

Abstract
Normal tissue injury from irradiation is an unfortunate consequence of radiotherapy. Technologic improvements have reduced the risk of normal tissue injury, however toxicity causing treatment breaks or long term side effects continue to occur in a subset of patients. The molecular events that lead to normal tissue injury are complex and span a variety of biologic processes, including oxidative stress, inflammation, depletion of injured cells, senescence, and elaboration of pro-inflammatory and pro-fibrogenic cytokines. This manuscript describes selected recent advances in normal tissue radiobiology.
Introduction
Normal tissue injury from irradiation is a key consideration for treatment of any condition with radiotherapy. The doses chosen for therapy are generally a compromise between the dose that effectively sterilizes cancer in the majority of patients and the dose that causes complications in the smallest subset. Although highly conformal therapies such as intensity modulated radiation therapy (IMRT) and stereotactic body radiation therapy combined with image guided treatments have made substantial progress in reducing the exposure of normal tissues to the prescription dose, toxicity continues to occur, albeit at a lower frequency. Understanding the molecular events responsible for the perpetuation of normal tissue injury is critical to developing methods to prevent, mitigate, and treat these toxicities.
Ionizing radiation results in the immediate generation of highly reactive free radicals with resulting rapid protein modifications and damage to DNA, RNA, and cell membranes1. Although a substantial amount of this damage may be repaired over time, it is increasingly recognized that these early events can stimulate changes in cellular function and inflammation that can persist over months to years2. Recent discoveries about the molecular events that drive normal tissue injury and the appreciation of the importance of tumor in this response have complemented the traditional radiobiological understanding of normal tissue injury. As it is impossible to review all of the potential mechanisms of radiation injury, this manuscript will summarize some of the recent advances in this context and highlight potential areas of opportunity for further preclinical and clinical exploration.
Oxidative Stress
Oxidative stress has long been thought to contribute to both acute and chronic radiation injury. Following the primary events of radiation exposure (hydrolysis of water and direct ionization), secondary reactions result in chronic elevation of reactive oxygen species (ROS), which are formed and propagated for extended periods of time3. In addition, ROS can be generated by a host of cell types in the setting of injury and inflammation, which can contribute to ongoing oxidative injury though generation of reactive nitrogen species (RNS)4. For example, injured endothelial cells, epithelial cells, and inflammatory cells can produce compounds such as superoxide and nitric oxide5, 6, which may further perpetuate local oxidative stress, resulting in ongoing injury after radiation exposure. Many attempts at minimizing injury from irradiation have focused on reducing oxidative stress by inhibiting the production of free radicals and delivering compounds capable of scavenging deleterious free radicals. Collectively, these studies have contributed greatly to our understanding of the mechanisms of normal tissue injury from irradiation.
One such radical, superoxide, is extremely toxic and is generated by NADPH oxidase in the process of normal metabolism, in the setting of immune response to pathogens, and as a consequence of injury, such as irradiation7. Superoxide can react directly with DNA or other cellular components to induce damage or can generate other harmful secondary radical species8. A large literature exists that has demonstrated delivery to or elaboration of superoxide dismutase (SOD), a detoxifying enzyme, in irradiated cells or tissues can reduce the deleterious effects of irradiation. Indeed, delivery of SOD and SOD modified to enhance delivery and stability have been successful in reducing the toxicity of radiation in clinical trials9, 10. Unfortunately, some studies have also failed to show a benefit and have reported allergic reactions11. Based on extensive preclinical data supporting SOD gene therapy as a potential therapeutic to prevent radiation injury12, alternative methods of SOD delivery, such as an SOD plasmid liposome, have successfully been delivered in Phase I trials13. The difficulty in effective and efficient delivery of SOD to irradiated tissue has spurred the development of alternative agents that can act similarly, also known as SOD mimetics. One concern with this approach is the possibility of tumor protection if the agent is not highly selective to normal tissue compared to tumor.
Indeed, delivery of agents capable of scavenging superoxide and ROS, such as genistein, AEOL 10150, EUK-189, and EUK-207, are capable of reducing tissue markers of oxidative stress and minimizing inflammation and radiation injury14–20. In general, these scavenging agents tend to reduce not only markers of oxidative stress in irradiated tissue, they also reduce inflammatory cell infiltration and pro-inflammatory, pro-fibrotic, and immunomodulatory cytokine expression14. In some cases, these global anti-inflammatory effects are capable of reducing what appears to be secondary injury, consistent with the hypothesis that ongoing inflammation can perpetuate continued injury in irradiated tissue14.
Although the delivery of mimetics is a promising strategy, the identification of redox sensors which can activate transcription of a wide range of genes important in the cellular response to oxidative stress has opened new potential opportunities for understanding radiation normal tissue injury and mitigating the deleterious effects of radiation. One such pathway is the Nrf2-ARE pathway. Nuclear factor erythroid 2-related factor 2 (Nrf2) is a transcription factor that regulates the expression of at least 200 genes, many of which are oxidative stress related proteins, including NADPH quinone oxidoreductase 121, hemoxygenase-1 (HO-1), SOD, and glutathione S-transferase proteins22–25. Loss or constitutive expression of Nrf2 can both be deleterious, suggesting that expression of Nrf2 must be balanced appropriately to the to the physiologic state26, 27.
Keap1 is a protein that binds to Nrf2 and facilitates its ubiquitination and proteolysis28. Interestingly, mutations in Keap1 and Nrf2 are frequently found in cancers and are associated with local recurrence after radiation and cellular radiation resistance29, 30. In the setting of radiation, NADPH oxidase activity is increased in lung tissue6, which is known to enhance, and in some settings be required for, Nrf2 expression31, 32, presumably reducing lung injury. Studies in Nrf2 deficient mice have demonstrated a substantial increase in late lung injury after localized irradiation33, 34. This injury is associated with an enhanced level of isoleuvoglandin (IsoLG) modification of proteins, an event that can alter protein function35. Interestingly, radiation also reduces Nrf2 expression in lung36, potentially further increasing the amount of IsoLG adducts. Recently published work has demonstrated that this enhanced IsoLG modification can result in enhanced apoptosis of pulmonary cells and can render collagen resistant to MMP-1 mediated degradation35. These findings not only provide a novel potential biomarker of injury, but also fundamentally alter our understanding of how redox sensitive pathways can cause perpetuation of injury long after the radiation exposure. Further, they provide an interesting rationale for the challenge in reversing established chronic radiation injury.
Metabolism
Metabolism is increasingly recognized as an important aspect of tissue homeostasis, which can be altered in the setting of malignancy. In general, tumor cells are metabolically heterogeneous and often times adaptable in their capacity to utilize various substrates as energy and carbon sources37, often utilizing anaerobic metabolism preferentially. Recently, it has become increasingly apparent that diseased tissue may also have metabolic derangements, partially due to hypoxia, inflammation, and the resulting activation of numerous pathways as a result that may also impact metabolism, such as HIF-1α and mTOR38. Indeed, hypoxia and oxidative stress in injured normal tissue after irradiation corresponds to enhanced HIF-1α signaling, which in turn parallels the induction of other known drivers of injury, such as TGFβ1 and VEGF39, 40. The presence of hypoxia in irradiated tissues has been confirmed in studies of irradiated human tissue that demonstrated morphologic evidence of radiation injury41.
Importantly, metabolism cannot be entirely separated from oxidative stress and hypoxia, as these three processes contribute to each other in many ways. For example, assessment of tetrahydrobiopterin (BH4), a non-enzymatic redox metabolite, has revealed reduced availability of BH4 after potentially damaging radiation exposures42. BH4 deficiency is thought to contribute to higher superoxide generation due to lack of BH4 complex formation with endothelial nitric oxide synthase43. As a result, tissue is subjected to enhanced oxidative stress and secondary formation of peroxynitrite44. This work demonstrates that metabolites may provide a window for mechanistic understanding of alterations in normal tissue and a potential marker of injury45.
The development of novel non-invasive methods to assess metabolism in living organisms has provided evidence that radiation can alter the metabolism of normal tissues. Magnetic resonance spectroscopy of hyperpolarized 13C pyruvate allows the non-invasive imaging of the fermentation of pyruvate to lactate. Using this approach, Thind et al46 demonstrated increased conversion of pyruvate to lactate in lung tissue irradiated to a fibrosis evoking dose compared to adjacent unirradiated normal tissue, consistent with altered metabolism as a consequence of injury.
Although metabolism plays a critical role in cancer and injury, it will be challenging to dissect the impact of metabolism when agents with multiple targets, including metabolism, are used to modify injury after irradiation. For example, mTOR, Akt, and other main signal transduction hubs play a large role in altering metabolism, but they may also alter a tremendous number of biological processes, presumably independent of metabolism. Assessment of specific inhibitors of metabolism currently being developed for cancer therapy in this context may provide interesting biologic information about metabolism in radiation injury and unique challenges and opportunities for intervention.
TGF-β and TNF-α signaling in normal tissue toxicity
TGF-β is a cytokine with a wide array of functions that is generally secreted by cells in a latent form. Activation of the latent peptide occurs after various stimuli, including ROS exposure47 and the actions of proteinases48, results in liberation of the active TGF-β protein and binding to one of several TGF-β receptors. Signaling through the TGF-β1 receptor is perpetuated by the Smad family of signaling proteins and transcription factors, eventually resulting in transcription and repression of an array of genes involved in cell proliferation, epithelial-mesenchymal transition, immune suppression, and inflammation49. Additional Smad independent TGF-β signaling pathways also exist, such as signaling via the RHO, ERK, JNK, and p38 MAP pathway49.
Radiation induces a dose and time dependent increase in TGF-β activity in tissue, with increased TGF-β expression detected within minutes to hours after irradiation50. Although TGF-β expression can return to baseline after this initial increase, it often rises again in irradiated tissue in the setting of chronic injury50. TGF-β is known to play a central role in a number of pathologic states characterized by tissue remodeling and inflammation, such as radiation injury. Indeed, data from human studies have suggested that plasma TGF-β is increased in settings associated with tissue injury, such as after thoracic radiotherapy. In this situation, plasma TGFβ concentrations may identify patients at low risk of radiation pneumonitis and late lung injury51–55, although controversy exists about whether an increase in TGF-β concentrations is an independent predictor of risk of lung injury56. Regardless, these data, a wealth of preclinical data, and other clinical series strongly support the importance of TGF-β in clinical radiation injury.
Agents targeting TGF-β signaling via targeting the TGF-β1 receptor or downstream intermediates have shown efficacy in reducing both acute and chronic radiation injury in preclinical models57–60. For late injury, the greatest degree of mitigation was observed with chronic dosing beginning immediately after exposure57. Similarly, delivery of halofuginone, a compound capable of inhibiting TGF-β1 receptor signaling, is capable of preventing radiation induced fibrosis of skin61 and lung62 while simultaneously enhancing tumor response to irradiation63. Although preclinical studies of TGF-β signaling inhibition have been promising, clinical translation in non-oncologic settings has been limited by concern for toxicity with chronic systemic administration of these inhibitors. These concerns are based on observation of inflammation, autoimmunity, and developmental defects in knockout mice64, 65 and additional toxicities observed in preclinical studies66, 67. Although these effects have not been seen in clinical trials of such agents, development of reversible cutaneous squamous cell carcinomas and keratoacanthomas has been observed68. The eventual importance of these agents in managing radiation injury remains uncertain given that available preclinical data suggests chronic dosing may be necessary for optimal effect.
Tumor necrosis factor alpha (TNF-α) is an inflammatory cytokine involved in the acute phase reaction, that is rapidly and persistently expressed in irradiated and adjacent tissue69. The delivery of antioxidants can, in some cases, prevent TNF-α elaboration and reduce radiation injury70. Deficiency of TNF-α in a lung injury model was sufficient to prevent symptoms of radiation pneumonitis70. TNF-α has also been implicated in radiation mucositis, enteritis, and dermatitis71–75. The clinical availability of agents targeting TNF-α to treat rheumatologic disorders provides a unique opportunity for clinical translation, and will likely enhance investigation into this molecule and its pathway.
Senescence in Radiation Injury
Another recent area of intense interest in radiation normal tissue injury is senescence of normal tissue stem cells. Senescence is a state of permanent growth arrest that occurs in several contexts, including as a normal part of development and aging. Replicative senescence occurs as the telomeres of dividing cells reach a limiting length after successive division, preventing further replication. In contrast, stress induced cellular senescence occurs after exposure to oxidative stress or DNA damaging stimuli, such as radiation. Recent evidence supports the concept that senescence plays an important role in radiation-induced normal tissue injury6, 76. Although many cell types appear to be susceptible to senescence after irradiation, the senescence that occurs in normal tissue stem cells may be particularly harmful due to the inability of the senescent stem cells to replace lethally damaged parenchymal cells after irradiation6, 76.
Although parenchymal depletion due to reduced regenerative capacity clearly plays an important role in the deleterious effects of senescence in normal tissue after irradiation, senescent cells are not merely inert bystanders6. A complex mixture of pro-inflammatory/immunomodulatory, pro-angiogenic, and mitogenic molecules are secreted by senescent cells, commonly referred to as the secretory profile of senescence (SASP)77. Importantly, several molecules of the SASP have been associated with normal tissue injury after irradiation, including IL-1, IL-6, TGFβ, EGF, VEGF, and TNF-α. Thus, senescent cells may further enhance or perpetuate normal tissue injury through the elaboration of these molecules with resulting paracrine effects such as inflammation, tissue remodeling, and secondary senescence and cell death.
Senescence of normal tissue stem cells may play a role in both the acute and late normal tissue responses to irradiation and have been identified in the intestine, skin, lung, kidney, bone marrow, mucosa, and many other organs76, 78, 79. At an organism level, accelerated aging and frailty has been appreciated as a consequence of cytotoxic therapies, such as irradiation, in preclinical studies and in patients76, 80–82. Although a number of agents have been identified that are capable of preventing senescence and preventing normal tissue injury, particularly in the setting of fibrosis6, 83, there are relatively few studies demonstrating that clearance of established senescence can reverse injury (treatment) or ameliorate progression (mitigation)76.
Macrophage Polarization and Radiation Injury
The field of immunology has developed at an explosive pace over recent years, and a growing appreciation of the immunologic effects of irradiation has been driven by the use of irradiation as an immune adjuvant in the context of cancer immunotherapy. Large volume irradiation has the greatest capacity for immunosuppression due to marrow suppression and a reduction of circulating or resident immune cells. In contrast, localized irradiation can induce a sterile inflammation (non-infectious) that is chronic in nature and can result in both acute and late toxicities.
Investigations of immunologic phenomenon and their contribution to normal tissue toxicity have focused on many organs, but perhaps most thoroughly in models of lung injury. For many years, it has been known that radiation drives an inflammatory response in the lung which can remain asymptomatic or progress to radiation pneumonitis and eventual radiation fibrosis. Indeed, radiation injury of the lung is generally treated with immunosuppressive glucocorticoids84. Although it is accepted that radiation lung injury is characterized by inflammatory infiltration, a more complete understanding of the complexities of the post-irradiation inflammatory state has been accumulating as interest in the intersection of radiation and immunologic phenomenon has expanded exponentially.
One of the major components of the accumulated inflammatory infiltration after irradiation in lung and other organs are macrophages85–88. Macrophages are known to play a key role in wound healing and repair, however they may also contribute to pathologic responses such as fibrosis89. Macrophages exhibit substantial plasticity, with the capacity to polarize into a state of activation and alternative activation depending on the surrounding microenvironment90, with resulting phenotypic and functional changes. The polarization of macrophages exists along a spectrum, with varying nomenclatures, however frequently is defined as classically activated (M1) and alternatively activated (M2). Alternatively activated macrophages, also known as regulatory or suppressive macrophages, can be generated after exposure to type 2 cytokines, such as IL-4 and IL-13. These macrophages can secrete IL-10 and act in an immunoregulatory, and in some cases, suppressive fashion. An additional subset of macrophages, termed profibrotic macrophages, that have been exposed to IL-4 and IL-13 secrete TGFβ and mitogenic factors89.
The relative numbers of phenotypically distinct macrophage subsets vary between fibrosis prone and fibrosis resistant strains of mice after irradiation86, with a notable accumulation of macrophages expressing markers of alternative activation, implicating type 2 inflammation as a driver of acute and late radiation lung injury. Studies of mice deficient in IL-4, a type 2 cytokine that is associated with alternatively activated macrophages, has suggested that the development of fibrosis observed after radiation is not dependent on IL-4, however the recovery of macrophage subpopulations and the phenotypes of accumulated macrophages can be substantially altered after irradiation in the absence of IL-487. In contrast, deficiency of IL13 through genetic manipulation or through systemic delivery of a therapeutic neutralizing antibody is sufficient to substantially reduce radiation fibrosis and the accumulation of alternatively activated macrophages in mouse models85. IL-13 deficiency reduces TGFβ activity and the expression of numerous downstream fibrosis associated genes in irradiated tissue85. It is not yet known if combined deficiency of IL-4 and IL-13 will further reduce the pneumonic changes after irradiation and further suppress late lung injury. Regardless, a number of agents targeting IL-13 are in late stage clinical trials for the treatment of asthma, autoimmune disorders, and allergic disease91, which may provide an opportunity for clinical translation in the setting of radiation injury once the optimal timing of delivery can be clarified.
Recently, additional pro-inflammatory feedback mechanisms, such as enhanced IFN-γ dependent inflammation, have been identified after suppression of IL-13 in fibrosis models, lending support to the notion that combined targeting of multiple pathways may be a critical component of effective therapy92. Further study of the role of type 2 inflammation on both the acute and late effects of irradiation is likely to clarify the extent to which agents and combination of agents targeting these processes may provide therapeutic opportunities. Because type 2 immunity has a known role in driving chronic inflammation in a number of pathologic conditions, such as asthma and idiopathic pulmonary fibrosis, drug development in this area is expanding, providing a tremendous opportunity for eventual clinical translation93.
T cells as a driver of radiation injury
Although T-cells do not account for a large proportion of the accumulated cells in irradiated tissues, they are capable of dramatically altering immune responses through effector mechanisms, and are known to drive chronic inflammatory states such as graft versus host disease and other automimmune phenomenon, resulting in pathologies strikingly similar to those observed after irradiation94. Indeed, T cell polarization and altered balance in T cell subsets has been implicated as a possible contributor to radiation injury94. Subsets of T lymphocytes are known to have differential sensitivities and recovery rates after irradiation, potentially leading to an altered balance of T cell subsets after irradiation95–98. Perhaps most importantly, selective depletion of T cell subsets has provided evidence that T cells can drive some aspects of radiation injury99–101.
Clinical data has suggested that the presence of CD4+ T cells have a higher risk of developing pneumonitis102, although from these studies it is not clear if CD4+ cells are a marker of radiation lung injury or drive the phenomenon. It is important to consider that CD4+ T helper (Th) cells can be further differentiated into several lineages, including the Th1, Th2, and Th17 lineage. Irradiation shifts inflammation towards a T helper 2 (Th2) dominant response and away from a Th1 response103, potentially activating damaging chronic inflammatory processes. Similarly, Th17 subsets and Treg subsets have been demonstrated to infiltrate irradiated lung tissue104. The importance of Th17 subsets in radiation injury is supported by the capacity of IL-17 targeted therapies to reduce the severity of radiation injury in animal models105.
Conceptually, these data are consistent with the above importance of type 2 inflammation, as IL-17 has been implicated in contributing to type 2 immune responses106. In contrast, depletion of Treg enhance Th17 responses, and mitigate lung injury after irradiation101. Clearly, given the complexity of Th17 responses, including the potential for a regulatory or pro-inflammatory component, there is still much to learn in terms of these immune subsets that may help modify toxicity of radiotherapy.
FLASH
Although dose rate has long been known to alter tumor and normal tissue responses to irradiation, some recent findings have fundamentally altered the concept of normal tissue effects from radiation. Studies of ultra-high dose rate irradiation (>40 Gy/second) delivered in short pulses, also known as FLASH irradiation, have suggested a remarkable tumor selectivity of cell killing107. The use of similar doses of FLASH and conventional irradiation (<0.03Gy/second) resulted in similar tumor responses. In contrast, normal lung tissue irradiated with FLASH irradiation did not demonstrate the typical rapid onset of apoptosis and TGFβ activation that was seen with conventional delivery. Further, mice treated with FLASH at doses as high as 17 Gy to the thorax did not develop pulmonary fibrosis. Although fibrosis was achieved in an attenuated form by giving 30 Gy whole thorax FLASH, this dose is rapidly fatal using conventional dosing methods.
The exact mechanism for the reduction in normal tissue injury using the FLASH approach is continuing to be studied. A reduction in apoptosis in multiple normal tissue cell types immediately after exposure with FLASH compared to the same conventional dose was appreciated107. Although delivery of TNFα prior to FLASH was capable of massively increasing apoptosis and pulmonary edema, it was not capable of inducing fibrosis, suggesting that normal tissue apoptotic sparing is not the primary mechanism of tumor selectivity of FLASH.
The ability to study and implement the FLASH phenomenon has been limited by the technical capacity to deliver such rapid dose pulses. Recently, a method for LINAC based FLASH was described, potentially allowing more widespread testing of the methods108. This has allowed confirmation of a normal tissue sparing effect of FLASH in an intestinal injury model109. Additional studies are anticipated to evaluate selectivity of FLASH in other tissues and to further understand the mechanism of preferential normal tissue sparing.
Conclusions
Radiation normal tissue injury is a complex process that spans acute and late injury in biologically diverse tissues. Classical radiobiological understanding of normal tissue injury has provided a framework into which molecular biologic, immunologic, and tissue homeostatic understanding is now being integrated. This enhanced understanding of radiation injury provides new possibilities for mitigation and therapeutic intervention. The development of novel assays and tools coupled with an exponentially increasing knowledge of the effects of radiation on the immune system suggests that numerous additional advances are on the horizon.
Figure 1. Senescence in radiation injury.
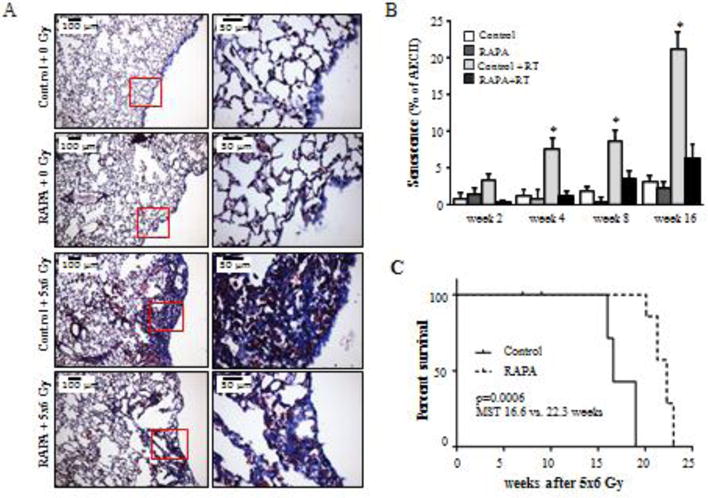
C57BL/6NCr Mice were exposed to 5×6 Gy of thoracic IR and treated with rapamycin or control diet. A) Masson trichrome staining of lung tissue at 16 weeks after IR. Collagen: blue, nuclei: purple, cytoplasm/epithelia: pink. B) The percentage of type II pneumocyte (AECII) cells staining for β-Gal activity at 2, 4, 8, and 16 weeks after IR was scored. A) Representative images at week 16 after IR. Percentage of AECII co-stained for Pro-surfactant-C (Pro-SP-C) at 16 weeks after IR. C) Kaplan-Meier survival analysis demonstrated that administration of rapamycin extended survival compared to mice receiving control diet after IR. Columns: mean, error bars: SD, brackets: p<0.05 by ANOVA. Reproduced with permission from110.
Figure 2. Pathways of radiation injury.
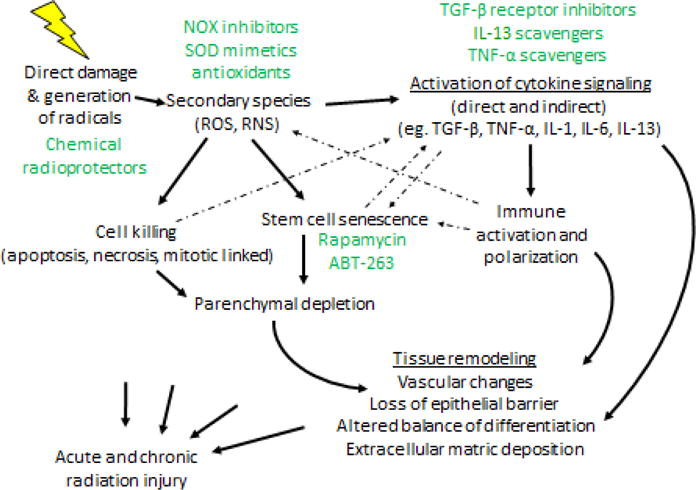
Radiation induces direct damage in normal tissues but also can generate free radicals, which can themselves cause injury or give rise to secondary species that may also cause injury. As a consequence, normal tissue cells are killed through a variety of mechanisms. Normal tissue stem cells may enter a state of senescence, which when combined with the initial cell killing, may result in parenchymal depletion and resulting tissue dysfunction. Simultaneously, ROS can initiate cytokine signaling, as can cell death and senescence. Collectively, these effects lead to immune activation and alterations in immune cell polarization, that can lead to chronic inflammation and further oxidative stress. Potential prevention, mitigation, and treatment approaches are highlighted in green.
Acknowledgments
This work was supported by the intramural program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Citrin D, Cotrim AP, Hyodo F. Radioprotectors and mitigators of radiation-induced normal tissue injury. Oncologist. 2010;15:360–71. doi: 10.1634/theoncologist.2009-S104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Martin M, Lefaix J, Delanian S. TGF-beta1 and radiation fibrosis: a master switch and a specific therapeutic target? Int J Radiat Oncol Biol Phys. 2000;47:277–90. doi: 10.1016/s0360-3016(00)00435-1. [DOI] [PubMed] [Google Scholar]
- 3.von Sontag C. The Chemical Basis of Radiation Biology. Francis Ta; London: 1987. [Google Scholar]
- 4.Khan MA, Van Dyk J, Yeung IW. Partial volume rat lung irradiation; assessment of early DNA damage in different lung regions and effect of radical scavengers. Radiother Oncol. 2003;66:95–102. doi: 10.1016/s0167-8140(02)00325-0. [DOI] [PubMed] [Google Scholar]
- 5.Choi SH, Kim M, Lee HJ. Effects of NOX1 on fibroblastic changes of endothelial cells in radiationinduced pulmonary fibrosis. Mol Med Rep. 2016;13:4135–42. doi: 10.3892/mmr.2016.5090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Citrin DE, Shankavaram U, Horton JA. Role of Type II Pneumocyte Senescence in Radiation-Induced Lung Fibrosis. J Natl Cancer Inst. 2013;105:1474–84. doi: 10.1093/jnci/djt212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hayyan M, Hashim MA, Al Nashef IM. Superoxide Ion: Generation and Chemical Implications. Chem Rev. 2016;116:3029–85. doi: 10.1021/acs.chemrev.5b00407. [DOI] [PubMed] [Google Scholar]
- 8.Hodgson EK, Fridovich I. The interaction of bovine erythrocyte superoxide dismutase with hydrogen peroxide: chemiluminescence and peroxidation. Biochemistry. 1975;14:5299–303. doi: 10.1021/bi00695a011. [DOI] [PubMed] [Google Scholar]
- 9.Huber W. Orgotein–(bovine Cu-Zn superoxide dismutase), an anti-inflammatory protein drug: discovery, toxicology and pharmacology. Eur J Rheumatol Inflamm. 1981;4:173–82. [PubMed] [Google Scholar]
- 10.Menander-Huber KB, Edsmyr F, Huber W. Orgotein (superoxide dismutase): a drug for the amelioration of radiation-induced side effects. A double-blind, placebo-controlled study in patients with bladder tumours. Urol Res. 1978;6:255–7. doi: 10.1007/BF00262630. [DOI] [PubMed] [Google Scholar]
- 11.Nielsen OS, Overgaard J, Overgaard M. Orgotein in radiation treatment of bladder cancer. A report on allergic reactions and lack of radioprotective effect. Acta Oncol. 1987;26:101–4. doi: 10.3109/02841868709091748. [DOI] [PubMed] [Google Scholar]
- 12.Greenberger J, Kagan V, Bayir H. Antioxidant Approaches to Management of Ionizing Irradiation Injury. Antioxidants (Basel) 2015;4:82–101. doi: 10.3390/antiox4010082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tarhini AA, Belani CP, Luketich JD. A phase I study of concurrent chemotherapy (paclitaxel and carboplatin) and thoracic radiotherapy with swallowed manganese superoxide dismutase plasmid liposome protection in patients with locally advanced stage III non-small-cell lung cancer. Hum Gene Ther. 2011;22:336–42. doi: 10.1089/hum.2010.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Calveley VL, Jelveh S, Langan A. Genistein can mitigate the effect of radiation on rat lung tissue. Radiat Res. 2010;173:602–11. doi: 10.1667/RR1896.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mahmood J, Jelveh S, Calveley V. Mitigation of radiation-induced lung injury by genistein and EUK-207. Int J Radiat Biol. 2011;87:889–901. doi: 10.3109/09553002.2011.583315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mahmood J, Jelveh S, Zaidi A. Mitigation of radiation-induced lung injury with EUK-207 and genistein: effects in adolescent rats. Radiat Res. 2013;179:125–34. doi: 10.1667/RR2954.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Para AE, Bezjak A, Yeung IW. Effects of genistein following fractionated lung irradiation in mice. Radiother Oncol. 2009;92:500–10. doi: 10.1016/j.radonc.2009.04.005. [DOI] [PubMed] [Google Scholar]
- 18.Rosenthal RA, Fish B, Hill RP. Salen Mn complexes mitigate radiation injury in normal tissues. Anticancer Agents Med Chem. 2011;11:359–72. doi: 10.2174/187152011795677490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rabbani ZN, Batinic-Haberle I, Anscher MS. Long-term administration of a small molecular weight catalytic metalloporphyrin antioxidant, AEOL 10150, protects lungs from radiation-induced injury. Int J Radiat Oncol Biol Phys. 2007;67:573–80. doi: 10.1016/j.ijrobp.2006.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rabbani ZN, Salahuddin FK, Yarmolenko P. Low molecular weight catalytic metalloporphyrin antioxidant AEOL 10150 protects lungs from fractionated radiation. Free Radic Res. 2007;41:1273–82. doi: 10.1080/10715760701689550. [DOI] [PubMed] [Google Scholar]
- 21.Venugopal R, Jaiswal AK. Nrf1 and Nrf2 positively and c-Fos and Fra1 negatively regulate the human antioxidant response element-mediated expression of NAD(P)H:quinone oxidoreductase1 gene. Proc Natl Acad Sci U S A. 1996;93:14960–5. doi: 10.1073/pnas.93.25.14960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Venugopal R, Jaiswal AK. Nrf2 and Nrf1 in association with Jun proteins regulate antioxidant response element-mediated expression and coordinated induction of genes encoding detoxifying enzymes. Oncogene. 1998;17:3145–56. doi: 10.1038/sj.onc.1202237. [DOI] [PubMed] [Google Scholar]
- 23.Soriano FX, Baxter P, Murray LM. Transcriptional regulation of the AP-1 and Nrf2 target gene sulfiredoxin. Mol Cells. 2009;27:279–82. doi: 10.1007/s10059-009-0050-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kobayashi M, Yamamoto M. Molecular mechanisms activating the Nrf2-Keap1 pathway of antioxidant gene regulation. Antioxid Redox Signal. 2005;7:385–94. doi: 10.1089/ars.2005.7.385. [DOI] [PubMed] [Google Scholar]
- 25.Chen XL, Kunsch C. Induction of cytoprotective genes through Nrf2/antioxidant response element pathway: a new therapeutic approach for the treatment of inflammatory diseases. Curr Pharm Des. 2004;10:879–91. doi: 10.2174/1381612043452901. [DOI] [PubMed] [Google Scholar]
- 26.Wakabayashi N, Itoh K, Wakabayashi J. Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nat Genet. 2003;35:238–45. doi: 10.1038/ng1248. [DOI] [PubMed] [Google Scholar]
- 27.Yoh K, Itoh K, Enomoto A. Nrf2-deficient female mice develop lupus-like autoimmune nephritis. Kidney Int. 2001;60:1343–53. doi: 10.1046/j.1523-1755.2001.00939.x. [DOI] [PubMed] [Google Scholar]
- 28.Tebay LE, Robertson H, Durant ST. Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease. Free Radic Biol Med. 2015;88:108–46. doi: 10.1016/j.freeradbiomed.2015.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jeong Y, Hoang NT, Lovejoy A. Role of KEAP1/NRF2 and TP53 Mutations in Lung Squamous Cell Carcinoma Development and Radiation Resistance. Cancer Discov. 2017;7:86–101. doi: 10.1158/2159-8290.CD-16-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stacy DR, Ely K, Massion PP. Increased expression of nuclear factor E2 p45-related factor 2 (NRF2) in head and neck squamous cell carcinomas. Head Neck. 2006;28:813–8. doi: 10.1002/hed.20430. [DOI] [PubMed] [Google Scholar]
- 31.Segal BH, Han W, Bushey JJ. NADPH oxidase limits innate immune responses in the lungs in mice. PLoS One. 2010;5:e9631. doi: 10.1371/journal.pone.0009631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sekhar KR, Crooks PA, Sonar VN. NADPH oxidase activity is essential for Keap1/Nrf2-mediated induction of GCLC in response to 2-indol-3-yl-methylenequinuclidin-3-ols. Cancer Res. 2003;63:5636–45. [PubMed] [Google Scholar]
- 33.Travis EL, Rachakonda G, Zhou X. NRF2 deficiency reduces life span of mice administered thoracic irradiation. Free Radic Biol Med. 2011;51:1175–83. doi: 10.1016/j.freeradbiomed.2011.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sekhar KR, Freeman ML. Nrf2 promotes survival following exposure to ionizing radiation. Free Radic Biol Med. 2015;88:268–74. doi: 10.1016/j.freeradbiomed.2015.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mont S, Davies SS, Roberts Second LJ. Accumulation of isolevuglandin-modified protein in normal and fibrotic lung. Sci Rep. 2016;6:24919. doi: 10.1038/srep24919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kalash R, Berhane H, Au J. Differences in irradiated lung gene transcription between fibrosis-prone C57BL/6NHsd and fibrosis-resistant C3H/HeNHsd mice. In Vivo. 2014;28:147–71. [PMC free article] [PubMed] [Google Scholar]
- 37.Martinez-Outschoorn UE, Peiris-Pages M, Pestell RG. Cancer metabolism: a therapeutic perspective. Nat Rev Clin Oncol. 2017;14:11–31. doi: 10.1038/nrclinonc.2016.60. [DOI] [PubMed] [Google Scholar]
- 38.Sun K, Tordjman J, Clement K. Fibrosis and adipose tissue dysfunction. Cell Metab. 2013;18:470–7. doi: 10.1016/j.cmet.2013.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rabbani ZN, Mi J, Zhang Y. Hypoxia inducible factor 1alpha signaling in fractionated radiation-induced lung injury: role of oxidative stress and tissue hypoxia. Radiat Res. 2010;173:165–74. doi: 10.1667/RR1816.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vujaskovic Z, Anscher MS, Feng QF. Radiation-induced hypoxia may perpetuate late normal tissue injury. Int J Radiat Oncol Biol Phys. 2001;50:851–5. doi: 10.1016/s0360-3016(01)01593-0. [DOI] [PubMed] [Google Scholar]
- 41.Westbury CB, Pearson A, Nerurkar A. Hypoxia can be detected in irradiated normal human tissue: a study using the hypoxic marker pimonidazole hydrochloride. Br J Radiol. 2007;80:934–8. doi: 10.1259/bjr/25046649. [DOI] [PubMed] [Google Scholar]
- 42.Pathak R, Pawar SA, Fu Q. Characterization of transgenic Gfrp knock-in mice: implications for tetrahydrobiopterin in modulation of normal tissue radiation responses. Antioxid Redox Signal. 2014;20:1436–46. doi: 10.1089/ars.2012.5025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Crabtree MJ, Tatham AL, Hale AB. Critical role for tetrahydrobiopterin recycling by dihydrofolate reductase in regulation of endothelial nitric-oxide synthase coupling: relative importance of the de novo biopterin synthesis versus salvage pathways. J Biol Chem. 2009;284:28128–36. doi: 10.1074/jbc.M109.041483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kohnen SL, Mouithys-Mickalad AA, Deby-Dupont GP. Oxidation of tetrahydrobiopterin by peroxynitrite or oxoferryl species occurs by a radical pathway. Free Radic Res. 2001;35:709–21. doi: 10.1080/10715760100301221. [DOI] [PubMed] [Google Scholar]
- 45.Pathak R, Cheema AK, Boca SM. Modulation of Radiation Response by the Tetrahydrobiopterin Pathway. Antioxidants (Basel) 2015;4:68–81. doi: 10.3390/antiox4010068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thind K, Chen A, Friesen-Waldner L. Detection of radiation-induced lung injury using hyperpolarized (13)C magnetic resonance spectroscopy and imaging. Magn Reson Med. 2013;70:601–9. doi: 10.1002/mrm.24525. [DOI] [PubMed] [Google Scholar]
- 47.Barcellos-Hoff MH, Dix TA. Redox-mediated activation of latent transforming growth factor-beta 1. Mol Endocrinol. 1996;10:1077–83. doi: 10.1210/mend.10.9.8885242. [DOI] [PubMed] [Google Scholar]
- 48.Yu Q, Stamenkovic I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 2000;14:163–76. [PMC free article] [PubMed] [Google Scholar]
- 49.Akhurst RJ, Hata A. Targeting the TGFbeta signalling pathway in disease. Nat Rev Drug Discov. 2012;11:790–811. doi: 10.1038/nrd3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rube CE, Uthe D, Schmid KW. Dose-dependent induction of transforming growth factor beta (TGF-beta) in the lung tissue of fibrosis-prone mice after thoracic irradiation. Int J Radiat Oncol Biol Phys. 2000;47:1033–42. doi: 10.1016/s0360-3016(00)00482-x. [DOI] [PubMed] [Google Scholar]
- 51.Anscher MS, Kong FM, Marks LB. Changes in plasma transforming growth factor beta during radiotherapy and the risk of symptomatic radiation-induced pneumonitis. Int J Radiat Oncol Biol Phys. 1997;37:253–8. doi: 10.1016/s0360-3016(96)00529-9. [DOI] [PubMed] [Google Scholar]
- 52.Anscher MS, Murase T, Prescott DM. Changes in plasma TGF beta levels during pulmonary radiotherapy as a predictor of the risk of developing radiation pneumonitis. Int J Radiat Oncol Biol Phys. 1994;30:671–6. doi: 10.1016/0360-3016(92)90954-g. [DOI] [PubMed] [Google Scholar]
- 53.Anscher MS, Kong FM, Andrews K. Plasma transforming growth factor beta1 as a predictor of radiation pneumonitis. Int J Radiat Oncol Biol Phys. 1998;41:1029–35. doi: 10.1016/s0360-3016(98)00154-0. [DOI] [PubMed] [Google Scholar]
- 54.Zhao L, Wang L, Ji W. Elevation of plasma TGF-beta1 during radiation therapy predicts radiation-induced lung toxicity in patients with non-small-cell lung cancer: a combined analysis from Beijing and Michigan. Int J Radiat Oncol Biol Phys. 2009;74:1385–90. doi: 10.1016/j.ijrobp.2008.10.065. [DOI] [PubMed] [Google Scholar]
- 55.Anscher MS, Marks LB, Shafman TD. Risk of long-term complications after TFG-beta1-guided very-high-dose thoracic radiotherapy. Int J Radiat Oncol Biol Phys. 2003;56:988–95. doi: 10.1016/s0360-3016(03)00184-6. [DOI] [PubMed] [Google Scholar]
- 56.De Jaeger K, Seppenwoolde Y, Kampinga HH. Significance of plasma transforming growth factor-beta levels in radiotherapy for non-small-cell lung cancer. Int J Radiat Oncol Biol Phys. 2004;58:1378–87. doi: 10.1016/j.ijrobp.2003.09.078. [DOI] [PubMed] [Google Scholar]
- 57.Anscher MS, Thrasher B, Zgonjanin L. Small molecular inhibitor of transforming growth factor-beta protects against development of radiation-induced lung injury. Int J Radiat Oncol Biol Phys. 2008;71:829–37. doi: 10.1016/j.ijrobp.2008.02.046. [DOI] [PubMed] [Google Scholar]
- 58.Rabender C, Mezzaroma E, Mauro AG. IPW-5371 Proves Effective as a Radiation Countermeasure by Mitigating Radiation-Induced Late Effects. Radiat Res. 2016;186:478–488. doi: 10.1667/RR14403.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Anscher MS, Thrasher B, Rabbani Z. Antitransforming growth factor-beta antibody 1D11 ameliorates normal tissue damage caused by high-dose radiation. Int J Radiat Oncol Biol Phys. 2006;65:876–81. doi: 10.1016/j.ijrobp.2006.02.051. [DOI] [PubMed] [Google Scholar]
- 60.Han G, Bian L, Li F. Preventive and therapeutic effects of Smad7 on radiation-induced oral mucositis. Nat Med. 2013;19:421–8. doi: 10.1038/nm.3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xavier S, Piek E, Fujii M. Amelioration of radiation-induced fibrosis: inhibition of transforming growth factor-beta signaling by halofuginone. J Biol Chem. 2004;279:15167–76. doi: 10.1074/jbc.M309798200. [DOI] [PubMed] [Google Scholar]
- 62.Calik M, Yavas G, Calik SG. Amelioration of radiation-induced lung injury by halofuginone: An experimental study in Wistar-Albino rats. Hum Exp Toxicol. 2016 doi: 10.1177/0960327116660753. [DOI] [PubMed] [Google Scholar]
- 63.Cook JA, Choudhuri R, Degraff W. Halofuginone enhances the radiation sensitivity of human tumor cell lines. Cancer Lett. 2010;289:119–26. doi: 10.1016/j.canlet.2009.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sanford LP, Ormsby I, Gittenberger-de Groot AC. TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development. 1997;124:2659–70. doi: 10.1242/dev.124.13.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Proetzel G, Pawlowski SA, Wiles MV. Transforming growth factor-beta 3 is required for secondary palate fusion. Nat Genet. 1995;11:409–14. doi: 10.1038/ng1295-409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Anderton MJ, Mellor HR, Bell A. Induction of heart valve lesions by small-molecule ALK5 inhibitors. Toxicol Pathol. 2011;39:916–24. doi: 10.1177/0192623311416259. [DOI] [PubMed] [Google Scholar]
- 67.Frazier K, Thomas R, Scicchitano M. Inhibition of ALK5 signaling induces physeal dysplasia in rats. Toxicol Pathol. 2007;35:284–95. doi: 10.1080/01926230701198469. [DOI] [PubMed] [Google Scholar]
- 68.Morris JC, Tan AR, Olencki TE. Phase I study of GC1008 (fresolimumab): a human anti-transforming growth factor-beta (TGFbeta) monoclonal antibody in patients with advanced malignant melanoma or renal cell carcinoma. PLoS One. 2014;9:e90353. doi: 10.1371/journal.pone.0090353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Johnston CJ, Piedboeuf B, Rubin P. Early and persistent alterations in the expression of interleukin-1 alpha, interleukin-1 beta and tumor necrosis factor alpha mRNA levels in fibrosis-resistant and sensitive mice after thoracic irradiation. Radiat Res. 1996;145:762–7. [PubMed] [Google Scholar]
- 70.Hill RP, Zaidi A, Mahmood J. Investigations into the role of inflammation in normal tissue response to irradiation. Radiother Oncol. 2011;101:73–9. doi: 10.1016/j.radonc.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.de Araujo AA, Varela H, de Medeiros CA. Azilsartan reduced TNF-alpha and IL-1beta levels, increased IL-10 levels and upregulated VEGF, FGF, KGF, and TGF-alpha in an oral mucositis model. PLoS One. 2015;10:e0116799. doi: 10.1371/journal.pone.0116799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Moura JF, Mota JM, Leite CA. A novel model of megavoltage radiation-induced oral mucositis in hamsters: Role of inflammatory cytokines and nitric oxide. Int J Radiat Biol. 2015;91:500–9. doi: 10.3109/09553002.2015.1021964. [DOI] [PubMed] [Google Scholar]
- 73.Zheng J, Wang J, Pouliot M. Gene expression profiling in non-human primate jejunum, ileum and colon after total-body irradiation: a comparative study of segment-specific molecular and cellular responses. BMC Genomics. 2015;16:984. doi: 10.1186/s12864-015-2168-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang Q, Zhu L, Wang G. Ionizing radiation promotes CCL27 secretion from keratinocytes through the cross talk between TNF-alpha and ROS. J Biochem Mol Toxicol. 2016 doi: 10.1002/jbt.21868. [DOI] [PubMed] [Google Scholar]
- 75.Citrin DE, Hitchcock YJ, Chung EJ. Determination of cytokine protein levels in oral secretions in patients undergoing radiotherapy for head and neck malignancies. Radiat Oncol. 2012;7:64. doi: 10.1186/1748-717X-7-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chang J, Wang Y, Shao L. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med. 2016;22:78–83. doi: 10.1038/nm.4010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Coppe JP, Desprez PY, Krtolica A. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5:99–118. doi: 10.1146/annurev-pathol-121808-102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Serra MP, Marongiu F, Sini M. Hepatocyte senescence induced by radiation and partial hepatectomy in rat liver. Int J Radiat Biol. 2014;90:876–83. doi: 10.3109/09553002.2014.922714. [DOI] [PubMed] [Google Scholar]
- 79.Iglesias-Bartolome R, Patel V, Cotrim A. mTOR inhibition prevents epithelial stem cell senescence and protects from radiation-induced mucositis. Cell Stem Cell. 2012;11:401–14. doi: 10.1016/j.stem.2012.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Arora M, Sun CL, Ness KK. Physiologic Frailty in Nonelderly Hematopoietic Cell Transplantation Patients: Results From the Bone Marrow Transplant Survivor Study. JAMA Oncol. 2016;2:1277–1286. doi: 10.1001/jamaoncol.2016.0855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ness KK, Krull KR, Jones KE. Physiologic frailty as a sign of accelerated aging among adult survivors of childhood cancer: a report from the St Jude Lifetime cohort study. J Clin Oncol. 2013;31:4496–503. doi: 10.1200/JCO.2013.52.2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Demaria M, O’Leary MN, Chang J. Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discov. 2017;7:165–176. doi: 10.1158/2159-8290.CD-16-0241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chung EJ, McKay-Corkum G, Chung S. Truncated Plasminogen Activator Inhibitor-1 Protein Protects From Pulmonary Fibrosis Mediated by Irradiation in a Murine Model. Int J Radiat Oncol Biol Phys. 2016;94:1163–72. doi: 10.1016/j.ijrobp.2015.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Graves PR, Siddiqui F, Anscher MS. Radiation pulmonary toxicity: from mechanisms to management. Semin Radiat Oncol. 2010;20:201–7. doi: 10.1016/j.semradonc.2010.01.010. [DOI] [PubMed] [Google Scholar]
- 85.Chung SI, Horton JA, Ramalingam TR. IL-13 is a therapeutic target in radiation lung injury. Sci Rep. 2016;6:39714. doi: 10.1038/srep39714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Groves AM, Johnston CJ, Misra RS. Whole-Lung Irradiation Results in Pulmonary Macrophage Alterations that are Subpopulation and Strain Specific. Radiat Res. 2015;184:639–49. doi: 10.1667/RR14178.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Groves AM, Johnston CJ, Misra RS. Effects of IL-4 on pulmonary fibrosis and the accumulation and phenotype of macrophage subpopulations following thoracic irradiation. Int J Radiat Biol. 2016;92:754–765. doi: 10.1080/09553002.2016.1222094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Horton JA, Hudak KE, Chung EJ. Mesenchymal stem cells inhibit cutaneous radiation-induced fibrosis by suppressing chronic inflammation. Stem Cells. 2013;31:2231–41. doi: 10.1002/stem.1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wynn TA, Vannella KM. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity. 2016;44:450–62. doi: 10.1016/j.immuni.2016.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Davies LC, Taylor PR. Tissue-resident macrophages: then and now. Immunology. 2015;144:541–8. doi: 10.1111/imm.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mitchell J, Dimov V, Townley RG. IL-13 and the IL-13 receptor as therapeutic targets for asthma and allergic disease. Curr Opin Investig Drugs. 2010;11:527–34. [PubMed] [Google Scholar]
- 92.Ramalingam TR, Gieseck RL, Acciani TH. Enhanced protection from fibrosis and inflammation in the combined absence of IL-13 and IFN-gamma. J Pathol. 2016;239:344–54. doi: 10.1002/path.4733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Parulekar AD, Diamant Z, Hanania NA. Role of biologics targeting type 2 airway inflammation in asthma: what have we learned so far? Curr Opin Pulm Med. 2017;23:3–11. doi: 10.1097/MCP.0000000000000343. [DOI] [PubMed] [Google Scholar]
- 94.Schaue D, McBride WH. T lymphocytes and normal tissue responses to radiation. Front Oncol. 2012;2:119. doi: 10.3389/fonc.2012.00119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.DeKruyff RH, Fang Y, Umetsu DT. IL-4-based helper activity of CD4+ T cells is radiation sensitive. Cell Immunol. 1995;160:248–56. doi: 10.1016/0008-8749(95)80035-h. [DOI] [PubMed] [Google Scholar]
- 96.Awwad M, North RJ. Sublethal, whole-body ionizing irradiation can be tumor promotive or tumor destructive depending on the stage of development of underlying antitumor immunity. Cancer Immunol Immunother. 1988;26:55–60. doi: 10.1007/BF00199848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Qu Y, Jin S, Zhang A. Gamma-ray resistance of regulatory CD4+CD25+Foxp3+ T cells in mice. Radiat Res. 2010;173:148–57. doi: 10.1667/RR0978.1. [DOI] [PubMed] [Google Scholar]
- 98.Qu Y, Zhang B, Liu S. 2-Gy whole-body irradiation significantly alters the balance of CD4+ CD25-T effector cells and CD4+ CD25+ Foxp3+ T regulatory cells in mice. Cell Mol Immunol. 2010;7:419–27. doi: 10.1038/cmi.2010.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wirsdorfer F, Cappuccini F, Niazman M. Thorax irradiation triggers a local and systemic accumulation of immunosuppressive CD4+ FoxP3+ regulatory T cells. Radiat Oncol. 2014;9:98. doi: 10.1186/1748-717X-9-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Xiong S, Pan X, Xu L. Regulatory T Cells Promote beta-Catenin–Mediated Epithelium-to-Mesenchyme Transition During Radiation-Induced Pulmonary Fibrosis. Int J Radiat Oncol Biol Phys. 2015;93:425–35. doi: 10.1016/j.ijrobp.2015.05.043. [DOI] [PubMed] [Google Scholar]
- 101.Xiong S, Guo R, Yang Z. Treg depletion attenuates irradiation-induced pulmonary fibrosis by reducing fibrocyte accumulation, inducing Th17 response, and shifting IFN-gamma, IL-12/IL-4, IL-5 balance. Immunobiology. 2015;220:1284–91. doi: 10.1016/j.imbio.2015.07.001. [DOI] [PubMed] [Google Scholar]
- 102.Martin C, Romero S, Sanchez-Paya J. Bilateral lymphocytic alveolitis: a common reaction after unilateral thoracic irradiation. Eur Respir J. 1999;13:727–32. doi: 10.1034/j.1399-3003.1999.13d05.x. [DOI] [PubMed] [Google Scholar]
- 103.Bass H, Mosmann T, Strober S. Evidence for mouse Th1- and Th2-like helper T cells in vivo. Selective reduction of Th1-like cells after total lymphoid irradiation. J Exp Med. 1989;170:1495–511. doi: 10.1084/jem.170.5.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cappuccini F, Eldh T, Bruder D. New insights into the molecular pathology of radiation-induced pneumopathy. Radiother Oncol. 2011;101:86–92. doi: 10.1016/j.radonc.2011.05.064. [DOI] [PubMed] [Google Scholar]
- 105.Wang BZ, Wang LP, Han H. Interleukin-17A antagonist attenuates radiation-induced lung injuries in mice. Exp Lung Res. 2014;40:77–85. doi: 10.3109/01902148.2013.872210. [DOI] [PubMed] [Google Scholar]
- 106.Allen JE, Sutherland TE, Ruckerl D. IL-17 and neutrophils: unexpected players in the type 2 immune response. Curr Opin Immunol. 2015;34:99–106. doi: 10.1016/j.coi.2015.03.001. [DOI] [PubMed] [Google Scholar]
- 107.Favaudon V, Caplier L, Monceau V. Ultrahigh dose-rate FLASH irradiation increases the differential response between normal and tumor tissue in mice. Sci Transl Med. 2014;6:245ra93. doi: 10.1126/scitranslmed.3008973. [DOI] [PubMed] [Google Scholar]
- 108.Schuler E, Trovati S, King G. Experimental Platform for Ultra-high Dose Rate FLASH Irradiation of Small Animals Using a Clinical Linear Accelerator. Int J Radiat Oncol Biol Phys. 2017;97:195–203. doi: 10.1016/j.ijrobp.2016.09.018. [DOI] [PubMed] [Google Scholar]
- 109.Schueler E, Trovati S, King G. TU-H-CAMPUS-TeP2-02: FLASH Irradiation Improves the Therapeutic Index Following GI Tract Irradiation. Med Phys. 2016;43:3783. [Google Scholar]
- 110.Chung EJ, Sowers A, Thetford A. Mammalian Target of Rapamycin Inhibition With Rapamycin Mitigates Radiation-Induced Pulmonary Fibrosis in a Murine Model. Int J Radiat Oncol Biol Phys. 2016;96:857–866. doi: 10.1016/j.ijrobp.2016.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]