

Abstract
Mice devoid of T, B, and NK cells distinguish between self and allogeneic non-self despite the absence of an adaptive immune system. When challenged with an allograft they mount an innate response characterized by accumulation of mature, monocyte-derived dendritic cells (DCs) that produce IL-12 and initiate graft rejection. The molecular mechanisms, however, by which the innate immune system detects allogeneic non-self to generate these DCs are not known. To address this question, we studied the innate response of Rag2−/−γc−/− mice, which lack T, B, NK cells, to grafts from allogeneic donors. We identified by positional cloning that donor polymorphism in the gene encoding signal regulatory protein alpha (SIRPα) is a key modulator of the recipient’s innate allorecognition response. Donors that differed from the recipient in one or both Sirpa alleles elicited an innate alloresponse. The response was mediated by binding of donor SIRPα to recipient CD47 and was modulated by the strength of the SIRPα-CD47 interaction. Therefore, sensing SIRPα polymorphism by CD47 provides a molecular mechanism by which the innate immune system distinguishes between self and allogeneic non-self independently of T, B, and NK cells.
Introduction
Recognition of allogeneic non-self by the mammalian immune system has been attributed to adaptive lymphoid cells that express re-arranging receptors for antigen (T and B lymphocytes) or to NK cells that express non-rearranging receptors but are activated in response to “missing self” (1, 2). The principal alloantigens recognized by lymphocytes and NK cells are the polymorphic major histocompatibility complex (MHC) molecules widely expressed on bodily tissues. Transplantation of tissues between MHC-mismatched individuals therefore triggers a potent lymphoid cell response that causes graft rejection.
Recent studies have shown that innate myeloid cells, which have elaborate systems for sensing microbial non-self, also engage in allorecognition (3). Upon encountering non-self cells or tissues transplanted from another individual, monocytes cause a delayed-type hypersensitivity (DTH)-like reaction or differentiate into potent antigen-presenting dendritic cells (DCs) independently of T, B, and NK cells (4–6). In one model (5), heart allografts transplanted to Rag2−/−γc−/− mice, which lack all lymphoid cells, were persistently infiltrated with mature, monocyte-derived DCs (mono-DCs) that expressed IL-12 and stimulated both T cell proliferation and IFN-γ production (5). In contrast, syngeneic grafts in the same mice harbored a smaller number of mono-DCs transiently and these DCs neither expressed IL-12 nor stimulated IFN-γ production by T cells. Only T cells activated by mono-DCs that were generated in response to allogeneic non-self caused graft rejection (5). Furthermore, mono-DCs that accumulated in the graft directly contributed to rejection by propagating the local effector T cell response (7). Other studies have shown that macrophages also respond to allogeneic non-self. Macrophages from mice primed with allogeneic cells subsequently killed cells grafted from the same donor, although this allotoxic response depended on CD4+ T lymphocyte help at the time of priming (8). Therefore, non-self sensing by innate myeloid cells is not restricted to microbes but extends to the recognition of allografts.
How monocytes or macrophages distinguish between self and allogeneic non-self is not known. Earlier studies suggested that activation of monocytes by allogeneic grafts is not dependent on MHC mismatch between donor and recipient but rather on mismatches elsewhere in the genome (4–6). This raised the possibility that genetically-defined, non-MHC determinants on donor cells control the host’s innate alloresponse. Identifying these determinants would provide fundamental insights into innate immune recognition mechanisms central to transplant rejection and the maternal immune response to the allogeneic fetus. We therefore embarked on a genetic mapping (positional cloning) study in the mouse that exploited differences in the magnitude of the monocyte response elicited by allografts from congenic, inbred donors. We report here that a key determinant of the innate immune response to allogeneic non-self is donor polymorphism in the immunoglobulin superfamily (IgSF) membrane protein SIRPα that is sensed by CD47 on recipient monocytes.
Results
Magnitude of the host innate alloresponse is influenced by the genetic background of the donor
To explore the mechanisms by which the innate immune system senses allogeneic grafts, we tested the innate response of lymphoid cell-deficient mice to grafts from genetically disparate donors. Bone marrow plugs from six common laboratory [DBA/2, non-obese diabetes resistant (NOR), C3H, FVB, BALB/c, and non-obese diabetic (NOD)] and three wild-derived [Pahari, Watkins Star Line B (WSB), and CAST] inbred mouse strains were transplanted individually under the kidney capsules of separate C57BL/B6 (B6) Rag2−/−γc−/− (BRG) recipients. BRG mice are devoid of lymphoid cell lineages and lack known T, B, and NK cell-mediated allorecognition. Control BRG mice were transplanted with bone marrow plugs from syngeneic B6 donors that were wild-type at the Rag2 and γc loci. Plugs were removed one week after transplantation and the number of infiltrating host monocyte-derived DCs (mono-DCs) quantified by flow cytometry as a measure of an innate alloresponse (5) (Supplementary Fig. 1). As shown in Fig. 1A, allogeneic grafts harbored significantly greater numbers of host mono-DCs than did syngeneic B6 grafts, with the largest mono-DC infiltrate observed in NOD allografts. In contrast, mono-DC infiltration of syngeneic NOD grafts transplanted to NOD.Rag2−/−γc−/− (NRG) recipients was minimal (Fig. 1B), indicating that the robust mono-DC infiltrate observed in NOD allografts is a true measure of an innate alloresponse. We also tested the host response to allogeneic (NOR, BALB/c, or NOD) and syngeneic (B6) bone marrow plugs transplanted simultaneously in the contralateral kidneys of the same BRG recipient. NOR mice are a recombinant inbred strain that is 88% identical by descent to NOD, including at the Mhc locus (9, 10). Allogeneic BALB/c but not allogeneic NOR grafts elicited greater mono-DC infiltration than syngeneic B6 grafts (Fig. 1C). In addition, NOD grafts displayed a much larger mono-DC infiltrate than did either B6 or NOR grafts (Fig. 1C), confirming that NOD donor tissue induces a robust innate alloresponse. The difference between NOD and NOR allografts was also observed in BALB/c.Rag2−/−γc−/− (CRG) recipients (Fig. 1D). Therefore, varying the genetic background of the donor modulates the host innate alloresponse, with NOD allografts inducing the strongest response while allografts from the closely related NOR mouse inducing a weak response.
Fig. 1. Magnitude of the host innate alloresponse is influenced by the genetic background of the donor.

Bone marrow plugs from different donors were transplanted individually under the kidney capsules of separate mice (A), or simultaneously in the contralateral kidneys of the same mouse (B, C). All recipients were B6.Rag2−/−γc−/− (BRG) unless otherwise stated. Donor strains are shown on the x-axis. Number of recipient monocyte-derived DCs (Mono-DC) infiltrating the grafts was determined 1 wk later as a measure of an innate alloresponse. (A) Responses of BRG recipients to allografts from 6 common (DBA.2, NOR, C3H, FVB, BALB/c, & NOD) and 3 wild-derived (Pahari, WSB, and CAST) inbred strains. Statistical significance shown is relative to B6. (B) NOD grafts transplanted to syngeneic NOD.Rag2−/−γc−/− mice (NOD to NRG) elicit a much weaker response than NOD grafts transplanted to allogeneic BRG mice (NOD to BRG). (C, D) Innate responses elicited by grafts from distinct donors transplanted under the contralateral kidney capsules of the same BRG recipient (C) or BALB/c.Rag2−/−γc−/−(CRG) recipient (D). n = 5–6 mice/group/experiment. Experiments were performed once or twice. Each dot represents an individual biological replicate. Bars are means. *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001; ns = not significant (unpaired, two-tailed t-test). BRG = B6.Rag2−/−γc−/−; NRG = NOD.Rag2−/−γc−/−; CRG = BALB/c.Rag2−/−γc−/−.
Innate alloresponse is determined by a single Mendelian locus in the donor not linked to the Mhc
To determine whether donor variables that influence the magnitude of the innate alloresponse are heritable, we crossed B6 Rag2−/−γc−/− (BRG) and NOD Rag2−/−γc−/− (NRG) mice and studied the response induced by F1 and F2 grafts transplanted to BRG recipients. As shown in Fig. 2A, grafts from F1 progeny induced an intermediate response (they were infiltrated with approximately half the number of host mono-DCs as NRG grafts), while responses elicited by F2 grafts segregated at a ~1:2:1 (7:15:8) ratio into weak (equivalent to BRG grafts), intermediate (equivalent to F1 grafts), and strong (equivalent to NRG grafts), respectively. These outcomes are consistent with the inheritance of a single Mendelian locus at which alleles exhibit an additive effect. Since all mice used in these experiments were on the Rag2−/−γc−/− background, the results also confirmed our previous demonstration that the alloresponse is not dependent on lymphoid cells in either the donor or the recipient (5).
Fig. 2. Innate alloresponse is determined by a single Mendelian locus in the donor not linked to the Mhc.

Bone marrow plugs were transplanted individually under the kidney capsules of separate mice. All recipients were B6.Rag2−/−γc−/− (BRG) except in D where they were BALB/c.Rag2−/−γc−/− (CRG). Donor strains are shown on the x-axis. Recipient Mono-DCs in grafts were measured as in Fig. 1. (A) Responses of BRG recipients to grafts from parental NRG and BRG strains or to grafts from (BRGxNRG)F1 and F2 generations. All donors and recipients were on the Rag2−/−γc−/− background. (B) Effect of donor-recipient non-MHC mismatch (BALB.B grafts) or MHC mismatch (B6.C grafts) on the innate alloresponse of BRG recipients. (C) Effects of donor MHCI-deficiency (NOD.scid.b2m−/− grafts) on the innate alloresponse of BRG recipients. (D) Effect of MHCII-deficiency (B6.MHCII−/− grafts) on the innate alloresponse of CRG recipients. n = 5–6 mice/group/experiment, except in F2 experiment (n = 30 mice transplanted in 2 separate batches). Experiments were performed once or twice. Each dot represents an individual biological replicate. Bars are means. *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001; ns = not significant (unpaired, two-tailed t-test).
The fact that NOR and NOD grafts elicited distinctly different responses (Fig. 1), despite sharing the same Mhc locus (9, 10), suggested that the locus controlling the magnitude of the alloresponse is not linked to the Mhc. This is supported by experiments in which we transplanted bone marrow plugs from BALB.B (H-2b) donors into BRG (H-2b) recipients, a donor-host combination that is genetically identical at the Mhc but disparate at non-Mhc loci. BALB.B grafts indeed exhibited significant mono-DC infiltration (Fig. 2B), comparable in magnitude to BALB/c grafts (Fig. 1A,B). Conversely, grafts from B6.C mice, which carry a distinct Mhc haplotype (H-2d) but are identical at all other loci to BRG recipients, elicited a weaker response than did BALB.B grafts (Fig. 2B), comparable to that of B6 grafts (Fig. 1). Therefore, mismatches at non-Mhc loci between donor and recipient are necessary and sufficient for triggering the innate alloresponse. We also observed that the alloresponse was not diminished when bone marrow plugs were derived from MHC I (β2-microglobulin)-deficient (NOD.Prkdcscid.b2m−/−) mice (Fig. 2C) compared to grafts from similarly lymphoid-deficient but MHC I-sufficient NRG mice (Fig. 2A). Similarly, MHC II expression was not required on the allografts because B6.Mhc2−/− and parental B6 allografts had equivalent mono-DC infiltration in CRG recipients (Fig. 2D). Thus, a single non-Mhc locus in the donor controls the magnitude of the innate alloresponse, and neither MHC-disparity between donor and recipient nor expression of MHC I or MHC II molecules on graft are necessary for eliciting the response.
Innate alloresponse maps to a small genomic region in the donor containing the Sirpa gene
The conspicuous difference in mono-DC infiltration between NOD and NOR grafts (Fig 1) provided the opportunity for fine genetic mapping of the innate alloresponse locus in the donor because of the availability of well-characterized NOD.NOR congenic mouse lines. NOD and NOR congenic mouse strains share approximately 88% genetic identity but differ in their susceptibilities to autoimmune diabetes. Subcongenic lines generated by introgressing genetic intervals from NOR to NOD mice have enabled the mapping of important diabetes susceptibility and immune response traits in the NOD mouse (10–13). One example is the superiority of the immune deficient NOD.Prkdcscid mouse as host for human hematopoietic stem cells compared to equally immune deficient NOR or B6 recipients. Genetic control of this trait was shown to be due to polymorphism at the gene encoding SIRPα in the recipient (14, 15). SIRPα is an immunoglobulin superfamily (IgSF) receptor expressed on myeloid cells, neurons, and other cell types (16, 17). SIRPα engagement by its ubiquitously expressed monomorphic ligand CD47 delivers an inhibitory signal that represses phagocytosis by macrophages and inhibits multiple aspects of DC activation (18–20). In some contexts, CD47-SIRPα signaling is bidirectional as SIRPα binding to CD47 triggers stimulatory signals in T cells and neurons (12, 21, 22). We therefore took a genetic approach to test whether donor SIRPα polymorphism controls the MHC-independent alloreactions we had observed. We used a series of congenic NOD strains carrying different NOR-derived genomic intervals of chromosome 2 that did or did not contain Sirpa as bone marrow donors for BRG recipients (12, 14). Bone marrow plugs from NOD.NOR-Ila-D2Gul482 (NOD.NOR-R7, referred to as R7) donors, which are identical to the parental NOD strain except for a ~2Mb segment that includes NOR-Sirpa (Fig. 3A), elicited an alloresponse indistinguishable from parental NOR grafts (Fig. 3B). In contrast, bone marrow from NOD.NOR-D2Gul169-D2Gul289 (NOD.NOR-R12, referred to as R12) mice that differ from the parental NOD strain in a different genomic segment than NOD.NOR-R7 and express the NOD-derived SIRPα (Fig. 3A), phenocopied parental NOD donors (Fig. 3B). This result was confirmed by showing that NOD and NOD.NOR-R12 bone marrow plugs transplanted into contralateral kidneys of the same BRG recipient displayed equivalent levels of mono-DC infiltration (Fig. 3C). Finally, we transplanted grafts from a B6.Rag2−/−γc−/− donor strain that is congenic for the NOD-derived Sirpa allele (BRGS) (15) and observed that these bone marrow plugs stimulated an innate alloresponse equivalent to NRG donor tissue (Fig. 3D). Therefore, the capacity of donor tissue to induce a robust innate alloresponse characterized by mono-DC infiltration of the graft mapped to a very small region in the mouse genome that encodes the polymorphic Sirpa gene.
Fig. 3. Innate alloresponse maps to a small genomic region in the donor containing the Sirpa gene.

Bone marrow plugs were transplanted individually under the kidney capsules of separate mice (B, D), or simultaneously in the contralateral kidneys of the same mouse (C). All recipients were B6.Rag2−/−γc−/−(BRG). Donor strains are shown on the x-axis. Recipient Mono-DCs in grafts were measured as in Fig. 1. (A) Mouse chromosome 2 region that differs between NOD.NOR-R7 (R7) and NOD.NOR-R12 (R12) congenic strains and contains the Sirpa locus. (B) Comparison of innate alloresponses elicited by NOR or NOD grafts to those elicited by congenic donors that carry the NOR (R7) or NOD (R12) Sirpa allele. (C) Innate alloresponses elicited by grafts from NOD and R12 congenic mice transplanted into the same BRG recipient. (D) Grafts from BRG mice congenic for the NOD Sirpa allele (BRGS) induce the same alloresponse as grafts from NOD.Rag2−/−γc−/− (NRG) mice in BRG recipients. n = 6–7 mice/group/experiment. Except for the BRGS group, experiments were performed twice. Each dot represents an individual biological replicate. Bars are means. **p<0.01; ****p<0.0001; ns = not significant (unpaired, two-tailed t-test).
Donor SIRPα binding to recipient CD47 is required for triggering the innate alloresponse
To determine whether the binding of SIRPα on donor tissue to its ligand CD47 on recipient cells is necessary for eliciting the innate alloresponse, BRG recipients of NOD grafts were treated with human CD47-Fc (hCD47-Fc), a fusion protein that binds NOD SIRPα on graft cells with high affinity and prevents it from binding to its native ligand mouse CD47 (14). Compared to isotype control human Fc protein (hIgG1 Fc), hCD47-Fc treatment inhibited the accumulation of mono-DCs in NOD grafts (Fig. 4A). Moreover, BRG recipients carrying a targeted deficiency in Cd47 (BRG.Cd47−/−) (23) did not respond to either NOD or BALB/c grafts (Fig. 4B). These data establish that SIRPα expressed on donor cells activates the host’s innate alloresponse by engaging CD47.
Fig. 4. Donor SIRPα binding to recipient CD47 is required for triggering the innate alloresponse.

(A) NOD grafts were transplanted to B6.Rag2−/−γc−/− (BRG) mice that received either hCD47-Fc, a decoy protein that binds to the NOD SIRPα variant and prevents it binding to mouse CD47, or isotype control Fc protein (hIgG1 Fc). Recipient Mono-DCs in grafts were measured as in Fig. 1. Control, untreated recipients (None) from prior experiments are shown for comparison. (B) NOD or BALB/c grafts were transplanted to B6.Rag2−/−γc−/−CD47−/− mice (BRG CD47−/−) and the innate alloresponse was compared to that of CD47-sufficient (BRG) recipients from prior experiments. (C, D) Grafts from wildtype (B6 and BALB/c) or mutant (B6 mSIRP-A) mice, which lack the intracellular signaling domain of SIRPα, were transplanted to BALB/c.Rag2−/−γc−/− (CRG) (C) or BRG (D) recipients to test the effect of removing SIRPα signaling from donor cells on the host response. (E) Proportion of mature (MHCIIhiCD80+) recipient or donor mono-DCs in NOD.Rag2−/−γc−/− (NRG) allografts transplanted to BRG mice. (F) Absolute number of mature host mono-DC in allogeneic (NRG) vs syngeneic (BRG) grafts transplanted to BRG recipients. n = 6 mice/group/experiment. Experiments were performed once or twice. Each dot represents an individual biological replicate. Bars are means. *p<0.05; **p<0.01; ****p<0.0001; ns = not significant (unpaired, two-tailed t-test).
We considered the possible confounder that donor SIRPα binding to CD47 on recipient mono-DCs could inhibit the phagocytic function of donor myeloid cells in the bone marrow graft, indirectly influencing the number of infiltrating mono-DCs. To test this possibility, we transplanted bone marrow plugs from B6.Sirpatm1 mice with a targeted deletion of the cytoplasmic region of SIRPα required for signaling (24). Allogeneic B6.Sirpatm1 bone marrow plugs transplanted to CRG recipients accumulated the same number of host mono-DCs as parental B6 grafts that express a full-length SIRPα protein (Fig. 4C). This could not be explained by inherent hyperactivity of cells in B6.Sirpatm1 bone marrow grafts because increased host mono-DC accumulation was not observed when these grafts were transplanted to syngeneic BRG mice (Fig. 4D). Therefore, eliminating the intracellular signaling function of SIRPα in donor cells did not influence the magnitude of the host innate alloresponse. We also ruled out the possibility that increased host mono-DC accumulation was an indirect consequence of donor monocyte activation by host cells (that is, a graft versus host reaction). As a measure of monocyte activation, we analyzed the maturation state of donor and host mono-DCs in NRG grafts transplanted to BRG mice. We found that only 2% of donor mono-DC had acquired a mature (MHCIIhiCD80+) phenotype compared to 52% of recipient mono-DCs (Fig. 4E). Moreover, the number of mature donor mono-DCs in NRG grafts did not exceed that in syngeneic BRG grafts (Fig. 4F). Therefore, the host innate alloresponse to bone marrow grafts is a consequence of donor SIRPα binding to recipient CD47 and not the indirect result of a graft versus host reaction.
Mouse SIRPα amino acid polymorphism modulates binding to CD47
The results presented so far support a model in which polymorphism in the extracellular domains of donor SIRPα is recognized by host CD47 to identify cells as self or non-self. To test this model further, we investigated the extent and location of amino acid polymorphisms in mouse SIRPα by aligning the predicted SIRPα protein sequences from 19 different mouse strains whose genomes had been sequenced (http://www.sanger.ac.uk/science/data/mouse-genomes-project). We found that amino acid variability is largely restricted to the extracellular region and is most frequent in the N-terminal, CD47-binding IgV domain (Fig. 5A, alignment shown in Supplemental Fig. 2) (25–27). Phylogenetic analysis revealed four unique IgV domains among the 13 common inbred mouse strains examined, while each of the six wild-derived mouse strains had its own unique IgV sequence (Fig. 5B). The latter suggests the presence of a considerable number of mouse SIRPα alleles in the wild. We also observed that NOD and CAST strains, which elicited the strongest innate alloresponses in BRG mice (Fig. 1A), have closely related IgV domains while NOR, which elicited a very weak alloresponse, shared the same IgV domain with B6 recipients (Fig. 5B & Supplemental Fig. 2). Since amino acid polymorphism in SIRPα’s IgV domain influences binding to CD47 (12), we asked whether NOD and CAST SIRPα variants share greater binding to CD47 than SIRPα variants of mouse strains that induce weaker alloresponses. To answer this question, we compared the binding of mouse CD47-Fc fusion protein (mCD47-Fc) to splenic monocytes from NOD and CAST mice to those from B6, BALB/c, and C3H mice. Splenic monocytes constitutively express high levels of SIRPα (28). As shown in Fig. 5C, mCD47-Fc displayed greater binding to NOD and CAST cells than B6, BALB/c, and C3H cells at each concentration tested. SIRPα expression was comparable on monocytes from all strains tested (Fig. 5D), indicating that dissimilarities in binding were not due to differences in SIRPα cell surface density. The data therefore suggest that donor SIRPα IgV domain polymorphism controls the host innate alloresponse by modulating the binding of SIRPα to CD47.
Fig. 5. Mouse SIRPα amino acid polymorphism modulates binding to CD47.

(A) Amino acid (AA) variability in mouse SIRPα protein based on alignment of sequences from 19 mouse strains. Variability was calculated for each position along the sequence and aligned against the denoted SIRPα protein domains. IgV domain (shaded in red) had the highest frequency of AA polymorphisms (number of vertical, blue lines) and contained polymorphisms with the greatest degree of variability (height of vertical, blue lines). (B) Phylogram representation of SIRPα IgV domain AA variation among the 19 mouse strains. Blue font = common inbred strains; red font = wild-derived inbred strains; bold font = mouse strains tested in Fig. 1; scale = proportion of AA substitutions; circles = mouse strains that share similar or identical CD47 IgV domains. (C) Binding of mCD47-Fc to splenic monocytes (Lin−CD11b+CD11c−F4/80− cells) from NOD and CAST compared to B6, BALB/c, and C3H mice. Serial dilution of mCD47-Fc shown on top. (D) SIRPα expression on monocytes from all strains tested. Histograms are representative of 2–3 biological replicates from 2 independent experiments (unpaired, two-tailed t-test).
Donor SIRPα polymorphism modulates monocyte proliferation
To further investigate the role of SIRPα polymorphism in monocyte activation, we immunized BRG mice with splenocytes from donors expressing SIRPα variants with different CD47-binding affinities and analyzed the proliferation of myeloid cells in the spleen 7 days later. Mice were injected with EdU 1-hr prior to spleen harvest to label dividing cells in situ. As shown in Fig. 6A, significant EdU uptake was observed only in Ly6Chi monocytes (and to a much lesser extent in mono-DCs) in mice immunized with allogeneic, but not syngeneic, splenocytes. Importantly, the magnitude of monocyte proliferation correlated with strength of SIRPα binding to CD47. Splenocytes from NOD mice, known to express a high affinity variant of SIRPα, induced the greatest proliferation; NOR splenocytes, which share the same lower affinity SIRPα allele with B6, induced the least; and BALB/c splenocytes gave an intermediate result (Fig. 6B). Monocyte proliferation was dramatically reduced in CD47-deficient recipients stimulated with NOD cells (Fig. 6B). Together, these data provide additional evidence that differential binding of donor SIRPα to CD47 modulates monocyte activation.
Fig. 6. Donor SIRPα polymorphism modulates monocyte proliferation.

(A) B6.Rag2−/−γc−/− (BRG) mice were immunized i.p. with irradiated allogeneic (BALB/c) or syngeneic (B6) splenocytes. Spleen cells were analyzed 1 wk later. Mice were pulsed with EdU 1 hr prior to spleen harvest. Representative flow plots of EdU staining of myeloid cell populations are shown. Arrows indicate EdU+ cell population. (B) BRG or BRG CD47−/− mice were stimulated as in A with splenocytes from mouse strains shown on x-axis and the proportion of EdU+ cells in Ly6Chi monocyte subset was determined and divided by proportion of EdU+ cells in mice immunized with syngeneic (B6) splenocytes to determine the Proliferation Index. n = 3 mice/group/experiment x 2 experiments except for B6 group where total n = 9. Each dot represents an individual biological replicate. Bars are means. *p<0.05; ***p<0.001; ns = not significant (unpaired, two-tailed t-test).
Innate alloactivation is triggered by mismatch between donor and recipient SIRPα
Since SIRPα and CD47 are co-expressed on both donor and host cells, we tested the hypothesis that transplantation of allogeneic grafts carrying non-self SIRPa allele(s) causes innate immune activation by disturbing the balance between activating and inhibitory signals mediated by CD47 and SIRPα in host monocytes, respectively. This hypothesis would predict that under steady state conditions or upon transplantation of a syngeneic graft, bidirectional interactions between CD47 and self-SIRPα are of equal affinity and therefore maintain monocytes in a quiescent state (Fig. 7A, upper panel). The data shown in Fig. 7B are consistent with the prediction as they demonstrate that syngeneic grafts lacking CD47 elicit robust mono-DC infiltration, comparable to that triggered by allogeneic grafts. In the case of allogeneic transplantation, on the other hand, the introduction of donor cells which express a non-self SIRPα variant with greater binding to CD47 than recipient SIRPα would be expected to upset the balance and cause monocyte activation (Fig. 7A, lower panel). This is most evident in the case of NOD- and CAST-derived grafts, which express SIRPα variants with greater binding to CD47 than B6 SIRPα (Fig. 5C) and elicit robust innate alloresponses in B6 (BRG) mice (Fig. 1A). The hypothesis would also predict that performing allogeneic transplantation in the opposite direction, such that donor SIRPα has weaker binding to CD47 than recipient SIRPα, should suppress the innate alloresponse. This prediction is borne out by the results shown in Fig. 7C: mono-DC accumulation in BRG grafts transplanted to NRG recipients was significantly less than that in NRG grafts transplanted to BRG recipients. Therefore, the experimental findings presented in this manuscript support a model in which the innate immune system senses allogeneic non-self by integrating activating and inhibitory signals delivered by CD47 and SIRPα, respectively.
Fig. 7. Innate alloactivation is triggered by mismatch between donor and recipient SIRPα.
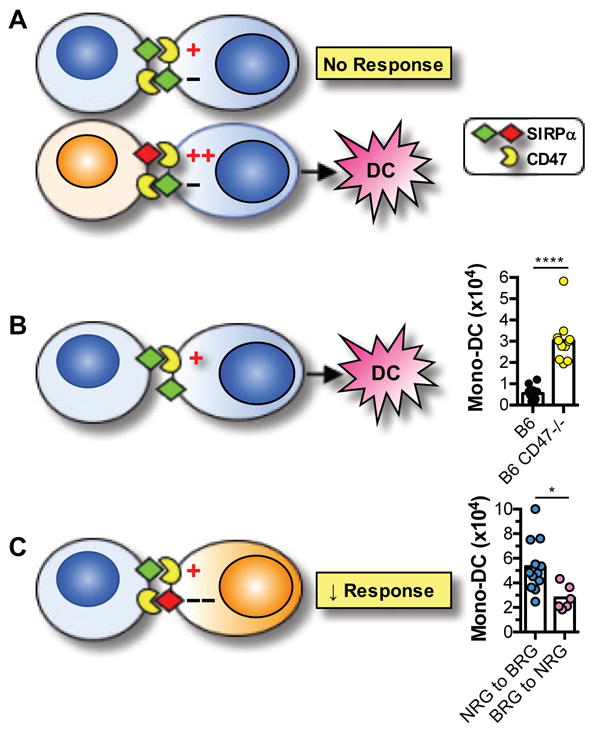
(A) Upper panel depicts balance between activating signals (+) mediated by CD47 and inhibitory signals (−) mediated by SIRPα in recipient monocytes in the syngeneic transplantation setting. Lower panel depicts the imbalance in the allogeneic setting when donor SIRPα (red) has greater affinity to CD47 than recipient SIRPα (green). Net result of this imbalance is recipient monocyte differentiation to mono-DC. (B) Imbalance is created if the syngeneic graft lacks CD47. Bar graph shows results from B6 CD47−/− and B6 wildtype grafts transplanted to separate B6.Rag2−/−γc−/− (BRG) recipients. Graft-infiltrating mono-DC were quantified as in Fig. 1. (C) Reversing direction of allotransplantation, such that donor SIRPα has weaker binding to CD47 than recipient SIRPα, inhibits the innate alloresponse. NRG = NOD.Rag2−/−γc−/−. Experimental data are shown in bar graph. n = 5–6 mice/group/experiment. Experiments were performed once or twice. ****p<0.0001; *p<0.05 (unpaired, two-tailed t-test).
Discussion
We have identified a mechanism by which the innate immune system distinguishes between self and allogeneic non-self that is distinct from allorecognition by T or NK cells. Unlike allorecognition by T cells, this innate mechanism is not linked to the Mhc and is not based on polymorphisms in both donor and recipient molecules. Instead, it is mediated by differential binding of a polymorphic ligand in the donor, SIRPα, to a monomorphic receptor in the recipient, CD47. The SIRPα-CD47 pathway resembles allorecognition by NK cells in that it relies on the integration of stimulatory and inhibitory signals by the responding cell (29), but differs from it in several aspects. First, monocytes utilize the same molecular pair (CD47 and SIRPα) that engage each other in opposite directions to deliver stimulatory and inhibitory signals, whereas NK cells rely on distinct sets of stimulatory and inhibitory ligand-receptor pairs. Second, monocytes sense allogeneic non-self through subtle variations in the binding of SIRPα to CD47, while NK cells sense missing self-MHC molecules. Third, unlike NK cell allorecognition, the monocyte allorecognition mechanism elucidated here is not dependent on MHC mismatch between the donor and recipient but instead on mismatch in a gene not linked to the MHC. Finally, allorecognition systems that pre-date the evolution of NK cells, T cells, and the MHC have been described in marine invertebrates (30). They too utilize polymorphic transmembrane proteins that contain Ig-like domains but have minimal homology to non-self recognition systems in vertebrates (31–33).
Inhibitory functions of SIRPα on myeloid cells have been well characterized (28, 34). SIRPα engagement by CD47 suppresses macrophage phagocytic function (18, 35) and DC maturation (20). CD47, in contrast, has stimulatory functions in immune cells. It provides costimulatory signals to T cells (21, 36) and enhances DC homeostasis and migration (37). Consistent with these functions, CD47−/− mice were refractory to the induction of autoimmune diseases; for example, experimental allergic encephalomyelitis (38), experimental colitis (39), and murine lupus (40). In addition, Wong et al. showed that enhanced binding of SIRPα to CD47 in the NOD mouse is a key determinant of the pathogenesis of autoimmune diabetes, likely due to the costimulatory actions of CD47 on T cells (12). Together, these data support our proposal that engagement of CD47 on recipient monocytes by SIRPα on graft cells provides an activation signal that causes monocyte proliferation and differentiation. A caveat to our study however is that it lacks direct proof that SIRPα polymorphism causes differential CD47 signaling in allostimulated monocytes. The intracellular signaling pathways triggered by CD47 in monocytes are not known and seem complex. Based on studies in non-myeloid cells, CD47 likely signals via Gi protein-dependent or independent pathways by associating with integrins in the cell membrane (41).
A role for CD47-SIRPα interactions in transplantation has been previously reported but only in the context of the long-held view that CD47 is a marker of self that, if altered or absent, triggers immune activation (28). Since CD47 is species-specific (42), xenografts activate host phagocytes because xenogeneic CD47 on the graft does not bind to host SIRPα (43). The phagocytic response however is significantly diminished if the graft is made to express host CD47 (44), or if the host carries a SIRPα variant that binds xenogeneic CD47 with high affinity. The latter is exemplified by the ease of acceptance of human stem cells by immune-deficient NOD mice because of the exceptional binding of NOD SIRPα to human CD47 (14, 15). CD47 expression on donor cells also plays a role in the alloimmune response. Wang et al. showed that the infusion of allogeneic spleen cells in mice results in the acceptance of a subsequent graft from the same donor but fails to do so if the spleen cells lacked CD47 due to activation of host DCs (45, 46). In contrast, our work has identified an alternate function of the CD47-SIRPα pathway whereby the polymorphic partner, SIRPα, serves as a marker of allogeneic non-self on donor tissues that is detected by CD47 on host monocytes.
Although we did not establish in this study a formal association between donor SIRPα polymorphism or SIRPα-CD47 interactions and allograft outcomes, several lines of evidence suggest that this pathway is important in both bone marrow and solid organ transplantation. For example, CD47−/− recipients were found to accept xenogeneic and allogeneic hematopoietic stem cells more readily than wildtype mice and to develop less graft versus host disease (GVHD) (23, 47). Conversely, CD47−/− donor cells failed to engraft because of increased phagocytosis by recipient macrophages (48). Similar to bone marrow grafts, heart allografts from NOD donors harbored significantly more mature mono-DCs than allografts from NOR mice (5). These DCs were particularly adept at driving type-1 T cell responses that drive allograft rejection (5, 7). Whether SIRPα polymorphism, which is also prevalent in the human population (14), modulates outcomes after bone marrow or solid organ transplantation remains to be seen. Another intriguing possibility is that SIRPα polymorphism may impact the maternal response to the allogeneic fetus or influence the pathogenesis of immune-mediated diseases of pregnancy. Therefore, exploring the role of SIRPα in natural (pregnancy) or artificial (transplantation) allogeneic encounters should yield important biological and clinical insights.
Materials and Methods
Study Design
We utilized a bone a marrow plug transplantation model to conduct a genetic mapping study. Six biological replicates (6 individual transplant recipients) per group were included in each experiment. Both sexes of mice were used. Experiments were repeated once in most instances resulting in a total of 12 biological replicates. Sample sizes were not based on power analysis but on our prior experience that 3–6 biological replicates are sufficient to discern statistically significant differences between groups using the same readout (number of infiltrating host mono-DC) (5). Sample size was not altered at any time during the course of the study. A biological replicate was excluded only if the mouse died before the bone marrow plug could be harvested on day 7 after transplantation. This exclusion criterion was established prospectively, it occurred very rarely, and we did not experience any technical problems in harvesting or analyzing the grafts that would have led to exclusion of a biological replicate. All data points were included and no outliers were excluded. All endpoints were prospectively selected. It was not possible to blind the study because of the need to identify donors and recipients and because in many instances the donor or recipient groups had different coat colors. The first investigator (HD or KIA) to analyze the flow cytometry data was not blinded, but randomly chosen subset of groups were re-analyzed blindly by either MHO or FGL.
Mice
C57BL/6J (B6), B6.SJL-Ptprca Pepcb/BoyJ (B6.CD45.1), BALB/cJ (BALB/c), BALB.B, B6.C-H2d/bByJ (B6.C), NOD/ShiLtJ (NOD), NOR/LtJ (NOR), DBA/2J (DBA), FVB/NJ (FVB), C3H/HeJ (C3H), Mus pahari/EiJ (Pahari), WSB-EiJ (WSB), CAST-EiJ (CAST), NOD.Cg-Rag1tm1Mom Il2rgtm1Wjl/SzJ (NOD.Rag2−/−γc−/− or NRG), C;129S4-Rag2tm1.1Flv Il2rgtm1.1Flv/J (BALB/c.Rag2−/−γc−/− or CRG), NOD.Cg-Prkdcscid B2mtm1Unc/J (NOD.Prkdcscid.b2m−/−), B6.129S-Rag2tm1Fwa Cd47tm1Fpl Il2rgtm1Wjl/J (B6. Rag2−/−γc−/−CD47−/−), and B6.129S2-H2dlAb1-Ea/J (B6.Mhc2−/−) mice were purchased from The Jackson Laboratory (JAX). B6-Rag2tm1Fwa II2rgtm1Wjl (B6.Rag2−/−γc−/− or BRG) were purchased from Taconic. B6.129P-Cx3cr1tm1Litt/J (B6 CX3CR1-eGFP CD45.2) (JAX) mice were bred on the Rag2−/−γc−/−background. C57BL/6.NOD-(D2Mit447-D2Mit338)Rag2−/−γc−/− (BRGS) congenic mice were a gift from Katsuto Takenaka and Koichi Akashi (Kyushu University, Fukuoka, Japan) (15). B6.Sirpatm1 mice, which lack the SIRPα intracytosolic domain, were provided by JSI and TM (24). All mice were maintained at the University of Pittsburgh animal facility under SPF conditions. All animal procedures were performed with approval of the IACUC at the University of Pittsburgh.
Surgical Procedures
Bone marrow plug transplantation was performed under the kidney capsule after isolating intact bone marrow plugs from donor femurs (49). Recipient mice were anesthetized and the kidney exposed via a small flank incision. A small incision was made in the kidney capsule, a pocket created with blunt forceps and a 4 mm bone marrow plug fragment was placed in the subcapsular pocket with vascular forceps.
Mouse Treatment
To block NOD SIRPα-CD47 interaction, mice received 250 μg recombinant human CD47 Fc chimera (hCD47-Fc) (R&D Systems) i.p. x4 every other day starting on day of transplantation. Control mice received equivalent dose of recombinant human IgG1 (hIgG1) Fc (R&D systems). Both reagents were provided in endotoxin-free PBS.
Mono-DC Analysis by Flow Cytometry
Bone marrow plug grafts were removed from the kidney capsule, homogenized using a GentleMACS tissue processor (Miltenyi), and digested at 37°C for 45 min in RPMI plus 10% FCS containing Collagenase IV (350 U/ml, Sigma-Aldrich) and DNAse I (20 ng/ml, Sigma-Aldrich). Leukocytes were isolated by gradient centrifugation using Lympholyte M (CedarLane Labs). Total recovered cells were counted using a hemocytometer before staining with antibodies. Fluorochrome- or biotin-tagged antibodies were purchased from BD Pharmingen, eBioscience, BioLegend or R&D Systems: CD90.2 (30-H12), CD45.1 (A20), CD45.2 (104), CD45R/B220 (RA3-6B2), CD49b (DX5), NK1.1 (PK136), F4/80 (BM8), CD11b (M1/70), CD11c (N418), Ly-6G (1A8), CD19 (1D3), MHCII (M5/114.15.2), CD80 (16-10A1), H2Kd (SF1-1.1.1), and H2kk (AF3-12.1.3). Fixable viability dye eFluor 506 was purchased from Affymetrix/eBiosciences. EdU uptake was analyzed using the Click-iT assay according to the manufacturer’s instructions (Thermo Fisher Scientific). Flow acquisition was performed on a LSR Fortessa flow cytometer (BD Biosciences), and data analyzed using Flowjo software (Treestar Corp.). Recipient and donor cells were distinguished using congenic markers (CD45.1/2), GFP expression, or H-2 expression (in case of BALB/c grafts).
CD47-SIRPα Binding Assay
Flow-based mouse CD47-Fc (mCD47-Fc) (R&D Systems) binding assay was performed as previously described with some modifications (12). Briefly, spleen cells from indicated mouse strains were blocked with anti-CD16/32 (BD Biosciences) followed by pre-clustering of SIRPα with unconjugated P84 antibody (eBiosciences). Cells were then incubated with serial dilution of mCD47-Fc for 30 minutes followed by secondary staining with anti-human IgG1-PE (R&D systems). Control cells were prepared in the same fashion except that hIgG1 Fc (R&D systems) was used instead of mCD47-Fc. Flow cytometry was performed to measure mCD47-Fc binding to monocyte subset (CD45+Lin−Ly6G−CD11b+CD11c−F480− cells). SIRPα expression was performed by staining cells with PE-conjugated P84 antibody (BD) under identical conditions as binding assay.
Mouse SIRPα Sequence Alignment
Whole mouse genomes were acquired from the Sanger Mouse Genome Project (http://www.sanger.ac.uk/science/data/mouse-genomes-project). A BLAST search was performed against the B6 Sirpa gene sequence to extract strain-specific Sirpa gene sequences from genomes of other strains. Gene sequences were translated to AA sequences and aligned to the B6 SIRPα sequence using CLC Genomics Workbench. Variability of amino acids along the SIRPα sequence was calculated as number of different residues/frequency of the most common residue - 1. This measure of variability was plotted against the length of the SIRPα amino acid sequence to identify the most variable regions. A phylogram of SIRPα IgV domain amino acid sequences was compiled in CLC Genomics Workbench to represent which strains were most similar or dissimilar from each other.
Statistical Analysis
All data points are shown in the graphs as scatterplots. Bars depict the means. GraphPad Prism 6 was used for statistical analyses. Statistical significance (p < 0.05) was calculated using the unpaired two-sided t-test and p-values are shown in the graphs. All comparisons found to be significant by t-test were also significant by the non-parametric Mann-Whitney test.
Supplementary Material
Fig. S1. Gating strategy for identifying recipient-derived, graft-infiltrating mono-DCs.
Fig. S2. Alignment of predicted SIRPα protein sequences from 19 mouse strains. Whole mouse genomes were acquired from the Sanger Mouse Genome Project. A BLAST search was performed against the B6 Sirpa sequence to extract strain-specific Sirpa sequences from multiple strains. Extracted sequences were translated to amino acid sequences and aligned to the B6 SIRPa sequence using CLC Genomics Workbench.
Supplementary Table 1. Raw Data File
Acknowledgments
The authors wish to thank Katsuto Takenaka and Koichi Akashi (Kyushu University Graduate School of Medical Sciences, Fukuoka, Japan) for providing femurs from BRGS mice, and Leo Buss (Yale University) for critical discussions. Femurs from BRGS and NOD.NOR congenic mice were provided from Kyushu University and University of Toronto, respectively, under material transfer agreements with the University of Pittsburgh.
Funding: This work was funded by NIH grant AI099465 and AI049466 (to FGL), the Frank & Athena Sarris Chair in Transplantation Biology (to FGL), Juvenile Diabetes Research Foundation, Genome Canada (administered through the Ontario Genomics Institute) (to JSD), the Canadian Institutes of Health Research (to JSD), and an American Heart Association grant 11SDG7230011 (to MHO). JSI was supported by grants 1R01HL112914 and 1R21EB017184 (NHLBI/NIH), the Institute for Transfusion Medicine, the Hemophilia Center of Western Pennsylvania and the Vascular Medicine Institute of the University of Pittsburgh School of Medicine.
Footnotes
Authors’ Contributions: HD, KIA, ALW, and SM-T performed experiments. AJF performed data analysis and participated in manuscript writing. HD and FGL carried out statistical analyses. MLT, DMR and WDS participated in research design and provided scientific advice. TM and JSI provided reagent (transgenic mouse) and scientific advice. MHO designed experiments, supervised and performed experiments and data analysis, and participated in manuscript writing. JSD and FGL conceived, designed and supervised project and wrote manuscript.
Competing Interests: JSI serves as chair of the scientific advisory boards of Tioma Therapeutics, Inc. (St. Louis, MO) and Radiation Control Technologies, Inc. (Jersey City, NJ).
References
- 1.Lakkis FG, Lechler RI. Origin and biology of the allogeneic response. Cold Spring Harbor perspectives in medicine. 2013;3 doi: 10.1101/cshperspect.a014993. pii: a014993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Karre K. Natural killer cell recognition of missing self. Nat Immunol. 2008;9:477–480. doi: 10.1038/ni0508-477. [DOI] [PubMed] [Google Scholar]
- 3.Oberbarnscheidt MH, Lakkis FG. Innate allorecognition. Immunol Rev. 2014;258:145–149. doi: 10.1111/imr.12153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zecher D, van Rooijen N, Rothstein D, Shlomchik W, Lakkis F. An innate response to allogeneic nonself mediated by monocytes. J Immunol. 2009;183:7810–7816. doi: 10.4049/jimmunol.0902194. [DOI] [PubMed] [Google Scholar]
- 5.Oberbarnscheidt MH, Zeng Q, Li Q, Dai H, Williams AL, Shlomchik WD, Rothstein DM, Lakkis FG. Non-self recognition by monocytes initiates allograft rejection. J Clin Invest. 2014;124:3579–3589. doi: 10.1172/JCI74370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chow KV, Delconte RB, Huntington ND, Tarlinton DM, Sutherland RM, Zhan Y, Lew AM. Innate Allorecognition Results in Rapid Accumulation of Monocyte-Derived Dendritic Cells. J Immunol. 2016;197:2000–2008. doi: 10.4049/jimmunol.1600181. [DOI] [PubMed] [Google Scholar]
- 7.Zhuang Q, Liu Q, Divito SJ, Zeng Q, Yatim KM, Hughes AD, Rojas-Canales DM, Nakao A, Shufesky WJ, Williams AL, Humar R, Hoffman RA, Shlomchik WD, Oberbarnscheidt MH, Lakkis FG, Morelli AE. Graft-infiltrating host dendritic cells play a key role in organ transplant rejection. Nature Communications. 2016;7 doi: 10.1038/ncomms12623. Article number: 12623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu W, Xiao X, Demirci G, Madsen J, Li XC. Innate NK cells and macrophages recognize and reject allogeneic nonself in vivo via different mechanisms. J Immunol. 2012;188:2703–2711. doi: 10.4049/jimmunol.1102997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prochazka M, Serreze DV, Frankel WN, Leiter EH. NOR/Lt mice: MHC-matched diabetes-resistant control strain for NOD mice. Diabetes. 1992;41:98–106. doi: 10.2337/diab.41.1.98. [DOI] [PubMed] [Google Scholar]
- 10.Serreze DV, Prochazka M, Reifsnyder PC, Bridgett MM, Leiter EH. Use of recombinant congenic and congenic strains of NOD mice to identify a new insulin-dependent diabetes resistance gene. J Exp Med. 1994;180:1553–1558. doi: 10.1084/jem.180.4.1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wicker LS, Chamberlain G, Hunter K, Rainbow D, Howlett S, Tiffen P, Clark J, Gonzalez-Munoz A, Cumiskey AM, Rosa RL, Howson JM, Smink LJ, Kingsnorth A, Lyons PA, Gregory S, Rogers J, Todd JA, Peterson LB. Fine mapping, gene content, comparative sequencing, and expression analyses support Ctla4 and Nramp1 as candidates for Idd5.1 and Idd5.2 in the nonobese diabetic mouse. J Immunol. 2004;173:164–173. doi: 10.4049/jimmunol.173.1.164. [DOI] [PubMed] [Google Scholar]
- 12.Wong AS, Mortin-Toth S, Sung M, Canty AJ, Gulban O, Greaves DR, Danska JS. Polymorphism in the innate immune receptor SIRPalpha controls CD47 binding and autoimmunity in the nonobese diabetic mouse. J Immunol. 2014;193:4833–4844. doi: 10.4049/jimmunol.1401984. [DOI] [PubMed] [Google Scholar]
- 13.Motta VN, Markle JG, Gulban O, Mortin-Toth S, Liao KC, Mogridge J, Steward CA, Danska JS. Identification of the inflammasome Nlrp1b as the candidate gene conferring diabetes risk at the Idd4.1 locus in the nonobese diabetic mouse. J Immunol. 2015;194:5663–5673. doi: 10.4049/jimmunol.1400913. [DOI] [PubMed] [Google Scholar]
- 14.Takenaka K, Prasolava TK, Wang JCY, Mortin-Toth SM, Khalouei S, Gan OI, Dick JE, Danska JS. Polymorphism in Sirpa modulates engraftment of human hematopoietic stem cells. Nat Immunol. 2007;8:1313–1323. doi: 10.1038/ni1527. [DOI] [PubMed] [Google Scholar]
- 15.Yamauchi T, Takenaka K, Urata S, Shima T, Kikushige Y, Tokuyama T, Iwamoto C, Nishihara M, Iwasaki H, Miyamoto T, Honma N, Nakao M, Matozaki T, Akashi K. Polymorphic Sirpa is the genetic determinant for NOD-based mouse lines to achieve efficient human cell engraftment. Blood. 2013;121:1316–1325. doi: 10.1182/blood-2012-06-440354. [DOI] [PubMed] [Google Scholar]
- 16.Barclay AN, Van den Berg TK. The interaction between signal regulatory protein alpha (SIRPalpha) and CD47: structure, function, and therapeutic target. Annu Rev Immunol. 2014;32:25–50. doi: 10.1146/annurev-immunol-032713-120142. [DOI] [PubMed] [Google Scholar]
- 17.Yao M, Rogers NM, Csanyi G, Rodriguez AI, Ross MA, St Croix C, Knupp H, Novelli EM, Thomson AW, Pagano PJ, Isenberg JS. Thrombospondin-1 activation of signal-regulatory protein-alpha stimulates reactive oxygen species production and promotes renal ischemia reperfusion injury. J Am Soc Nephrol. 2014;25:1171–1186. doi: 10.1681/ASN.2013040433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oldenborg PA, Zheleznyak A, Fang YF, Lagenaur CF, Gresham HD, Lindberg FP. Role of CD47 as a marker of self on red blood cells. Science. 2000;288:2051–2054. doi: 10.1126/science.288.5473.2051. [DOI] [PubMed] [Google Scholar]
- 19.Latour S, Tanaka H, Demeure C, Mateo V, Rubio M, Brown EJ, Maliszewski C, Lindberg FP, Oldenborg A, Ullrich A, Delespesse G, Sarfati M. Bidirectional negative regulation of human T and dendritic cells by CD47 and its cognate receptor signal-regulator protein-alpha: down-regulation of IL-12 responsiveness and inhibition of dendritic cell activation. J Immunol. 2001;167:2547–2554. doi: 10.4049/jimmunol.167.5.2547. [DOI] [PubMed] [Google Scholar]
- 20.Yi T, Li J, Chen H, Wu J, An J, Xu Y, Hu Y, Lowell CA, Cyster JG. Splenic Dendritic Cells Survey Red Blood Cells for Missing Self-CD47 to Trigger Adaptive Immune Responses. Immunity. 2015;43:764–775. doi: 10.1016/j.immuni.2015.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seiffert M, Brossart P, Cant C, Cella M, Colonna M, Brugger W, Kanz L, Ullrich A, Buhring HJ. Signal-regulatory protein alpha (SIRPalpha) but not SIRPbeta is involved in T-cell activation, binds to CD47 with high affinity, and is expressed on immature CD34(+)CD38(−) hematopoietic cells. Blood. 2001;97:2741–2749. doi: 10.1182/blood.v97.9.2741. [DOI] [PubMed] [Google Scholar]
- 22.Toth AB, Terauchi A, Zhang LY, Johnson-Venkatesh EM, Larsen DJ, Sutton MA, Umemori H. Synapse maturation by activity-dependent ectodomain shedding of SIRPalpha. Nature neuroscience. 2013;16:1417–1425. doi: 10.1038/nn.3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lavender KJ, Pang WW, Messer RJ, Duley AK, Race B, Phillips K, Scott D, Peterson KE, Chan CK, Dittmer U, Dudek T, Allen TM, Weissman IL, Hasenkrug KJ. BLT-humanized C57BL/6 Rag2−/−gammac−/−CD47−/− mice are resistant to GVHD and develop B- and T-cell immunity to HIV infection. Blood. 2013;122:4013–4020. doi: 10.1182/blood-2013-06-506949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Inagaki K, Yamao T, Noguchi T, Matozaki T, Fukunaga K, Takada T, Hosooka T, Akira S, Kasuga M. SHPS-1 regulates integrin-mediated cytoskeletal reorganization and cell motility. EMBO J. 2000;19:6721–6731. doi: 10.1093/emboj/19.24.6721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu Y, O’Connor MB, Mandell KJ, Zen K, Ullrich A, Buhring HJ, Parkos CA. Peptide-mediated inhibition of neutrophil transmigration by blocking CD47 interactions with signal regulatory protein alpha. J Immunol. 2004;172:2578–2585. doi: 10.4049/jimmunol.172.4.2578. [DOI] [PubMed] [Google Scholar]
- 26.Nakaishi A, Hirose M, Yoshimura M, Oneyama C, Saito K, Kuki N, Matsuda M, Honma N, Ohnishi H, Matozaki T, Okada M, Nakagawa A. Structural insight into the specific interaction between murine SHPS-1/SIRP alpha and its ligand CD47. J Mol Biol. 2008;375:650–660. doi: 10.1016/j.jmb.2007.10.085. [DOI] [PubMed] [Google Scholar]
- 27.Hatherley D, Graham SC, Turner J, Harlos K, Stuart DI, Barclay AN. Paired receptor specificity explained by structures of signal regulatory proteins alone and complexed with CD47. Mol Cell. 2008;31:266–277. doi: 10.1016/j.molcel.2008.05.026. [DOI] [PubMed] [Google Scholar]
- 28.Barclay AN, van den Berg TK. The interaction between signal regulatory protein alpha (SIRPa) and CD47: structure, function, and therapeutic target. Annu Rev Immunol. 2014;32:25–50. doi: 10.1146/annurev-immunol-032713-120142. [DOI] [PubMed] [Google Scholar]
- 29.Lanier LL. NK cell recognition. Annu Rev Immunol. 2005;23:225–274. doi: 10.1146/annurev.immunol.23.021704.115526. [DOI] [PubMed] [Google Scholar]
- 30.Rosengarten RD, Nicotra ML. Model systems of invertebrate allorecognition. Curr Biol. 2011;21:R82–92. doi: 10.1016/j.cub.2010.11.061. [DOI] [PubMed] [Google Scholar]
- 31.Nicotra ML, Powell AE, Rosengarten RD, Moreno M, Grimwood J, Lakkis FG, Dellaporta SL, Buss LW. A hypervariable invertebrate allodeterminant. Curr Biol. 2009;19:583–589. doi: 10.1016/j.cub.2009.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rosa SF, Powell AE, Rosengarten RD, Nicotra ML, Moreno MA, Grimwood J, Lakkis FG, Dellaporta SL, Buss LW. Hydractinia allodeterminant alr1 resides in an immunoglobulin superfamily-like gene complex. Curr Biol. 2010;20:1122–1127. doi: 10.1016/j.cub.2010.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Voskoboynik A, Newman AM, Corey DM, Sahoo D, Pushkarev D, Neff NF, Passarelli B, Koh W, Ishizuka KJ, Palmeri KJ, Dimov IK, Keasar C, Fan HC, Mantalas GL, Sinha R, Penland L, Quake SR, Weissman IL. Identification of a colonial chordate histocompatibility gene. Science. 2013;341:384–387. doi: 10.1126/science.1238036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Murata Y, Kotani T, Ohnishi H, Matozaki T. The CD47-SIRPalpha signalling system: its physiological roles and therapeutic application. J Biochem. 2014;155:335–344. doi: 10.1093/jb/mvu017. [DOI] [PubMed] [Google Scholar]
- 35.Jaiswal S, Jamieson CH, Pang WW, Park CY, Chao MP, Majeti R, Traver D, van Rooijen N, Weissman IL. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell. 2009;138:271–285. doi: 10.1016/j.cell.2009.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reinhold MI, Lindberg FP, Kersh GJ, Allen PM, Brown EJ. Costimulation of T cell activation by integrin-associated protein (CD47) is an adhesion-dependent, CD28-independent signaling pathway. J Exp Med. 1997;185:1–11. doi: 10.1084/jem.185.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hagnerud S, Manna PP, Cella M, Stenberg A, Frazier WA, Colonna M, Oldenborg PA. Deficit of CD47 results in a defect of marginal zone dendritic cells, blunted immune response to particulate antigen and impairment of skin dendritic cell migration. J Immunol. 2006;176:5772–5778. doi: 10.4049/jimmunol.176.10.5772. [DOI] [PubMed] [Google Scholar]
- 38.Han MH, Lundgren DH, Jaiswal S, Chao M, Graham KL, Garris CS, Axtell RC, Ho PP, Lock CB, Woodard JI, Brownell SE, Zoudilova M, Hunt JF, Baranzini SE, Butcher EC, Raine CS, Sobel RA, Han DK, Weissman I, Steinman L. Janus-like opposing roles of CD47 in autoimmune brain inflammation in humans and mice. J Exp Med. 2012;209:1325–1334. doi: 10.1084/jem.20101974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fortin G, Raymond M, Van VQ, Rubio M, Gautier P, Sarfati M, Franchimont D. A role for CD47 in the development of experimental colitis mediated by SIRPalpha+CD103- dendritic cells. J Exp Med. 2009;206:1995–2011. doi: 10.1084/jem.20082805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shi L, Bian Z, Chen CX, Guo YN, Lv Z, Zeng C, Liu Z, Zen K, Liu Y. CD47 deficiency ameliorates autoimmune nephritis in Fas(lpr) mice by suppressing IgG autoantibody production. J Pathol. 2015;237:285–295. doi: 10.1002/path.4574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brown EJ, Frazier WA. Integrin-associated protein (CD47) and its ligands. Trends Cell Biol. 2001;11:130–135. doi: 10.1016/s0962-8924(00)01906-1. [DOI] [PubMed] [Google Scholar]
- 42.Subramanian S, Boder ET, Discher DE. Phylogenetic divergence of CD47 interactions with human signal regulatory protein alpha reveals locus of species specificity. Implications for the binding site. J Biol Chem. 2007;282:1805–1818. doi: 10.1074/jbc.M603923200. [DOI] [PubMed] [Google Scholar]
- 43.Ide K, Wang H, Tahara H, Liu J, Wang X, Asahara T, Sykes M, Yang Y-G, Ohdan H. Role for CD47-SIRPalpha signaling in xenograft rejection by macrophages. Proc Natl Acad Sci USA. 2007;104:5062–5066. doi: 10.1073/pnas.0609661104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tena A, Kurtz J, Leonard DA, Dobrinsky JR, Terlouw SL, Mtango N, Verstegen J, Germana S, Mallard C, Arn JS, Sachs DH, Hawley RJ. Transgenic expression of human CD47 markedly increases engraftment in a murine model of pig-to-human hematopoietic cell transplantation. Am J Transplant. 2014;14:2713–2722. doi: 10.1111/ajt.12918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang H, Wu X, Wang Y, Oldenborg PA, Yang YG. CD47 is required for suppression of allograft rejection by donor-specific transfusion. J Immunol. 2010;184:3401–3407. doi: 10.4049/jimmunol.0901550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang Y, Wang H, Bronson R, Fu Y, Yang YG. Rapid dendritic cell activation and resistance to allotolerance induction in anti-CD154-treated mice receiving CD47-deficient donor-specific transfusion. Cell Transplant. 2014;23:355–363. doi: 10.3727/096368912X661346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chhabra A, Ring AM, Weiskopf K, Schnorr PJ, Gordon S, Le AC, Kwon HS, Ring NG, Volkmer J, Ho PY, Tseng S, Weissman IL, Shizuru JA. Hematopoietic stem cell transplantation in immunocompetent hosts without radiation or chemotherapy. Sci Transl Med. 2016;8:351ra105. doi: 10.1126/scitranslmed.aae0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Blazar BR, Lindberg FP, Ingulli E, Panoskaltsis-Mortari A, Oldenborg PA, Iizuka K, Yokoyama WM, Taylor PA. CD47 (integrin-associated protein) engagement of dendritic cell and macrophage counterreceptors is required to prevent the clearance of donor lymphohematopoietic cells. J Exp Med. 2001;194:541–549. doi: 10.1084/jem.194.4.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Varas F, Grande T, Ramirez A, Bueren JA. Implantation of bone marrow beneath the kidney capsule results in transfer not only of functional stroma but also of hematopoietic repopulating cells. Blood. 2000;96:2307–2309. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Gating strategy for identifying recipient-derived, graft-infiltrating mono-DCs.
Fig. S2. Alignment of predicted SIRPα protein sequences from 19 mouse strains. Whole mouse genomes were acquired from the Sanger Mouse Genome Project. A BLAST search was performed against the B6 Sirpa sequence to extract strain-specific Sirpa sequences from multiple strains. Extracted sequences were translated to amino acid sequences and aligned to the B6 SIRPa sequence using CLC Genomics Workbench.
Supplementary Table 1. Raw Data File