

Abstract
Cell therapies suffer from poor survival post-transplant due to placement into hostile implant sites characterized by host immune response and innate production of high levels of reactive oxygen species (ROS). We hypothesized that cellular encapsulation within an injectable, antioxidant hydrogel would improve viability of cells exposed to high oxidative stress. To test this hypothesis, we applied a dual thermo- and ROS-responsive hydrogel comprising the ABC triblock polymer poly[(propylene sulfide)-block-(N,N-dimethyl acrylamide)-block-(N-isopropylacrylamide)] (PPS135-b-PDMA152-b-PNIPAAM225, PDN). The PPS chemistry reacts irreversibly with ROS such as hydrogen peroxide (H2O2), imparting inherent antioxidant properties to the system. Here, PDN hydrogels were successfully integrated with type 1 collagen to form ROS-protective, composite hydrogels amenable to spreading and growth of adherent cell types such as mesenchymal stem cells (MSCs). It was also shown that, using a control hydrogel substituting nonreactive polycaprolactone in place of PPS, the ROS-reactive PPS chemistry is directly responsible for PDN hydrogel cytoprotection of both MSCs and insulin-producing β-cell pseudo-islets against H2O2 toxicity. In sum, these results establish the potential of cytoprotective, thermogelling PDN biomaterials for injectable delivery of cell therapies.
Keywords: : antioxidant, cell-delivery, hydrogel, thermoresponsive
Introduction
Cell therapies are promising for many biomedical applications such as tissue regeneration, immunomodulation, and replacement of lost cell function.1–5 Stem cells, both terminally differentiated and undifferentiated, have the potential to comprise impactful therapies that promote myocardial, cutaneous, or orthopedic tissue repair.6–10 In particular, it has been demonstrated that undifferentiated mesenchymal stem cells (MSCs) have immunomodulatory functions through the secretion of paracrine factors11 that can positively direct tissue regeneration. Other diseases, such as type 1 diabetes (T1D), are characterized by loss of a specific functional cell type (β-cells that produce insulin); T1D could potentially be cured by the transplantation and long-term survival of donated pancreatic islets12 or delivery of stem cell-derived, insulin-producing cells13 into diabetic patients.
While cell therapies have a broad range of potential applications, survival of the delivered cells is hampered by the host's innate and acquired immune response14,15 and minimal cellular engraftment and retention at the implantation site.16,17 These translational difficulties and the subsequent limited success of cell delivery in the clinic has motivated the development of next generation biomaterials to improve cell-based therapies.
Polymeric hydrogels are particularly advantageous for cell encapsulation and delivery due to their hydrated state and ability to be engineered with multiple functions such as cell adhesiveness and degradability. Furthermore, injectable hydrogels that undergo in situ gelation through temperature change,18 ultraviolet (UV) irradiation,19 shear forces,20 or host–guest interactions21 offer a strategy for mixing cells with gel precursors before minimally invasive injection and gelation.
Poly(N-isopropylacrylamide) (PNIPAAM) has been studied extensively as an injectable, thermogelling material due to its distinguishing lower critical solution temperature (LCST) behavior at around 34°C,18 allowing for thermogelation between ambient and physiological temperatures. However, hydrogels synthesized from PNIPAAM homopolymers are limited as cell delivery vehicles because they can undergo syneresis (hydrophobic expulsion of liquid as they thermoform),18 are minimally biodegradable, and do not provide recognizable extracellular matrix cues for cellular attachment.22
To leverage the LCST behavior of PNIPAAM in a more cytocompatible format, we recently developed an ABC triblock polymer, poly[(propylene sulfide)-block-(N,N-dimethyl acrylamide)-block-(N-isopropylacrylamide)] (PPS135-b-PDMA152-b-PNIPAAM225, PDN), which forms an injectable, cell-protective hydrogel.18 Mechanistically, the hydrophobic PPS “A” block triggers micelle formation in aqueous solution, the hydrophilic PDMA “B” block stabilizes the hydrophilic corona and prevents syneresis of the assembled gels, and the PNIPAAM “C” block endows thermal gelation properties at temperatures consistent with PNIPAAM homopolymer. The core-forming PPS component enables loading of hydrophobic drugs and is also sensitive to reactive oxygen species (ROS); oxidation of sulfides to sulfones and sulfoxides causes PPS to become more hydrophilic,23 driving micellar disassembly, hydrogel degradation, and controlled release of encapsulated drugs.24
High, localized concentrations of ROS, or oxidative stress, are produced at sites of biomaterial implantation25,26 and can lead to detrimental, cytotoxic effects such as irreparable DNA/protein modification and the triggering of bystander cell apoptosis.27 As such, oxidative stress can cause failure of cellular therapies.28 PPS-containing PDN hydrogels have been shown to minimize the toxicity of hydrogen peroxide (H2O2) when overlaid onto NIH 3T3 mouse fibroblasts grown in two-dimensional (2D) tissue culture plates.18 This result motivates the current exploration of PDN hydrogels for encapsulation and delivery of more therapeutically relevant cell types such as human mesenchymal stem cells (hMSCs) and pancreatic islets in a three-dimensional (3D) format that is more relevant to in vivo cell delivery.
One of the challenges of application of PDN hydrogels for cell delivery is that they do not feature intrinsic cellular adhesion motifs that can support long-term viability of adherent cell types. Previous reports have demonstrated that natural extracellular matrix components (i.e., collagen, hyaluronic acid, fibronectin, etc.) can be homogenously incorporated into PNIPAAM-based materials to promote cell adhesion with minimal impact on overall hydrogel LCST behavior.22 This significantly improves the cell adhesive properties of the hydrogel matrix, and produces comparable results to growth in the natural material alone.22 In particular, type 1 collagen (T1C) is one of the most abundant structural proteins found in almost all tissue and promotes robust cellular adhesion.29 Similar to PNIPAAM-based polymers, T1C solutions also undergo thermoresponsive gel formation,30 therefore making incorporation of T1C into PDN hydrogels an attractive strategy for increasing the cellular adhesion capacity of these materials. Herein, we have extended the utility and retained the injectability of PDN hydrogels by incorporating collagen into these materials to improve the adhesion, growth, and proliferation of both adherent and nonadherent cells in 3D culture. Furthermore, we explored the potential of PDN hydrogels to protect both the suspension culture of therapeutically relevant insulin-producing MIN6 pseudo-islets (PIs) and adherent hMSCs from cytotoxic levels of ROS. To our knowledge, this work represents the first successful demonstration of long-term 3D encapsulation and ROS protection of therapeutic cells within antioxidant, injectable hydrogels.
Materials
Normal cell medium (NCM) was prepared from Gibco (Grand Island, NY) 1 × Dulbecco's modified Eagle's medium (DMEM) with 4.5 g/L d-Glucose, l-Glutamine, 25 mM HEPES, and supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (P/S). Normal MSC medium (NMM) was prepared from Gibco 1 × MEM Alpha with l-Glutamine, Ribonucleosides, and Deoxyribonucleosides (Ref. No. 12571-063), and supplemented with 15% FBS and 1% P/S. Imaging cell medium (ICM) was prepared from Gibco Fluorobrite DMEM with high d-Glucose and 3.7 g/L sodium bicarbonate, and supplemented with 10% FBS. RatCol, rat tail type I collagen was purchased from Advanced Biomatrix (Carlsbad, CA). Mouse insulinoma pancreatic β-cells (MIN6) were a generous gift from the David Jacobson Laboratory at Vanderbilt University. Human bone marrow-derived hMSCs were purchased from Lonza (Walkersville, MD). Unless otherwise mentioned, all other materials were purchased from Sigma-Aldrich Corp. (St. Louis, MO).
Methods
Synthesis of PPS135-b-PDMA152-b-PNIPAAM225 (PDN) triblock copolymer
The PDN triblock polymer was synthesized as previously described.18 Briefly, hydroxyl functional poly(propylene sulfide) (PPS135-OH) was prepared via anionic ring-opening polymerization of propylene sulfide (135 mmol, 9.99 g, 10.56 mL) using 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU; 3 mmol, 0.46 g, 0.45 mL) as a base and 1-butane thiol (1 mmol, 0.122 g, 0.138 mL) as an initiator in tetrahydrofuran (THF, 10 mL) solvent at ambient temperature followed by quenching of the polymerization after 2 h with 2-iodo ethanol. The polymerization mixture was purified by three precipitations into cold methanol. The resultant PPS135-OH (0.6 mmol, 6.0 g) polymer was then conjugated to the reversible addition−fragmentation chain transfer (RAFT) chain transfer agent (CTA) 4-cyano-4-(ethylsulfanyltiocarbonyl) sulfanylpentanoic acid (ECT, 2.4 mmol, 0.628 g) using Steglich esterification coupling with dicyclohexylcarbodiimide (DCC, 2.4 mmol, 0.495 g) and 4-dimethylaminopyridine (DMAP, 0.18 mmol, 0.021 g) for 24 h at room temperature to yield PPS135-ECT as a RAFT macro CTA.
The obtained PPS135-ECT in dichloromethane (DCM) was purified through three precipitations into cold methanol. To prepare the diblock copolymer PPS135-b-PDMA152, N,N-dimethylacrylamide (DMA, 0.445 mL, 4.5 mmol) was RAFT polymerized from the PPS macro-CTA (0.3 g, 0.03 mmol) in 1,4-dioxane at 35°C for 16 h using the radical initiator 2,2′-azobis(4-methoxy-2,4-dimethylvaleronitrile) (V-70) (0.92 mg, 0.003 mmol) at a CTA:initiator molar ratio of 10:1. The polymer was purified by precipitation into a 10-fold excess of cold diethyl ether. Finally, the diblock copolymer, PPS135-b-PDMA152 (0.015 mmol, 0.37 g), was used for RAFT polymerization of N-isopropyl acrylamide (NIPAAM, 2.7 mmol, 0.30 g) in 1,4-dioxane using V-70 (0.0015 mmol, 0.46 mg) as an initiator at 35°C for 15 h at 5:1 ratio of CTA to V-70 to yield PPS135-b-PDMA152-b-PNIPAAM225 (PDN) triblock copolymer.
The crude mixture was purified by precipitation into cold diethyl ether. The precipitated polymer was dissolved into methanol and dialyzed against water for 48 h before lyophilization to yield the final product. The PDN triblock copolymer and precursor polymers at each step of synthesis were analyzed by 1H nuclear magnetic resonance (NMR, 400 MHz, CDCl3), and product molecular weight was determined using gel permeation chromatography (GPC; Agilent Technologies, Santa Clara, CA) run with a mobile phase of N,N-dimethylformamide (DMF) with 100 mM LiBr and in-line light scattering (Wyatt Technology Corporation, Santa Barbara, CA) and refractive index (RI) detectors.
Synthesis of PCL85-b-PDMA150-b-PNIPAAM150 (CDN)
The synthesis of PCL85-b-PDMA150-b-PNIPAAM150 was performed using a modified protocol from the synthesis of the PDN polymer. In the first step, hydroxyl functional PCL85-OH was synthesized by the ring opening polymerization of ɛ-caprolactone (64 mmol, 7.23 g) using benzyl alcohol (0.8 mmol, 0.086 g) as an initiator and Sn(Oct)2 (0.04 mmol, 0.014 g, 0.014 mL) as the catalyst for 2 h at 140°C. The crude polymer was dissolved in DCM and purified through precipitation into cold diethyl ether. The resulting PCL85-OH (0.6 mmol, 6 g) was conjugated with ECT (2.4 mmol, 0.63 g) at room temperature, using DCC (0.49 g, 2.4 mmol)/DMAP (0.021 g, 0.18 mmol) esterification for 24 h to obtain PCL85-ECT as a RAFT macro CTA. The crude polymer in DCM was purified by three precipitations into cold methanol.
The PCL85-ECT (5 g, 0.5 mmol) macro CTA was used to RAFT polymerize DMA (75 mmol, 7.425 g) using V-70 (0.025 mmol, 0.0077 g) as an initiator (5:1 ratio of macro CTA to V-70) in a 1,4-dioxane/DCM solvent mixture at 35°C for 16 h to prepare PCL85-b-DMA150. The resultant crude diblock copolymer solution was purified by precipitation into cold diethyl ether. In the final step, the PCL85-b-DMA150 (0.2 mmol, 5 g) macro CTA was used to polymerize NIPAAM (45 mmol, 5.08 g) using V-70 (0.02 mmol, 0.0061 g) as an initiator (5:1 ratio CTA to V-70) in 1,4-dioxane solvent at 35°C for 15 h to yield PCL85-b-PDMA150-b-PNIPAAM150 (CDN). The crude polymerization mixture was purified through precipitation into a 10-fold excess of cold diethyl ether. The vacuum-dried polymer was dissolved into methanol, dialyzed against water for 48 h, and then lyophilized. The structure and molecular weight of the lyophilized CDN triblock copolymer and precursors at each synthetic step were characterized by 1H NMR and GPC, respectively.
Preparation and rheological characterization of polymer-collagen composites
To select the appropriate concentration of collagen to maximize growth and viability of adherent cell types within our hydrogels, we used fibroblasts as a model cell type (Supplementary Fig. S1 and Supplementary Data; Supplementary Data are available online at www.liebertpub.com/tea).
To determine the storage modulus (G′) of our materials, lyophilized PDN polymer was weighed and dissolved in either 0.9 mL of phosphate-buffered saline (PBS) or a solution of 0.2 wt. % acid-solubilized rat tail, T1C to prepare two different polymer solutions at 3 or 5 wt. % PDN polymer. The polymers were allowed to dissolve for 48 h at 4°C, permitting polymer micellization. For collagen-only control gels, 950 μL of 0.2 wt. % T1C was pipetted into a separate tube, and 50 μL of the included neutralization solution was added to the tube. Five hundred microliters of each solution was dispensed onto 25 mm aluminum rheological base plates and allowed to gel for 1 h in a humidified incubator at 37°C. Each plate was then transferred to a rheometer (AR 2000 × Rheometer; TA Instruments, New Castle, DE), with the solidified hydrogel being compressed between the parallel plates at a gap distance of 1000 μm. The G′ values were then measured over a frequency range of 0.1–100 rad/s at a constant strain of 1% and a constant temperature of 37°C.
To demonstrate the injectability and thermogelation behavior of our PDN hydrogel+collagen composites, lyophilized PDN polymer was weighed and dissolved in 950 μL of 0.2 wt. % acid-solubilized T1C to prepare two aliquots of 3 wt. % PDN polymer solutions. The polymers were allowed to dissolve for 48 h at 4°C, permitting polymer micellization. The first polymer/collagen sample was then mixed with 50 μL of the included neutralization solution, and 500 μL of the solution was immediately dispensed onto a 25 mm aluminum rheological base plate attached to the rheometer. The solution was then compressed at 1000 μm gap distance. The dynamic viscosity was then measured over a strain rate range of 100–6000/s (the upper machine limit) at a constant temperature of 20°C.
Next, the second polymer/collagen solution sample was similarly neutralized and dispensed onto the base plate using the same solution volume and plate gap distance. To demonstrate the injectability of the material, the sample's viscosity was measured at 20°C, 6000/s shear rate for 30 s. The shear rate was then stepped immediately down to 0.100/s, while temperature was held at 20°C for the next 30 s. Temperature was then stepped up to 37°C while shear rate was held at 0.100/s, and measurements were continued for the next 60 s.
Scanning electron microscopy of polymer-collagen composites
PDN or CDN polymer solutions, with and without collagen, or collagen-alone solutions were prepared as described above. One hundred microliters of each solution was dispensed into microcentrifuge tubes (Fisherbrand Polyethylene Sample Vials with Hinged Cap, Size 1A; Fisher Scientific, Hampton, NH) and placed in an incubator at 37°C for 30 min. Following gelation, the tubes were maintained at 37°C using a hot plate, and the gel samples were carefully covered with 500 μL of 37°C PBS before being incubated at 37°C overnight.
To prepare the collagen-only and PDN/CDN hydrogel+collagen composites for scanning electron microscopy (SEM), the PBS solution covering the gels was aspirated and replaced with prewarmed 2.5% glutaraldehyde in PBS for fixation. The tubes were kept at 37°C for 3 h. The glutaraldehyde solution was removed, and the gels were successively incubated at 37°C with solutions of 50%, 60%, 70%, 80%, 90%, and 100% ethanol for 30 min each. The materials were carefully cut out of the tubes and critical point dried against liquid CO2 for 5 h. The materials were then bisected and placed onto carbon tape on SEM posts to enable imaging of the gels' internal morphology. For the PDN-only and CDN-only hydrogels, the PBS solution covering the gel was aspirated, and the gel was flash frozen in liquid nitrogen to preserve the thermoresponsive architecture. The materials were quickly cut out of the tubes and placed onto carbon tape on precooled SEM posts in a cooler filled with dry ice before being lyophilized. All SEM samples were gold sputter coated and imaged using a ZEISS MERLIN SEM (Carl Zeiss Microscopy, LLC, ZEISS Group, Thornwood, NY).
Human mesenchymal stem cell culture and protection from hydrogen peroxide-induced cell death
Bone marrow-derived hMSCs below passage 8 were seeded in a tissue culture flask at a density of ∼1500 cells/cm2 and cultured in NMM. Media were changed every 4 days until the cells reached ∼90% confluence. Cells were then dissociated with 0.25% Trypsin-EDTA and used directly for hydrogel experimentation. Polymer solutions of 0.2 wt. % T1C, 3 wt. % CDN with 0.2 wt. % T1C, or 3 wt. % PDN with 0.2 wt. % T1C were prepared as described above.
Harvested hMSCs (500,000 cells/tube) were pelleted and then dispersed into 1 mL of each respective polymer solution. The cell-polymer solutions were then dispensed into six separate wells of a black-walled 96-well plate at a volume of 75 μL/well (37,500 cells per gel sample). The plate was then incubated at 37°C, 5% CO2 for 30 min to allow gel formation. After the gels were solidified, the plate was placed on a hot plate at 37°C to ensure that the thermally responsive gels remained intact. Next, 150 μL of 37°C NMM, with or without 200 μM H2O2, was added on top of each gel (all treatments done in triplicate). The plate was incubated at 37°C, 5% CO2 for 24 h.
After 24 h, each well was incubated with 200 μL of freshly prepared 0.5 mg/mL collagenase P (Roche Life Sciences, Pleasanton, CA), and the plates were placed on an orbital shaker (100 rpm) in an incubator at 37°C for 15 min. Each plate was then placed on ice for 15 min, permitting the thermoresponsive materials to de-gel. The solutions from each well were placed into 0.6 mL microcentrifuge tubes and centrifuged at 300 g for 5 min. The supernatant was then carefully aspirated from each tube, and 200 μL of CellTiter-Glo solution (CellTiter-Glo Luminescent Cell Viability Assay Kit; Promega, Madison, WI) was used to resuspend the cells. Solutions were pipetted back into the respective wells of the original plate and mixed. After 10 min, total luminescent flux of each well was read for 2 s using an IVIS imaging system (IVIS Lumina Series III, Caliper LifeSciences; Perkin Elmer, Waltham, MA).
To establish the ROS-protective capacity of the PDN hydrogel+collagen groups over longer time points, the previous experiment was repeated using MSC-laden PDN hydrogel+collagen composites seeded in three separate plates with and without 200 μM H2O2 in NMM. Media with and without H2O2 was replaced every 48 h, and viability screening was performed as described above on plates at 2-, 4-, and 6-day time points.
Confocal microscopy of hydrogel-encapsulated human mesenchymal stem cells
Cells were dissociated and suspended into the polymer solutions (0.2 wt. % T1C, 3 wt. % CDN with 0.2 wt. % T1C, or 3 wt. % PDN with 0.2 wt. % T1C) as described above. Thirty μL of the polymer/cell solutions were then dispensed into the well of a 35 mm glass bottom plate with a 7 mm inset (35 mm dish, no. 0 coverslip, 7 mm glass diameter, uncoated; MatTek Corporation, Ashland, MA) and allowed to solidify for 30 min in an incubator at 37°C, 5% CO2. The gelled material was overlaid with 2 mL of ICM with or without 200 μM H2O2. Cells were then incubated for 24, 48, or 144 h. After incubation, cells were stained with 3 μM calcein-AM (green stain, indicating live cells) and 3 μM ethidium homodimer (red stain, indicating dead cells). Cells were imaged with a confocal scanning laser microscope (Nikon Eclipse Ti Microscope with D-Eclipse C1 laser; Nikon Instruments, Inc., Melville, NY) using a 488 nm (green) and 532 nm (red) laser line on a heated stage set to 37°C. Z-stacks of each sample were taken and averaged 4 × per frame to minimize light-scattering and background noise caused by the opacity of the materials. Maximum intensity projections were acquired, and the resulting images were processed by splitting the green and red channels from the maximum intensity projections. The red channels were thresholded, converted to binary, and then background subtracted before being overlaid above the green channel images.
MIN6 culture and PI generation
Mouse insulinoma pancreatic β-cells (MIN6) below passage 10 and stably transduced to express luciferase and mApple fluorescent protein were seeded into a tissue culture flask at a density of 1.5 × 104 cells/cm2 and cultured at 37°C, 5% CO2 for ∼4 days or until confluent. Next, the cells were prepared into PI aggregates based upon previously reported methods.31 The cells were dissociated with 0.25% trypsin for 5 min, suspended in NCM, and centrifuged at 300 g for 5 min to form a pellet. The media was aspirated, and the pelleted cells were resuspended in NCM to a density of 5 × 105 cells/mL. Fifteen mL of the cell solution was pipetted into a T-75 flask, which was placed on an orbital shaker within an incubator at 70 rpm (to prevent adherence). After 48 h, the cells were viewed using an inverted optical microscope to confirm PI formation.
Protection of PIs from hydrogen peroxide-induced cell death
First, 50 mg of lyophilized PDN or CDN polymer was weighed and dissolved in 1 mL of sterile PBS (5 wt. % polymer solutions) and allowed to dissolve for 48 h at 4°C. Fully aggregated PIs (∼7.5 × 107 MIN6 cells in about 3,000 aggregates) were pipetted into a 15 mL conical tube and centrifuged at 50 g for 1 min to form a cell pellet. The media was aspirated, and the PIs were carefully resuspended in 5 mL of NCM using a wide-orifice pipette tip, dispensed evenly into five, 2-mL microcentrifuge tubes, and centrifuged again at 50 g for 1 min. The media was aspirated and replaced with 1 mL of (1) 5 wt. % PDN, (2) 5 wt. % CDN, or (3) PBS. PIs were then carefully and homogenously resuspended into the overlaid solution using a wide-orifice pipette. Next, each solution was dispensed into six wells per group of a black-walled 96-well plate at a volume of 75 μL/well to embed ∼200 PIs per gel sample. The plate was then incubated at 37°C, 5% CO2 for 30 min to allow gel formation.
After 30 min, the plate was placed on a 37°C hot plate, and each well was given 150 μL of 37°C NCM containing 0.45 mg/mL d-Luciferin monopotassium salt. After 10 min, the plate was placed on a heated stage set to 37°C in an IVIS imaging system, and total luminescent flux was read for 2 min. The plate was then transferred back to the hot plate, and the supernatant was replaced with 150 μL of prewarmed 37°C PBS. This washing step was repeated two times more to ensure removal of the d-Luciferin monopotassium salt.
Next, 150 μL was aspirated from each well and replaced with NCM with or without 100 μM H2O2 such that each group and treatment was performed in triplicate. The plate was then incubated at 37°C, 5% CO2 for 24 h. After 24 h of incubation, each well was washed as described above and replaced with NCM containing 0.45 mg/mL d-Luciferin monopotassium salt. Luminescence readings were taken again, the wells were rewashed, and new solutions of NCM with or without 100 μM H2O2 were replaced into each respective well. Solutions were replaced every 24 h, and measurements were taken three more times up until 120 h after the initiation of the experiment. Based on using vital bioluminescence as a readout, we were able to take longitudinal data on the same samples over time. To confirm the veracity of this approach, a cell number versus bioluminescence curve was generated, validating that bioluminescence readings directly correlate to cell number for the MIN6 cells (Supplementary Fig. S2 and Supplementary Data).
Statistical analysis
Data are expressed as mean ± standard error of the mean. All statistical analyses were performed by one-way analysis of variance (ANOVA), and post hoc Sidak's multiple comparisons were tested via GraphPad Prism 6 software. For all tests, p < 0.05 was considered statistically significant.
Results
PDN and CDN polymer synthesis and hydrogel formation
ABC triblock polymers, PPS135-b-PDMA152-b-PNIPAAM225 (PDN, Fig. 1A) and PCL85-b-PDMA150-b-PNIPAAM150 (CDN), were successfully synthesized and characterized by NMR and GPC (Supplementary Figs. S3 and S4 and Supplementary Data). Polymeric solutions in water visibly gel when heated from ambient to physiologic temperature as demonstrated in Figure 1B. Furthermore, acid-disassociated T1C added to the PDN micelle solution can be neutralized and seamlessly incorporated into the thermogelling PDN hydrogels (Fig. 1B). SEM images of hydrogels made from T1C alone, PDN alone, CDN alone, or the polymers combined with T1C suggest that there is homogenous integration of the collagen network into the thermoresponsive polymers, forming uniform material composites (Fig. 1C).
FIG. 1.

(A) Chemical structure of PPS135-b-PDMA152-b-PNIPAAM225, PDN hydrogel. (B) Vial inversion demonstrates thermal gelation of the hydrogel at 37°C. At 22°C, PDN hydrogel with (bottom images) or without (top images) collagen is a liquid. Both self-assemble into gels when heated. (C) The collagen network is homogeneously incorporated into the PDN and CDN polymer hydrogels, forming composite materials. Scale bar: 20 μm. (Ci) Collagen only. (Cii) 3 wt. % PDN hydrogel only. (Ciii) Collagen with 3 wt. % PDN hydrogel. (Civ) 3 wt. % CDN hydrogel. (Cv) 3 wt. % CDN hydrogel and collagen. PPS135-b-PDMA152-b-PNIPAAM225, PDN, poly[(propylene sulfide)-block-(N,N-dimethyl acrylamide)-block-(N-isopropylacrylamide)].
Rheometric characterization of hydrogels
To assess the mechanical properties of these materials, rheometric characterization of hydrogel storage moduli (G′) was performed across a range of frequencies at 37°C. As shown in Figure 2, 2 mg/mL collagen gels provided similar mechanical properties to a 3 wt. % PDN hydrogel (an average G′ value of ∼50 Pa). As seen in previous work,18 increasing the wt. % of PDN from 2.5 to 5 increased the stiffness of the hydrogels to 100–200 Pa. Interestingly, the addition of collagen (2 mg/mL) to a 3 wt. % PDN solution significantly increased G′ values relative to collagen or PDN alone (Fig. 2), and the composite materials had similar mechanical characteristics to those shown in the 5 wt. % PDN-only hydrogels (G′ value of ∼200 Pa).
FIG. 2.

Rheometric analysis of the PDN hydrogel at 37°C with or without collagen addition demonstrates that incorporation of the collagen into the 3 wt. % PDN hydrogel increases the storage modulus of the composite material to relative equivalence with hydrogels made solely with 5% PDN polymer. Color images available online at www.liebertpub.com/tea
To assess the utility of the PDN hydrogel+collagen composite as an injectable substrate, rheometric flow tests were performed to measure dynamic viscosity as a function of shear rate and temperature. The viscosity of the PDN+collagen solution at 20°C (liquid state) is substantially decreased as the shear rate is increased from 100 to 6000/s (Fig. 3A). As demonstrated in Figure 3B, the viscosity is ∼0.002 Pa × s at 20°C and a 6000/s shear rate, but at the same temperature and a lower shear rate of 0.100/s, the viscosity increases to ∼2.0 Pa × s. While maintaining the shear rate at 0.100/s and increasing the temperature to 37°C the viscosity of the composite material increases to ∼200 Pa × s indicating gelation.
FIG. 3.

(A) The PDN hydrogel+collagen composites have shear-thinning behavior at 20°C as demonstrated by a substantial decrease in viscosity over a shear sweep from 0.100/s to 6000/s. (B) The PDN hydrogel+collagen composite is injectable. At high shear rate (6000/s) and low temperature (20°C), the viscosity of the composite is very low, comparable to a liquid. At low shear rate (0.100/s) and low temperature, the viscosity recovers. At low shear rate and physiological temperature (37°C), the viscosity undergoes an ∼100-fold increase, indicative of gelation. Color images available online at www.liebertpub.com/tea
A related study was done to confirm that the PDN materials could be utilized as an injectable vehicle for cell delivery. In this additional proof-of-concept experiment, fibroblasts were used as a model cell type (Supplementary Fig. S5 and Supplementary Data). Cells were suspended in the PDN+collagen precursor solution and injected through a 16G needle onto a plate warmed to physiologic temperature; the resultant cell-laden gels were then overlaid with media. The hydrogel encapsulated cells remained viable and exclusively associated with the hydrogel material after 24 h.
Protection of hMSCs from ROS-induced toxicity
Mixing hMSCS into PDN±collagen precursor solutions resulted in the formation of a cell-laden hydrogel (Fig. 4A). As shown in Figure 4B, hMSCs encapsulated in hydrogels solely comprised of PDN possessed a rounded morphology, indicating the cells' inability to either migrate through or adhere to the material substrate. However, cells seeded in T1C hydrogels featured robust attachment and spreading through the material. Consequently, cells in the PDN hydrogel+collagen composite were able to spread to a level comparable to that seen in the T1C-only hydrogels, demonstrating the cell adhesiveness imparted to these hybrid materials with the inclusion of collagen.
FIG. 4.

(A) Scheme depicting the formation of the cell-encapsulating hydrogel. PDN micelles, precollagen solution, and hMSCs are mixed together and then incubated at 37°C to prepare an ROS-protective, cell-adherent construct. (B) After 24 h in culture, Calcein-AM (green, live stain)/Ethidium Homodimer (red, dead stain) staining reveals hMSC adherence to the composite PDN hydrogel when collagen is incorporated. Cells in the PDN hydrogel alone were unable to spread and adhere, indicating that the inclusion of collagen contributes to a more suitable environment for the encapsulation of adherent hMSCs. Images taken with 20 × objective lens. hMSCs cultured in 3D in (Bi.) Three wt. % PDN hydrogel alone; (Bii) collagen alone, or (Biii) 3 wt. % PDN hydrogel with collagen (scale bar = 500 μm). 3D, three-dimensional; hMSCs, human mesenchymal stem cells; ROS, reactive oxygen species. Color images available online at www.liebertpub.com/tea
To demonstrate the ROS-detoxifying effect of PPS-based hydrogels, hMSCs were encapsulated in collagen gels, PDN hydrogels with 2 mg/mL T1C, and CDN hydrogels with 2 mg/mL T1C. The control CDN hydrogels were developed to provide thermogelling, cell-encapsulating material that does not contain the ROS-reactive, antioxidant PPS component. After 24 h of culture in normal MSC media (n = 5 gels for all groups), viability as determined by luminescence measurements of ATP content through a Cell TiterGlo assay showed that there were no significant differences between the number of viable encapsulated hMSCs in the three hydrogel formulations (Fig. 5). However, when 200 μM H2O2 was added to provide an oxidative stress-mimicking environment, the PDN hydrogel protected the cells from H2O2-induced cell death, while the other two groups displayed a statistically decreased number of viable cells. Importantly, there was no significant difference in viable cell number for normal MSC media versus the 200 μM H2O2 media for cells encapsulated within the PDN hydrogels (Fig. 5). Representative confocal microscopy maximum intensity z-stack images taken after the cells were cultured for 48 h confirm the results found in the quantitative measurements of cell number (Fig. 6).
FIG. 5.

Adherent hMSCs encapsulated in collagen, CDN hydrogel+collagen composites, or PDN hydrogel+collagen composites for 24 h maintain similar levels of viability in normal media. However, media supplemented with 200 μM H2O2 causes significant cytotoxicity in collagen and CDN hydrogel+collagen composite materials, while the PDN hydrogel+collagen composite protects adherent hMSCs from H2O2-induced cell death (*p < 0.05, ns p > 0.05, n = 5, data expressed as mean ± standard error of the mean). H2O2, hydrogen peroxide. Color images available online at www.liebertpub.com/tea
FIG. 6.
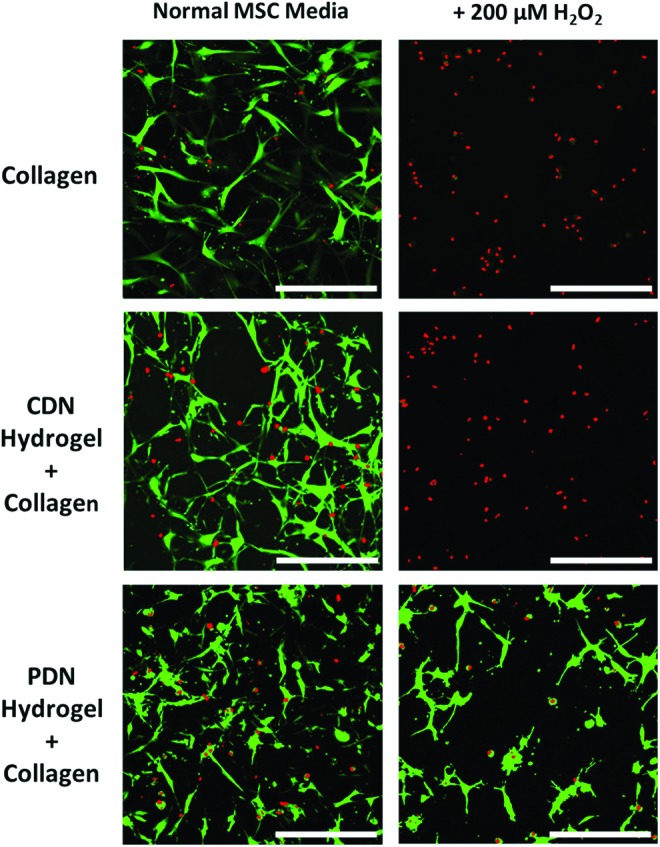
Confocal microscopy images of hydrogel-encapsulated hMSCs at 24 h confirm that all gels maintain similar levels of cell viability in normal media, while only PDN hydrogels protects adherent hMSCs from H2O2-induced cell death. Images taken with 20 × objective lens. Cells were stained with Calcein-AM (green, live stain)/Ethidium Homodimer (red, dead stain) (scale bar = 500 μm). Color images available online at www.liebertpub.com/tea
As assessed by calcein-AM staining of live cells within the three formulations, embedded hMSCs possessed a similar density and spread morphology when treated with NMM. However, for cells in both the collagen-only and the CDN+collagen groups, media containing 200 μM H2O2 was cytotoxic as visualized by red ethidium homodimer staining. Conversely, cells encapsulated within PDN+collagen show a much higher ratio of live/dead cells following 200 μM H2O2 treatment in agreement with the quantitative findings in Figure 5, demonstrating the antioxidative behavior of PDN+collagen composite materials. Furthermore, the viability of hMSCs encapsulated in PDN hydrogel+collagen was maintained for at least 4 days in the presence of cytotoxic H2O2 concentrations (Fig. 7A). After 6 days and reapplication of H2O2 every 2 days, there was a statistically significant decrease in the viability of hydrogel-encapsulated hMSCs versus day 2 levels. However, ∼80% of viable cells were still maintained at day 6, in agreement with qualitative live/dead staining images of hydrogel encapsulated (Fig. 7B).
FIG. 7.

(A) hMSCs encapsulated within PDN hydrogel+collagen composites maintain ∼80% viability over 6 days of in vitro culture when treated with 200 μM H2O2 every 2 days in comparison to normal MSC media controls (*p < 0.05, n = 3, data expressed as mean ± standard error of the mean, normalized to gel-encapsulated cells in normal MSC media). (B) Confocal z-stack images of gel-encapsulated hMSCs in media± three doses 200 μM H2O2 at day 6 taken with 20 × objective lens. Cells were stained with Calcein-AM (green, live stain)/Ethidium Homodimer (red, dead stain). Scale bar = 500 μm. MSC, mesenchymal stem cell. Color images available online at www.liebertpub.com/tea
Protection of pseudo-islet, suspension culture from ROS
PIs created from MIN6 cell aggregates (Fig. 8A) were cultured within 5 wt. % PDN and CDN hydrogels or on 2D tissue culture polystyrene for 4 days. When the cells were overlaid with normal cell media, there were no significant differences between the groups, indicating that the PDN hydrogels are not cytotoxic to PIs in comparison to 2D culture (Fig. 8B). However, when 100 μM H2O2 was added to the cell media to mimic in vivo oxidative stress, PIs encapsulated in PDN hydrogels had a statistically higher number of viable cells than PIs cultured in CDN gels or on tissue culture polystyrene (TCPS). The CDN material control gels also had higher viable cell signal than TCPS PIs, indicating that thermogels without inherent antioxidant capacity have some nonspecific protective effect (Fig. 8B). These data demonstrate that PDN hydrogels effectively protect PIs from cytotoxic levels of ROS and that cell aggregates (and potentially other nonadherent cells types) can be effectively delivered/protected by PDN hydrogels without the addition of cell-adhesive T1C.
FIG. 8.

(A) Scheme for preparation of pseudo-islet cell aggregates from a high-density MIN6 cell suspension. (B) PDN hydrogels protect nonadherent MIN6 aggregates from H2O2-induced cell death. When cultured in normal cell media, differences between cellular viability in the different substrates were insignificant, indicating that the hydrogels are not cytotoxic to the pseudo-islets. When 100 μM H2O2 was added to the cell media, the PDN hydrogel significantly protected viability of cultured cells compared to CDN and TCPS, while the CDN provided an intermediate level of protection relative to PDN and TCPS. (*p < 0.05 PDN versus TCPS and CDN, ┼p < 0.05, CDN vs. TCPS, n = 3, data expressed as mean ± standard error of the mean.) TCPS, tissue culture polystyrene. Color images available online at www.liebertpub.com/tea
Discussion
Hydrogels fabricated from PDN polymers feature effective thermogelation at 37°C, are noncytotoxic, possess an inherent cell-protective antioxidative capacity, and can serve as depots for local drug delivery in vivo.18 It was hypothesized that these thermoresponsive materials would also be effective vehicles for 3D cell encapsulation and in vivo delivery applications due to their gelation characteristics and antioxidative qualities.
Though encapsulation of nonadherent cell aggregates is feasible with the initial PDN hydrogel design, this material lacks inherent cell adhesion motifs, motivating the exploration of a composite architecture that integrates a naturally derived substrate to promote attachment. Other NIPAAM-based thermogels have been fabricated to include natural components such as hyalauronic acid,32 gelatin,33 and T1C22; these composites demonstrate improved local adhesion of cells, enhanced integration of peripheral tissue with the material substrate, and boost revascularization of the transplant. In particular, T1C is a ubiquitous component of the extracellular matrix and features integrin binding domains that permit cell spreading within a 3D microenvironment.34 To this end, PDN hydrogel+collagen composites were successfully fabricated and were subsequently evaluated for mechanical robustness, cell adhesion, and ROS-detoxification.
As an important and highly relevant benchmark control to better elucidate that the PPS chemistry was responsible for this ROS-detoxification effect, the CDN hydrogel was employed. By using the CDN hydrogel as our control, we were able to test the hypothesis that the primary ROS protective function of PDN hydrogels is achieved by the inclusion of the propylene sulfide monomer into the backbone of the PDN hydrogel rather than any chemical or structural components affiliated with other portions of the polymer.
Through a qualitative initial comparison of PDN hydrogel+collagen composites with PDN-only gels, the gelation properties of PDN polymer-only hydrogels were preserved in the new hybrid materials. (Fig. 1B). As assessed by SEM, the PDN hydrogel+collagen composites uniformly incorporate the fibrillar network of polymerized T1C into the more globular structure of the polymeric PDN materials to form a homogenous, composite hydrogel featuring both synthetic and naturally derived components (Fig. 1C).
Natural materials often lack robust mechanical properties, and composites consisting of natural and synthetic components can be utilized to enhance strength of biomaterials.22,35 The inclusion of the collagen into the PDN hydrogel increased the storage modulus of the composite relative to PDN alone (Fig. 2), achieving G′ values (around 200 Pa), which favorably compares with the stiffness of native soft tissues.36,37 Through a more rigorous quantitative assessment, the PDN hydrogel+collagen composite was shown to be intrinsically shear-thinning at 20°C (Fig. 3A). At 20°C and a high shear rate (6000/s, the upper machine limit) the viscosity approaches a minimum of ∼0.002 Pa × s, comparable to and on the same order of magnitude as the viscosity of water (∼0.001 Pa × s).38 The maximum shear rate used is approximately one order of magnitude less than the experimentally measured maximum (26,800/s) of flow through a 28G syringe needle at 1000 μL/min,39 though lower shear rates can be attained with both lower gauge (larger diameter) needles and decreased flow rates. Since a low viscosity was achieved at 6000/s, we expect our PDN hydrogel+collagen composite material to display low viscosity at shear rates relevant to syringe or cathetar injection of cell-polymer solutions before in situ gelation.
For the PDN hydrogel+collagen composites at 20°C (liquid state), the shear thinning behavior is exemplified by the rapid, three orders of magnitude-increase in viscosity as the shear rate is decreased from 6000/s to 0.100/s (Fig. 3B). By increasing the temperature to 37°C while maintaining the 0.100/s shear rate, the hydrogel viscosity increased another two orders of magnitude, indicative of the thermogelling behavior of the material (Fig. 3B). These experiments collectively exhibit the injectability of these materials due to their shear-thinning behavior at ambient temperature while also conclusively demonstrating their potential as in situ gelling materials due to the LCST-mediated increase in viscosity at body temperature.
For most cell delivery applications, adherent cells grown in a 3D material can more readily spread and migrate through a matrix when they can proteolytically cleave the material substrate.40 The inclusion of collagen within these hydrogels provides both motifs for cellular adhesion and the opportunity to remodel the substrate through cell-produced metalloproteinases. Consequently, adherent hMSCs seeded in hydrogels comprised solely of PDN polymer were incapable of spreading within these materials and showed a rounded morphology (Fig. 4Bi) while cells grown in collagen hydrogels were able to spread through the matrix (Fig. 4Bii). When T1C was integrated into the PDN hydrogels, the hMSC morphology was similar to that of cells grown in collagen-only hydrogels (Fig. 4Biii), and the relative number of viable cells supported by composites (either PDN or CDN) was similar to that of collagen-only gels or control, 2D culture conditions. The need to include an adhesive component such as collagen to support adherent cell types is also analogous to the strategies employed with commonly utilized polyethylene glycol (PEG) hydrogels. For example, incorporation of arginylglycylaspartic acid (RGD) tripeptide (an integrin binding domain) into PEG hydrogels is necessary to promote robust cell-spreading and adhesion.41 In sum, these data indicate that integration of collagen as an adhesion motif within PDN hydrogels renders these materials more suitable for the delivery of adherent cell types.
Long-term engraftment of patient-implanted cells is hindered by a number of difficulties1,2 including oxidative stress produced by the host's innate immune response, with H2O2 serving as a key cytotoxic component implicated with reperfusion injury at transplantation sites.42–44 In excess, H2O2 triggers cell apoptotic mechanisms via lipid peroxidation of the cellular membrane45 and irreversible damage to proteins and DNA.46 Wong et al. reported that an antioxidant biomaterial scaffold composed of the carbohydrate glucan pullalan improved MSC survival in the presence of H2O2 in vitro and in vivo.47,48 However, their system is prefabricated into a specific geometry and therefore noninjectable, limiting its utility for some applications. In this work, we have shown that the cytotoxic effects of H2O2 can be mitigated by encapsulating adherent MSCs in injectable PDN hydrogel+collagen composites (Fig. 5). The composite PDN gels, featuring ROS-sensitive PPS in the polymer structure,18,23,24 absorb H2O2 and prevent cell death of encapsulated MSCs in contrast to cells in collagen gels. Moreover, hydrogels made with the thermoresponsive CDN polymers comprised of ROS-insensitive PCL instead of PPS do not protect encapsulated cells from H2O2-mediated toxicity, indicating that the PPS component is the main driver of the retention in cellular viability. These results were qualitatively confirmed in confocal microscopy imaging of Live/Dead stained cells encapsulated in the hydrogel formulations with or without H2O2 treatment (Fig. 6).
The ROS-detoxifying behavior of PDN hydrogel+collagen composites was also sustained over an extended time frame as the viability of encapsulated hMSCs was maintained at 80% or higher for at least 6 days (Fig. 7), whereas the other nonantioxidant treatment groups saw minimal protection even after 24 or 48 h in culture (Figs. 5 and 6). In all, the PDN hydrogels+collagen composites feature a superior injectable cell delivery format and protect encapsulated cells from cytotoxic levels of H2O2.
We have also shown in proof-of-concept experiments that the PDN hydrogels provide protection to model PIs, suggesting future utilization of this system for delivery of insulin-producing cell therapies to cure T1D. Because these cells, like the cells that comprise native islets, are adhered to one another, they are less dependent on adhesion to an extracellular matrix. Thus, the requirement of adhesion proteins for cell spreading is not crucial, and the cells can survive for longer periods in the PDN-only gels.
For islets transplanted into patients in the clinic, many islets experience hypoxia-induced intracellular ROS generation that often causes a significant loss in islet mass.49 Furthermore, most islets naturally produce lower amounts of antioxidant enzymes compared with other tissue types, making islets acutely susceptible to ROS-mediated toxicity.50 Therefore, many groups have explored strategies for delivering islets while simultaneously detoxifying ROS. Weaver and Stabler encapsulated PIs in composite microparticles composed of an alginate bulk matrix and interstitial cerium oxide nanoparticles to preserve islets metabolic activity even in the presence of 10 mM superoxide.51 Additionally, Dang et al. previously explored the use of encapsulating islets in microcapsules alongside antioxidant drugs, showing a significant improvement in islet graft survival and function upon implantation.52
In contrast to these and other previously developed systems, we have prepared a material that inherently functions as an antioxidant, and consequently, the complexity of material drug loading and tailoring release kinetics do not need to be considered. In this study, we showed that the hydrogel-encapsulated PIs maintained their viability (∼100% or greater of the initial cell number) on par with PIs on 2D TCPS for 4 days of culture (Fig. 8B). However, with the addition 100 μM H2O2 only the PDN hydrogel maintained full viability of the PIs (Fig. 8B). The toxicity of PIs in CDN hydrogels also provides further validation that the specific ROS-reactive PPS chemistry mediates protection from ROS, though the control CDN materials do act as a nonspecific barrier to limit ROS-mediated toxicity and preserve some encapsulated PI viability.
Though the PPS component of the injectable hydrogel system is primarily used to neutralize cytotoxic levels of ROS in the presented work, the degradability of the PDN hydrogels in the presence of ROS is also a key component in this system.18 As demonstrated by other groups,53,54 tissue remodeling and local vascularization of the islet-delivering biomaterial implant is critical for oxygenating the implanted cells, improving the glucose transaction efficiency, and producing/delivering therapeutic levels of insulin. We anticipate that the antioxidant PDN hydrogels will protect encapsulated PIs from initial immune-generated ROS levels before being gradually remodeled into a well-vascularized construct for improved islet engraftment and survival postimplantation.
As the current system does not provide longstanding, physical isolation from the immune system, we also intend to explore the use of islet surface coatings and/or immunomodulatory drugs that can be hydrophobically loaded in the micellar core of these materials to protect islets from longer term rejection by the acquired immune response.
Conclusion
In summary, we have validated the utility of our previously described antioxidant PDN hydrogel chemistry18 for use in 3D culture of therapeutically relevant cell types. Furthermore, PDN hydrogels have been proven to provide cell-protective benefits against cytotoxic levels of oxidative stress induced by H2O2 in both adherent (hMSC) and suspension/aggregate (MIN6 PIs) cell models. It was found that spreading and growth of adherent cells within PDN gels benefits from integration of native matrix proteins such as T1C and that PDN hydrogel+collagen composites harness the beneficial cell adhesive properties of T1C while maintaining the thermogelling and ROS-protective capacity of the PDN hydrogels. These results motivate preclinical exploration of PDN hydrogels for hMSC delivery in models of tissue regeneration and for delivery of primary pancreatic islets or other engineered insulin-producing cells in models of diabetes.
Supplementary Material
Acknowledgments
Experiments were performed in part using resources provided and maintained by VUMC Cell Imaging Shared Resource (CISR) (supported by NIH grants CA68485, DK20593, DK58404, HD1502, DK59637, and Ey008126) and the Vanderbilt Institute of Nanoscale Science and Engineering (VINSE). Funding was provided in part by the National Institute of Health Integrated Training in Engineering and Diabetes Training Grant (NIH 1T32DK101003-01A1) and the National Science Foundation CAREER Award (DMR) (BMAT 1349604).
Disclosure Statement
No competing financial interests exist.
References
- 1.Kean T.J., Lin P., Caplan A.I., and Dennis J.E. MSCs: delivery routes and engraftment, cell-targeting strategies, and immune modulation. Stem Cells Int 2013, 2013. Article ID: 732742, 13 pages [Online.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Orive G., Santos E., Pedraz J.L., and Hernández R.M. Application of cell encapsulation for controlled delivery of biological therapeutics. Adv Drug Deliv Rev 67–68, 3, 2014 [DOI] [PubMed] [Google Scholar]
- 3.Malafaya P.B., Silva G.A., and Reis R.L. Natural-origin polymers as carriers and scaffolds for biomolecules and cell delivery in tissue engineering applications. Adv Drug Deliv Rev 59, 207, 2007 [DOI] [PubMed] [Google Scholar]
- 4.Mooney D.J., and Vandenburgh H. Cell delivery mechanisms for tissue repair. Cell Stem Cell 2, 205, 2008 [DOI] [PubMed] [Google Scholar]
- 5.Gill S., and June C.H. Going viral: chimeric antigen receptor T-cell therapy for hematological malignancies. Immunol Rev 263, 68, 2015 [DOI] [PubMed] [Google Scholar]
- 6.Griffin M.D., Ritter T., and Mahon B.P. Immunological aspects of allogeneic mesenchymal stem cell therapies. Hum Gene Ther 21, 1641, 2010 [DOI] [PubMed] [Google Scholar]
- 7.Pittenger M.F., and Martin B.J. Mesenchymal stem cells and their potential as cardiac therapeutics. Circ Res 95, 9, 2004 [DOI] [PubMed] [Google Scholar]
- 8.Lam M.T., Nauta A., Meyer N.P., Wu J.C., and Longaker M.T. Effective delivery of stem cells using an extracellular matrix patch results in increased cell survival and proliferation and reduced scarring in skin wound healing. Tissue Eng Part A 19, 738, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bianco P., and Robey P.G. Stem cells in tissue engineering. Nature 414, 118, 2001 [DOI] [PubMed] [Google Scholar]
- 10.Fisher J.P., Jo S., Mikos A.G., and Reddi A.H. Thermoreversible hydrogel scaffolds for articular cartilage engineering. J Biomed Mater Res A 71, 268, 2004 [DOI] [PubMed] [Google Scholar]
- 11.Parekkadan B., and Milwid J.J. Mesenchymal stem cells as therapeutics. Annu Rev Biomed Eng 12, 87, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shapiro A.M.J., et al. . Islet transplantation in seven patients with type 1 diabetes mellitus using a glucocorticoid-free immunosuppressive regimen. N Engl J Med 343, 230, 2000 [DOI] [PubMed] [Google Scholar]
- 13.Assady S., et al. . Insulin production by human embryonic stem cells. Diabetes 50, 1691, 2001 [DOI] [PubMed] [Google Scholar]
- 14.Tilney N.L. Transplantation and its biology: from fantasy to routine. J Appl Physiol 89, 1681, 2000 [DOI] [PubMed] [Google Scholar]
- 15.Wood K.J., and Goto R. Mechanisms of rejection: current perspectives. Transplantation 93, 1, 2012 [DOI] [PubMed] [Google Scholar]
- 16.Hou D., et al. . Radiolabeled cell distribution after intramyocardial, intracoronary, and interstitial retrograde coronary venous delivery: implications for current clinical trials. Circulation 112, 150, 2005 [DOI] [PubMed] [Google Scholar]
- 17.Blocklet D., et al. . Myocardial homing of nonmobilized peripheral-blood CD34+ cells after intracoronary injection. Stem Cells 24, 333, 2006 [DOI] [PubMed] [Google Scholar]
- 18.Gupta M.K., et al. . Cell protective, ABC triblock polymer-based thermoresponsive hydrogels with ROS-triggered degradation and drug release. J Am Chem Soc 136, 14896, 2014 [DOI] [PubMed] [Google Scholar]
- 19.Burdick J.A., and Anseth K.S. Photoencapsulation of osteoblasts in injectable RGD-modified PEG hydrogels for bone tissue engineering. Biomaterials 23, 4315, 2002 [DOI] [PubMed] [Google Scholar]
- 20.Park H., Kang S.W., Kim B.S., Mooney D.J., and Lee K.Y. Shear-reversibly crosslinked alginate hydrogels for tissue engineering. Macromol Biosci 9, 895, 2009 [DOI] [PubMed] [Google Scholar]
- 21.Koopmans C., and Ritter H. Formation of physical hydrogels via host-guest interactions ofβ-cyclodextrin polymers and copolymers bearing adamantyl groups. Macromolecules 41, 7416, 2008 [Google Scholar]
- 22.Barnes A.L., Genever P.G., Rimmer S., and Coles M.C. Collagen–poly(N-isopropylacrylamide) hydrogels with tunable properties. Biomacromolecules 17, 723, 2016 [DOI] [PubMed] [Google Scholar]
- 23.Napoli A., Valentini M., Tirelli N., Müller M., and Hubbell J. a. Oxidation-responsive polymeric vesicles. Nat Mater 3, 183, 2004 [DOI] [PubMed] [Google Scholar]
- 24.Gupta M.K., Meyer T.A., Nelson C.E., and Duvall C.L. Poly(PS-b-DMA) micelles for reactive oxygen species triggered drug release. J Control Release 162, 591, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anderson J.M., Rodriguez A., and Chang D.T. Foreign body reaction to biomaterials. Semin Immunol 20, 86, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu W.F., et al. . Real-time in vivo detection of biomaterial-induced reactive oxygen species. Biomaterials 32, 1796, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lopez-Neblina F., Toledo A.H., and Toledo-Pereyra L.H. Molecular biology of apoptosis in ischemia and reperfusion. J Invest Surg 18, 335, 2005 [DOI] [PubMed] [Google Scholar]
- 28.Ingulli E. Mechanism of cellular rejection in transplantation. Pediatr Nephrol 25, 61, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yeung T., et al. . Effects of substrate stiffness on cell morphology, cytoskeletal structure, and adhesion. Cell Motil Cytoskeleton 60, 24, 2005 [DOI] [PubMed] [Google Scholar]
- 30.Lapworth J.W., Hatton P.V., Goodchild R.L., and Rimmer S. Thermally reversible colloidal gels for three-dimensional chondrocyte culture. J R Soc Interface 9, 362, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jia D., Dajusta D., and Foty R.A. Tissue surface tensions guide in vitro self-assembly of rodent pancreatic islet cells. Dev Dyn 236, 2039, 2007 [DOI] [PubMed] [Google Scholar]
- 32.Ohya S., Nakayama Y., and Matsuda T. Thermoresponsive artificial extracellular matrix for tissue engineering: hyaluronic acid bioconjugated with poly(N-isopropylacrylamide) grafts. Biomacromolecules 2, 856, 2001 [DOI] [PubMed] [Google Scholar]
- 33.Chen J.-P., Leu Y.-L., Fang C.-L., Chen C.H., and Fang J.-Y. Thermosensitive hydrogels composed of hyaluronic acid and gelatin as carriers for the intravesical administration of cisplatin. J Pharm Sci 100, 655, 2011 [DOI] [PubMed] [Google Scholar]
- 34.Knight C.G., et al. . The collagen-binding A-domains of integrins alpha(1)beta(1) and alpha(2)beta(1) recognize the same specific amino acid sequence, GFOGER, in native (triple-helical) collagens. J Biol Chem 275, 35, 2000 [DOI] [PubMed] [Google Scholar]
- 35.Burdick J.A., Chung C., Jia X., Randolph M.A., and Langer R. Controlled degradation and mechanical behavior of photopolymerized hyaluronic acid networks. Biomacromolecules 6, 386, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dikovsky D., Bianco-Peled H., and Seliktar D. Defining the role of matrix compliance and proteolysis in three-dimensional cell spreading and remodeling. Biophys J 94, 2914, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Engler A.J., Sen S., Sweeney H.L., and Discher D.E. Matrix elasticity directs stem cell lineage specification. Cell 126, 677, 2006 [DOI] [PubMed] [Google Scholar]
- 38.Kestin J., Sokolov M., and Wakeham W.A. Viscosity of liquid water in the range −8C to 150C. J Phys Chem Ref Data 7, 941, 1978 [Google Scholar]
- 39.Aguado B.A., et al. . Improving viability of stem cells during syringe needle flow through the design of hydrogel cell carriers. Tissue Eng Part A 18, 806, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Raeber G.P., Lutolf M.P., and Hubbell J.A. Molecularly engineered PEG hydrogels: a novel model system for proteolytically mediated cell migration. Biophys J 89, 1374, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Phelps E.A., et al. . Maleimide cross-linked bioactive PEG hydrogel exhibits improved reaction kinetics and cross-linking for cell encapsulation and in situ delivery. Adv Mater 24, 64, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zeng W., et al. . Antioxidant treatment enhances human mesenchymal stem cell anti-stress ability and therapeutic efficacy in an acute liver failure model. Sci Rep 5, 11100, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kunduzova O.R., Bianchi P., Parini A., and Cambon C. Hydrogen peroxide production by monoamine oxidase during ischemia/reperfusion. Eur J Pharmacol 448, 225, 2002 [DOI] [PubMed] [Google Scholar]
- 44.Pendergrass K.D., et al. . Acute preconditioning of cardiac progenitor cells with hydrogen peroxide enhances angiogenic pathways following ischemia-reperfusion injury. Stem Cells Dev 22, 2414, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sevanian A., and Ursini F. Lipid peroxidation in membranes and low-density lipoproteins: similarities and differences. Free Radic Biol Med 29, 306, 2000 [DOI] [PubMed] [Google Scholar]
- 46.Elliott R.M., Astley S.B., Southon S., and Archer D.B. Measurement of cellular repair activities for oxidative DNA damage. Free Radic Biol Med 28, 1438, 2000 [DOI] [PubMed] [Google Scholar]
- 47.Wong V.W., et al. . Pullulan hydrogels improve mesenchymal stem cell delivery into high-oxidative-stress wounds. Macromol Biosci 11, 1458, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wong V.W., Rustad K.C., Galvez M.G., Neofytou E., Glotzbach J.P., Januszyk M., Major M.R., Sorkin M., Longaker M.T., Rajadas J., and Gurtner G.C. Engineered pullulan–collagen composite dermal hydrogels improve early cutaneous wound healing. Tissue Eng Part A 17, 631, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Monfared S.S.M.S., Larijani B., and Abdollahi M. Islet transplantation and antioxidant management: a comprehensive review. World J Gastroenterol 15, 1153, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lenzen S., and Drinkgern J. Low antioxidant enzyme gene expression in pancreatic islets compared with various other mouse tissues. Free Radic Biol Med 20, 463, 1996 [DOI] [PubMed] [Google Scholar]
- 51.Weaver J.D., and Stabler C.L. Antioxidant cerium oxide nanoparticle hydrogels for cellular encapsulation. Acta Biomater 16, 136, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dang T.T., et al. . Enhanced function of immuno-isolated islets in diabetes therapy by co-encapsulation with an anti-inflammatory drug. Biomaterials 34, 5792, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dufour J.M., et al. . Development of an ectopic site for islet transplantation, using biodegradable scaffolds. Tissue Eng 11, 1323, 2005 [DOI] [PubMed] [Google Scholar]
- 54.Phelps E.A., Headen D.M., Taylor W.R., Thulé P.M., and García A.J. Vasculogenic bio-synthetic hydrogel for enhancement of pancreatic islet engraftment and function in type 1 diabetes. Biomaterials 34, 4602, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.