized ameloblasts on the labial aspect of the incisor (Goodwin et al. 2014). However, a mouse with a BrafL597V gain-of-function mutation (Braf GOF; Andreadi et al. 2012) has normal incisor development and maintenance. These specific Hras GOF and Braf GOF alleles lead to many features that are characteristic of the human conditions Costello syndrome and cardiofaciocutaneous syndrome, respectively, including abnormal craniofacial and dental development, and thus serve as models of these syndromes. Taken together, these mouse models reveal that dysregulation of RTK and Ras signaling perturbs the mouse laCL, yet little is known about the effect on downstream components of the Ras pathway in the laCL.
Here, we studied the role of Ras signaling in the mouse incisor with the Fgf3−/−;Fgf10+/−, Spry2+/−;Spry4−/−, Hras GOF, and Braf GOF mouse models as well as inhibitors of the MAPK and PI3K pathways. Utilizing these models of Ras dysregulation, we determined that MAPK and PI3K regulate DESC activity and T-A cell proliferation and that pERK levels affect enamel formation. Finally, our data reveal crosstalk between the MAPK and PI3K pathways in the laCL.
Materials and Methods
Mouse Lines
Wild-type mice on mixed background and mouse lines carrying mutant alleles of BrafL597V, HrasG12V, Fgf3−, and Fgf10− on mixed background and Spry4− and Spry2− on Friend virus B-type inbred (FVBN) background were generated and genotyped. Eight- to 10-wk-old adult mice were used in all experiments, and littermate wild-type mice were used as controls. All experiments involving mice were conducted in accordance with protocols approved by the University of California, San Francisco Institutional Animal Care and Use Committee.
Tissue Preparation and Immunofluorescence
After CO2 euthanasia and cervical dislocation, adult mice were perfused with 4% paraformaldehyde. Mandibles were dissected and fixed in 4% paraformaldehyde overnight at 4 °C and then decalcified in 0.5M ethylenediaminetetraacetic acid (EDTA) for 2 wk at room temperature, dehydrated, embedded in paraffin, and cut in 2- to 4-μm serial sagittal sections.
Immunofluorescence was performed following standard protocols with antibodies against pERK (4370, 1:200; Cell Signaling), pMEK (9121, 1:200; Cell Signaling), pAKT (135650, 1:200; Santa Cruz), γTubulin (T5326, 1:500; Sigma-Aldrich), PH3 (H0412, 1:800; Sigma-Aldrich), Ki67 (9106, 1:400; Thermo Fisher Scientific), and BrdU (6326, 1:500; Abcam). Click-iT EdU Imaging kit (C10337; Invitrogen) was used to stain EdU-incorporated cells according to a standard protocol. Sections were mounted with mounting medium with DAPI (H-1200; Vector).
Inhibitor Treatment
The MEK inhibitor PD0325901 (Pfizer) and PI3K inhibitor GDC0941 (Genentech) were formulated in 0.5% (w/v; hydroxypropyl) methyl cellulose (HPMT; Sigma-Aldrich). PD0325901 was administered by oral gavage at 12.5 mg/kg (Trejo et al. 2012) and GDC0941 at 75 mg/kg (Wullschleger et al. 2012) once per day for 5 to 9 d.
Proliferation Analysis
Each mouse was injected via intraperitoneal injection with 250 μg of EdU (Invitrogen) 2 h prior to euthanasia, and 4-μm serial sagittal sections were EdU stained and imaged at 20× on a Leica upright microscope. Every other serial section was selected to quantify the percentage of proliferative EdU+ cells in the T-A region with ImageJ software, and the average was calculated. Continuous sections (4 μm) were stained with antibody against Ki67 and imaged at 20×. The number of Ki67+ cells in the proximal laCL on each section was quantified with ImageJ, and the average was determined.
Quantification of Ameloblasts Generated in 3 d
BrdU (500 μg) was injected intraperitoneal 3 d prior to euthanasia so that dividing cells exiting the IEE would incorporate BrdU. EdU (250 μg) was then injected intraperitoneal 2 h prior to euthanasia, which labeled the cycling T-A cells to define the T-A region. The length of the preameloblast/ameloblast region between the most distal BrdU- and EdU-labeled cells was measured with ImageJ software.
Statistical Analysis
The statistical difference in T-A proliferation, DESC proliferation, and length of the preameloblast/ameloblast region among control, MEKi-treated, and PI3Ki-treated mice was tested with analysis of variance. Student’s t test was used to compare the Braf GOF and littermate control mice.
Results
MAPK and PI3K Pathways Are Active in the laCL of the Mouse Incisor
Immunofluorescence performed on wild-type mice revealed that the MAPK and PI3K pathways are both active in the laCL (Fig. 1C–E′). We began by examining phosphorylation of ERK, a downstream component of the MAPK pathway, as phospho-ERK (pERK) is considered to be the active form of the protein. Low levels of pERK were detected in the putative DESC compartment, composed of the OEE and SR, and high levels were present in the T-A and ameloblast regions, but no pERK was detected in the preameloblast region between the T-A and ameloblasts (Fig. 1C, C′). The pERK staining was cytoplasmic in all cell types (Appendix Fig. 1A′, A′′). The upstream MAPK signaling component MEK was also phosphorylated (pMEK) in the DESC and T-A compartments (Fig. 1D, D′). In the OEE, pMEK staining was detected at the membrane of cells contacting the SR (Appendix Fig. 1B′). In the T-A and SR regions, there was evenly distributed, weak membrane staining, in addition to intense nuclear staining in several cells (Appendix Fig. 1B′′). The PI3K downstream effector AKT was phosphorylated in the DESC compartment and T-A region (Fig. 1E, E′). There was generalized weak cytoplasmic and bright punctate staining in the T-A and DESC regions, and bright nuclear phospho-AKT (pAKT) staining was evident in several cells in the T-A region (Appendix Fig. 1C′, C′′).
MEK and AKT Are Phosphorylated in Proliferating Cells in the laCL
As a first step in determining whether the MAPK and PI3K pathways play a role in proliferation in the laCL, we performed immunofluorescence utilizing an antibody against phosphorylated histone H3 (PH3) to label cells undergoing division (Hans and Dimitrov 2001). In the laCL, proliferating cells, as marked by an antibody against PH3, were present in the DESC and T-A regions, with the majority of proliferating cells residing in the T-A region and relatively few in the DESC compartment (Fig. 2A1). To determine whether MEK and AKT were phosphorylated in proliferating cells, we performed pMEK, pAKT, and PH3 staining on adjacent 2-μm sections. Since mammalian cells are typically ~15 to 20 μm in diameter with nuclei of approximately ~10 μm in diameter, adjacent 2-μm sections were used to capture different sections of the same cells. We found that MEK was phosphorylated in the nucleus of some but not all PH3+ mitotic cells (Fig. 2A1, A2). Furthermore, MEK and AKT were both phosphorylated in some of the same mitotic cells on adjacent sections in the T-A region, suggesting that phosphorylation of MEK and AKT regulates the proliferating T-A cells (Fig. 2B1, B2).
Figure 2.

Co-localization of pMEK and pAKT in proliferating cells in the laCL. (A1) PH3+ cells (white arrowheads) were present in the T-A region. (A2) Serial section (2 μm) adjacent to panel A1 showed MEK phosphorylation in some of the same PH3+ cells (white arrowheads). (A2′) Magnified view shows pMEK staining in the cytoplasm of the PH3+ dividing cell in A1′ (green arrowhead). (B1, B2) Immunofluorescence on adjacent 2-μm serial sections showed nuclear co-localization of pMEK and pAKT in the T-A region (white arrowheads). There was also phosphorylation of AKT and MEK in the IEE, OEE, and SR. (C) Immunofluorescence showed co-localization of pMEK and γTu staining; pMEK and γTu staining overlapped at opposite poles in the nucleus of 1 dividing cell (orange arrowheads). There was also isolated γTu staining that did not overlap with pMEK (white arrowhead). (D, D′, D′′) Immunofluorescence showed co-localization of pAKT and γTu in the nucleus of some cells in the T-A region (white arrowheads; D′) and closely associated but not overlapping staining of pAKT and γTu (red and white arrowheads; D′′). IEE, inner enamel epithelium; laCL, labial cervical loop; OEE, outer enamel epithelium; SR, stellate reticulum; T-A, transit-amplifying.
In addition to the nuclear pMEK and pAKT staining in the mitotic cells in the T-A region, we detected 1 to 2 bright foci of pMEK and pAKT staining that costained with gamma tubulin (γTu; Fig. 2C–D′′), which labels the centrosomal apparatus required for chromosome separation during mitosis. pMEK appeared to co-localize with γTu (Fig. 2C), and pAKT staining showed a co-localization pattern similar to pMEK in some cells (Fig. 2D, D′). In other cells, punctate pAKT staining surrounded by dots of γTu staining was observed (Fig. 2D′′). The close association among pMEK, pAKT, and γTu points to a role for MAPK and PI3K signaling in cell division in the proliferative cells of the laCL.
Modulation of Activators and Antagonists of the RTK Signaling Pathway Results in Changes in MAPK Signaling
To provide insight into MAPK and PI3K signaling in the laCL, we utilized several models with dysregulated RTK and Ras signaling. We focused on phosphorylation of ERK and MEK because differential AKT phosphorylation was difficult to assess due to high variability of antibody staining (data not shown). Inactivation of both alleles of a single Fgf gene, Fgf3, did not noticeably affect pERK and pMEK staining as compared with control (Fig. 3A–D); however, deleting both alleles of Fgf3 and one of Fgf10 (Fgf3−/−;Fgf10+/−) resulted in a malformed laCL and a clear decrease in phosphorylation of ERK and MEK (Fig. 3E, F). We next examined mice that were homozygous null at the Spry4 locus and found that these animals did not have noticeably altered levels of pERK and pMEK (Fig. 3G, H). However, when a combination of Sprouty genes were deleted in Spry2+/−;Spry4−/− mutants, we observed an extensive increase in pERK and pMEK staining in the proximal laCL (Fig. 3I, J), indicating increased MAPK signaling.
Figure 3.
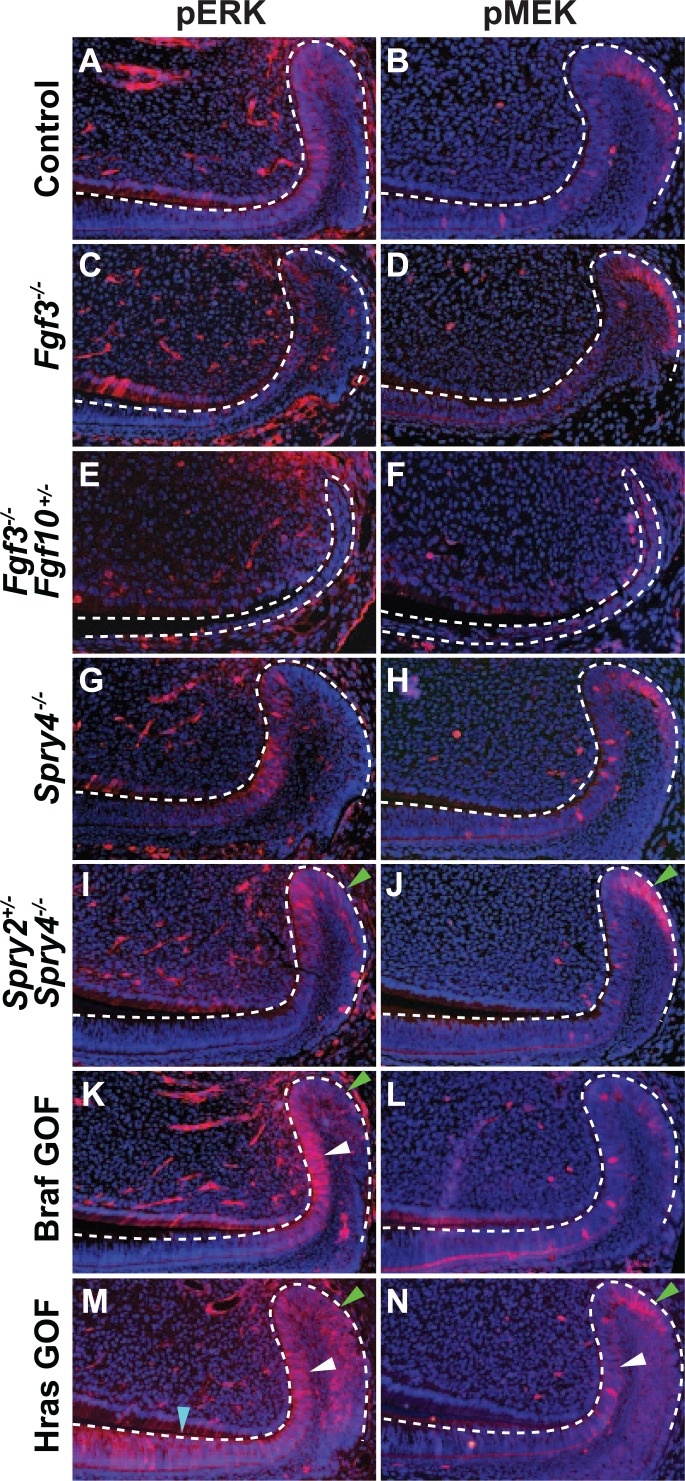
Phosphorylation of ERK and MEK in the laCL of mouse models with dysregulated RTK signaling. Compared with control (A, B), pERK and pMEK staining was similar in the Fgf3−/− mutant mouse laCL (C, D). In the Fgf3−/−;Fgf10+/− mouse, the levels of ERK and MEK phosphorylation were greatly reduced, and the laCL was small and malformed (E, F). While deletion of Spry4−/− did not noticeably affect pERK and pMEK staining (G, H) compared with control, the Spry2+/−;Spry4−/− mutant showed increased levels of ERK and MEK phosphorylation in the DESC compartment (SR and OEE) as marked by green arrows (I, J). In the BrafL597V gain-of-function mutation (Braf GOF) mouse, pERK staining was increased in the T-A and DESC regions as marked by white and green arrows, respectively, (K) while pMEK staining did not differ significantly (L) compared with control. In HrasG12V gain-of-function mutation (Hras GOF) mutants, phosphorylation of MEK and ERK was increased in the T-A (white arrowhead) and OEE (green arrowhead) regions (M, N) compared with control. pERK staining was also increased in the pre-am region (blue arrowhead; M). DESC, dental epithelial stem cell; laCL, labial cervical loop; OEE, outer enamel epithelium; pre-am, preameloblast; SR, stellate reticulum; T-A, transit-amplifying.
In the Braf GOF mutant, pERK staining was moderately increased in the T-A and DESC regions (Fig. 3K); however, there did not appear to be a difference in pMEK staining (Fig. 3L). In Hras GOF mutants, pERK and pMEK staining was increased in the DESC and T-A compartments (Fig. 3M, N), and pERK staining was present in the preameloblast region (Fig. 3M), where it is not observed in wild type.
Inhibition of the PI3K Pathway Results in an Increase in MAPK Signaling through pERK in the laCL
To further functionally analyze MAPK and PI3K signaling in the laCL, we utilized MEK and PI3K inhibitors. Inhibiting MEK phosphorylation with the MEK inhibitor PD0325901 (MEKi) for 5 to 9 d (mean = 7 d) resulted in a decrease in pERK staining in the proximal laCL, particularly in the IEE, OEE, and SR; however, pERK staining was still detected in the T-A region with MEKi treatment (Fig. 4B, B′). Interestingly, inhibiting PI3K activity with the PI3K inhibitor GDC0941 (PI3Ki) for the same amount of time resulted in a distinct increase in pERK staining in the proximal laCL, including the IEE, OEE, and SR (Fig. 4C, C′).
Figure 4.

Effect of MEK inhibition (MEKi) and PI3K inhibition (PI3Ki) on the labial cervical loop. (A–C) pERK staining in the labial cervical loop after oral administration of inhibitors for 7 d. (A′–C′) High-magnification images of the area of interest outlined by green dashed line in panels A–C. MEKi caused a decrease in ERK phosphorylation, particularly in the OEE and IEE (B, B′). Conversely, PI3Ki led to an increase in ERK phosphorylation, particularly in the OEE, IEE, and SR (C, C′). (D–F) Increased number of EdU+ cells was observed in the T-A region (between white arrowheads) with MEKi (E) or PI3Ki (F) as compared with control (D). (G) Quantification of the percentage of EdU+ cells in the T-A region. (H, I) Mice were coinjected with BrdU for 3 d and EdU for 2 h, and the length of the pre-am/am region was determined (between white arrowheads; I). The length of the pre-am/am region increased with MEKi treatment, but PI3Ki treatment did not affect pre-am/am length (H). am, ameloblast; IEE, inner enamel epithelium; OEE, outer enamel epithelium; pre-am, preameloblast; SR, stellate reticulum; T-A, transit-amplifying. Error bars represent SEM; ** P < 0.01.
Since the MAPK and PI3K signaling pathways are active in mitotic cells in the laCL (Fig. 2), we analyzed the effect of MEK and PI3K inhibition on proliferation in the laCL. With MEKi and PI3Ki treatment, T-A proliferation was increased, as shown by an increase in the percentage of EdU-labeled proliferative cells in the T-A region (marked by white arrows) after MEK and PI3K inhibition as compared with control (Fig. 4D–G).
To determine if the increased proliferation after MEK and PI3K inhibition in the T-A region resulted in an increase in the number of ameloblasts generated, we carried out cell migration assays. BrdU was injected 3 d prior to euthanasia to label cells migrating from the IEE. Then, 2 h prior to euthanasia, mice were injected with EdU to label the T-A region. The distance was measured between the most distal BrdU-labeled ameloblast and the end of the T-A region labeled by the most distal EdU-labeled cell. This distance constitutes the preameloblast/ameloblast region, and since the cells form a single layer on the labial aspect of the incisor, the length of the preameloblast/ameloblast region correlates with the number of ameloblasts produced in 3 d. MEKi treatment resulted in an approximately 30% increase in the length of the preameloblast/ameloblast region, suggesting an increase in the number of ameloblasts produced compared with control (Fig. 4H, I). PI3Ki treatment did not affect the length of the preameloblast/ameloblast region and thus had no effect on the number of ameloblasts generated (Fig. 4H, I).
The proportion of EdU+ proliferative cells in the T-A region was similar with MEK and PI3K inhibition, yet the number of am generated was higher in the MEKi- versus PI3Ki-treated samples. One possible explanation for this discrepancy could be a difference in number of DESCs entering the T-A region due to a change in proliferation in the DESC compartment. To test this hypothesis, we measured proliferation in the proximal laCL with Ki67 staining but found that the number of Ki67+ cells in the DESC region was similar with MEK and PI3K inhibition as compared with control (Appendix Fig. 1A1–B). Thus, increased DESC proliferation did not account for the increased number of ameloblasts generated in the MEKi-treated laCL.
The Braf GOF model showed a moderate increase in pERK staining, suggesting that Braf activation leads to phosphorylation of ERK in the Braf GOF model (Fig. 3K). Similar to the PI3Ki-treated mice, Braf GOF mice had increased proliferation in the T-A region but not DESC compartment, with no change in the length of the preameloblast/ameloblast region (Appendix Fig. 2A–E). These findings provide further evidence that inhibition of PI3K results in upregulation of ERK phosphorylation in the laCL.
Discussion
In the work reported here, we found that the MAPK and PI3K signaling pathways are active in the laCL, and our data suggest that, in the laCL, MAPK and PI3K regulate DESC activity, proliferation in the T-A region, and enamel formation. First, our data suggest that MAPK signaling plays a role in regulation of DESC activity. Inhibition of MEK and PI3K increased proliferation in the T-A region. With higher T-A proliferation, one would expect more ameloblasts to be produced; however, only MEK inhibition, not PI3K inhibition, resulted in an increase in the number of ameloblasts produced when compared with control. One potential explanation for this finding is that the DESCs were less proliferative with PI3K inhibition versus MEK inhibition; however, we found that DESC proliferation with MEK and PI3K inhibition did not differ significantly from control. Another possibility is that fewer cells exit the DESC niche with PI3K inhibition, a condition in which ERK phosphorylation is increased, compared with MEK inhibition. Whether the number of DESCs entering the T-A region was affected with MEK inhibition is not clear based on our data; thus, we propose that one hypothesis to test in the future would be that the MAPK pathway negatively regulates the decision of the DESCs to exit the niche, with high ERK activity encouraging DESCs to remain in the niche and pursue the stem cell fate and with low ERK activity driving DESCs to exit the niche and differentiate.
Progenitor cells exit the DESC compartment and proliferate in the T-A region. MEK and PI3K inhibition both resulted in increased proliferation in the T-A region, suggesting the MAPK and PI3K pathways negatively regulate T-A cell proliferation. This finding differs from our previous work, which showed a decrease in T-A proliferation with MEKi and PI3Ki treatment (Goodwin et al. 2014); however, the mice were treated for different lengths of time in these experiments (previously 21 d vs. currently 7 d). Long-term treatment with inhibitors may lead to negative feedback loops that modulate the direct effect of pERK on T-A proliferation. Similarly, increased pERK in the Braf GOF mutant resulted in increased proliferation in the T-A region, which could also result from negative feedback loops. A large body of work has explored negative feedback loops in the Ras pathway; for example, ERK negatively regulates the upstream factors MEK and SOS (Langlois et al. 1995; Dong et al. 1996; Eblen et al. 2004). The regulatory networks activated in the context of MEK inhibition or Braf activation are incompletely understood and an important area for study in the future.
As cells exit the T-A region, they differentiate into ameloblasts that produce enamel. Deletion of Fgf3 alone was insufficient to cause notable changes in phosphorylation of MAPK, but combined inactivation of Fgf3 and Fgf10 in the Fgf3−/−;Fgf10+/− mutant resulted in a clear decrease in pERK and pMEK staining. Similarly, deleting Spry4 did not obviously affect MAPK signaling; however, in the Spry2+/−;Spry4−/− mutant, staining of pMEK and pERK was significantly increased. As previously reported, the Fgf3−/−;Fgf10+/− mutant has a small, malformed cervical loop and no enamel formation (Wang et al. 2007), whereas enamel forms on the labial and lingual aspects of the incisor of the Spry2+/−; Spry4−/− mutant (Klein et al. 2008; Boran et al. 2009). Thus, the levels of pMEK and pERK appear highly correlated to the enamel phenotype, since decreased pMEK and pERK are detected in genotypes, leading to absence of enamel, while increased pMEK and pERK are detected in genotypes, leading to ectopic enamel formation.
Furthermore, the location and level of MAPK signaling are correlated to enamel phenotype, since the Hras GOF mouse (with increased ERK phosphorylation in the DESC, T-A, preameloblast, and ameloblast regions) resulted in thin, hypomineralized enamel while the Braf GOF mouse (with only a moderate increase in pERK expression in the DESC, T-A, and ameloblast but not preameloblast compartments) had no enamel defect (Goodwin et al. 2014). Thus, MAPK, not PI3K, signaling has a dosage-dependent effect on enamel phenotype, primarily through ERK activation.
Analysis of Ras signaling in the laCL revealed interactions between the MAPK and PI3K pathways. We found that short-term PI3Ki treatment of wild-type mice led to pERK upregulation in the DESC, T-A, and ameloblast regions. PI3K is an upstream regulator of ERK in the mouse laCL, and previous studies have shown crosstalk between the MAPK and PI3K pathways through AKT inhibition of Raf (Guan et al. 2000; Reusch et al. 2001; Mabuchi et al. 2002; El-Naggar et al. 2009). Additionally, PI3K inhibitors have been shown to activate MEK and ERK signaling in cancer models (reviewed in Rozengurt et al. 2014). More work is necessary to understand the complexities of the crosstalk between the MAPK and PI3K pathways in the laCL.
Thus, MAPK and PI3K are active in the DESC compartment, and our data suggest that pERK may play a role in DESC differentiation but not proliferation and that T-A proliferation is sensitive to pERK levels. Additionally, ERK phosphorylation in the laCL must be maintained in a specific range, since dental epithelium exposed to high or low levels of pERK does not produce proper enamel. Finally, inhibition of the PI3K pathway resulted in increased ERK phosphorylation in the laCL. These data further our understanding of downstream Ras signaling in the laCL, increasing the foundation of knowledge on which future regenerative therapies will be based.
Author Contributions
X. Zheng, contributed to design, data acquisition, analysis, and interpretation, drafted and critically revised the manuscript; A.F. Goodwin, contributed to conception, data acquisition, analysis, and interpretation, drafted and critically revised the manuscript; H. Tian, contributed to data acquisition, analysis, and interpretation, critically revised the manuscript; A.H. Jheon, contributed to data analysis and interpretation, drafted the manuscript; O.D. Klein, contributed to conception, design, data analysis, and interpretation, drafted and critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work.
Supplementary Material
Acknowledgments
We thank Sarah Alto and Rebecca d’Urso for assistance with the mouse colony, Dr. Amnon Sharir and members of the Klein laboratory for helpful discussion, Genentech for providing the PI3K inhibitor, and Dr. Martin McMahon for providing the MEK inhibitor.
Footnotes
A supplemental appendix to this article is available online.
This work was funded in part by the National Institutes of Health (R35-DE026602 to O.D. Klein) and the American Association of Orthodontists Foundation (Postdoctoral Fellowship Award to A.F. Goodwin).
The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
References
- Andreadi C, Cheung LK, Giblett S, Patel B, Jin H, Mercer K, Kamata T, Lee P, Williams A, McMahon M, et al. 2012. The intermediate-activity (L597V)BRAF mutant acts as an epistatic modifier of oncogenic RAS by enhancing signaling through the RAF/MEK/ERK pathway. Genes Dev. 26(17):1945–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biehs B, Hu JK, Strauli NB, Sangiorgi E, Jung H, Heber RP, Ho S, Goodwin AF, Dasen JS, Capecchi MR, et al. 2013. BMI1 represses Ink4a/Arf and Hox genes to regulate stem cells in the rodent incisor. Nat Cell Biol. 15(7):846–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boran T, Peterkova R, Lesot H, Lyons DB, Peterka M, Klein OD. 2009. Temporal analysis of ectopic enamel production in incisors from sprouty mutant mice. J Exp Zool B Mol Dev Evol. 312(5):473–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Mitsutake N, LaPerle K, Akeno N, Zanzonico P, Longo VA, Mitsutake S, Kimura ET, Geiger H, Santos E, et al. 2009. Endogenous expression of Hras(G12V) induces developmental defects and neoplasms with copy number imbalances of the oncogene. Proc Natl Acad Sci U S A. 106(19):7979–7984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Maximy AA, Nakatake Y, Moncada S, Itoh N, Thiery JP, Bellusci S. 1999. Cloning and expression pattern of a mouse homologue of drosophila sprouty in the mouse embryo. Mech Dev. 81(1–2): 213–216. [DOI] [PubMed] [Google Scholar]
- Dong Chen, Waters SB, Holt KH, Pessin JE. 1996. SOS phosphorylation and disassociation of the Grb2-SOS complex by the ERK and JNK signaling pathways. J Biol Chem. 271(11):6328–6332. [DOI] [PubMed] [Google Scholar]
- Eblen ST, Slack-Davis JK, Tarcsafalvi A, Parsons JT, Weber MJ, Catling AD. 2004. Mitogen-activated protein kinase feedback phosphorylation regulates MEK1 complex formation and activation during cellular adhesion. Mol Cell Biol. 24(6):2308–2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Naggar S, Liu Y, Dean DC. 2009. Mutation of the Rb1 pathway leads to overexpression of mTor, constitutive phosphorylation of Akt on serine 473, resistance to anoikis, and a block in c-Raf activation. Mol Cell Biol. 29(21):5710–5717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin AF, Tidyman WE, Jheon AH, Sharir A, Zheng X, Charles C, Fagin JA, McMahon M, Diekwisch TG, Ganss B, et al. 2014. Abnormal Ras signaling in Costello syndrome (CS) negatively regulates enamel formation. Hum Mol Genet. 23(3):682–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan KL, Figueroa C, Brtva TR, Zhu T, Taylor J, Barber TD, Vojtek AB. 2000. Negative regulation of the serine/threonine kinase B-Raf by Akt. J Biol Chem. 275(35):27354–27359. [DOI] [PubMed] [Google Scholar]
- Hans F, Dimitrov S. 2001. Histone H3 phosphorylation and cell division. Oncogene. 20(24):3021–3027. [DOI] [PubMed] [Google Scholar]
- Harada H, Kettunen P, Jung HS, Mustonen T, Wang YA, Thesleff I. 1999. Localization of putative stem cells in dental epithelium and their association with Notch and FGF signaling. J Cell Biol. 147(1):105–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada H, Toyono T, Toyoshima K, Ohuchi H. 2002. FGF10 maintains stem cell population during mouse incisor development. Connect Tissue Res. 43(2–3): 201–204. [DOI] [PubMed] [Google Scholar]
- Juuri E, Saito K, Ahtiainen L, Seidel K, Tummers M, Hochedlinger K, Klein OD, Thesleff I, Michon F. 2012. Sox2+ stem cells contribute to all epithelial lineages of the tooth via Sfrp5+ progenitors. Dev Cell. 23(2):317–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein OD, Lyons DB, Balooch G, Marshall GW, Basson MA, Peterka M, Boran T, Peterkova R, Martin GR. 2008. An FGF signaling loop sustains the generation of differentiated progeny from stem cells in mouse incisors. Development. 135(2):377–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langlois WJ, Sasaoka T, Saltiel AR, Olefsky JM. 1995. Negative feedback regulation and desensitization of insulin- and epidermal growth factor-stimulated p21ras activation. J Biol Chem. 270(43):25320–25323. [DOI] [PubMed] [Google Scholar]
- Lapthanasupkul P, Feng J, Mantesso A, Takada-Horisawa Y, Vidal M, Koseki H, Wang L, An Z, Miletich I, Sharpe PT. 2012. Ring1a/b polycomb proteins regulate the mesenchymal stem cell niche in continuously growing incisors. Dev Biol. 367(2):140–153. [DOI] [PubMed] [Google Scholar]
- Mabuchi S, Ohmichi M, Kimura A, Hisamoto K, Hayakawa J, Nishio Y, Adachi K, Takahashi K, Arimoto-Ishida E, Nakatsuji Y, et al. 2002. Inhibition of phosphorylation of BAD and Raf-1 by Akt sensitizes human ovarian cancer cells to paclitaxel. J Biol Chem. 277(36):33490–33500. [DOI] [PubMed] [Google Scholar]
- Minowada G, Jarvis LA, Chi CL, Neubuser A, Sun X, Hacohen N, Krasnow MA, Martin GR. 1999. Vertebrate Sprouty genes are induced by FGF signaling and can cause chondrodysplasia when overexpressed. Development. 126(20):4465–4475. [DOI] [PubMed] [Google Scholar]
- Parsa S, Kuremoto K, Seidel K, Tabatabai R, Mackenzie B, Yamaza T, Akiyama K, Branch J, Koh CJ, Al Alam D, et al. 2010. Signaling by FGFR2b controls the regenerative capacity of adult mouse incisors. Development. 137(22):3743–3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reusch HP, Zimmermann S, Schaefer M, Paul M, Moelling K. 2001. Regulation of Raf by Akt controls growth and differentiation in vascular smooth muscle cells. J Biol Chem. 276(36):33630–33637. [DOI] [PubMed] [Google Scholar]
- Rozengurt E, Soares HP, Sinnet-Smith J. 2014. Suppression of feedback loops mediated by PI3K/mTOR induces multiple overactivation of compensatory pathways: an unintended consequence leading to drug resistance. Mol Cancer Ther. 13(11):2477–2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidel K, Ahn CP, Lyons D, Nee A, Ting K, Brownell I, Cao T, Carano RA, Curran T, Schober M, et al. 2010. Hedgehog signaling regulates the generation of ameloblast progenitors in the continuously growing mouse incisor. Development. 137(22):3753–3761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trejo CL, Juan J, Vincent S, Sweet-Cordero A, McMahon M. 2012. MEK1/2 inhibition elicits regression of autochthonous lung tumors induced by KRASG12D or BRAFV600E. Cancer Res. 72(12):3048–3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XP, Suomalainen M, Felszeghy S, Zelarayan LC, Alonso MT, Plikus MV, Maas RL, Chuong CM, Schimmang T, Thesleff I. 2007. An integrated gene regulatory network controls stem cell proliferation in teeth. PLoS Biol. 5(6):e159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wullschleger S, Garcia-Martinez JM, Duce SL. 2012. Quantitative MRI establishes the efficacy of PI3K inhibitor (GDC-0941) multi-treatments in PTEN-deficient mice lymphoma. Anticancer Res. 32(2):415–420. [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Feng J, Seidel K, Shi S, Klein O, Sharpe P, Chai Y. 2014. Secretion of shh by a neurovascular bundle niche supports mesenchymal stem cell homeostasis in the adult mouse incisor. Cell Stem Cell. 14(2):160–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.