

ABSTRACT
Development of cisplatin-resistance is an obstacle in non-small cell lung cancer (NSCLC) therapeutics. To investigate which molecules are associated with cisplatin-resistance, we analyzed expression profiles of several DNA repair and anti-apoptosis associated molecules in parental (A549P and H157P) and cisplatin-resistant (A549CisR and H157CisR) NSCLC cells. We detected constitutively upregulated nuclear ATM and cytosolic Mcl-1 molcules in cisplatin-resistant cells compared with parental cells. Increased levels of phosphorylated ATM (p-ATM) and its downstream molecules, CHK2, p-CHK2, p-53, and p-p53 were also detected in cisplatin-resistant cells, suggesting an activation of ATM signaling in these cells. Upon inhibition of ATM and Mcl-1 expression/activity using specific inhibitors of ATM and/or Mcl-1, we found significantly enhanced cisplatin-cytotoxicity and increased apoptosis of A549CisR cells after cisplatin treatment. Several A549CisR-derived cell lines, including ATM knocked down (A549CisR-siATM), Mcl-1 knocked down (A549CisR-shMcl1), ATM/Mcl-1 double knocked down (A549CisR-siATM/shMcl1) as well as scramble control (A549CisR-sc), were then developed. Higher cisplatin-cytotoxicity and increased apoptosis were observed in A549CisR-siATM, A549CisR-shMcl1, and A549CisR-siATM/shMcl1 cells compared with A549CisR-sc cells, and the most significant effect was shown in A549CisR-siATM/shMcl1 cells. In in vivo mice studies using subcutaneous xenograft mouse models developed with A549CisR-sc and A549CisR-siATM/shMcl1 cells, significant tumor regression in A549CisR-siATM/shMcl1 cells-derived xenografts was observed after cisplatin injection, but not in A549CisR-sc cells-derived xenografts. Finally, inhibitor studies revealed activation of Erk signaling pathway was most important in upregulation of ATM and Mcl-1 molcules in cisplatin-resistant cells. These studies suggest that simultaneous blocking of ATM/Mcl-1 molcules or downstream Erk signaling may recover the cisplatin-resistance of lung cancer.
KEYWORDS: ATM, cisplatin-resistance, cisplatin-sensitivity, Mcl-1, non-small cell lung cancer
Introduction
Lung cancer is a predominant cause of cancer death in both men and women.1 Lung cancer is heterogeneous and histologically divided into 2 types: small cell lung carcinomas (SCLCs) and non-small cell lung carcinomas (NSCLCs). NSCLCs comprise 85% of lung cancer cases2 and constitute a heterogeneous population of squamous, adenocarcinoma, and large cell carcinomas.1 Despite recent progress, lung cancer therapeutic outcomes remain unsatisfactory.
Platinum-based drugs, particularly cis-diammine-dichloroplatinum (II) (cisplatin, DDP), are used in the treatment of many cancers, including lung cancer. Cisplatin treatment initially seems successful, but often chemoresistance develops and therapy fails.3,4 Cisplatin treatment induces DNA lesions, which can lead to cell cycle arrest, DNA repair, senescence, and apoptotic death, which is collectively called DNA damage response (DDR).4-6 Lai et al.7 found an enhancement of DNA repair activity in the cisplatin-resistant cell lines and suggested that this enhancement played the major role in the cisplatin resistance phenotype.
Ataxia-telangiectasia-mutated (ATM) is a member of the phosphatidylinositol 3-kinase-related kinase (PIKK) family of Ser/Thr protein kinases.8 It mediates cell cycle arrest, anti-apoptosis, and facilitates DNA repair via activation of its downstream molecules, such as checkpoint 2 protein (CHK2) and p53.9 Constitutive activation of the ATM pathway in 5-azacytidine-resistant myeloid leukemia cell lines has been reported10 and sensitization of radioresistant breast cancer cells by inhibiting ATM activation has also been reported.11
Myeloid cell leukemia 1 (Mcl-1) is an anti-apoptotic member of the B-cell lymphoma 2 (Bcl-2) family of apoptosis-regulating proteins.12 Mcl-1 regulates cell survival and sensitivity to diverse apoptotic stimuli in lung cancer cells.13 Along with its roles in differentiation and apoptosis, Mcl-1 is also known to delay cell-cycle progression through interactions with cyclin dependent kinase 1 (CDK-1),14 proliferating cell nuclear antigen (PCNA)15 and CHK-1.16 Under resting conditions, Mcl-1 is localized to various cellular membranes, including the mitochondria17 and nuclear envelope,18 but is predominantly located in the cytosolic compartment as a heterodimer with Bax.19 However, upon stimulation with drugs or irradiation, Mcl-1 is reported to be translocated to the nucleus and involved in DNA repair.14,16 The importance of Mcl-1 in the DDR pathway has been emphasized recently, and circumventing drug resistance by targeting Mcl-1 has also been reported.20
In this study, we analyzed expression profiles of several DNA repair and anti-apoptosis associated molecules in parental (A549P and H157P) and cisplatin-resistant (A549CisR and H157CisR) NSCLC cells, and found ATM and Mcl-1 molcules are significantly upregulated in cisplatin-resistant NSCLC cells. We tested the effects of simultaneous blocking upregulation/activation of these 2 molcules on increasing cisplatin-sensitivity of cisplatin-resistant NSCLC cells in in vitro and in vivo studies.
Results
Constitutively upregulated ATM and Mcl-1 molcules in cisplatin-resistant NSCLC cells
Two cisplatin-resistant NSCLC cell lines, A549CisR and H157CisR, were developed by treating parental A549 (A549P) and H157 (H157P) cells with increasing doses of cisplatin over 6 months.21 These cells showed 5–8 fold higher IC50 values than parental cells depending on passage numbers, as continuous culture of these cells with cisplatin increased the IC50 values (Fig. 1A). We first investigated cytosolic and nucleic basal levels of several key molecules associated with DNA repair and anti-apoptosis in A549P/A549CisR and H157P/H157CisR cell sets. These molecules include ATM,9 DNA-dependent protein kinase (DNA-PK),22 and poly (ADP-ribose) polymerase (PARP)-1,23 Ku70,24 BRCA1,25 bcl-2,26 and Mcl-1.13 We detected significantly upregulated basal levels of ATM (in nucleus) and Mcl-1 (in cytosol) in A549CisR and H157CisR cells compared with parental cells (Fig. 1B). The ATM, CHK2, and Mcl-1 levels in nucleus of A549P and A549CisR cells were further investigated after cisplatin stimulation (different cisplatin concentrations were used for A549P and A549CisR cells according to cisplatin-cytotoxicity tests). As shown in Fig. 1C, there was cisplatin-induced upregulation of these molecules in A549P cells, but not in A549CisR cells. It is interesting to note that the basal levels (without cisplatin treatment) of these molecules in A549CisR and H157CisR cells were higher than the cisplatin-treated A549P cells (Fig. 1C). This result suggests that the ATM and Mcl-1 are constitutively upregulated in cisplatin-resistant cells.
Figure 1.
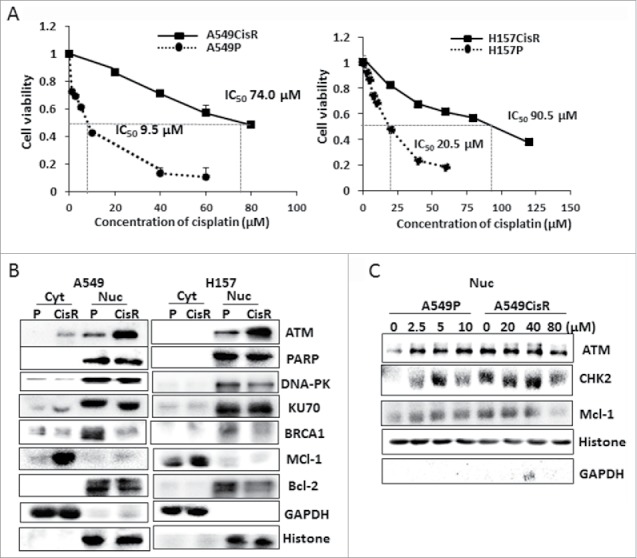
ATM and Mcl-1 expression in parental and cisplatin-resistant lung cancer cells. A. Cisplatin-cytotoxicity tests of A549P/H157CisR and H157P/H157CisR cells. A549CisR and H157CisR cells were obtained by continuous treatment of cells with increasing dose of cisplatin. Cell cytotoxicities of A549P vs. A549CisR and H157P vs. H157CisR cells to varied concentrations of cisplatin were analyzed in MTT assay. B. Western blot analysis. Cytosolic and nucleic cell extracts were obtained from parental (A549P and H157P) and cisplatin-resistant cells (A549CisR and H157CisR) and western blot analyses were performed using antibodies of indicated molecules. C. Western blot analysis. Cytosolic and nucleic cell extracts of A549P and A549CisR were obtained after treatment with cisplatin (near IC50 value of each cell line) for 48 hours and used in Western blot analyses.
ATM-CHK2-p53 signaling axis is constitutively activated in cisplatin-resistant cells
To answer whether not only the upregulation of ATM molecule, but also ATM kinase activity is increased in cisplatin-resistant cells, phosphorylated ATM (p-ATM) levels in A549P/A549CisR and H157P/H157CisR cell sets were compared. As shown in Fig. 2A, higher p-ATM levels were detected in A549CisR and H157CisR cells than in parental cells. Higher levels of the 2 well-known ATM substrates, CHK2 and p5327 were also detected in A549CisR and H157CisR cells than in parental cells (Fig. 2B). In addition, higher expression of the ATM-CHK2-p53 signaling axis downstream molecules, such as Mediator of DNA damage checkpoint 1 (MDC1)28 and p21,29,30 were further detected in A549CisR and H157CisR cells than in parental cells (Fig. 2C). These results indicated that ATM signaling is also constitutively activated in cisplatin-resistant cells. The upregulation of p-ATM and p-p53 in A549CisR and H157CisR cells compared with parental cells were also observed in immunofluorescence (IF) staining (Fig. 2D).
Figure 2.
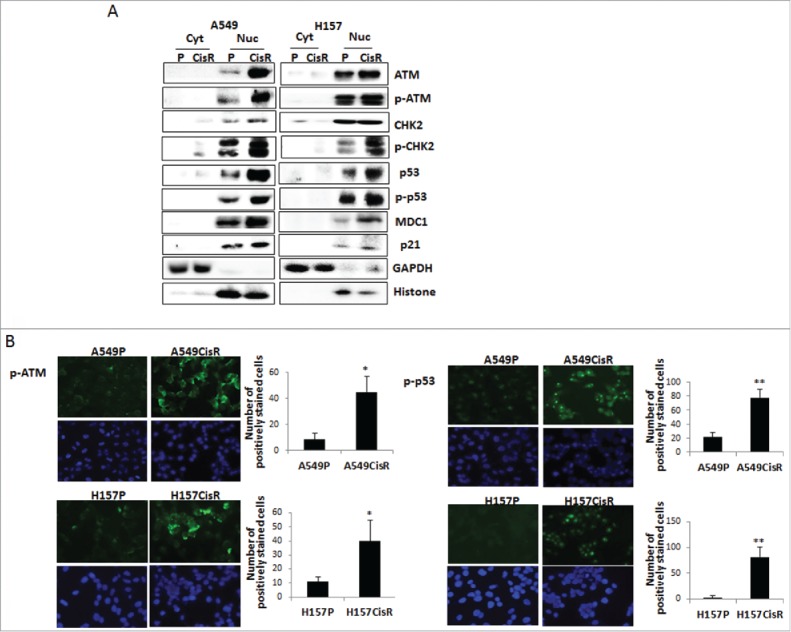
Investigations on ATM downstream signaling in A549P/A549CisR and H157P/H157CisR cells. A. Western blot analysis. Cytosolic and nucleic cell extracts were obtained from A549P/A549CisR and H157P/H157CisR cells (non-cisplatin treated) and used in Western blot analyses using antibodies of indicated molecules. B. IF staining. Cells (A549P/A549CisR and H157P/H157CisR) were plated in chamber slides (without treating with cisplatin) and IF staining was performed using antibodies of p-ATM and p-p53. Quantitation shown on right. Error bars and significance values were obtained by counting positively stained cells in one randomly chosen area of 3 different stainings. Magnification, 20X. *p < 0.05, **p < 0.01
Inhibition of ATM and Mcl-1 increased cisplatin-sensitivity and induced apoptotic death of A549CisR cells
We next investigated whether blocking of ATM and/or Mcl-1 expression/activation increases sensitivity of cisplatin-resistant cells to cisplatin and results in more apoptotic death of these cells after cisplatin treatment.
In cisplatin-cytotoxicity tests (Fig. 3A), increased cisplatin-cytotoxicity was observed when A549CisR cells were treated with (same concentrations of) inhibitors of ATM kinase (CP466722)31 and/or Mcl-1 (UMI77).32 The most significant inhibitory effect was observed when 2 inhibitors were used together (Fig. 3B). Moreover, a more significant inhibitory effect was observed in A549CisR cells than in A549P cells showing 39.3% IC50 reduction (from 14.0 μM to 8.5 μM) in A549P cell vs. 48.6% IC50 reduction (from 72.5 μM to 35.0 μM) in A549CisR cell (Fig. 3B). This result suggests that the upregulation/activation of ATM and Mcl-1 molcules is critical in protecting cisplatin-resistant cells from apoptosis, so we speculate that simultaneously targeting these 2 molcules may be an effective way to increase cisplatin-sensitivity of these cells.
Figure 3.
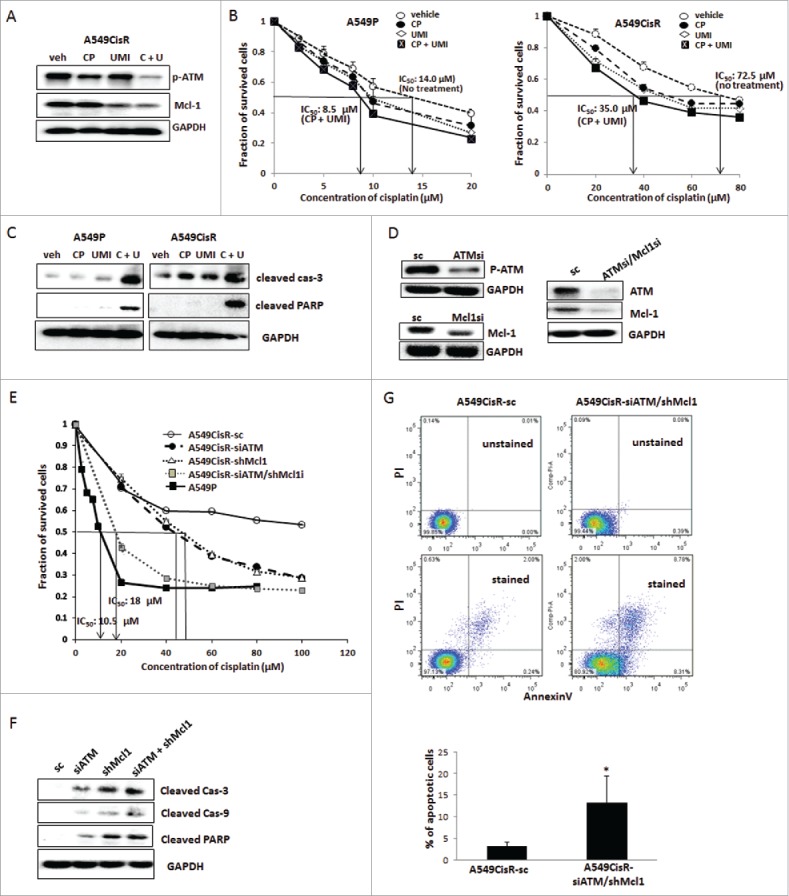
Test effects of selective inhibitors or knockdown strategy of ATM and/or Mcl-1 molcules in altering cisplatin-sensitivity and apoptotic death of cisplatin-resistant cells upon cisplatin treatment. A. Western blot analysis showing inhibition of ATM signaling and Mcl-1 expression in A549CisR cells upon incubation with ATM/Mcl-1 inhibitor(s) (veh; vehicle, CP; CP466722, UMI; UMI77, C + U, combined use of CP466722 plus UM177). B. Cisplatin-cytotoxicity tests of A549P and A549CisR in the absence and presence of ATM/Mcl-1 inhibitor(s). Cell survival upon treatment with varied concentrations of cisplatin was analyzed in MTT assay. C. Western blot analysis of apoptotic markers in A549P and A549CisR cells upon cisplatin treatment, in the absence and presence of ATM/Mcl-1 inhibitors. Cell (A549P and A549CisR) were treated with cisplatin (10 μM and 30 μM cisplatin were used for treating A549P and A549CisR cells, respectively) for 48 hours, in the absence and presence of inhibitors, and cell extracts were obtained. Expression of apoptotic markers were examined in Western blot analyses. D. Western blot analysis testing ATM and Mcl-1 levels in A549CisR-sc, A549CisR-siATM, A549CisR-shMcl1, and A549CisR-siATM/shMcl1 cells. Cell extracts were obtained from A549CisR-sc, A549CisR-ATMsi, A549CisR-Mcl1si, and A549CisR-ATMsi/Mcl1si cells and ATM-1 and Mcl-1 levels were examined by Western blot analysis. E. Cisplatin-cytotoxicity tests of A549CisR-sc, A549CisR-siATM, A549CisR-shMcl1, and A549CisR-siATM/shMcl1 cells. Cells were treated with indicated concentration of cisplatin and cell survival was analyzed by MTT assay. F. Western blot analyses investigating expression of the apoptotic markers A549CisR-sc, A549CisR-siATM, A549CisR-shMcl1, and A549CisR-siATM/shMcl1 cells upon cisplatin treatment. Cell extracts were obtained from and expression of apoptotic markers were examined by Western blot analyses. G. Flow cytometric analysis of apoptotic cells. A549CisR-sc and A549CisR-siATM/shMcl1 cells were treated with cisplatin (30 μM) for 24 hours and apoptotic cells (%) were analyzed in AnnexinV based flow cytometric analyses. The graph shown right indicates average values obtained from 3 independent experiments. *p < 0.05
In Western blot analyses investigating expression levels of apoptosis markers, increased expression of cleaved caspase 3 and cleaved PARP were detected in both A549P and A549CisR cells after addition of inhibitors (each alone or in combination) into the culture, and a more significant effect was observed in A549CisR cells than in A549P cells. As with the cisplatin-sensitivity experiment result, the most significant increase was observed when the 2 inhibitors were used together (Fig. 3C).
Knockdown of ATM and Mcl-1 increased cisplatin-sensitivity of A549CisR cells and resulted in apoptotic death.
To further investigate the effect of blocking ATM and/or Mcl-1 on increasing cisplatin-sensitivity of cisplatin-resistant NSCLC cells, several A549CisR cells-derived lines, such as ATM knocked down (549CisR-siATM), Mcl-1 knocked down (A549CisR-shMcl1), ATM/Mcl-1 double knocked down (A549CisR-siATM/shMcl1), and sc control cells (A549CisR-sc), were developed by lentiviral transduction. Knockdown of ATM and/or Mcl-1 in these cell lines were confirmed in Western blot analysis (Fig. 3D).
In cisplatin-cytotoxicity tests, increased cisplatin-cytotoxicities were observed in 549CisRsiATM, A549shMcl1, and A549CisR-siATM/shMcl1 cells compared with A549CisR-sc cells, and the most significant cisplatin-sensitivity increase was observed in A549CisR-siATM/shMcl1 cells (Fig. 3E). We observed decreased IC50 value of 17 μM in A549CisR-siATM/shMcl1 cells, which is close to parental A549 cells IC50 value (10.5 μM).
When we treated these cells with cisplatin, increased expression of the apoptotic markers cleaved caspase 3 and cleaved PARP were detected in 549CisR-siATM, A549shMcl1 cells compared with A549CisR-sc cells, and the most significant increase was observed in A549CisR-siATM/shMcl1 cells (Fig. 3F). Consistently, flow cytometric analysis detecting annexin V-bound apoptotic cells revealed a higher percentage of apoptotic cells in A549CisR-siATM/shMcl1 cells than in A549CisR-sc cells upon cisplatin treatment (Fig. 3G).
In vivo mice studies showing the higher degree of cisplatin-treatment-induced tumor regression in A549CisR-siATM/shMcl1 xenografts than in A549CisR-sc xenografts.
To confirm the in vitro results in in vivo mice studies, subcutaneous xenograft mouse models were developed by injecting A549CisR-siATM/shMcl1 and A549CisR-sc cells into nude mice (n = 10 each group, 2 tumor sites/mouse). When tumors developed to 300–350 mm3, mice were divided into 2 subgroups and i.p. injected with either vehicle or cisplatin (5 mg/kg) (n = 5/sub-group) for 3 weeks. Higher concentrations of cisplatin than the previous experiment (3 mg/kg)(21) was used considering that the tumors were derived from cisplatin-resistant cells. Immunohistochemical (IHC) staining of tumor tissues of each group showed significantly lower expression of ATM and Mcl-1 in A549CisR-siATM/shMcl1 cells-derived xenografts than in A549CisR-sc cells-derived xenografts, confirming the ATM/Mcl-1 knockdown in tumor tissues of A549CisR-sc cells-derived xenografts (Fig. 4A).
Figure 4.
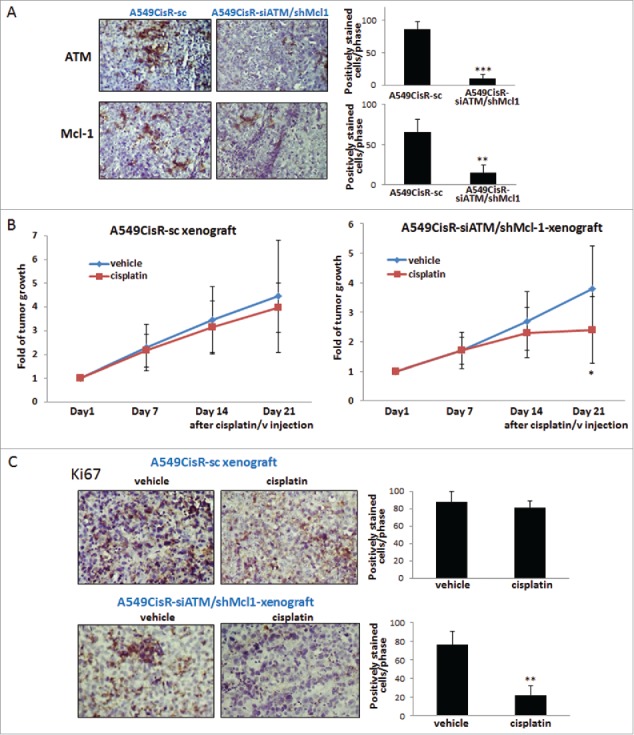
In vivo mice studies. A. IHC staining of tumor tissues (ATM/Mcl-1). Tumor tissues of each mouse group were obtained at sacrifice of mice and 5 μm tissues sections were used in staining using antibodies of ATM and Mcl-1. Quantitation of positively stained cells was shown on right. B. Graphs showing tumor regression upon cisplatin injection into tumor bearing mice. Mice bearing A549CisR-sc and A549CisR-siATM/shMcl1 cells derived tumors were treated with cisplatin (5 mg/kg, vehicle as control) and tumor volume at indicated time points were measured. C. IHC staining of tumor tissues. Tumor tissues of vehicle or cisplatin treated mice in A549CisR-sc and A549CisR-siATM/shMcl1 xenografts were obtained and 5 μm tissues sections were used in staining using Ki67 antibody. Quantitation of positively stained cells was shown on right. Error bars and significance values in IHC staining (A and C) were obtained by counting positively stained cells in one randomly chosen area of slides of 3 different stains. Magnification, 40X. *p < 0.05, **p < 0.01.
We observed significantly higher tumor regression in A549CisR-siATM/shMcl1 cells-derived xenografts at 21 days after cisplatin-treatment than in A549CisR-sc cells-derived xenografts (Fig. 4B). IHC staining of tumor tissues obtained from the cisplatin/vehicle treated mice of each group was also performed. We observed significantly reduced numbers of Ki67 positively stained cells in tumor tissues of cisplatin-treated A549CisR-siATM/shMcl1 xenografts after cisplatin injection than in tissues of A549CisR-sc xenografts (Fig. 4C).
Erk and MAPK signaling pathway is responsible for upregulation/activation of ATM/Mcl-1 in cisplatin-resistant NSCLC cells.
It was reported that ATM activation can be mediated by Akt and Erk1/2 pathways.33 It was also suggested that the DDR activated MAPK signaling is ATM-dependent,34 although whether ATM activation, in turn, is regulated by MAPK pathway remains unknown. Meanwhile, the mTOR pathway was reported to negatively regulate the ATM signaling.35
To investigate whether activation of these signaling pathways is responsible for constitutive expression of ATM/Mcl-1 activation of these signaling pathways in parental and cisplatin-resistant cells was analyzed. We found higher levels of p-Akt, p-Erk, and p-MAPK were detected in A549CisR and H157CisR cells than in parental cells (Fig. 5A).
Figure 5.
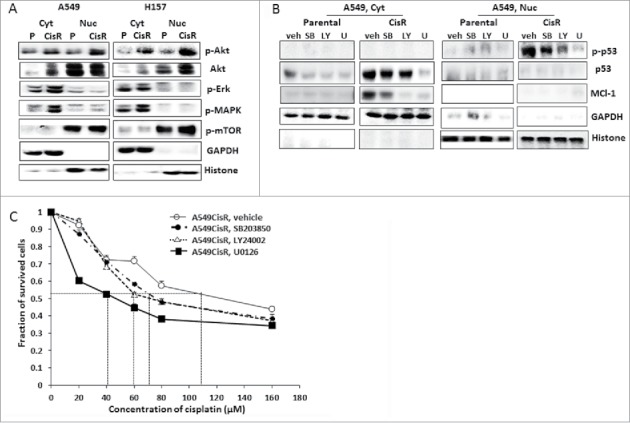
Investigation of signaling pathways responsible for the upregulated ATM/Mcl-1 in cisplatin-resistant cells. A. Western blot analysis investigating signaling pathways responsible for upregulated ATM/Mcl-1 in cisplatin-resistant cells. Cytosolic and nucleic cell extracts were obtained from parental (A549P and H157P) and cisplatin-resistant cells (A549CisR and H157CisR) and western blot analyses were performed using antibodies of indicated signaling molecules. B. Western blot analysis of ATM/Mcl-1 in A549CisR and H157CisR cells upon treatment with inhibitors of each signaling pathway. Cytosolic and nucleic cell extracts were obtained from parental (A549P and H157P) and cisplatin-resistant cells (A549CisR and H157CisR), in the presence of inhibitors of each signaling pathway and levels of ATM and Mcl-1 were analyzed in Western blot analyses. (V: vehicle, SB; SB203850, LY; LY24002, U; U0126) C. Cisplatin-cytotoxicity test of A549P and A549CisR in the presence of inhibitors of each signaling pathway. Cells were treated with indicated concentration of cisplatin, in the presence of inhibitors of each signaling pathway and cell survival was analyzed in MTT assay. Different cisplatin concentration was used in parental cisplatin-resistant cells according to the IC50 value of each cell line.
A549CisR and H157CisR cells were then cultured in the presence of inhibitor of these signaling pathways; LY294002 (10 µM), SB203580 (10 µM), and U0126 (10 µM) that inhibit the Akt, MAPK, and MEK/Erk pathways, respectively, with cisplatin. As shown in Fig. 5B, adding inhibitors of Erk1/2 and MAPK signaling pathways into the A549CisR cell culture reduced the levels of ATM and Mcl-1 molcules, indicating an implication of these signaling pathways in upregulation/activation of ATM/Mcl-1 in cisplatin-resistant cells.
We then tested whether inhibiting these signaling pathways influences A549CisR cells' sensitivity to cisplatin. Cisplatin-cytotoxicity tests showed that addition of the Erk1/2 pathway inhibitor led A549CisR cells to significantly sensitize to cisplatin while other inhibitors did not show significant effects (Fig. 5C), suggesting that the Erk signaling pathway may be most responsible for constitutive expression of ATM/Mcl-1 in cisplatin-resistant cells.
Discussion
Increased DNA repair and anti-apoptosis is the most critical molecular mechanism that leads to cisplatin-resistance.21,36 In comparing the expression of several key molecules in DNA repair and anti-apoptotic pathways in cisplatin-resistant and parental cells, we detected dramatic upregulation of ATM and Mcl-1 molcules in cisplatin-resistant cells. We included PARP and DNA-PK in analyses since recent evidence indicates an implication of these molecules in drug resistance.22,23 However, we could not detect upregulation of these molecules in cisplatin-resistant NSCLC cell lines. As the molecular signature defining the resistant phenotype varies between tumors,37 the profile we observed in cisplatin-resistant NSCLC cells may be a lung cancer-specific phenomenon. However, further studies using more NSCLC cell lines are necessary to draw a clear conclusion.
In addition to the expression level increase, we found constitutive activation of ATM signaling in cisplatin-resistant cells, so activation of the ATM-CHK2-p53 signaling axis in these cells was further detected. A549 cells were reported to have wild-type p5338 while H157 cells express mutated p53 molecules.39 Interestingly, we observed p53 activation in both A549 and H157 cells derived cisplatin-resistant cells. As the major factor affecting the loss of apoptotic function is suggested to be p53 gene mutation,37 it was interesting to note that activation of the ATM-CHK2-p53 axis in cisplatin-resistant NSCLC cells is independent from the mutation status of p53. However, whether the cisplatin-resistant cell lines have the same p53 mutation status as parental cells have not been identified, so further investigation is necessary.
The ATM-CHK2 and ATR-CHK1 axis are well-known signaling cascades activated by DNA double-strand breaks (DSBs) and single-stranded breaks (SSB), respectively,40 although cross-talk between these 2 pathways have been suggested.41 Whether ATR-CHK1 is also activated in cisplatin-resistant cells needs to be addressed in the future.
We found Mcl-1 was also dramatically upregulated in cisplatin-resistant cells. Although we observed nuclear Mcl-1 in cisplatin-resistant cells, not in parental cells in IF staining (data not shown), a high level of nuclear Mcl-1 was not detected in Western blot analysis, indicating that the Mcl-1 nuclear translocation process may be transient. Further investigation needs to be performed to conclude this issue since the nuclear Mcl-1 role in DNA repair process is emerging.16 Specifically, the nuclear Mcl-1 role in ATR mediated CHK1 phosphorylation has been suggested.42 It was also suggested that Ku70 co-localizes with Mcl-1 in mitochondria, endoplasmic reticulum, and nucleus in lung cancer cells and controls apoptosis by stabilizing and protecting Mcl-1 from ubiquitination.43 So, whether Mcl-1 binds with Ku70 in the nucleus of cisplatin-resistant NSCLC cells also needs to be studied.
In in vitro testing the inhibitory effects of ATM and Mcl-1 on increasing cisplatin-sensitivity and inducing apoptotic death of cisplatin-resistant cells upon cisplatin treatment, we applied 2 approaches: inhibitor studies and utilization of knocked down cell lines. Both approaches exhibited similar effects; significantly increased cisplatin-sensitivity after inhibition/knockdown of the 2 molcules was most significant when simultaneously inhibited (Fig. 3). The in vivo mice studies results confirmed the in vitro result. Although there was a big individual mouse variation, we detected significantly higher tumor regression in A549CisR-siATM/shMcl1 cells-derived xenografts after cisplatin injection, but not A549CisR-sc cells-derived xenografts.
In investigating an upstream signaling molecule that could be responsible for upregulated ATM/Mcl-1 molcules in cisplatin-resistant NSCLC cells, we discovered that inhibition of the Erk1/2 pathway reduced ATM/Mcl-1 levels in cisplatin-resistant cells, and induced cisplatin-sensitivity increase. This result is consistent with previous reports suggesting that inhibition of Erk1/2 activity increased sensitivity to cisplatin.44 We also observed activation of Akt pathway since Akt activation of ATM33 and increased cisplatin-cytotoxicity of glioblastoma cells upon Akt inhibition have been reported.45 However, inhibiting Akt pathway did not significantly induce cisplatin-sensitivity of cisplatin-resistant cells. Therefore, we suggest that the strategy of targeting the Erk1/2 pathway may be an alternative way of replacing the ATM/Mcl-1 targeting strategy in increasing cisplatin-sensitivity.
ATM is considered an attractive target for anticancer chemo- and radiosensitization.46 For example, the strategy of blocking ATM or DNA-PK, either alone or in combination with cisplatin has been suggested for personalized therapy in treating breast cancer.47 So, preclinical evaluation of several ATM inhibitors has been performed. Among those, the ATM inhibitor KU59403 showed promising results in its potency, selectivity, and solubility.46
UMI77 is reported to be a selective Mcl-1 inhibitor, showing selectivity over other members of the Bcl-2 family.32 However, this drug has not been used in clinical trials yet although the BH3-mimetics ABT-199 or ABT-263, which are inhibitors of whole Bcl-1 family, have been applied in clinical trials to target Mcl-1.48
Clinical application of combined use of a Mcl-1 inhibitor and an ATM inhibitor may be a long-term project, but this study provides a solid basis and potential for using this strategy to circumvent the cisplatin-resistance obstacle in lung cancer therapeutics.
Methods and materials
Cell culture
A549 and H157 cell lines were purchased from the American Type Culture Collection (ATCC, Manassas, VA) and cultured in RPMI 1640 containing 10% FBS. All cells were maintained in a humidified 5% CO2 environment at 37°C. For inhibition of ATM and/or Mcl-1, the inhibitors of ATM (CP466722, 10 μM) and Mcl-1 (UMI-77, 10 μM) (both from Selleckchem) were added into the culture. For inhibition of signaling pathways, LY294002 (10 µM, Sigma, 440202), SB203580 (10 µM, Sigma, 559387), AG490 (5 µM, Sigma, T3434), and U0126 (10 µM, Cell Signaling, 9903) that inhibit the Akt, MAPK, JAK/Stat3, and MEK/Erk pathways, respectively, were added into the culture together with cisplatin.
Development of cisplatin-resistant cell lines
Parental A549 (A549P) and H157 (H157P) cells were continuously treated with a gradually increased dose of cisplatin over 6 months according to the method described by Barr et al.49 Briefly, cells were treated with 1 µM cisplatin for 72 hours and cells were allowed to recover for the following 72 hours. After repeating one more cycle at 1 µM cisplatin concentration, the cells were then treated with 2 µM cisplatin in the following 2 cycles. This procedure was continued with increasing cisplatin concentration up to 30 µM. During the cisplatin-resistance induction procedure, the IC50 values of every 5 passage cells were accessed in comparison with those of passage number matched parental cells. The cisplatin-resistant cell lines obtained by this method were maintained in growth media containing 10 µM cisplatin.
Development of ATM knocked down, Mcl-1 knocked down and ATM/Mcl-1 double knocked down cell lines by lentiviral transduction
The lentiviral vector containing the siRNA sequence to ATM (Human) was obtained from ABM (i001550a) (piLenti-siRNA-GFP) (target sequence, CATCTAGATCGGCATTCAG) and the Mcl-1shRNA (lenti) that contained 3 target-specific constructs that encode 19–25 nt (plus hairpin) shRNA designed to knock down gene expression was purchased from Santa Cruz (sc-35877-V). Each vector was transfected into 293T cells with a mixture of lentiviral vector, psPAX2 (virus-packaging plasmid), and pMD2G (envelope plasmid) (4:3:2 ratio) using PolyFect Transfection reagent (Qiagen, 301105). For generation of ATM/Mcl-1 double knocked down cells, the lentivirus constructs carrying both siRNA to ATM and shRNA to Mcl-1 (1:1) were used in transfection. After infection procedure to A549CisR and H157CisR cells, the media containing the virus was replaced with normal culture media, and maintained under normal cell culture conditions. The ATM and/or Mcl-1 knocked down cells and sc cells were selected by adding puromycin (2 μg/ml) (Millipore, 540411) into the culture and after selection, cells were maintained in media containing 0.1 μg/ml puromycin.
Cisplatin-cytotoxicity test
Cisplatin-cytotoxicity was analyzed by MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide, 5 mg/ml, Sigma, M2128) assay. Cells were seeded on 96-well plates (7 × 103 cells/well) and treated with various concentrations of cisplatin for 48 hours. MTT test was then performed and absorbance at 490 nm was measured. Cell viability was calculated using the formula: OD sample/OD blank control × 100. Triplicate experiments were performed and average values with mean ± SEM were represented.
Flow cytometric analysis of Apoptosis
Cells (A549CisR-sc and A549CisR-siATM/shMcl1) were seeded on 24-well plates (2 × 104 cells/well), treated with cisplatin (30 μM) for 24 hours, and their apoptotic death rates were analyzed by the AnnexinV based apoptosis kit (eBioscience, 88–8007–72) according to the manufacturer's instructions.
Immunofluoresence (IF) staining
Cells were mounted on chamber slide, fixed, and stained with antibodies of p-ATM (Ser 1981, Millipore 05–740), and p-p53 (Ser 20, Ameritech, ATB-P1263). After reaction with Alexa flour 488 goat anti-rabbit secondary antibody (Life Technologies, A11034), images were recorded using a fluorescent microscope (Zeiss).
In vivo mice studies
The A549CisR-sc and A549CisR-siATM/shMcl1 cells (1 × 106/site) were subcutaneously injected into flanks of 8-week-old female nude mice (NCI) (10 mice per group, total 20 mice). Tumor volumes (V), calculated by V = (L × S2)/2 formula (L and S present long and short axis of tumors, respectively) were recorded twice a week. When tumor volumes reached about 300–350 mm3, cisplatin (5 mg/kg) or vehicle (control) were i.p. injected into each group mice (n = 5, each group) twice per week for 3 weeks. Tumor growth following cisplatin (or vehicle) treatment was monitored until the end of treatment. Mice in all groups were killed at the end of treatment and tumor tissues were obtained and processed for staining. All animal studies were performed under the supervision and guidelines of the University of Rochester Medical Center's Animal Care and Use Committee.
Isolation of total, cytoplasmic, and nuclear extracts
To obtain total cell extracts, cells were lysed in RIPA buffer (50 mM Tris-Cl at pH 7.5, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 1 mM EDTA, 1 μg/mL leupeptin, 1 μg/mL aprotinin, 0.2 mM PMSF). For isolation of cytoplasmic and nuclear extracts, cells were harvested and incubated in buffer A containing 10 mM HEPES, pH 7.5, 10 mM KCl, 2 mM MgCl2, 1% NP40, 5 mM EDTA, and protease inhibitor for 20 min, and centrifuged (500 g, 5 min). The resulting supernatants were designated as cytosolic fraction. The nuclear pellet was resuspended in the same buffer A, supplemented with 500 mM NaCl, 25% glycerol, and kept on ice for 30 minutes. Samples were centrifuged (12000 g, 5 min), and the supernatant obtained were used as nuclear extracts.
Western blot analysis
Proteins (20–40 µg) were separated on 8–10% SDS/PAGE gel and then transferred onto PVDF membranes (Millipore, IPVH00010). After blocking procedure, membranes were incubated with primary antibodies (1:1000), HRP-conjugated secondary antibodies (1:5000), and protein bands were visualized in Imager (Bio-Rad) using ECL system (Thermo Fisher Scientific, 34095). Antibodies used were: ATM (Millipore, 07–1286), p-ATM (Millipore, 05–740), p-p53 (Ameritech, ATB-P1263), histone (Bioss USA, bs0439R), and p-mTOR (Millipore, 09–345), p21 (Santa Cruz, sc397), Akt (Santa Cruz, sc8312), p-Akt (Cell Signaling, 9271), bcl-2 (Santa Cruz, sc492), p53 (Santa Cruz, sc65334), and PARP (Santa Cruz. sc7150), DNA-PK (Ameritech, ATB-T1387), KU70 (Abgent, AP6775C), BRCA1 (Abbiotec, 200160), CHK2 (Abgent, AP4999a), MDC1 (Bioss USA, bs-2400R), and p-CHK2 (Ameritech, ATB-P1237), GAPDH (Cell Signaling, 2118S), Mcl-1 (Cell Signaling, 5354S), p-Erk (Cell Signaling, 4695), p-MAPK (Cell Signaling, 9101S), cleaved cas-3 (Cell Signaling, 9664S), cleaved cas-9 (Cell Signaling, 9505P), and cleaved-PAR (Cell Signaling, 5625).
Histology and immunohistochemistry
Tumor tissues obtained from animal studies were fixed in 10% (v/v) formaldehyde in PBS, embedded in paraffin, and cut into 5-µm sections. Tumor tissue sections were deparaffinized in xylene solution, rehydrated, and processed for immunostaining using the IHC kit (Santa Cruz, SC2018). Antibodies of Ki67 (Cell Signaling, 8D5), Mcl-1 (Cell Signaling, 5453S), and ATM (Abcam, ab78)) (antibodies dilutions at 1:50 to 1:250 dilution per manufacturer's suggestions) were applied in staining. For Ki67 staining, the antigen retrieval process was performed in 10 mM citric buffer, pH 6.0 for 20 minutes using a cooker before staining. After staining, tissues were counterstained by hematoxylin.
Statistics
The data values were presented as the mean ± SEM. Differences in mean values between the 2 groups were analyzed by 2-tailed Student's t test. p ≤ 0.05 was considered statistically significant.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgment
We thank Laura Finger for assistance with manuscript preparation.
Funding
Meaghan's Hope.
References
- 1.Cersosimo RJ. Lung cancer: a review. Am J Health-System Pharmacy: AJHP: Official J Am Society Health-System Pharmacists 2002; 59(7):611-42. Epub 2002/04/12; PMID:11944603 [DOI] [PubMed] [Google Scholar]
- 2.Parsons A, Daley A, Begh R, Aveyard P. Influence of smoking cessation after diagnosis of early stage lung cancer on prognosis: systematic review of observational studies with meta-analysis. BMJ 2010; 340:b5569. Epub 2010/01/23; PMID:20093278; https://doi.org/ 10.1136/bmj.b5569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chang A. Chemotherapy, chemoresistance and the changing treatment landscape for NSCLC. Lung Cancer 2011; 71(1):3-10; PMID:20951465; https://doi.org/ 10.1016/j.lungcan.2010.08.022 [DOI] [PubMed] [Google Scholar]
- 4.Galluzzi L, Vitale I, Michels J, Brenner C, Szabadkai G, Harel-Bellan A, Castedo M, Kroemer G. Systems biology of cisplatin resistance: past, present and future. Cell Death Disease 2014; 5:e1257; PMID:24874729; https://doi.org/ 10.1038/cddis.2013.428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O, Castedo M, Kroemer G. Molecular mechanisms of cisplatin resistance. Oncogene 2012; 31(15):1869-83; PMID:21892204; https://doi.org/ 10.1038/onc.2011.384 [DOI] [PubMed] [Google Scholar]
- 6.Huen MS, Chen J. The DNA damage response pathways: at the crossroad of protein modifications. Cell Res 2008; 18(1):8-16; PMID:18087291; https://doi.org/ 10.1038/cr.2007.109 [DOI] [PubMed] [Google Scholar]
- 7.Lai SL, Hwang J, Perng RP, Whang-Peng J. Modulation of cisplatin resistance in acquired-resistant nonsmall cell lung cancer cells. Oncol Res 1995; 7(1):31-8; PMID:7549042 [PubMed] [Google Scholar]
- 8.Weber AM, Ryan AJ. ATM and ATR as therapeutic targets in cancer. Pharmacol Therapeutics 2015; 149:124-38; PMID:25512053; https://doi.org/ 10.1016/j.pharmthera.2014.12.001 [DOI] [PubMed] [Google Scholar]
- 9.Shiloh Y, Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol 2013; 14(4):197-210; PMID:23486281; https://doi.org/ 10.1038/nrm3546 [DOI] [PubMed] [Google Scholar]
- 10.Imanishi S, Umezu T, Ohtsuki K, Kobayashi C, Ohyashiki K, Ohyashiki JH. Constitutive activation of the ATM/BRCA1 pathway prevents DNA damage-induced apoptosis in 5-azacytidine-resistant cell lines. Biochem Pharmacol 2014; 89(3):361-9; PMID:24680865; https://doi.org/ 10.1016/j.bcp.2014.03.008 [DOI] [PubMed] [Google Scholar]
- 11.Zhang T, Shen Y, Chen Y, Hsieh JT, Kong Z. The ATM inhibitor KU55933 sensitizes radioresistant bladder cancer cells with DAB2IP gene defect. Int J Radiation Biol 2015; 91(4):368-78; PMID:25585815; https://doi.org/ 10.3109/09553002.2015.1001531 [DOI] [PubMed] [Google Scholar]
- 12.Thomas LW, Lam C, Edwards SW. Mcl-1; the molecular regulation of protein function. FEBS Letters 2010; 584(14):2981-9; PMID:20540941; https://doi.org/ 10.1016/j.febslet.2010.05.061 [DOI] [PubMed] [Google Scholar]
- 13.Song L, Coppola D, Livingston S, Cress D, Haura EB. Mcl-1 regulates survival and sensitivity to diverse apoptotic stimuli in human non-small cell lung cancer cells. Cancer Biol Therapy 2005; 4(3):267-76; PMID:15753661; https://doi.org/ 10.4161/cbt.4.3.1496 [DOI] [PubMed] [Google Scholar]
- 14.Jamil S, Sobouti R, Hojabrpour P, Raj M, Kast J, Duronio V. A proteolytic fragment of Mcl-1 exhibits nuclear localization and regulates cell growth by interaction with Cdk1. Biochem J 2005; 387(Pt 3):659-67; PMID:15554878; https://doi.org/ 10.1042/BJ20041596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fujise K, Zhang D, Liu J, Yeh ET. Regulation of apoptosis and cell cycle progression by MCL1. Differential role of proliferating cell nuclear antigen. J Biol Chem 2000; 275(50):39458-65; PMID:10978339; https://doi.org/ 10.1074/jbc.M006626200 [DOI] [PubMed] [Google Scholar]
- 16.Jamil S, Stoica C, Hackett TL, Duronio V. Mcl-1 localizes to sites of DNA damage and regulates DNA damage response. Cell Cycle 2010; 9(14):2843-55; PMID:20647761; https://doi.org/ 10.4161/cc.9.14.12354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Akgul C, Moulding DA, White MR, Edwards SW. In vivo localisation and stability of human Mcl-1 using green fluorescent protein (GFP) fusion proteins. FEBS Letters 2000; 478(1-2):72-6; PMID:10922472; https://doi.org/ 10.1016/S0014-5793(00)01809-3 [DOI] [PubMed] [Google Scholar]
- 18.Yang T, Kozopas KM, Craig RW. The intracellular distribution and pattern of expression of Mcl-1 overlap with, but are not identical to, those of Bcl-2. J Cell Biol 1995; 128(6):1173-84; PMID:7896880; https://doi.org/ 10.1083/jcb.128.6.1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Epling-Burnette PK, Zhong B, Bai F, Jiang K, Bailey RD, Garcia R, Jove R, Djeu JY, Loughran TP Jr, Wei S. Cooperative regulation of Mcl-1 by Janus kinase/stat and phosphatidylinositol 3-kinase contribute to granulocyte-macrophage colony-stimulating factor-delayed apoptosis in human neutrophils. J Immunol 2001; 166(12):7486-95; PMID:11390502; https://doi.org/ 10.4049/jimmunol.166.12.7486 [DOI] [PubMed] [Google Scholar]
- 20.Pei XY, Dai Y, Felthousen J, Chen S, Takabatake Y, Zhou L, Youssefian LE, Sanderson MW, Bodie WW, Kramer LB, et al.. Circumvention of Mcl-1-dependent drug resistance by simultaneous Chk1 and MEK1/2 inhibition in human multiple myeloma cells. PloS One 2014; 9(3):e89064; PMID:24594907; https://doi.org/ 10.1371/journal.pone.0089064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Duan S, Tsai Y, Keng P, Chen Y, Lee SO, Chen Y. IL-6 signaling contributes to cisplatin resistance in non-small cell lung cancer via the up-regulation of anti-apoptotic and DNA repair associated molecules. Oncotarget 2015; 6(29):27651-60; PMID:26313152; https://doi.org/ 10.18632/oncotarget.4753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yoon JH, Ahn SG, Lee BH, Jung SH, Oh SH. Role of autophagy in chemoresistance: regulation of the ATM-mediated DNA-damage signaling pathway through activation of DNA-PKcs and PARP-1. Biochem Pharmacol 2012; 83(6):747-57; PMID:22226932; https://doi.org/ 10.1016/j.bcp.2011.12.029 [DOI] [PubMed] [Google Scholar]
- 23.Michels J, Vitale I, Galluzzi L, Adam J, Olaussen KA, Kepp O, Senovilla L, Talhaoui I, Guegan J, Enot DP, et al.. Cisplatin resistance associated with PARP hyperactivation. Cancer Res 2013; 73(7):2271-80; PMID:23554447; https://doi.org/ 10.1158/0008-5472.CAN-12-3000 [DOI] [PubMed] [Google Scholar]
- 24.Lee KJ, Saha J, Sun J, Fattah KR, Wang SC, Jakob B, et al.. Phosphorylation of Ku dictates DNA double-strand break (DSB) repair pathway choice in S phase. Nucleic Acids Res 2016; 44(4):1732-45; PMID:26712563; https://doi.org/26180923 10.1093/nar/gkv1499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lohse I, Borgida A, Cao P, Cheung M, Pintilie M, Bianco T, Holter S, Ibrahimov E, Kumareswaran R, Bristow RG, et al.. BRCA1 and BRCA2 mutations sensitize to chemotherapy in patient-derived pancreatic cancer xenografts. Br J Cancer 2015; 113(3):425-32; PMID:26180923; https://doi.org/ 10.1038/bjc.2015.220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leisching G, Loos B, Botha M, Engelbrecht AM. Bcl-2 confers survival in cisplatin treated cervical cancer cells: circumventing cisplatin dose-dependent toxicity and resistance. J Translational Med 2015; 13:328; PMID:26474854; https://doi.org/ 10.1186/s12967-015-0689-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sun Y, Xu Y, Roy K, Price BD. DNA damage-induced acetylation of lysine 3016 of ATM activates ATM kinase activity. Mol Cell Biol 2007; 27(24):8502-9; PMID:17923702; https://doi.org/ 10.1128/MCB.01382-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eliezer Y, Argaman L, Kornowski M, Roniger M, Goldberg M. Interplay between the DNA damage proteins MDC1 and ATM in the regulation of the spindle assembly checkpoint. J Biol Chem 2014; 289(12):8182-93; PMID:24509855; https://doi.org/ 10.1074/jbc.M113.532739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Buscemi G, Ricci C, Zannini L, Fontanella E, Plevani P, Delia D. Bimodal regulation of p21(waf1) protein as function of DNA damage levels. Cell Cycle 2014; 13(18):2901-12; PMID:25486478; https://doi.org/ 10.4161/15384101.2014.946852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gaul L, Mandl-Weber S, Baumann P, Emmerich B, Schmidmaier R. Bendamustine induces G2 cell cycle arrest and apoptosis in myeloma cells: the role of ATM-Chk2-Cdc25A and ATM-p53-p21-pathways. J Cancer Res Clin Oncol 2008; 134(2):245-53; PMID:17653574; https://doi.org/ 10.1007/s00432-007-0278-x [DOI] [PubMed] [Google Scholar]
- 31.Rainey MD, Charlton ME, Stanton RV, Kastan MB. Transient inhibition of ATM kinase is sufficient to enhance cellular sensitivity to ionizing radiation. Cancer Res 2008; 68(18):7466-74; PMID:18794134; https://doi.org/ 10.1158/0008-5472.CAN-08-0763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abulwerdi F, Liao C, Liu M, Azmi AS, Aboukameel A, Mady AS, Gulappa T, Cierpicki T, Owens S, Zhang T, et al.. A novel small-molecule inhibitor of Mcl-1 blocks pancreatic cancer growth in vitro and in vivo. Mol Cancer Therapeutics 2014; 13(3):565-75; PMID:24019208; https://doi.org/ 10.1158/1535-7163.MCT-12-0767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hein AL, Ouellette MM, Yan Y. Radiation-induced signaling pathways that promote cancer cell survival (review). Int J Oncol 2014; 45(5):1813-9; PMID:25174607; https://doi.org/ 10.3892/ijo.2014.2614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim WJ, Rajasekaran B, Brown KD. MLH1- and ATM-dependent MAPK signaling is activated through c-Abl in response to the alkylator N-methyl-N′-nitro-N′-nitrosoguanidine. J Biol Chem 2007; 282(44):32021-31; PMID:17804421; https://doi.org/ 10.1074/jbc.M701451200 [DOI] [PubMed] [Google Scholar]
- 35.Shen C, Houghton PJ. The mTOR pathway negatively controls ATM by up-regulating miRNAs. Proc Natl Acad Sci U S A 2013; 110(29):11869-74; PMID:23818585; https://doi.org/ 10.1073/pnas.1220898110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Basu A, Krishnamurthy S. Cellular responses to Cisplatin-induced DNA damage. J Nucleic Acids 2010; 2010:201367; PMID:20811617; https://doi.org/ 10.4061/2010/201367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Siddik ZH. Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene 2003; 22(47):7265-79; PMID:14576837; https://doi.org/ 10.1038/sj.onc.1206933 [DOI] [PubMed] [Google Scholar]
- 38.Goodrum FD, Ornelles DA. p53 status does not determine outcome of E1B 55-kgdalton mutant adenovirus lytic infection. J Virol 1998; 72(12):9479-90; PMID:9811681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sun SY, Yue P, Wu GS, El-Deiry WS, Shroot B, Hong WK, Lotan R. Implication of p53 in growth arrest and apoptosis induced by the synthetic retinoid CD437 in human lung cancer cells. Cancer Res 1999; 59(12):2829-33; PMID:10383141 [PubMed] [Google Scholar]
- 40.Smith J, Tho LM, Xu N, Gillespie DA. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv Cancer Res 2010; 108:73-112; PMID:21034966; https://doi.org/ 10.1016/B978-0-12-380888-2.00003-0 [DOI] [PubMed] [Google Scholar]
- 41.Reinhardt HC, Aslanian AS, Lees JA, Yaffe MB. p53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage. Cancer Cell 2007; 11(2):175-89; PMID:17292828; https://doi.org/ 10.1016/j.ccr.2006.11.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jamil S, Mojtabavi S, Hojabrpour P, Cheah S, Duronio V. An essential role for Mcl-1 in ATR-mediated CHK1 phosphorylation. Mol Biol Cell 2008; 19(8):3212-20; PMID:18495871; https://doi.org/ 10.1091/mbc.E07-11-1171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang B, Xie M, Li R, Owonikoko TK, Ramalingam SS, Khuri FR, Curran WJ, Wang Y, Deng X. Role of Ku70 in deubiquitination of Mcl-1 and suppression of apoptosis. Cell Death Differentiation 2014; 21(7):1160-9; PMID:24769731; https://doi.org/ 10.1038/cdd.2014.42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Persons DL, Yazlovitskaya EM, Cui W, Pelling JC. Cisplatin-induced activation of mitogen-activated protein kinases in ovarian carcinoma cells: inhibition of extracellular signal-regulated kinase activity increases sensitivity to cisplatin. Clin Cancer Res 1999; 5(5):1007-14; PMID:10353733 [PubMed] [Google Scholar]
- 45.Carminati PO, Donaires FS, Marques MM, Donadi EA, Passos GA, Sakamoto-Hojo ET. Cisplatin associated with LY294002 increases cytotoxicity and induces changes in transcript profiles of glioblastoma cells. Mol Biol Reports 2014; 41(1):165-77; PMID:24218165; https://doi.org/ 10.1007/s11033-013-2849-z [DOI] [PubMed] [Google Scholar]
- 46.Batey MA, Zhao Y, Kyle S, Richardson C, Slade A, Martin NM, Lau A, Newell DR, Curtin NJ. Preclinical evaluation of a novel ATM inhibitor, KU59403, in vitro and in vivo in p53 functional and dysfunctional models of human cancer. Mol Cancer Therapeutics 2013; 12(6):959-67; PMID:23512991; https://doi.org/ 10.1158/1535-7163.MCT-12-0707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Albarakati N, Abdel-Fatah TM, Doherty R, Russell R, Agarwal D, Moseley P, Perry C, Arora A, Alsubhi N, Seedhouse C, et al.. Targeting BRCA1-BER deficient breast cancer by ATM or DNA-PKcs blockade either alone or in combination with cisplatin for personalized therapy. Mol Oncol 2015; 9(1):204-17; PMID:25205036; https://doi.org/ 10.1016/j.molonc.2014.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cang S, Iragavarapu C, Savooji J, Song Y, Liu D. ABT-199 (venetoclax) and BCL-2 inhibitors in clinical development. J Hematol Oncol 2015; 8:129; PMID:26589495; https://doi.org/ 10.1186/s13045-015-0224-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barr MP, Gray SG, Hoffmann AC, Hilger RA, Thomale J, O'Flaherty JD, Fennell DA, Richard D, O'Leary JJ, O'Byrne KJ. Generation and characterisation of cisplatin-resistant non-small cell lung cancer cell lines displaying a stem-like signature. PloS One 2013; 8(1):e54193; PMID:23349823; https://doi.org/ 10.1371/journal.pone.0054193 [DOI] [PMC free article] [PubMed] [Google Scholar]