

Abstract
A 38-year-old man presented with excessive height gain and progressive enlargement of the extremities since childhood. This was compounded by lower limb deformities over the past 5 years. On examination, his height was 196 cm, he had macroglossia, acral enlargement, seborrhoea, hyperhidrosis—suggesting acrogigantism. He had facial asymmetry, wind-swept deformity of lower limbs and a café-au-lait macule over his trunk. Investigations revealed normal-sized pituitary gland with dysplastic cranial bones. Isotope bone scintigraphy was suggestive of polyostotic fibrous dysplasia. A diagnosis of McCune-Albright syndrome was made and trans-sphenoidal hypophysectomy was undertaken. He had persistent hypophosphataemia. Tubular reabsorption of phosphate adjusted for glomerular filtration rate was low and serum FGF-23 level was high. Ga-DOTATATE scintigraphy showed somatostatin-receptor expression in all the dysplastic lesions. FGF-23 produced by the bony lesions could counteract the phosphate-retaining effect of GH excess resulting in hypophosphataemia, which further worsened following hypophysectomy.
Keywords: pituitary disorders, calcium and bone
Background
Acromegaly is a disorder characterised by chronic growth hormone (GH) hypersecretion. GH excess leads to overproduction of insulin-like growth factor 1 (IGF-1), resulting in somatic overgrowth, physical disfigurement, metabolic comorbidities and early mortality. GH, acting directly on the proximal convoluted tubule, causes increased renal reabsorption of phosphate. Hence, as many as 55% of patients with acromegaly have elevated serum phosphate levels. Hyperphosphataemia correlates with disease activity and may, in fact, be an indicator for a low likelihood of disease remission.1
Herein, we report a case of a 38-year-old acrogiant presenting with multiple bony deformities. Contrast-enhanced MRI sella revealed normal anterior pituitary and dysplastic cranial bones. Bone scan revealed polyostotic fibrous dysplasia (FD). He was diagnosed as having McCune-Albright syndrome (MAS). His serum phosphate levels were persistently low. This was coupled with low Tmp/GFR (tubular reabsorption of phosphate adjusted for glomerular filtration rate) and elevated serum FGF-23 level. The phosphatonin was being produced by the dysplastic bony lesions. Hypophosphataemia worsened following trans-sphenoidal hypophysectomy as GH levels were reduced.
Case presentation
A 38-year-old man presented to our institute complaining of deformities in his legs, pain in his knees and difficulty in walking. He also complained of excessive height gain since childhood, progressive increase in the size of footwear and worsening facial deformity. He denied any history of headache or visual disturbance; neither was there any history suggestive of precocious puberty nor renal stone disease. His history was unremarkable and none of his family members had any such complaints.
Examination revealed a tall stature (height=196 cm), ‘coarse’ look, facial asymmetry (figure 1), prognathism, macroglossia and widely spaced upper incisors. He had seborrhoea, sweaty palms and a sonorous voice. In addition, he had large hands and feet, wind-swept deformity of the lower limbs (figure 1) and a small café-au-lait macule with a ‘coast of Maine’ appearance over his back on the right side.
Figure 1.
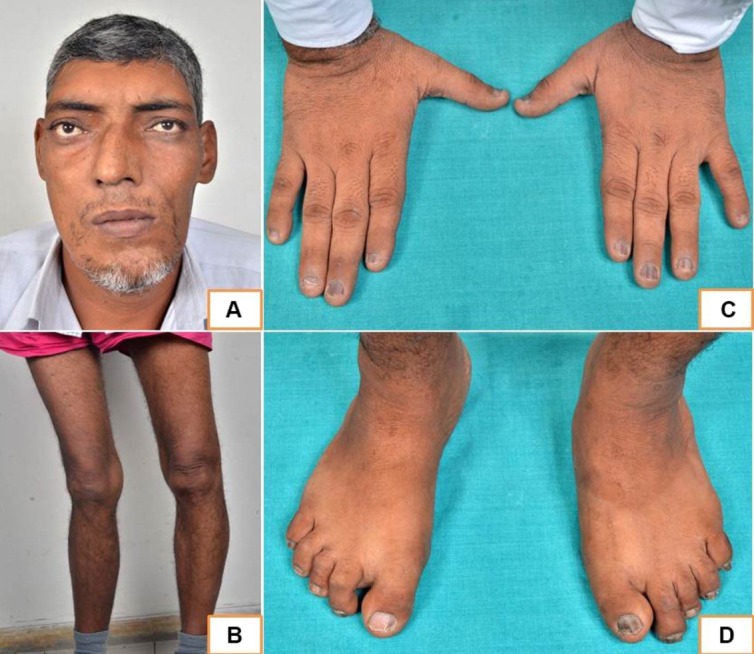
Clinical photograph showing coarse facies with facial asymmetry (A), wind-swept deformity of lower limbs (B) and acral enlargement (C and D).
Investigations
Complete blood count showed haemoglobin 10.9 g/dL, total leucocyte count 6.4×109/L and platelet count 201×109/L. Renal and liver function tests were normal. His blood glucose was elevated with a glycated haemoglobin of 6.7%. He had hypophosphataemia (serum phosphate=1.8 mg/dL), normocalcaemia, normomagnesaemia and elevated serum alkaline phosphatase levels (up to one and half times elevated). He was vitamin D sufficient (serum 25(OH) vitamin D=34.0 ng/mL) and had a serum intact parathyroid hormone (PTH) level of 16.2 pg/mL. TmP/GFR was low (0.18 mg/dL, normal range 3.09–4.18 mg/dL), suggesting renal phosphate wasting. Serum FGF-23 level was high (316.9 RU/mL, normal range 0.0–150.0 RU/mL), implying FGF-23-mediated phosphaturia.
His serum IGF-1 level was elevated. Basal GH level was high (87 ng/mL) and was non-suppressible following an oral glucose load. Rest of the anterior pituitary profile revealed euthyroidism (T4=7.2 µg/dL, range 4.8–12.7 µg/dL), hypocortisolism (08:00: cortisol=165 nmol/L, range 171–536 nmol/L), hypogonadotropic hypogonadism [testosterone=2.08 nmol/L (range 9.9–27.8 nmol/L), luteinizing hormone=1.91 mIU/mL (range 1.7–8.6 mIU/mL), follicle-stimulating hormone=3.79 mIU/mL (range 1.5–12.4 mIU/mL)] and hyperprolactinaemia (serum prolactin=561.4 ng/mL, range 4.0–15.2 ng/mL).
Electrocardiography of the patient revealed sinus bradycardia with occasional ventricular ectopics. Two-dimensional echocardiography revealed concentric left ventricular hypertrophy with an ejection fraction of 60%. Radiograph of the knees showed similar lesions involving the right femur and tibia with narrowing of the joint space. Non-contrast CT of the skull revealed ground-glass appearance of the frontal bone and the right greater wing of sphenoid (figure 2). Contrast-enhanced MRI sella showed normal pituitary gland with underlying dysplastic sphenoid bone. There was no visible focal lesion in the pituitary gland (figure 3). Tc99m-methylene diphosphonate bone scintigraphy revealed dysplastic lesions involving bones on the right side of the body, notably the right half of frontal bone, right half of sphenoid, right mandible, right ilium, femur and tibia. Ga68-DOTATATE scintigraphy showed somatostatin receptor expressing dysplastic skeletal lesions (figure 4). Ultrasonography of the neck showed thyromegaly and that of scrotum revealed bilateral testicular microlithiasis.
Figure 2.

Radiograph of the lower limbs showing mixed sclerotic and lytic lesions involving the right femur and tibia suggestive of fibrous dysplasia. (B and C) Non-contrast CT scan of the head (bone window) showing ‘ground-glass’ appearance of the greater wing of sphenoid (right sided) and the right ethmoid bone suggestive of fibrous dysplasia.
Figure 3.

MRI sella precontrast (A) and postcontrast (B) showing normal pituitary gland (marked in blue arrow) and underlying solid-cystic dysplastic sphenoid bone (marked in red arrow).
Figure 4.

Bone scintigraphy images showing increased osteoblastic activity in multiple bones, predominantly on the right side of the body suggestive of polyostotic fibrous dysplasia. (B and C) Ga68-DOTATATE positron-emission tomography CT images showing somatostatin-receptor expression in the dysplastic bones of the cranium.
Differential diagnosis
The clinical and biochemical profile of the patient was consistent with acrogigantism. However, bony deformities and facial asymmetry are rather uncommon in classical acromegaly, and their presence should make the physician think of two possibilities: multiple endocrine neoplasia type 1 (MEN1) and MAS. Bony deformity in MEN1 and MAS is explained by the presence of brown tumours and FD, respectively. Our patient did not have primary hyperparathyroidism as was evidenced by repeatedly normal serum calcium levels. On the other hand, the presence of polyostotic FD, endocrinopathy (in the form of acrogigantism) and café-au-lait macule made a clinical diagnosis of MAS more likely. Hypophosphataemia, on the other hand, could be explained by the release of FGF-23 from the dysplastic bony lesions. Low serum phosphate in MAS could also be explained by renal phosphate wasting secondary to constitutive PTH-coupled Gsα activation in the proximal renal tubule.
Treatment
Patient was started on oral hydrocortisone supplementation at admission. In view of ventricular ectopics, he was put on oral metoprolol with which his heart rate was regularised. Oral metformin was required for control of blood glucose. He underwent endoscopic endonasal trans-sphenoidal surgery. Dysplastic sphenoid bone made access to the sella difficult; hence, sphenoidectomy had to be done. Intraoperatively, no definite pituitary adenoma could be visualised, so near total hypophysectomy was done. Sellar floor was sealed with abdominal fat graft. Surprisingly, histopathological examination of the resected tissue showed pituitary adenoma surrounded by normal pituitary tissue with focal invasion of the bony sellar floor at places. For polyostotic FD, he was given intravenous zoledronate.
Outcome and follow-up
On postoperative day 1, his serum GH had decreased from 87 to 12.8 ng/mL. His glycaemic control improved and he was taken-off oral metformin. However, his hypophosphataemia worsened and on postoperative day 3, serum phosphate level had reached a nadir of 1.2 mg/dL for which he required oral phosphate and active vitamin D supplementation. Phosphate was supplemented in the form of sodium phosphate granules at a dose of 20 mg/kg/day. Oral hydrocortisone replacement was continued.
He was followed up after 3 months. His random serum GH was still high (15.7 ng/mL) implying non-remission. Hence, he was started on oral cabergoline at a dose of 0.25 mg/day. Serum phosphate was 2.8 mg/dL on supplementation. He came for his second follow-up after another 3 months. His serum IGF-1 levels had normalised; however, random GH level was high (28.6 ng/mL). Contrast-enhanced MRI sella was repeated which showed residual pituitary gland. He is subsequently planned for gamma-knife therapy to the pituitary bed.
Discussion
MAS is a complex genetic syndrome that affects bones, skin and/or endocrine organs. The syndrome was originally described by Dr McCune and Dr Albright as a classical triad of polyostotic FD, café-au-lait macule and precious puberty.2 FD is the most common manifestation (98% of patients) followed by café-au-lait macule. Precocious puberty is the most prevalent endocrine manifestation and it tends to be more common in females than in males.3 Common to our knowledge is the fact that postzygotic GNAS mutation (which codes for Gsα subunit) is responsible for the clinical manifestations of MAS. Most notably, constitutive activation of Gsα coupled to PTH receptor in bones leads to FD, while that coupled to melanocyte stimulating hormone receptor in skin results in café-au-lait macules. In lieu of the wide range of manifestations encountered in MAS, the clinically relevant definition of MAS was broadened to include any two of the three features: FD, café-au-lait macule, at least one hyperfunctioning endocrinopathy.4
Acromegaly is seen in about 20% of patients with MAS.5 The oldest association between acromegaly and MAS comes from autopsy study performed on Mr Thomas Hasler, popularly known as ‘The Tegernsee Giant’. Mr Thomas had been growing unusually rapidly since the age of 9 years and had a premature death at the age of 25 years when he had already attained an astonishing height of 235 cm. Radiological examination revealed a multicystic appearance involving all the skull bones and left-sided tibia, fibula, femur, humerus, metacarpal and iliac bones. The bones on the right half of the body were unremarkable. The sella turcica appeared widened. Histological examination of a bony tissue specimen from the skull revealed features of FD.6
Acromegaly in MAS differs from “classical’ acromegaly in multiple aspects. First, patients with MAS tend to be younger at the time of diagnosis of acromegaly (20–30 years vs 40–50 years in classical acromegaly). Second, hyperprolactinaemia is more common in MAS-associated acromegaly than the classical variant (70%–90% vs 30%–40%). Third, on sellar imaging, a pituitary adenoma can be visualised in as many as 95% of cases of classical acromegaly. On the contrary, in MAS-associated acromegaly, a definite adenoma can be seen in only 50% of patients; however, with advancement in imaging techniques, the number has increased to 70%.7 Our index patient was young, had hyperprolactinaemia and no visible adenoma on sellar imaging. In addition, response to standard treatment differs between the two groups. While trans-sphenoidal resection of pituitary adenoma remains the treatment of choice in classical acromegaly with a remission rate of 75%, the same does not hold true for MAS-associated acromegaly. Trans-sphenoidal surgery in MAS is technically difficult due to the presence of dysplastic bones at the skull base. Absence of adenoma on preoperative imaging makes surgery all the more difficult and hence total hypophysectomy remains the best option. Even then the remission rate is a mere 20%; however, with recent neuro-navigation-guided technique, higher remission rates have been achieved. Response to medical therapies is similarly low in the MAS group as compared with the classical group.5 7
Hypophosphataemia in MAS is secondary to renal-phosphate wasting. Though phosphaturia occurs in about 50% of patients with MAS, clinically significant hypophosphataemia is rare.8 It is mediated by FGF-23 produced by the dysplastic bony lesions. Serum levels of FGF-23 positively correlate with disease activity and frank hypophosphataemia is seen only in patients with significant FD burden. In addition to increased FGF-23 production, processing of the phosphatonin is altered in MAS. Gsα activation leads to cyclic adenosine monophosphate mediated decrease in the activity of the enzyme that glycosylates FGF-23, thereby rendering it prone to cleavage by furin. This leads to a proportionally greater increase in the circulating levels of C-terminal peptide relative to intact FGF-23.9 Phosphaturia in MAS could also result from constitutive activation of PTH-coupled Gsα in the proximal renal tubule which would impair phosphate reabsorption. Nevertheless, whatever may be the underlying mechanism, hypophosphataemia increases the risk of fractures and needs prompt treatment.
GH excess in MAS tends to have a dual effect on serum phosphate level. GH per se increases proximal tubular reabsorption of phosphate and leads to hyperphosphataemia. At the same time, GH excess can lead to further growth of FD lesions in MAS, which in turn would increase the circulating levels of FGF-23 and contribute to hypophosphataemia.
In our index patient, MAS was characterised by polyostotic FD, functioning endocrinopathy in the form of acrogigantism and café-au-lait macule. In addition, he had thyromegaly and testicular microlithiasis, features well described in MAS. He had hypophosphataemia with elevated FGF-23 levels and a substantial FD burden. Features of osteomalacia that were present in our patient were bone pains and lower limb bony deformities. Following surgery, as the GH levels declined, the phosphate retaining effect of GH was abolished and as expected, hypophosphataemia worsened.
Learning points.
Growth hormone (GH) excess is usually associated with hyperphosphataemia; low serum phosphate in the presence of GH excess should make one think of McCune-Albright syndrome (MAS).
Bony deformities in a patient with acromegaly suggest either multiple endocrine neoplasia type 1 or MAS.
Hypophosphataemia in MAS can either be due to FGF-23 production by the dysplastic skeletal lesions or constitutive activation of parathyroid hormone-coupled Gsα in the proximal renal tubule leading to phosphaturia.
Footnotes
Contributors: RP prepared the manuscript. PD helped in managing the patient. KKM operated on the patient. AB conceptualized the manuscript and provided overall guidance.
Competing interests: None declared.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Yalin GY, Tanrikulu S, Gul N, et al. Utility of baseline serum phosphorus levels for predicting remission in acromegaly patients. J Endocrinol Invest 2017;40:867–74. 10.1007/s40618-017-0657-3 [DOI] [PubMed] [Google Scholar]
- 2.Albright F, Butler AM, Hampton AO, et al. Syndrome characterized by osteitis fibrosa disseminata, areas of pigmentation and endocrine dysfunction, with precocious puberty in females. N Engl J Med Overseas Ed 1937;216:727–46. 10.1056/NEJM193704292161701 [DOI] [Google Scholar]
- 3.Collins MT, Singer FR, Eugster E. McCune-Albright syndrome and the extraskeletal manifestations of fibrous dysplasia. Orphanet J Rare Dis 2012;7 Suppl 1:S4 10.1186/1750-1172-7-S1-S4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dumitrescu CE, Collins MT. McCune-Albright syndrome. Orphanet J Rare Dis 2008;3:12 10.1186/1750-1172-3-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Salenave S, Boyce AM, Collins MT, et al. Acromegaly and McCune-Albright syndrome. J Clin Endocrinol Metab 2014;99:1955–69. 10.1210/jc.2013-3826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nerlich A, Peschel O, Löhrs U, et al. Juvenile gigantism plus polyostotic fibrous dysplasia in the Tegernsee giant. Lancet 1991;338:886–7. 10.1016/0140-6736(91)91542-3 [DOI] [PubMed] [Google Scholar]
- 7.Yao Y, Liu Y, Wang L, et al. Clinical characteristics and management of growth hormone excess in patients with McCune-Albright syndrome. Eur J Endocrinol 2017;176:295–303. 10.1530/EJE-16-0715 [DOI] [PubMed] [Google Scholar]
- 8.Riminucci M, Collins MT, Fedarko NS, et al. FGF-23 in fibrous dysplasia of bone and its relationship to renal phosphate wasting. J Clin Invest 2003;112:683–92. 10.1172/JCI18399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robinson C, Collins MT, Boyce AM. Fibrous dysplasia/McCune-Albright syndrome: clinical and translational perspectives. Curr Osteoporos Rep 2016;14:178–86. 10.1007/s11914-016-0317-0 [DOI] [PMC free article] [PubMed] [Google Scholar]