

Summary
Nuclear receptor (NR) transcription factors bind various coreceptors, small molecule ligands, DNA response element sequences, and transcriptional coregulator proteins to affect gene transcription. Small molecule ligands and DNA are known to influence receptor structure, coregulator protein interaction, and function; however, little is known on the mechanism of synergy between ligand and DNA. Using quantitative biochemical, biophysical, and solution structural methods, including 13C-detected NMR and hydrogen/deuterium exchange (HDX) mass spectrometry, we show that ligand and DNA cooperatively recruit the intrinsically disordered Steroid Receptor Coactivator-2 (SRC-2/TIF2/GRIP1/NCoA-2) receptor interaction domain (RID) to peroxisome proliferator-activated receptor gamma-retinoid X receptor alpha (PPARγ-RXRα) heterodimer and reveal the binding determinants of the complex. Our data reveal a thermodynamic mechanism by which DNA binding propagates a conformational change in PPARγ-RXRα, stabilizes the receptor ligand-binding domain dimer interface, and impacts ligand potency and cooperativity in NR coactivator recruitment.
Keywords: allostery, ligand binding, NMR spectroscopy, hydrogen/deuterium exchange (HDX) mass spectrometry, peroxisome proliferator-activated receptor, retinoid X receptor, nuclear receptor, cooperativity, stabilization, transcription
eTOC Blurb
de Vera et al. combine biophysical and atomic-resolution solution structural methods to show that ligand and DNA binding to PPARγ-RXRα heterodimer cooperatively enhances recruitment of the SRC-2 coactivator, thereby revealing a complex allosteric communication pathway integrating signals from ligand, DNA, and coactivator.
Introduction
Nuclear receptors (NRs) function as transcriptional protein scaffolds exerting powerful influences on all aspects of physiology. NRs recruit transcriptional coregulator proteins and other chromatin remodeling machinery to target gene promoters to facilitate chromatin reactions such as histone acetylation and deacetylation to epigenetically regulate transcription (Xu et al., 2009). NRs are modular domain proteins containing an N-terminal disordered regulatory region, followed by a DNA-binding domain (DBD), and a C-terminal ligand-binding domain (LBD) containing the activation function-2 (AF–2) coregulator interaction surface (Rastinejad et al., 2013). Coregulator proteins are classified as coactivators or corepressors depending on their ability to promote or repress gene transcription. Coactivators interact with NRs using a flexible, intrinsically disordered region called the Receptor Interaction Domain (RID) typically containing three to four LXXLL sequence ″NR box″ binding motifs connected by disordered linker regions (Millard et al., 2013). Natural and synthetic ligands, including FDA-approved drugs, bind to the NR LBD and affect NR activity through stabilizing specific LBD conformations that influence interactions with coregulators (Gronemeyer et al., 2004). NRs also bind DNA response elements of similar but varying sequences located in receptor target gene promoter regions via their DBD that also influence NR structure and activity (Meijsing et al., 2009; Schone et al., 2016; Watson et al., 2013).
Whereas ligand binding is known to affect coregulator/receptor interaction, the impact of DNA binding is less clear. Furthermore, the complex relationship between ligand and DNA binding on coregulator/receptor interaction is also poorly understood. No large LBD structural changes have been observed in crystal structures of isolated NR LBDs compared to DNA-bound full-length receptor complexes (Chandra et al., 2008; Chandra et al., 2013; Helsen and Claessens, 2014; Lou et al., 2014). However, studies on glucocorticoid receptor (GR) have revealed that DNA can act as an allosteric nuclear receptor ligand, functioning in part by affecting the conformation of the DBD (Meijsing et al., 2009; Schone et al., 2016; Watson et al., 2013). Extending this concept beyond the DBD, hydrogen/deuterium exchange mass spectrometry (HDX-MS) studies on the vitamin D receptor (VDR)-RXRα heterodimer have shown that DNA binding can impact the conformation of surfaces remote from the DBD, including the receptor dimerization and AF-2 coregulator interaction surfaces located in the LBD (Zhang et al., 2011). Quantitative studies on other NRs provide support for this, where DNA and coregulator cooperatively bind to the thyroid receptor (TR) and progesterone receptor (PR) (Heneghan et al., 2007; Putcha and Fernandez, 2009). This indicates a potential for ligand and DNA to cooperatively affect coactivator interaction, and while some studies have probed features of this relationship (Bain et al., 2014; Bain et al., 2012; De Angelis et al., 2015; Heneghan et al., 2007; Putcha and Fernandez, 2009), relatively little is known on the structural mechanism of this synergy.
Here we focused on a coactivator/NR complex that integrates multiple allosteric binding components to determine if and how coactivator binding is influenced by ligand and DNA. Peroxisome proliferator-activated receptor γ (PPARγ) functions as a NR heterodimer with a cognate receptor, retinoid X receptor α (RXRα), to recruit coactivators such as Steroid Receptor Coactivator-2 (SRC-2; also called TIF2, GRIP1, and NCoA-2) to target gene promoters. SRC-2 influences a PPARγ-driven gene transcription program resulting in the differentiation of mesenchymal stem cells into adipocytes (Hartig et al., 2011; Picard et al., 2002). A crystal structure of the intact PPARγ-RXRα heterodimer complex bound to DNA, small molecule ligands (rosiglitazone and 9-cis-retinoic acid; Rosi and 9cRA, respectively), and a peptide derived from an “NR box ”peptide present in the SRC-2 RID revealed the LXXLL-containing peptide can bind to the AF-2 surface of both receptors (Chandra et al., 2008). However, a low-resolution structural analysis using small-angle X-ray scattering (SAXS) suggested that the entire SRC-2 RID flexibly interacts with PPARγ-RXRα through asymmetric binding only to PPARγ using one LXXLL motif (Osz et al., 2012b). To explore this interesting observation, we used biochemical, biophysical, and atomic-resolution solution structural methods to probe the interaction specificity of PPARγ-RXRα with SRC-2 RID, and the effect of binding receptor ligands and DNA. We show that SRC-2 RID binds to PPARγ-RXRα using three LXXLL “NR box ”motifs, and using specificity assays we show that RXRα plays a dominant role over PPARγ in the interaction, although both receptors contribute to the interaction with SRC-2 RID through different thermodynamic mechanisms. Moreover, our studies reveal a role for allostery in the cooperative binding of ligand and DNA to PPARγ-RXRα in the interaction with SRC-2 RID, whereby a conformational change mediated upon binding DNA stabilizes the LBD dimerization surface, reduces the interaction conformational entropy of the complex to afford higher affinity binding of SRC-2 RID, and affects ligand potency for SRC-2 RID recruitment.
Results
SRC-2 RID interacts with PPARγ-RXRα using three LXXLL motifs
To extend the previously reported low-resolution SAXS studies (Osz et al., 2012b), we performed solution NMR spectroscopy to probe the interaction surface of the PPARγ-RXRα LBD heterodimer on the SRC-2 RID at the atomic level. Preliminary 2D [1H,15N]-HSQC NMR analysis of our SRC-2 RID construct, which contains three LXXLL motifs and a C-terminal extension containing a related LEEIL motif, revealed very poor spectral dispersion and missing peaks that hindered the interaction analysis (Figure 1A). We therefore utilized 13C-detected NMR methods (Felli and Pierattelli, 2012), which alleviated issues of poor spectral dispersion and rapid exchange of amide protons with solvent that are often problematic with disordered proteins such as SRC-2 RID (Figure S1). 2D [13C,15N]-CON (Figure 1B) and [13C,15N]-intraCAN (Figure 1C) spectra afforded dispersed spectra that revealed significant changes upon addition of PPARγ-RXRα, including observation of 16 proline residues in SRC-2 RID that cannot be observed in 2D [1H,15N]-HSQC NMR data. Using 2D and 3D 13C-detected NMR methods, we obtained backbone chemical shift assignments for 213 of 235 residues (~91%) of SRC-2 RID.
Figure 1. Structural mapping the PPARγ-RXRα LBD heterodimer interaction surface on SRC-2 RID.

(A–D) 2D (A) 1H–detected HSQC, (B) 13C-detected CON, and (C) 13C-detected intraCAN NMR experiments of [13C,15N]-labeled SRC-2 RID. (D) Change in relative CON and intraCAN peak intensity upon addition of unlabeled PPARγ-RXRα LBD heterodimer. (E) Differential HDX-MS analysis of SRC-2 RID ± PPARγ-RXRα LBD heterodimer. See also Figure S1.
To structurally map the regions within SRC-2 RID affected upon complex formation, we used NMR chemical shift footprinting by comparing 2D [13C,15N]-CON and [13C,15N]-intraCAN NMR data with and without the PPARγ-RXRα LBD heterodimer. We showed previously that PPARγ and RXRα form a complete heterodimeric complex (Kojetin et al., 2015). Addition of PPARγ-RXRα LBD heterodimer resulted in significant peak line broadening and disappearance of NMR peaks for residues within, flanking, and linking all three LXXLL motifs in SRC-2 RID (Figure 1D). This indicates that all three LXXLL motifs are likely in direct contact with PPARγ-RXRα. In contrast, the C-terminal extension containing the LEEIL motif showed only a moderate decrease in peak intensity, likely due to only a modest increase in local rotational correlation time (τc) for this region; this indicates it is extended away from, but flexibly attached to, the 57 kDa PPARγ-RXRα LBD heterodimer (82 kDa complex). Although the 13C-detected NMR data provide superior dispersion for SRC-2 RID relative to traditional 1H-detected NMR, the data still suffered from significant line broadening upon complex formation.
We validated the NMR results with HDX-MS structural footprinting using a time window expansion method that uses acidic pH conditions to allow studying HDX in the millisecond time scale, which is needed to observe interactions with disordered proteins (Goswami et al., 2013). Protection from solvent exchange was observed for regions surrounding two LXXLL motifs, NR boxes 1 and 3 (Figure 1E), in agreement with the NMR studies, indicating multiple regions of SRC-2 RID are involved in the interaction with PPARγ-RXRα. NR box 2 and the linker regions are not perturbed in the HDX-MS data, but residues in these regions show changes in the NMR data. However, NR box 2 contains a histidine residue, and the acidic conditions under which the HDX experiment is performed significantly decreases the binding affinity of this motif (Figure S2). This pH-dependent weakening of SRC-2 NR box 2 affinity provides a rationale for the lack of protection for this region in SRC-2 RID at acidic pH. Furthermore, because NR box 2 is the highest affinity LXXLL-containing motif in SRC-2 RID (vide infra), the acidic pH conditions necessary for this HDX experiment may have limited the overall degree of protection from solvent exchange observed in SRC-2 RID upon binding PPARγ-RXRα LBD heterodimer.
Effect of ligands and contributions of individual SRC-2 LXXLL motifs
The combined NMR and HDX-MS data reveal that all three LXXLL motifs in SRC-2 RID are involved in binding PPARγ-RXRα. To determine the contributions of each receptor, we performed fluorescence polarization (FP) assays in the absence and presence of receptor-specific agonist ligands (Rosi and 9cRA). Although both ligands decreased the SRC-2 RID interaction dissociation constant (Kd) (Figure 2A), RXRα agonist (9cRA) had a greater impact on decreasing the Kd over PPARγ agonist (Rosi), indicating that RXRα is dominant over PPARγ in the interaction of the heterodimer with SRC-2 RID (Figure 2B). We next performed FP to determine the binding contributions of each SRC-2 LXXLL motif (Figure 2B). The apo-heterodimer showed the highest affinity (lowest Kd) for the second LXXLL motif (NR box 2 peptide) with an overall affinity rank ordering NR box 2, NR box 3, and NR box 1. In all cases, ligand binding decreased the peptide interaction Kd. Again, RXR agonist (9cRA) had a greater effect on lowering the Kd, in particular for NR boxes 2 and 3, whereas PPARγ agonist (Rosi) contributed to a much lesser degree. We also performed the assay using a peptide derived from the related C-terminal LEEIL motif in SRC-2 RID (NR box 4), and consistent with the NMR chemical shift footprinting data, no binding was observed.
Figure 2. Ligand and LXXLL motif contributions to receptor interaction specificity.

(A) FP assay of SRC-2 RID binding to PPARγ-RXRα LBD heterodimer ± ligands. Data plotted as the average and s.e.m. of two experimental replicates and fit to a one-site specific binding equation. (B–G) Kd values from FP analysis of SRC-2 RID and individual LXXLL peptides to PPARγ-RXRα LBD heterodimer or each receptor alone ± ligands; data representative of two independent measurements. (H) Thermodynamic parameters from ITC analysis of SRC-2 RID binding to PPARγ-RXRα LBD heterodimer ± ligands; data representative of two independent measurements. See also Figure S2 and S3.
To determine the contribution of each receptor, we performed FP assays to monitor the recruitment of the peptides to PPARγ LBD and RXRα LBD individually. Relative to the heterodimer data, PPARγ showed higher interaction Kd values and binding of Rosi had only a modest effect in decreasing the Kd values (Figure 2C). In contrast, RXRα showed a lower interaction Kd for the peptides and binding of 9cRA substantially lowered the Kd values (Figure 2D). This further confirms a dominant role for RXRα over PPARγ in binding SRC-2 RID. Studies with the SRC-2 NR box 4 peptide revealed no binding as expected.
The results indicating a dominant role for RXRα over PPARγ are consistent with PDBePISA analysis of the buried surface area of SRC-2 NR box 2 peptide bound to the AF-2 surface of PPARγ-RXRα (PDB 3DZY), which reports a more negative ΔG for the interaction with RXRα (−8.9 kcal/mol) vs. PPARγ (−7.5 kcal/mol). Although our data appear to contradict a previous assertion that SRC-2 NR box 3 interacts with PPARγ but not RXRα within the context of the PPARγ-RXRα heterodimer (Osz et al., 2012b), this peptide binding preference was inferred from a study in the absence of RXRα (Li et al., 2008). Moreover, prior work showed that RXR ligands induce recruitment of p160 coactivators, such as SRC-2, to PPARγ-RXRα (IJpenberg et al., 2004; Yang et al., 2000), whereas PPARγ ligands induce the binding of TRAP220. Consistent with this, we confirmed that a peptide derived from TRAP220 is more effectively recruited to PPARγ-RXRα by Rosi (Figure 2E) and displays higher affinity for PPARγ (Figure 2F) than RXRα (Figure 2G).
We performed isothermal titration calorimetry (ITC) to determine the thermodynamic contributions of each receptor-specific ligand on the interaction with SRC-2 RID. The ITC data revealed a 1:1 binding stoichiometry of SRC-2 RID to the PPARγ-RXRα LBD heterodimer with or without ligands (Figure S3) consistent with published analytical ultracentrifugation data (Osz et al., 2012b). Thermodynamic parameters derived from the data (Figure 2H) revealed a more favorable enthalpic (ΔH) component for the 9cRA-bound complex, indicating hydrogen bond formation between SRC-2 RID and RXRα is a strong contributor to the interaction. In contrast, the thermodynamic signature for the Rosi-bound complex revealed a more favorable entropic (ΔS) component, indicating a change in conformation or solvation entropy of PPARγ is a major contributor to SRC-2 RID interaction.
Ligand binding modulates the interaction of SRC-2 RID and PPARγ-RXRα
We used differential HDX-MS analysis to structurally confirm the biochemical interaction data demonstrating that ligand binding to RXRα is the primary driver of SRC-2 RID recruitment to PPARγ-RXRα. Addition of SRC-2 RID to PPARγ-RXRα in the absence of ligand induced a similar, modest protection from solvent exchange in the AF-2 coregulator interaction surface of both PPARγ and RXRα, including helix 3 and helix 12 (Figure 3A), indicating binding of SRC-2 RID to both receptors. Protection from exchange is also observed at the helix 11 heterodimerization surface, suggesting SRC-2 RID binding may compact the heterodimer or this could also indicate a direct interaction of SRC-2 RID with this surface. Addition of PPARγ agonist (Rosi) resulted in relatively minor changes in solvent exchange relative to the apo-heterodimer (Figure 3B). In contrast, addition of RXR agonist (9cRA) resulted in a large protection from solvent exchange in the RXRα helix 11 heterodimerizaton surface and helix 12/AF-2 coregulator binding surface (Figure 3C). A modest but notable decrease in solvent exchange is also observed in the PPARγ AF-2 surface. In the presence of both ligands, the protection from HDX is similar to the 9cRA-bound form (Figure 3D). Differential 13C-detected NMR analysis of the same liganded states revealed subtle chemical shift changes (Figure S4) for peaks corresponding to residues in loop regions between LXXLL motifs and the C-terminal extension containing the LEEIL motif that do not interact with PPARγ-RXRα. This indicates ligand binding to PPARγ-RXRα can allosterically affect the conformation of SRC-2 loop regions between interacting LXXLL motifs, as well as the C-terminal extended region that does not directly interact with PPARγ-RXRα.
Figure 3. Structural mapping the SRC-2 RID interaction surface on PPARγ-RXRα LBD heterodimer.

(A–D) Differential HDX-MS analysis of PPARγ-RXRα LBD ± SRC-2 RID ± receptor-specific ligands shown as the change in deuterium uptake mapped onto the crystal structure of the PPARγ-RXRα LBD heterodimer (PDB 1FM9). (E–L) Deuterium uptake plots for helix 12 peptides in PPARγ (E–H) and RXRα (I–L). Deuterium uptake curves in the absence of ligand are displayed as grey circles. See also Figure S2, S4, and S5.
To gain insight into the contributions of each receptor, we analyzed deuterium uptake curves for peptides corresponding to helix 12 of PPARγ (Figure 3E – H) and RXRα (Figure 3I – L). As depicted in the differential HDX plots (Figure 3A – D), the magnitude of change in deuterium uptake for PPARγ helix 12 upon binding SRC-2 RID was relatively similar in the apo and Rosi-bound states (Figure 3E – H). However, in the absence of SRC-2 RID, Rosi binding resulted in significant protection from solvent exchange in PPARγ helix 12 (Figure 3E,F). In contrast, 9cRA binding caused a slight deprotection from solvent exchange in RXRα helix 12 (Figure 3K,L), consistent with previous studies (Xia et al., 2011; Yan et al., 2004; Zhang et al., 2011), and a large change in solvent protection upon binding SRC-2 RID compared to the apo-form (Figure 3I,K,L). These HDX-MS data combined with the NMR chemical shift and HDX footprinting performed on SRC-2 RID reveal that both PPARγ and RXRα are structurally affected by binding multiple LXXLL motifs within SRC-2 RID. These data also confirm that RXRα and 9cRA play a dominant role over PPARγ and Rosi in driving SRC-2 binding to PPARγ-RXRα. Furthermore, the helix 12 deuterium uptake curves relate well to the ITC thermodynamic parameters discussed above. Namely, binding of 9cRA resulted in favorable change in interaction enthalpy, indicative of hydrogen bond formation and agrees with HDX-MS data that revealed a large change in RXRα helix 12 solvent protection when SRC-2 RID binds to the heterodimer bound to 9cRA. In contrast, binding of Rosi contributes a favorable change in entropy, which is consistent with the HDX-MS data that showed a large protection in PPARγ helix 12 solvent exchange (i.e., it stabilizes helix 12) when Rosi binds to the heterodimer in the absence of SRC-2 RID. There are two caveats to the relatively minor change in solvent exchange that occurs in PPARγ helix 12 upon binding SRC-2 RID. First, it is possible our protein could be bound to bacterial lipids, which can be pulled-down and retained during expression and purification. However, HDX-MS analysis of protein subjected to delipidation revealed no significant difference in solvent exchange relative to non-delipidated protein (Figure S5). Second, Rosi binding to PPARγ causes significant protection from solvent exchange in the absence of SRC-2 RID, which could limit the window to observe additional protection upon binding SRC-2 RID. In contrast, the increase in solvent exchange caused when 9cRA binds to RXRα could provide a more sensitive window to observe protection from solvent exchange upon binding SRC-2 RID.
DNA binding influences SRC-2 RID interaction
We next determined the impact of DNA binding on the interaction of full-length PPARγ-RXRα and SRC-2 RID. We selected three putative PPAR response element (PPRE) DNA sequences from the promoter regions of the PPARγ target genes SULT2A1, APOCIIIB, and Resistin (Heinaniemi et al., 2007). Using FP assays, we observed that the PPRE binding affinities to PPARγ-RXRα are strengthened in the presence of SRC-2 RID (Figure 4A,B), which indicates cooperativity between DNA and coregulator binding. We explored this further using FP assays to determine if SRC-2 RID binding affinity to PPARγ-RXRα is affected upon binding DNA. In all cases, SRC-2 RID binds with higher affinity to PPARγ-RXRα complexed to DNA (Figure 4C,D). In fact, the rank order of DNA binding affinity of PPARγ-RXRα when bound to SRC-2 RID (Figure 4B, triangles), but not in its absence (Figure 4B, circles), matches the SRC-2 RID binding affinity ranking when PPARγ-RXRα is bound to DNA (Figure 4D, squares).
Figure 4. Cooperativity of DNA and SRC-2 RID on binding to full-length PPARγ-RXRα.

(A) FP assay to determine FITC-labeled PPRE DNA binding affinity ± SRC-2 RID; (B) fitted Kd values. (C) FP assay to determine FITC-labeled SRC-2 RID binding affinity ± PPRE DNA sequences; (D) fitted Kd values. In (A,C), data plotted as the average and s.e.m. of two experimental replicates and fit to a one-site specific binding equation. In (B,D), data represent the mean and s.e.m. of two independent measurements. (E) Thermodynamic parameters from ITC analysis of SRC-2 RID binding to full-length PPARγ-RXRα LBD ± SULT2A1 DNA; data representative of two independent measurements. See also Figure S6.
To determine the thermodynamic contributions of DNA binding on the interaction with SRC-2 RID, we performed ITC analysis where SRC-2 RID was titrated into full-length PPARγ-RXRα bound to Rosi and 9cRA in the absence or presence of SULT2A1 DNA (Figure S6). The fitted ITC parameters (Figure 4E) reveal 1:1 binding stoichiometry and a more favorable entropic component for the DNA-bound complex, indicating a reduction in conformational or solvation entropy factor (TΔS) affords the more favorable change in Gibbs free energy (ΔG) upon binding SRC-2 RID relative to the complex not bound to DNA.
DNA-induced allosteric communication pathway in PPARγ-RXRα
The ITC data above indicates DNA binding may stabilize regions of PPARγ-RXRα (i.e., a favorable change in conformational entropy) resulting in higher affinity binding of SRC-2 RID. We performed differential HDX-MS analysis to structurally explore how DNA binding affects the conformation of PPARγ-RXRα/SRC-2 RID complex. For this analysis, we mapped the HDX information onto PPARγ-RXRα structural models representing the compact structural conformation observed via crystallography (Figure 5A – C) (Chandra et al., 2008) and the elongated structural conformation observed via solution SAXS structural methods (Figure 5D – F) (Osz et al., 2012b). Binding of Rosi and 9cRA to PPARγ-RXRα resulted in robust protection from solvent exchange within the ligand-binding pocket, helix 11 heterodimer surface, and helix 12/AF-2 surface of each receptor (Figure 5A,D). We next analyzed the effect of SRC-2 RID binding to ligand-bound PPARγ-RXRα; similar to the LBD studies above, protection from solvent exchange was observed in helix 12 of the PPARγ LBD and more robustly on helix 3 and 12 of the RXRα LBD (Figure 5B,E). Finally, we analyzed the effect of SULT2A1 PPRE DNA binding to the ligand-bound PPARγ-RXRα/SRC-2 RID complex, which revealed robust protection from solvent exchange in several regions (Figure 5C,F–H), including the DNA-binding domains of PPARγ and RXRα; helix6/β-strand surface region within the ligand-binding pocket of the PPARγ LBD; and helix 11 in the PPARγ LBD, which comprises the dimerization surface with RXRα. The latter two observations, which reveal DNA binding functions as an allosteric “ligand ”that stabilizes the PPARγ LBD, is consistent with the ITC data showing that DNA binding affords a favorable change in entropy driving higher affinity interaction with SRC-2 RID.
Figure 5. Structural mapping of ligand, SRC-2 RID, and DNA binding to full-length PPARγ-RXRα.

Differential HDX-MS analysis of full-length PPARγ-RXRα, plotted on (A–C) a “compact” crystallized (PDB 3DZY) and (D–F) “elongated” solution conformation: (A,D) ± receptor-specific ligands (9cRA and Rosi); (B,E) bound to ligands ± SRC-2 RID; and (C,F) bound to ligand and SRC-2 RID ± SULT2A1 DNA. 9cRA and Rosi shown as peach and purple spheres, respectively; DNA and SRC-2 NR box 2 coactivator peptides from the crystal structure are shown in black. Red arrows (C,F) denote a DNA-induced conformational change via (C) the RXRα DBD/PPARγ LBD interaction surface or (F) a long-range allosteric conformational change. (G,H) Deuterium uptake plots for peptides affected by DNA binding in the (G) RXRα DBD and (H) PPARγ LBD. Data represent the mean and s.d. of three experimental replicates.
When these data are considered with both the compact and elongated PPARγ-RXRα structural conformations, two possible mechanisms for the allosteric conformational change emerge. In the compact conformation, DNA binding could induce a global conformational change (Figure 5C; red arrow), transmitting information from DNA to the RXRα DBD (Figure 5G) to the PPARγ LBD (Figure 5H)—perhaps mediated through the hydrogen bond interaction between PPARγ Glu379 (helix 6) and RXRα Tyr189 (DBD) observed in the crystal structure (Chandra et al., 2008)—thereby affecting coregulator interaction. In the elongated conformation (Figure 5F), DNA binding may stabilize the linker region that connects the DBDs and LBDs; thus, stabilization of the PPARγ LBD heterodimer interface (Figure 5H) could also occur through an allosteric mechanism, since the LBDs do not interact with DNA. In this case, the protection from solvent exchange observed in the helix 6/β-strand surface of the PPARγ LBD (Figure 5H) could occur through affecting the conformation of Rosi, which interacts with the helix 6/β-strand surface, or through an allosteric conformational change as helix 6 is near the helix 11 dimer interface.
Synergy between ligand and DNA on SRC-2 RID interaction
To determine if ligand binding can synergize with DNA binding to affect the interaction of PPARγ-RXRα with SRC-2 RID, we performed time-resolved fluorescence resonance energy transfer (TR-FRET) assays where Rosi or 9cRA was titrated alone, or in the presence of a saturating amount of the coreceptor ligand (20 µM). We monitored effects on ligand potency (EC50 value), cooperativity of ligand and DNA in recruiting SRC-2 RID (Hill slope), and efficacy of SRC-2 RID recruitment (TR-FRET window). TR-FRET experiments comparing ligand binding to the PPARγ-RXRα LBD heterodimer (Figure 6A,B) and full-length PPARγ-RXRα in the absence of DNA (Figure 6A,C) or in the presence of a scrambled non-PPRE control DNA sequence (Figure 6A,D) revealed similar ligand potency, cooperativity, and efficacy of SRC-2 RID recruitment. However, in the presence of a PPRE DNA sequence, widespread changes in the TR-FRET profiles were observed (Figure 6A,E–G) where the ligands exhibited higher potency, positive cooperativity (Hill slopes > 1), and more efficacious recruitment of SRC-2 RID to DNA-bound PPARγ-RXRα that is due in part to the tighter SRC-2 RID binding affinity in the presence of DNA (Figure 5). The increase in TR-FRET window could also indicate DNA binding induces a conformational change in PPARγ-RXRα that brings the disordered N-terminal activation function-1 (AF-1) region, which contains an N-terminal His-tag and TR-FRET donor antibody, within closer distance to FITC-labeled SRC-2 RID bound to the LBD.
Figure 6. Cooperativity of ligand and DNA on SRC-2 RID binding to PPARγ-RXRα.

(A) Summary of the TR-FRET analysis including EC50 values, Hill slope, and TR-FRET window. Data represent the mean and s.e.m. of two independent measurements. Data in the absence or presence of DNA are boxed as groups, respectively, for comparison. (B–G) TR-FRET assays of SRC-2 RID binding to (B) PPARγ-RXRα LBD heterodimer or (C–G) full-length PPARγ-RXRα under the conditions indicated. Data plotted as the average and s.e.m. of two experimental replicates and fit to a variable slope sigmoidal dose response equation.
Although DNA binding in general influences the TR-FRET profiles, the magnitude and direction of the changes occur in a DNA-dependent and coreceptor ligand-dependent manner. In the absence of DNA, Rosi was more potent when RXRα is bound to 9cRA (Figure 6A; grey circles vs. green circles); but in contrast, 9cRA was less potent when PPARγ is bound to Rosi (Figure 6A; orange circles vs. blue circles). DNA binding results in higher Rosi potency (Figure 6A; grey circles vs. grey squares), a change that is muted when RXRα is bound to 9cRA (Figure 6A; grey circles and squares vs. green circles and squares). In contrast, DNA binding has a small effect on 9cRA potency alone (Figure 6A; orange circles vs. orange squares); however, in the presence of Rosi and DNA, a large change in 9cRA potency is observed (Figure 6A; blue circles vs. blue squares). Overall, these data demonstrate a synergistic role for binding PPRE DNA and ligands on the recruitment of SRC-2 RID to PPARγ-RXRα.
Discussion
Crystallography studies have defined the structure of NR DBDs and contacts made between the DBD and DNA response element sequences; and the structure of NR LBDs and contacts made when bound to small molecule ligands. These structural studies along with biochemical studies defined a “charge clamp ”structural mechanism by which ligand binding influences interaction with small coregulator LXXLL peptides (Gronemeyer et al., 2004). Structural studies of full-length NRs bound to DNA, ligands, and either coregulator peptides or RIDs via crystallography (Chandra et al., 2008; Chandra et al., 2013; Helsen and Claessens, 2014; Lou et al., 2014), cryo-electron microscopy (Maletta et al., 2014; Orlov et al., 2012; Yi et al., 2015), and small-angle neutron scattering (SANS) and SAXS (Bernardes et al., 2012; Osz et al., 2012b; Rochel et al., 2011) have revealed unique quaternary architectures. Yet, there remains some poorly understood aspects about the structure and function of NRs. Coregulator RIDs contain multiple LXXLL motifs connected by disordered linker regions; these regions are thought to be important for NR interaction, NR selectivity, binding, and a full transcriptional response (Chang et al., 1999; Darimont et al., 1998; McInerney et al., 1998), but very little is known structurally about their interaction with NRs. NR can form heterodimers to integrate signaling through ligand binding to one or both LBDs. Are the mechanisms by which their ligands influence coactivator binding similar or different? Structures of individual DBD and LBD domains show no significant structural changes relative to full-length complex bound to DNA (Helsen and Claessens, 2014). Although the structure of intact PPARγ-RXRα indicates there may be no direct modulation of coactivator binding to the LBD from other NR domains (Chandra et al., 2008), HDX-MS studies revealed that DNA binding can induce conformational changes in the NR LBD upon binding DNA (Bernardes et al., 2012; Zhang et al., 2011). Furthermore, the structural mechanism by which ligand and DNA binding to NRs may synergize to bind coregulator proteins remains unclear.
Our atomic-resolution solution-state structural methods along with biochemical and biophysical approaches reveal the complex interplay between ligand, SRC-2 RID, and DNA binding to PPARγ-RXRα. Our structural studies show that all three LXXLL motifs within the SRC-2 RID are involved or affected in the interaction with PPARγ-RXRα and that ligand binding to RXRα is the primary driver of SRC-2 RID recruitment. 9cRA binding to RXRα dominates over Rosi binding to PPARγ in enhancing the affinity of SRC-2 RID; however, both 9cRA and Rosi contribute to enhance the SRC-2 RID interaction through different thermodynamic mechanism. This is corroborated by studies showing that ligand binding to RXRα but not PPARγ significantly enhances binding of p160 coactivators including SRC-2 to PPARγ-RXRα (IJpenberg et al., 2004; Yang et al., 2000). Collectively, our data suggest a model (Figure 7) by which SRC-2 RID is recruited to PPARγ-RXRα primarily by ligand binding to RXRα and “clamps ”down on both proteins within the context of the heterodimer. For example, one of the LXXLL motifs binds to the RXRα AF-2 surface, most likely NR box 2 as it has the highest affinity to RXRα; a second LXXLL motif binds more weakly to the PPARγ AF-2 surface; and the third LXXLL motif interacts perhaps transiently with a region somewhere on the heterodimer LBD surface. However, other explanations could account for the fact that all three SRC-2 RID LXXLL motifs are affected upon binding PPARγ-RXRα. First, we could be observing heterogeneous binding of SRC-2 RID to PPARγ-RXRα. For example, a crystal structure of HNF4α bound to the PGC-1α coactivator RID containing three LXXLL motifs revealed only a small portion of an LXXLL motif bound to the AF-2 surface (Rha et al., 2009). However, analysis of the electron density indicated the LXXLL motif can be modeled as an average of more than one LXXLL motif. It is similarly possible that in our solution structural studies, we are observing an averaging of different SRC-2 RID binding modes to PPARγ-RXRα. Another possibility is that SRC-2 RID exists as semi-compact, molten globule-like domain and that only binds to RXRα, either through one LXXLL motif or all three via averaging as described above. In this case, it is possible that the SRC-2 RID-induced binding changes in PPARγ observed in the HDX-MS data are allosteric conformational effects. However, this is unlikely because it is not well supported by our NMR and HDX-MS results, and because our FP data show that SRC-2 LXXLL peptides have reasonable affinity (25–80 µM) for PPARγ, we would expect to observe direct binding-induced structural effects.
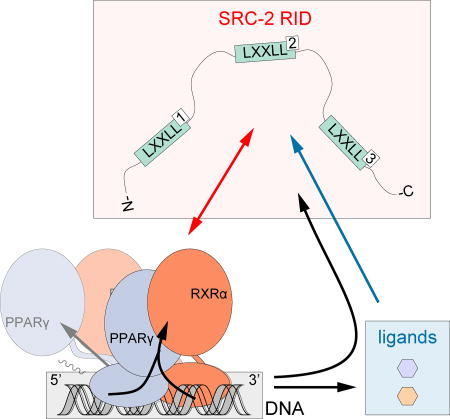
Figure 7. Structural cooperativity model between ligand, DNA, and coregulator binding to PPARγ-RXRα.

Schematic illustrations summarizing the data presented in this study related to the (A) compact crystalized conformation and (B) elongated solution conformation. SRC-2 RID NR box 2 motif binds with highest affinity (dark red arrows) and is more significantly influenced by the RXRα agonist 9cRA (dark blue arrows) than the PPARγ agonist Rosi. DNA binding influences the conformation of PPARγ-RXRα (black arrows), SRC-2 RID affinity, and ligand potency.
Our work contributes to a growing number of studies revealing the complex nature of coactivator binding to NRs. Studies on a related coactivator, SRC-1, revealed it only binds to the RAR NR within the context of the RAR-RXR heterodimer (Rochel et al., 2011) or one monomer within RAR or ER homodimers (Osz et al., 2012a); a similar one monomer interaction was also observed for PGC-1α binding to ERRα and ERRγ homodimers (Takacs et al., 2013). However, SRC-1 can be recruited to a CAR-RXR heterodimer though interactions with both proteins (Pavlin et al., J. Biol. Chem, 2014), likely because each NR binds to SRC-1 LXXLL motifs with relatively high affinity (Wright et al., Biochemistry, 2007). These interaction stoichiometry differences could manifest from several things. For example, the RID sequence identity within three related SRC coactivators varies around 50% (Mukherjee et al., 2007); such differences in amino acid sequence will obviously influence coactivator-NR interactions. Furthermore, though most NR AF-2 surfaces contain conserved “charge clamp ”residues important for binding LXXLL motifs, other sequence differences will also impact binding specificity and affinity. This could manifest in stoichiometry differences allowing two different coactivators to bind separate proteins in a NR heterodimer. Future work that includes a systematic biophysical comparison on how different coactivator proteins interact with different NRs will likely reveal even more insight into the complexities NR-coregulator interactions.
Differences in NR DNA response element sequence are thought to propagate an allosteric conformational change, initiating in the NR DBD and functionally transmitted to other domains of full-length NRs (Meijsing et al., 2009). This model, structurally validated using HDX-MS for VDR-RXRα where DNA binding impacted the conformation of the AF-2 surface in the LBD (Zhang et al., 2011), suggests that DNA binding could also influence coregulator/NR interactions. We show here that DNA binding to full-length PPARγ-RXRα stabilizes the conformation of the PPARγ LBD dimerization surface that interacts with the RXRα LBD and influences coregulator binding affinity and ligand potency. Our data also show that differences in DNA sequence not only affects DNA binding affinity to PPARγ-RXRα but SRC-2 RID binding affinity as well, and vice versa. These observations, which show the cooperativity between ligand, DNA, and coregulator binding to PPARγ-RXRα, indicate that the DNA sequence to which the NR is bound impacts coregulator affinity and ligand potency, which could play a role in driving promoter-specific NR activation profiles. Importantly, these DNA binding-induced effects are compatible with both the compact and elongated conformations of PPARγ-RXRα derived from crystallography and solution structural studies, respectively (Figure 7). Namely, through an allosteric pathway via the RXRα DBD-PPARγ LBD interaction (Figure 5C) or through stabilizing the conformational mobility of the LBDs (Figure 5F) and flexible linker with the DBDs.
Relatively few studies have probed the thermodynamic consequences of ligand and DNA binding on coregulator/NR interactions. DNA binding was shown to affect binding of SRC-1 LXXLL peptide to thyroid receptor (TR), RXR, and TR-RXR heterodimers in part through a favorable change in conformational entropy (Putcha and Fernandez, 2009) and that peptide binding results in higher DNA binding affinity and vice versa. A similar binding cooperativity was observed for progesterone receptor (PR) binding to DNA and SRC-2 coactivator RID (Heneghan et al., 2007). Additionally, studies on GR have shown that DNA binding affinity is the dominant contributor to DNA sequence-specific transcriptional activity (Bain et al., 2014; Bain et al., 2012), and it has been hypothesized that ligand and coactivator binding may play an important role in increasing the residence time of receptors on DNA to influence unique transcriptional responses (De Angelis et al., 2015). Thus, a growing body of work suggests there may be a universal cooperativity mechanism for affecting coregulator/NR interactions in a ligand-, DNA-, and coregulator-specific manner on different target gene promoters of varying response element sequence. How then can these structural and biophysical studies be translated to cells to decipher allosteric functional roles of DNA and ligand on coregulator-NR interaction and transcription on the genome level? In the case of PPARγ and RXRα, chromatin immunoprecipitation coupled to high-throughput DNA sequencing (ChIP-seq) studies have discovered genomic PPRE sites occupied by PPARγ-RXRα heterodimers and individual PPARγ or RXRα monomers (Nielsen et al., 2008). By extending these types of studies, it could be possible to make quantitative-to-genome-wide connections by coupling the quantitative data to receptor and coregulator occupancy on target gene promoters in ChIP-seq data, for example as performed for PPARγ and Med1 (Haakonsson et al., 2013), along with gene expression analysis methods (Gerhold et al., 2002; Sears et al., 2007) in the presence or absence of receptor-specific ligands.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to and will be fulfilled by the Lead Contact, Douglas Kojetin (dkojetin@scripps.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Bacterial Strains Used for Protein Expression
Proteins were expressed in Escherichia coli BL21(DE3) cells using autoinduction ZY media or M9 minimal media supplemented with NMR isotopes. Human SRC-2 RID, residues 624–829; PPARγ LBD, residues 231–505, isoform 2 numbering; and RXRα LBD, residues 223– 462; full-length PPARγ (residues 1–505; isoform 2); and full-length RXRα (1–462); subcloned in pET-46 Ek/LIC plasmid (Novagen) were expressed in Escherichia coli BL21(DE3) cells in autoinduction media as TEV-cleavable hexahistidine-tagged fusion proteins using protocols previously described (Hughes et al., 2012; Hughes et al., 2014; Kojetin et al., 2015). SRC-2 RID used in NMR studies was expressed in M9 media supplemented with 15NH4Cl and 13C-glucose.
METHOD DETAILS
Protein Purification
Cell lysate was applied to a Ni-NTA affinity column (HisTrap HP, GE Healthcare Life Sciences); proteins in wash buffer 50 mM Tris pH 7.4, 500 mM NaCl, 5 mM TCEP) were eluted against a 500 mM imidazole gradient, followed by size-exclusion chromatography (HiLoad 16/60 Superdex 75 pg or 200 pg, GE Healthcare Life Sciences). The final buffer consisted of 20 mM KPO4 (pH 8.0), 50 mM KCl, 5 mM TCEP, 0.5 mM EDTA, and 2.5 mg/mL cOmplete Mini protease inhibitor cocktail (Roche). When working with full-length PPARγ and RXRα, 0.5 mM potassium citrate was used in place of EDTA to prevent chelation of Zn2+ from the zinc finger DNA-binding domains.
Ligands and Other Reagents
Rosiglitazone (Rosi) and 9-cis-retinoic acid (9cRA) were purchased from commercial sources (Sigma and Cayman) and prepared as 20 mM stocks in DMSO-d6. Unlabeled and 5′-FITC-labeled 20-nt PPRE duplex sequences were obtained from Integrated DNA Technologies for Resistin (5'-TGGAAAGAGGAGAAAGTTCT-3'), APOCIIIB (5′-AGGGCGCTGGGCAAAGG TCA-3′), SULT2A1 (5'-GTAAAATAGGTGAAAGGTAA-3'), and scrambled DNA sequence for negative control (5'-AGAGCATTCCGTTCGAGCAT-3'). PPRE DNA duplexes were annealed at 95°C for 3 minutes and slowly cooled to room temperature overnight. N-terminus FITC-labeled peptides were obtained from LifeTein for SRC-2 NR box 1 (DSKGQTKLLQLLTTKSDQM); SRC-2 NR box 2 (LKEKHKILHRLLQDSSSPV); SRC-2 NR box 3 (KKKENALLRYLLDKDDTKD); SRC-2 NR box 4 (GDQPGSELDNLEEILDDLQN SQLPQL); and TRAP220 NR box 2 (NTKNHPMLMNLLKDNPAQD).
NMR Spectroscopy
NMR data were acquired at 298K on a 700 MHz Bruker NMR instrument equipped with a QCI cryoprobe. 2D and 3D 13C-detected NMR experiments were performed on 400 µM [13C, 15N]-SRC-2 RID, including normal and amino acid-selective 2D (HaCa)CON experiments; 2D (HCaCO)N(CO)Ca (inter-CAN); and 3D (H)CbCaCON, 3D (H)CbCaNCO and 3D (H)CaNCaCO. When mapping interactions with LBDs, NMR data were acquired using a 5-fold molar excess of unlabeled LBDs to ensure complete complex formation, with or without 1:1 Rosi and/or 9cRA with respect to LBDs. Data were processed using Topspin 3.0 (Bruker BioSpin) and analyzed with NMRViewJ (OneMoon Scientific, Inc.).
HDX-MS
Solution-phase amide hydrogen/deuterium exchange (HDX) experiments were performed using a fully automated system described previously (Chalmers et al., 2006). Five microliters of a 10 µM protein solution in the absence or presence of 10× ligand or 2–3× biomolecule (protein or DNA) was mixed with 20 µL of D2O containing HDX buffer (20 mM KPO4 at pH 7.4, 50 mM KCl) and incubated for 10, 30, 60, 900, and 3600 s at 4 °C. Low pH HDX experiments were carried out in a similar fashion using pH 6 D2O buffer (Goswami et al., 2013). Following on exchange, forward or back exchange was minimized and protein was denatured by addition of 25 µL of quench solution [0.1% v/v trifluoroacetic acid (TFA) in 3 M urea]. Samples were then passed through an immobilized pepsin column that was prepared in-house (Chalmers et al., 2007) at 200 µL/min (0.1% v/v TFA, 15 °C) and the resulting peptides were trapped on a C8 column (Hypersil Gold, Thermo Fisher). The bound peptides were then gradient eluted (4–40% CH3CN w/v and 0.3% formic acid) across a 2×50 mm C18 high performance liquid chromatography column (Hypersil Gold, Thermo Fisher) for 5 min at 4 °C. The eluted peptides were then subjected to electrospray ionization (ESI) directly coupled with a high resolution Orbitrap mass spectrometer (Exactive, Thermo Fisher). HDX analyses were performed in triplicate with single preparations of each purified protein/complex. The intensity weighted mean m/z centroid value of each peptide envelope was calculated and subsequently converted into a percentage of deuterium incorporation using HDX Workbench software (Pascal et al., 2012).
ITC
Isothermal titration calorimetry (ITC) experiments were performed using a MicroCal iTC200 microcalorimeter (MicroCal Inc., Northampton, MA) using the iTC200 software (v 1.24.2) for instrument control and data acquisition. SRC-2 RID, PPARγ-RXRα LBD heterodimer with or without 1:1 molar ratio of Rosi and/or 9cRA, and full-length PPARγ-RXRα with or without PPRE DNA were separately contained in 200 µL dialysis tubes and co-dialyzed against 1× dialysis buffer consisting of 20 mM KPO4 (pH 8.0), 50 mM KCl, and 5 mM TCEP at 4 °C to minimize buffer mismatch. SRC-2 RID (600–850 µM) was titrated in 20 injections (0.4-µL pre-injection followed by nineteen 2 µL injections) to 60 µM protein (PPARγ-RXRα LBD heterodimer or full-length PPARγ-RXRα) in the sample cell. Injection duration was set to 7.82 s with a 3-min interval between injections into the sample cell. Mixing was carried out at 25 °C with reference power and rotational stirring set at 5 µcal/s and 1000 rpm, respectively. Data fitting binding parameters were obtained through curve fitting on integration of enthalpy peaks using ORIGIN (version 7.0). Calorimetric data were corrected from background signals generated by heat effects of injection, mixing, hydration, and dilution. Background controls included titrations of buffer to buffer and SRC-2 RID to buffer.
FP Assay
SRC-2 RID was labeled with fluorescein isothiocyanate isomer I (FITC) from a 25 mM FITC stock in DMSO; 20 µM protein was incubated with 80-fold molar excess of FITC in the dark at RT for 1.5 h in buffer containing 20 mM KPO4 (pH 8.0), 50 mM KCl, 5 mM TCEP and 100 mM NaHCO3. The sample was subsequently buffer-exchanged against a buffer containing 20 mM KPO4 (pH 8.0), 50 mM KCl, 5 mM TCEP and 0.01% Tween-20 to remove excess FITC and residual DMSO. Fluorescence polarization (FP) assay was performed on a 384-well Greiner black opaque plate using a SpectraMax M5 plate reader at excitation and emission wavelengths of 485 nm and 538 nm, respectively. The concentration of FITC-labeled SRC-2 RID and FITC-labeled peptide was fixed at 250 nM; FITC-labeled PPRE DNA concentration was fixed at 25 nM. The concentration of PPARγ-RXRα LBD heterodimer, with or without Rosi and/or 9cRA, was varied from 30 nM to 80 µM; doubly-liganded full-length PPARγ-RXRα was varied from 2 nM to 5 µM. Data were analyzed using GraphPad Prism (FP signal in millipolarization units vs. ligand concentration).
TR-FRET Assay
The time-resolved fluorescence resonance energy transfer (TR-FRET) assay was performed using 4 nM His-tagged PPARγ-RXRα LBD heterodimer or full-length PPARγ-RXRα, 1.6 µM FITC-labeled SRC-2 RID, 2 nM LanthaScreen® Elite Tb-anti-HIS Antibody (Thermo Fisher Scientific); in 1× assay buffer (20 mM KPO4 pH 8.0, 50 mM KCl, 0.5 mM citrate, 5 mM TCEP, and 0.01% Tween-20); with or without 20 µM Rosi or 9cRA. Full-length PPARγ-RXRα was incubated with or without 20 µM of PPRE DNA duplex (Resistin, SULT2A, or APOCIIIB) or scrambled DNA as negative control; see above for sequences. Samples were incubated for 30 min in the dark at RT, aliquoted onto a 384-well Greiner black plate, and Rosi or 9cRA (both in DMSO) were subsequently titrated to final ligand concentrations ranging from 10.2 pM to 40 µM with a final DMSO concentration of 1%. The plate was read at RT with a BioTek Synergy Neo multimode plate reader; Tb donor was excited at 340 nm, its emission was measured at 495 nm, and acceptor FITC emission was measured at 520 nm. Data were analyzed using GraphPad Prism (TR-FRET fluorescence ratio at 520 nm/495 nm vs. ligand concentration). Comparative analysis was performed using two-way ANOVA and Tukey’s multiple comparisons test.
QUANTIFICATION AND STATISTICAL ANALYSIS
GraphPad Prism (v 7.0) software was used to determine average values, standard errors, and standard deviations. For each figure, number of experimental replicates and other information relevant for assessing the accuracy and precision of the analysis are included in the accompanying legend. For HDX analysis, statistical significance for the differential HDX data is determined by an unpaired t-test for each time point within HDX Workbench software (Pascal et al., 2012).
DATA AND SOFTWARE AVAILABILITY
The NMR chemical shift assignments for SRC-2 RID have been deposited in the Biological Magnetic Resonance Data Bank (BMRB) under ID code 27104.
Supplementary Material
Highlights.
Structural and biophysical analysis of SRC-2 RID/PPARγ-RXRα interaction.
Ligand binding to RXRα dominates over PPARγ to enhance interaction with SRC-2 RID.
DNA binding to PPARγ/RXRα influences SRC-2 RID affinity and vice versa.
Ligand and DNA cooperatively enhance SRC-2 RID binding to PPARγ/RXRα.
Acknowledgments
We thank Wolfgang Bermel (Bruker Biospin) for providing 13C-detected NMR pulse sequences and advice, Mark Rance (U. Cincinnati) for a critical reading of the manuscript, and Natacha Rochel (IGBMC) for providing the elongated PPARγ-RXRα structural model. This work was supported in part by the James and Esther King Biomedical Research Program, Florida Department of Health (1KN-09); the William R. Kenan, Jr. Charitable Trust (TSRI High School Student Summer Internship Program); National Institutes of Health (NIH) grants DK101871 (DJK), DK105825 (PRG), F32DK097890 (TH), K99DK103116 (TH), and F32DK108442 (RB); and American Heart Association (AHA) fellowship awards 12POST12050025 (TH) and 16POST27780018 (RB).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
I.M.S.dV. designed research, performed NMR experiments and assays, and analyzed data. J.Z., S.N., D.P.M, and P.R.G. designed, performed, analyzed, and interpreted HDX-MS experiments. T.H, R.B., J.S., P.M.-T., W.J.G. Jr. prepared samples and assisted with experiments. X.K. assisted with and performed NMR experiments. D.J.K. designed research, performed NMR experiments, and analyzed data. I.M.S.dV. and D.J.K. wrote the manuscript with input from all authors.
References
- Bain DL, Connaghan KD, Maluf NK, Yang Q, Miura MT, De Angelis RW, Degala GD, Lambert JR. Steroid receptor-DNA interactions: toward a quantitative connection between energetics and transcriptional regulation. Nucleic Acids Res. 2014;42:691–700. doi: 10.1093/nar/gkt859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bain DL, Yang Q, Connaghan KD, Robblee JP, Miura MT, Degala GD, Lambert JR, Maluf NK. Glucocorticoid receptor-DNA interactions: binding energetics are the primary determinant of sequence-specific transcriptional activity. J Mol Biol. 2012;422:18–32. doi: 10.1016/j.jmb.2012.06.005. [DOI] [PubMed] [Google Scholar]
- Bernardes A, Batista FA, de Oliveira Neto M, Figueira AC, Webb P, Saidemberg D, Palma MS, Polikarpov I. Low-resolution molecular models reveal the oligomeric state of the PPAR and the conformational organization of its domains in solution. PLoS One. 2012;7:e31852. doi: 10.1371/journal.pone.0031852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busby SA, Chalmers MJ, Griffin PR. Improving digestion efficiency under H/D exchange conditions with activated pepsinogen coupled columns. Int J Mass spectrom. 2007;259:130–139. [Google Scholar]
- Chalmers MJ, Busby SA, Pascal BD, He Y, Hendrickson CL, Marshall AG, Griffin PR. Probing protein ligand interactions by automated hydrogen/deuterium exchange mass spectrometry. Anal Chem. 2006;78:1005–1014. doi: 10.1021/ac051294f. [DOI] [PubMed] [Google Scholar]
- Chalmers MJ, Busby SA, Pascal BD, Southern MR, Griffin PR. A two-stage differential hydrogen deuterium exchange method for the rapid characterization of protein/ligand interactions. J Biomol Tech. 2007;18:194–204. [PMC free article] [PubMed] [Google Scholar]
- Chandra V, Huang P, Hamuro Y, Raghuram S, Wang Y, Burris TP, Rastinejad F. Structure of the intact PPAR-gamma-RXR-alpha nuclear receptor complex on DNA. Nature. 2008;456:350–356. doi: 10.1038/nature07413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra V, Huang P, Potluri N, Wu D, Kim Y, Rastinejad F. Multidomain integration in the structure of the HNF-4alpha nuclear receptor complex. Nature. 2013;495:394–398. doi: 10.1038/nature11966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C, Norris JD, Gron H, Paige LA, Hamilton PT, Kenan DJ, Fowlkes D, McDonnell DP. Dissection of the LXXLL nuclear receptor-coactivator interaction motif using combinatorial peptide libraries: discovery of peptide antagonists of estrogen receptors alpha and beta. Mol Cell Biol. 1999;19:8226–8239. doi: 10.1128/mcb.19.12.8226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darimont BD, Wagner RL, Apriletti JW, Stallcup MR, Kushner PJ, Baxter JD, Fletterick RJ, Yamamoto KR. Structure and specificity of nuclear receptor-coactivator interactions. Genes Dev. 1998;12:3343–3356. doi: 10.1101/gad.12.21.3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Angelis RW, Maluf NK, Yang Q, Lambert JR, Bain DL. Glucocorticoid Receptor-DNA Dissociation Kinetics Measured in Vitro Reveal Exchange on the Second Time Scale. Biochemistry. 2015;54:5306–5314. doi: 10.1021/acs.biochem.5b00693. [DOI] [PubMed] [Google Scholar]
- Felli IC, Pierattelli R. Recent progress in NMR spectroscopy: toward the study of intrinsically disordered proteins of increasing size and complexity. IUBMB Life. 2012;64:473–481. doi: 10.1002/iub.1045. [DOI] [PubMed] [Google Scholar]
- Gampe RT, Jr, Montana VG, Lambert MH, Miller AB, Bledsoe RK, Milburn MV, Kliewer SA, Willson TM, Xu HE. Asymmetry in the PPARgamma/RXRalpha crystal structure reveals the molecular basis of heterodimerization among nuclear receptors. Mol Cell. 2000;5:545–555. doi: 10.1016/s1097-2765(00)80448-7. [DOI] [PubMed] [Google Scholar]
- Gerhold DL, Liu F, Jiang G, Li Z, Xu J, Lu M, Sachs JR, Bagchi A, Fridman A, Holder DJ, et al. Gene Expression Profile of Adipocyte Differentiation and Its Regulation by Peroxisome Proliferator-Activated Receptor-gamma Agonists. Endocrinology. 2002;143:2106–2118. doi: 10.1210/endo.143.6.8842. [DOI] [PubMed] [Google Scholar]
- Goswami D, Devarakonda S, Chalmers MJ, Pascal BD, Spiegelman BM, Griffin PR. Time window expansion for HDX analysis of an intrinsically disordered protein. J Am Soc Mass Spectrom. 2013;24:1584–1592. doi: 10.1007/s13361-013-0669-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gronemeyer H, Gustafsson J-Å, Laudet V. Principles for modulation of the nuclear receptor superfamily. Nat Rev Drug Discov. 2004;3:950–964. doi: 10.1038/nrd1551. [DOI] [PubMed] [Google Scholar]
- Haakonsson AK, Stahl Madsen M, Nielsen R, Sandelin A, Mandrup S. Acute genome-wide effects of rosiglitazone on PPARgamma transcriptional networks in adipocytes. Mol Endocrinol. 2013;27:1536–1549. doi: 10.1210/me.2013-1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartig SM, He B, Long W, Buehrer BM, Mancini MA. Homeostatic levels of SRC-2 and SRC-3 promote early human adipogenesis. J Cell Biol. 2011;192:55–67. doi: 10.1083/jcb.201004026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinaniemi M, Uski JO, Degenhardt T, Carlberg C. Meta-analysis of primary target genes of peroxisome proliferator-activated receptors. Genome Biol. 2007;8:R147. doi: 10.1186/gb-2007-8-7-r147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helsen C, Claessens F. Looking at nuclear receptors from a new angle. Mol Cell Endocrinol. 2014;382:97–106. doi: 10.1016/j.mce.2013.09.009. [DOI] [PubMed] [Google Scholar]
- Heneghan AF, Connaghan-Jones KD, Miura MT, Bain DL. Coactivator assembly at the promoter: efficient recruitment of SRC2 is coupled to cooperative DNA binding by the progesterone receptor. Biochemistry. 2007;46:11023–11032. doi: 10.1021/bi700850v. [DOI] [PubMed] [Google Scholar]
- Hughes TS, Chalmers MJ, Novick S, Kuruvilla DS, Chang MR, Kamenecka TM, Rance M, Johnson BA, Burris TP, Griffin PR, et al. Ligand and receptor dynamics contribute to the mechanism of graded PPARγ agonism. Structure. 2012;20:139–150. doi: 10.1016/j.str.2011.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes TS, Giri PK, de Vera IM, Marciano DP, Kuruvilla DS, Shin Y, Blayo AL, Kamenecka TM, Burris TP, Griffin PR, et al. An alternate binding site for PPARγ ligands. Nat Commun. 2014;5:3571. doi: 10.1038/ncomms4571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IJpenberg A, Tan NS, Gelman L, Kersten S, Seydoux J, Xu J, Metzger D, Canaple L, Chambon P, Wahli W, et al. In vivo activation of PPAR target genes by RXR homodimers. EMBO J. 2004;23:2083–2091. doi: 10.1038/sj.emboj.7600209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojetin DJ, Matta-Camacho E, Hughes TS, Srinivasan S, Nwachukwu JC, Cavett V, Nowak J, Chalmers MJ, Marciano DP, Kamenecka TM, et al. Structural mechanism for signal transduction in RXR nuclear receptor heterodimers. Nat Commun. 2015;6:8013. doi: 10.1038/ncomms9013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Kovach A, Suino-Powell K, Martynowski D, Xu HE. Structural and biochemical basis for the binding selectivity of peroxisome proliferator-activated receptor gamma to PGC-1alpha. J Biol Chem. 2008;283:19132–19139. doi: 10.1074/jbc.M802040200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou X, Toresson G, Benod C, Suh JH, Philips KJ, Webb P, Gustafsson JA. Structure of the retinoid X receptor alpha-liver X receptor beta (RXRalpha-LXRbeta) heterodimer on DNA. Nat Struct Mol Biol. 2014;21:277–281. doi: 10.1038/nsmb.2778. [DOI] [PubMed] [Google Scholar]
- Maletta M, Orlov I, Roblin P, Beck Y, Moras D, Billas IM, Klaholz BP. The palindromic DNA-bound USP/EcR nuclear receptor adopts an asymmetric organization with allosteric domain positioning. Nat Commun. 2014;5:4139. doi: 10.1038/ncomms5139. [DOI] [PubMed] [Google Scholar]
- McInerney EM, Rose DW, Flynn SE, Westin S, Mullen TM, Krones A, Inostroza J, Torchia J, Nolte RT, Assa-Munt N, et al. Determinants of coactivator LXXLL motif specificity in nuclear receptor transcriptional activation. Genes Dev. 1998;12:3357–3368. doi: 10.1101/gad.12.21.3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijsing SH, Pufall MA, So AY, Bates DL, Chen L, Yamamoto KR. DNA binding site sequence directs glucocorticoid receptor structure and activity. Science. 2009;324:407–410. doi: 10.1126/science.1164265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millard CJ, Watson PJ, Fairall L, Schwabe JW. An evolving understanding of nuclear receptor coregulator proteins. J Mol Endocrinol. 2013;51:T23–36. doi: 10.1530/JME-13-0227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee A, Amato P, Allred DC, DeMayo FJ, Lydon JP. Steroid receptor coactivator 2 is required for female fertility and mammary morphogenesis: insights from the mouse, relevance to the human. Nuclear receptor signaling. 2007;5:e011. doi: 10.1621/nrs.05011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen R, Pedersen TA, Hagenbeek D, Moulos P, Siersbaek R, Megens E, Denissov S, Borgesen M, Francoijs KJ, Mandrup S, et al. Genome-wide profiling of PPARgamma:RXR and RNA polymerase II occupancy reveals temporal activation of distinct metabolic pathways and changes in RXR dimer composition during adipogenesis. Genes Dev. 2008;22:2953–2967. doi: 10.1101/gad.501108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlov I, Rochel N, Moras D, Klaholz BP. Structure of the full human RXR/VDR nuclear receptor heterodimer complex with its DR3 target DNA. EMBO J. 2012;31:291–300. doi: 10.1038/emboj.2011.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osz J, Brelivet Y, Peluso-Iltis C, Cura V, Eiler S, Ruff M, Bourguet W, Rochel N, Moras D. Structural basis for a molecular allosteric control mechanism of cofactor binding to nuclear receptors. Proc Natl Acad Sci U S A. 2012a;109:E588–594. doi: 10.1073/pnas.1118192109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osz J, Pethoukhov MV, Sirigu S, Svergun DI, Moras D, Rochel N. Solution Structures of PPARgamma2/RXRalpha Complexes. PPAR Res. 2012b;2012:701412. doi: 10.1155/2012/701412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascal BD, Willis S, Lauer JL, Landgraf RR, West GM, Marciano D, Novick S, Goswami D, Chalmers MJ, Griffin PR. HDX workbench: software for the analysis of H/D exchange MS data. J Am Soc Mass Spectrom. 2012;23:1512–1521. doi: 10.1007/s13361-012-0419-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard F, Gehin M, Annicotte J, Rocchi S, Champy MF, O’Malley BW, Chambon P, Auwerx J. SRC-1 and TIF2 control energy balance between white and brown adipose tissues. Cell. 2002;111:931–941. doi: 10.1016/s0092-8674(02)01169-8. [DOI] [PubMed] [Google Scholar]
- Putcha BD, Fernandez EJ. Direct interdomain interactions can mediate allosterism in the thyroid receptor. J Biol Chem. 2009;284:22517–22524. doi: 10.1074/jbc.M109.026682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rastinejad F, Huang P, Chandra V, Khorasanizadeh S. Understanding nuclear receptor form and function using structural biology. J Mol Endocrinol. 2013;51:T1–T21. doi: 10.1530/JME-13-0173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rha GB, Wu G, Shoelson SE, Chi YI. Multiple binding modes between HNF4alpha and the LXXLL motifs of PGC-1alpha lead to full activation. J Biol Chem. 2009;284:35165–35176. doi: 10.1074/jbc.M109.052506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochel N, Ciesielski F, Godet J, Moman E, Roessle M, Peluso-Iltis C, Moulin M, Haertlein M, Callow P, Mely Y, et al. Common architecture of nuclear receptor heterodimers on DNA direct repeat elements with different spacings. Nat Struct Mol Biol. 2011;18:564–570. doi: 10.1038/nsmb.2054. [DOI] [PubMed] [Google Scholar]
- Schone S, Jurk M, Helabad MB, Dror I, Lebars I, Kieffer B, Imhof P, Rohs R, Vingron M, Thomas-Chollier M, et al. Sequences flanking the core-binding site modulate glucocorticoid receptor structure and activity. Nat Commun. 2016;7:12621. doi: 10.1038/ncomms12621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sears DD, Hsiao A, Ofrecio JM, Chapman J, He W, Olefsky JM. Selective modulation of promoter recruitment and transcriptional activity of PPARgamma. Biochem Biophys Res Commun. 2007;364:515–521. doi: 10.1016/j.bbrc.2007.10.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takacs M, Petoukhov MV, Atkinson RA, Roblin P, Ogi FX, Demeler B, Potier N, Chebaro Y, Dejaegere A, Svergun DI, et al. The asymmetric binding of PGC-1alpha to the ERRalpha and ERRgamma nuclear receptor homodimers involves a similar recognition mechanism. PLoS One. 2013;8:e67810. doi: 10.1371/journal.pone.0067810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson LC, Kuchenbecker KM, Schiller BJ, Gross JD, Pufall MA, Yamamoto KR. The glucocorticoid receptor dimer interface allosterically transmits sequence-specific DNA signals. Nat Struct Mol Biol. 2013;20:876–883. doi: 10.1038/nsmb.2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia G, Boerma LJ, Cox BD, Qiu C, Kang S, Smith CD, Renfrow MB, Muccio DD. Structure, energetics, and dynamics of binding coactivator peptide to the human retinoid X receptor alpha ligand binding domain complex with 9-cis-retinoic acid. Biochemistry. 2011;50:93–105. doi: 10.1021/bi101288y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Wu RC, O’Malley BW. Normal and cancer-related functions of the p160 steroid receptor co-activator (SRC) family. Nat Rev Cancer. 2009;9:615–630. doi: 10.1038/nrc2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan X, Broderick D, Leid ME, Schimerlik MI, Deinzer ML. Dynamics and ligand-induced solvent accessibility changes in human retinoid X receptor homodimer determined by hydrogen deuterium exchange and mass spectrometry. Biochemistry. 2004;43:909–917. doi: 10.1021/bi030183c. [DOI] [PubMed] [Google Scholar]
- Yang W, Rachez C, Freedman LP. Discrete roles for peroxisome proliferator-activated receptor gamma and retinoid X receptor in recruiting nuclear receptor coactivators. Mol Cell Biol. 2000;20:8008–8017. doi: 10.1128/mcb.20.21.8008-8017.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi P, Wang Z, Feng Q, Pintilie GD, Foulds CE, Lanz RB, Ludtke SJ, Schmid MF, Chiu W, O’Malley BW. Structure of a biologically active estrogen receptor-coactivator complex on DNA. Mol Cell. 2015;57:1047–1058. doi: 10.1016/j.molcel.2015.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Chalmers MJ, Stayrook KR, Burris LL, Wang Y, Busby SA, Pascal BD, Garcia-Ordonez RD, Bruning JB, Istrate MA, et al. DNA binding alters coactivator interaction surfaces of the intact VDR-RXR complex. Nat Struct Mol Biol. 2011;18:556–563. doi: 10.1038/nsmb.2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.