

Abstract
Angioedema is the swelling of mucosal and sub-mucosal tissue. Typically, it manifests as the swelling of the face, lips, and tongue. Angioedema can be severe and life threatening when it involves the respiratory tract. Drug induced allergic angioedema and drug induced non-allergic angioedema differ in their mediator, their clinical presentations, and their management. In drug induced non-allergic angioedema, symptoms are resistant to antihistamine and corticosteroid treatment. The aim of the analysis was to identify which medications are associated with drug-induced non-allergic angioedema and to understand the mechanism of action via which of these medication cause angioedema.
Keywords: angioedema, hereditary angioedema, non-allergic, causality, drug hypersensitivity, drug induced reactions, urticarial, hypersensitivity, female
Introduction and background
Angioedema is the swelling of mucosa and submucosal tissue. Typically, it manifests as the swelling of the face, lips, and tongue. Angioedema can be severe and life threatening when it involves the respiratory tract [1]. Drugs are second to food as the most common cause of angioedema cases seen in the emergency department [2]. Drugs may induce two different types of angioedema; allergic and non-allergic angioedema [3]. Studies have shown that to date, most physicians in the emergency department do not recognize the specific type of angioedema presenting in a patient, and they don’t treat the angioedema episode considering the difference in management that each type of angioedema implies [4]. Indeed, drug induced allergic angioedema and drug induced non-allergic angioedema differ in their mediator, their clinical presentations, and their management.
Drug induced allergic angioedema is a type I hypersensitivity and mediated by histamine [5]. In type I hypersensitivity, the medication cross link with immunoglobulin E (IgE) antibody bound on the surface of mast cells which results in the release of histamine [6]. Clinically, drug induced allergic angioedema will present with the rapid onset of swelling of mucosa and submucosa tissues. The patient will also have a typical urticarial rash. Symptoms will respond quickly to antihistamine, epinephrine and corticosteroid treatment [1].
Drug induced non-allergic angioedema is mediated by bradykinin [7]. In drug induced non-allergic angioedema the urticarial rash is absent. In addition, the onset is more progressive as compared to histamine mediated angioedema. Symptoms may subside in three to five days [7]. In drug induced non-allergic angioedema, symptoms are resistant to antihistamine and corticosteroid treatment, symptoms resolve only after drug discontinuation [8].
Because bradykinin mediated angioedema is under recognized, poorly managed, and the patients have an unfavorable outcome, this article will review drug induced non-allergic angioedema. The drugs covered in this article are selected as examples of key targets in the kallikrein-kiting system (KKS), and because together they cover the entire KKS from bradykinin synthesis to bradykinin receptors.
Article published on PubMed index journal between 1978 and 2017 and related to the topic of interest were included in the study. The keywords used to search the article included “angioedema”, “non-allergic”, “causality”,“ drug hypersensitivity”, “histamine antagonists”, “urticaria”, “bradykinin”, “epidemiology”, “drug combinations” and “gender”. A total of 138 articles were reviewed and analyzed. Out of 138 articles; 40 articles were found to be pertinent to our study and were included in the study.
Review
The kallikrein-kinin cascade and its interactions with the renin-angiotensin-aldosterone and the complement systems
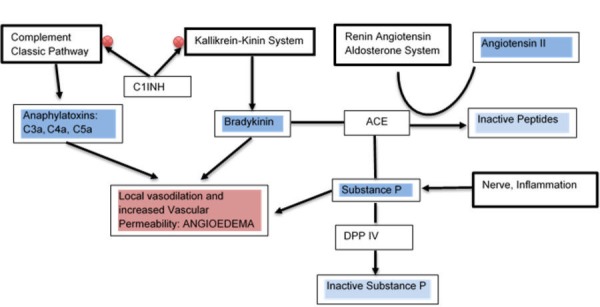
The KKS is a cascade of proteolytic enzymes that release vasoactive peptides. Plasma kallikrein cleaves human high molecular weight kininogen and releases bradykinin [9]. Bradykinin stimulates beta-2 adrenergic (B2) receptors which result in the release of nitric oxide and prostacyclin [10-11]. Nitric oxide and prostacyclin release result in local vasodilation and increased vascular permeability which leads to the development of angioedema [12]. The KKS antagonizes the renin angiotensin aldosterone system (RAAS) in its vascular effect [13]. The KKS and RAAS are coupled by the angiotensin converting enzyme (ACE). The ACE degrades bradykinin in the KKS and synthesizes Angiotensin II from Angiotensin I in the RAAS [14].
Another cascade of proteolytic enzymes is the complement system. Once activated, the complement system will produce anaphylatoxins (C3a, C4a, and C5a) and membrane attack complex. Anaphylatoxins and bradykinin have a similar mechanism of action. They increase local vascular permeability and cause vasodilation. The membrane attack complex also causes cell lysis [15-17].
The KKS is also coupled to the complement system by the C1 Inhibitor (C1INH). The C1INH is a serine protease that inhibits the KKS via the inactivation of factor XIIa and kallikrein and inhibits the complement system via the inactivation of C1r and C1s of the classical pathway [18-20]. Interaction of KKS, RAAS and complement system in the development of angioedema has been illustrated in Figure 1.
Figure 1. Interaction of kallikrein-kiting system (KKS), renin angiotensin aldosterone system (RAAS) and complement system in the development of angioedema.
Simvastatin and bradykinin type two receptors
A 75-year-old African American female patient with type 2 diabetes and dyslipidemia was admitted to the hospital with recurrent night episodes of facial, lip and tongue swelling. The patient denies any rash during these episodes and mentioned that self-medication with diphenhydramine did not relieve her symptoms. The patient was hemodynamically stable. The patient's normal level of C4, C1 esterase inhibitors, and C1q binding assay ruled out the diagnosis of hereditary angioedema. Her medications were reviewed, Simvastatin dose was increased each night from 10 to 20 mg, nine months prior to admission. During this period, episodes of angioedema woke the patient up almost each night. Simvastatin was considered as the possible trigger of these episodes of angioedema and was discontinued. Oxygen saturation was monitored continuously. The patient facial, lip and tongue swelling resolved over the next 36 hours without the use of any further corticosteroid treatment. In the following six months, the patient reported only one episode of daytime angioedema but no new episode of nighttime angioedema [21]. In this case report, bradykinin is most likely the mediator of angioedema episode because of at least four facts; there was no urticarial rash, the relatively long delay of angioedema onset after the simvastatin administration, the lack of improvement of the swelling with diphenhydramine, and the resolution of angioedema after simvastatin discontinuation.
At least two mechanisms have been identified in Statins induced non-allergic angioedema. Firstly, statins increase expression of bradykinin type 2 receptors on endothelial cells. In an experience where human coronary endothelial cells were cultured, Lovastatin upregulated bradykinin type two receptors. Secondly, statins may also potentiate the action of bradykinin on its receptors. Both mechanisms can make a patient susceptible to develop angioedema with circulating level of bradykinin through the increased release of prostacyclin and nitric oxide [12, 22].
Sitagliptin, angiotensin converting enzyme and dipeptidyl peptidase IV
A case reported concluded that Sitagliptin is associated with drug induced non-allergic angioedema. In this case report a 79-year old female with type 2 diabetes previously taking detemir insulin, atorvastatin 20 mg, and Irbesartan 150 mg was started on sitagliptin due to the high glycated hemoglobin (HbA1c) which was 8.1%. The patient developed angioedema with swelling of her lips, tongue, and mouth 14 days after starting sitagliptin. Sitagliptin was discontinued and remission was observed within a few days without supportive medication. Ten days later, the sitagliptin was re-started. The patient developed angioedema again two days later. Sitagliptin was discontinued permanently. This case describes the scenario of an oral drug challenge to confirm non-allergic angioedema: the urticarial rash is not present, symptoms are only induced by the administration of sitagliptin and they have a delayed onset. The discontinuation of the drug led to resolution of all symptoms [23].
Sitagliptin is a dipeptidyl peptidase IV (DPP-IV) inhibitor. The DPP-IV inactivates the incretins glucose-dependent insulinotropic polypeptide and glucagon-like peptide [24]. As a result, DPP-IV improves glycemic control in type 2 diabetes mellitus [25]. In addition to incretins, DPP-IV also inactivates substance P, a vasoactive peptide that increases vascular permeability by its action on neurokinin receptor 1 (NK1) [26]. Substance P is involved in ACE inhibitor (ACEI) associated angioedema. Studies have demonstrated that Substance P levels are increased during ACEI associated angioedema [27]. Also, infusion of bradykinin or substance P caused tracheal edema in the rate [28]. Both bradykinin and substance P are degraded by ACE. During ACE inhibition, accumulation of Substance P doesn’t cause angioedema because it is inactivated by DPP-IV. Thus, the addition of DPP-IV inhibitor such as Sitagliptin may cause angioedema in susceptible patients [29-30].
Vildagliptin, another DPP-IV inhibitor has not been associated with angioedema when it is taken alone. It has been shown to causes angioedema in the patients who are also taking ACEI concomitantly [31]. This latter finding confirms the contribution of substance P in the pathogenesis of angioedema.
Risperidone and C1 inhibitor
To date, only four cases of risperidone have been reported to be associated with angioedema [32-35]. Among them, a case-report described an episode of angioedema that occurred in a 30-year-old female treated for the schizoaffective disorder. The patient developed facial and periorbital edema without urticaria two weeks after risperidone was started at 6 mg dose. When the dose was halved, symptoms subsided, but the mental status deteriorated. Risperidone was then increased to 6 mg/day but resulted in the recurrence of the facial and periorbital edema. Risperidone was stopped and the facial and periorbital edema resolved completely over two weeks. The Immunology laboratory test results showed a normal level of C3 and low levels of C4 and C1 esterase inhibitor [34]. A similar case of angioedema was reported in a 38-year-old female patient with amphetamine induced mood disorder who was admitted for aggressiveness, irritability, and grandiose thoughts. Risperidone was started at 2 mg and then increased to 6 mg/day. Nine days after the treatment, the patient developed facial and periorbital swelling. Laboratory showed normal levels of C3, C4, and C1 esterase inhibitor. Risperidone was discontinued; low dose hydrocortisone and hydroxyzine were prescribed. The swelling resolved completely within four days. The clinical pattern of angioedema episodes in these cases supports a non-allergic form of angioedema because there was no urticaria. In addition, the delayed onset of symptoms after medication was administrated, and the progressive recovery is less likely to be the manifestation of a type I hypersensitivity reaction.
The chronology of symptoms in both cases is more consistent with bradykinin-mediated angioedema. Finally, the complete resolution of symptoms after the discontinuation of risperidone supports bradykinin-mediated angioedema. Risperidone causes non-allergic angioedema by two mechanisms. The first mechanism is the suppression of C1INH [34]. This is supported by the low level of C1INH, normal level of C3 and low-level C4 found during these two case reports. Normally, C1INH inhibits the complement system and the KKS. The C1INH suppression will increase spontaneous activation of the classical pathway of the complement and anaphylatoxins production (C3a, C4a, C5a) [18]. Anaphylatoxins will cause vascular permeability and vasodilation [16]. The C1INH inhibition will also leave the KKS unopposed with increased synthesis of bradykinin. Both actions will result in angioedema. The second mechanism is the aggregation of bradykinin. In this case, the patient will have a normal level of C1INH, C3, and C4 (32). Bradykinin triggers the release of nitric oxide and prostacyclin by endothelial cells. Nitric Oxide and prostacyclin will increase vascular permeability and will lead to the development of angioedema [12].
Summary of mechanism of action of drug induced non-allergic angioedema
To our knowledge, drugs can target three different sites of actions in the KKS to cause bradykinin mediated angioedema:
1. The synthesis of bradykinin with C1 INH suppression: Risperidone increases bradykinin production by inhibiting the C1 INH. The same mechanism also increases complement activity
2. The response to bradykinin: Bradykinin receptors type 2: Statins for example increase blood gene BR2 expression and their sensitivity to bradykinin
3. The metabolism of bradykinin and substance P with ACE and DPP-IV suppression: The action of ACEI on ACE and Sitagliptin on DPP-IV inhibits Substance P degradation and inactivation respectively. In addition, ACEI also decreases the metabolism of bradykinin
Management
Drug induced non-allergic angioedema (bradykinin mediated angioedema) will present with some characteristic features in the clinical presentation. It is important to distinguish non-allergic from allergic angioedema in order to treat the patient accordingly. The speed of onset is characteristic. Histamine mediated angioedema starts less than an hour after the exposure and bradykinin mediated angioedema may take several hours to days to develop. Urticaria is never present in bradykinin mediated angioedema. Abdominal manifestations are more common in drug induced non-allergic angioedema than in drug induced allergic angioedema [36].
Respiratory tract involvement is uncommon in bradykinin mediated angioedema. The most important action to take when a drug induced non-allergic angioedema is suspected is to discontinue the offending drug [7-8]. In Histamine induced angioedema, symptoms resolve within 24 hours. Symptoms last longer in bradykinin mediated angioedema and can last up to five days [37]. Antihistamine, epinephrine, and corticosteroid have not shown to decrease the duration of symptoms [38]. Supportive care is provided if the respiratory tract is involved.
Although laboratory results are not available early enough to guide the treatment, it is important to measure C4 level and tryptase level to rule out other etiologies and to ensure proper follow-up. The C4 level is always decreased in hereditary angioedema. The diagnosis of hereditary angioedema is more likely in the absence of obvious etiology. Tryptase is elevated in allergic angioedema [37-38]. The patient who does not have any involvement of the tongue, larynx or any other airway compromise can be discharged after 12 to 24 hours of observation.
A patient who had life threatening episode of angioedema should discontinue the medication indefinitely. the blood results can be evaluated in an outpatient setting with the primary care physician. If hereditary angioedema (HAE) is suspected, the patient should be referred to an immunologist for further evaluation and prophylactic treatment [39-40].
Conclusions
Drug induced angioedema is a common scenario in the emergency department. It can be a life threatening when airways are involved. Many physicians approached it like an allergic reaction. There is a need to increase awareness about drug induced non-allergic angioedema as its management is different from allergic angioedema. In the case of the nonallergic reaction, stopping the offending agent is the most important action to take.
The content published in Cureus is the result of clinical experience and/or research by independent individuals or organizations. Cureus is not responsible for the scientific accuracy or reliability of data or conclusions published herein. All content published within Cureus is intended only for educational, research and reference purposes. Additionally, articles published within Cureus should not be deemed a suitable substitute for the advice of a qualified health care professional. Do not disregard or avoid professional medical advice due to content published within Cureus.
Footnotes
The authors have declared that no competing interests exist.
References
- 1.Angioedema. Kaplan AP, Greaves MW. J Am Acad Dermatol. 2005;53:373–388. doi: 10.1016/j.jaad.2004.09.032. [DOI] [PubMed] [Google Scholar]
- 2.Angioedema: Clinical and etiological aspects. Kulthanan K, Jiamton S, Boochangkool K, et al. Clin Dev Immunol. 2007;2007:1–6. doi: 10.1155/2007/26438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Plasma bradykinin in angio-oedema. Nussberger J, Cugno M, Amstutz C, et al. Lancet. 1998;351:1693–1697. doi: 10.1016/S0140-6736(97)09137-X. [DOI] [PubMed] [Google Scholar]
- 4.Are physicians aware of the side effects of angiotensin-converting enzyme inhibitors?. A questionnaire survey in different medical categories. Lombardi C, Crivellaro M, Dama A, et al. Chest. 2005;128:976–979. doi: 10.1378/chest.128.2.976. [DOI] [PubMed] [Google Scholar]
- 5.Angioneurotic oedema with tadalafil: A rare case report. Raj R, Sidhu BS. Indian J Psychiatry. 2006;48:263–264. doi: 10.4103/0019-5545.31562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pathogenesis of drug allergy--current concepts and recent insights. Schnyder B, Brockow K. Clin Exp Allergy. 2015;45:1376–1383. doi: 10.1111/cea.12591. [DOI] [PubMed] [Google Scholar]
- 7.Development and validation of the angiotensin-converting enzyme inhibitor (ACEI) induced angioedema investigator rating scale and proposed discharge criteria. Bonner N, Panter C, Kimura A, et al. BMC Health Serv Res. 2017;17:366. doi: 10.1186/s12913-017-2274-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clinical immunology review series: An approach to the patient with angio-oedema. Grigoriadou S, Longhurst HJ. Clin Exp Immunol. 2009;155:367–377. doi: 10.1111/j.1365-2249.2008.03845.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ferritin binds to light chain of human H-kininogen and inhibits kallikrein-mediated bradykinin release. Parthasarathy N, Torti SV, Torti FM. Biochem J. 2002;365:279–286. doi: 10.1042/BJ20011637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Inflammatory muscle pain is dependent on the activation of kinin B₁ and B₂ receptors and intracellular kinase pathways. Meotti FC, Campos R, da Silva K, et al. Br J Pharmacol. 2012;166:1127–1139. doi: 10.1111/j.1476-5381.2012.01830.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bradykinin receptor ligands: Therapeutic perspectives. Marceau F, Regoli D. Nat Rev Drug Discov. 2004;3:845–852. doi: 10.1038/nrd1522. [DOI] [PubMed] [Google Scholar]
- 12.Bradykinin B2 receptor knockout mice are protected from thrombosis by increased nitric oxide and prostacyclin. Shariat-Madar Z, Mahdi F, Warnock M, et al. Blood. 2006;108:192–199. doi: 10.1182/blood-2006-01-0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.The plasma kallikrein-kinin system counterbalances the renin-angiotensin system. Schmaier AH. J Clin Invest. 2002;109:1007–1009. doi: 10.1172/JCI15490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fragment-based design for the development of N-domain-selective angiotensin-1-converting enzyme inhibitors. Douglas RG, Sharma RK, Masuyer G, et al. Clin Sci. 2014;126:305–313. doi: 10.1042/CS20130403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Comparative genomics reveal that host-innate immune responses influence the clinical prevalence of Legionella pneumophila serogroups. Khan MA, Knox N, Prashar A, et al. PLoS One. 2013 doi: 10.1371/journal.pone.0067298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.The complement system: history, pathways, cascade and inhibitors. Nesargikar PN, Spiller B, Chavez R. Eur J Microbiol Immunol. 2012;2:103–111. doi: 10.1556/EuJMI.2.2012.2.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fc microparticles can modulate the physical extent and magnitude of complement activity. Holt BA, Bellavia MC, Potter D, et al. Biomater Sci. 2017;5:463–474. doi: 10.1039/c6bm00608f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Regulation of complement and contact system activation via C1 inhibitor potentiation and factor XIIa activity modulation by Sulfated Glycans - structure-activity relationships. Schoenfeld AK, Lahrsen E, Alban S. PLoS One. 2016 doi: 10.1371/journal.pone.0165493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cross-talk between the complement and the kinin system in vascular permeability. Bossi F, Peerschke EI, Ghebrehiwet B, et al. Immunol Lett. 2011;140:7–13. doi: 10.1016/j.imlet.2011.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Biochemical characterization of a novel high-affinity and specific plasma kallikrein inhibitor. Kolte D, Bryant J, Holsworth D, et al. Br J Pharmacol. 2011;162:1639–1649. doi: 10.1111/j.1476-5381.2010.01170.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Simvastatin: A risk factor for angioedema? Nisly SA, Kara A, Knight TB. J Pharm Technol. 2013;29:149–152. [Google Scholar]
- 22.Lovastatin induces the expression of bradykinin type 2 receptors in cultured human coronary artery endothelial cells. Liesmaa I, Kokkonen JO, Kovanen PT, et al. J Mol Cell Cardiol. 2007;43:593–600. doi: 10.1016/j.yjmcc.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 23.Angio-oedema induced by dual dipeptidyl peptidase inhibitor and angiotensin II receptor blocker: A first case report. Skalli S, Wion-Barbot N, Baudrant M, et al. Diabet Med. 2010;27:486–487. doi: 10.1111/j.1464-5491.2010.02973.x. [DOI] [PubMed] [Google Scholar]
- 24.Assessment of saxagliptin efficacy: Meta-analysis of 14 phase 2 and 3 clinical trials. Sjöstrand M, Wei C, Cook W, et al. Diabetes Ther. 2017;8:587–599. doi: 10.1007/s13300-017-0261-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dipeptidyl peptidase-4 and kidney fibrosis in diabetes. Shi S, Koya D, Kanasaki K. Fibrogenesis Tissue Repair. 2016;9:1. doi: 10.1186/s13069-016-0038-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Neurokinin mediation of edema and inflammation. Campos MM, Calixto JB. Neuropeptides. 2000;34:314–322. doi: 10.1054/npep.2000.0823. [DOI] [PubMed] [Google Scholar]
- 27.Association of angiotensin-converting enzyme inhibitor-associated angioedema with transplant and immunosuppressant use. Byrd JB, Grice WA, Stone E, et al. Allergy. 2010;65:1381–1387. doi: 10.1111/j.1398-9995.2010.02398.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.The effect of acute angiotensin-converting enzyme and neutral endopeptidase 24.11 inhibition on plasma extravasation in the rat. Sulpizio AC, Pullen MA, Edwards RM, et al. J Pharmacol Exp Ther. 2004;309:1141–1147. doi: 10.1124/jpet.103.064105. [DOI] [PubMed] [Google Scholar]
- 29.Dipeptidyl peptidase iv in angiotensin-converting enzyme inhibitor–associated angioedema. Byrd JB, Touzin K, Sile S, et al. Hypertension. 2008;51:141–147. doi: 10.1161/HYPERTENSIONAHA.107.096552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dipeptidyl peptidase-IV inhibitor use associated with increased risk of ACE inhibitor-associated angioedema. Brown NJ, Byiers S, Carr D, et al. Hypertension. 2009;54:516–523. doi: 10.1161/HYPERTENSIONAHA.109.134197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dipeptidyl peptidase-4 inhibitors and angioedema: a class effect? Saisho Y, Itoh H. Diabet Med. 2013;30:149–150. doi: 10.1111/dme.12134. [DOI] [PubMed] [Google Scholar]
- 32.Angioedema - an unusual serious side effect of risperidone injection. Güneş F, Batgi H, Akbal A, et al. Clin Toxicol. 2013;51:122–123. doi: 10.3109/15563650.2013.765010. [DOI] [PubMed] [Google Scholar]
- 33.Self-limiting atypical antipsychotics-induced edema: Clinical cases and systematic review. Umar MU, Abdullahi AT. Indian J Psychol Med. 2016;38:182–188. doi: 10.4103/0253-7176.183089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Risperidone-induced recurrent giant urticaria. Mishra B, Saddichha S, Kumar R, et al. Br J Clin Pharmacol. 2007;64:558–559. doi: 10.1111/j.1365-2125.2007.02909.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Acquired angioedema induced by risperidone. Talaei A, Faraji Rad S, Moghani BM, et al. Iran J Psychiatry Behav Sci. 2016;10:4807. [Google Scholar]
- 36.Abdominal attacks and treatment in hereditary angioedema with C1-inhibitor deficiency. Rubinstein E, Stolz LE, Sheffer AL, et al. BMC Gastroenterol. 2014;14:71. doi: 10.1186/1471-230X-14-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Diagnosis and treatment of bradykinin-mediated angioedema: outcomes from an angioedema expert consensus meeting. Craig TJ, Bernstein JA, Farkas H, et al. Int Arch Allergy Immunol. 2014;165:119–127. doi: 10.1159/000368404. [DOI] [PubMed] [Google Scholar]
- 38.Angioedema in the emergency department: A practical guide to differential diagnosis and management. Bernstein JA, Cremonesi P, Hoffmann TK, et al. Int J Emerg Med. 2017;10:15. doi: 10.1186/s12245-017-0141-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.A consensus parameter for the evaluation and management of angioedema in the emergency department. Moellman JJ, Bernstein JA, Lindsell C, et al. Acad Emerg Med. 2014;21:469–484. doi: 10.1111/acem.12341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Predicting airway risk in angioedema: staging system based on presentation. Ishoo E, Shah UK, Grillone GA, et al. Otolaryngol Head Neck Surg. 1999;121:263–268. doi: 10.1016/S0194-5998(99)70182-8. [DOI] [PubMed] [Google Scholar]