

Abstract
The aim of this study was to obtain data about the epidemiology of the different types of mucopolysaccharidoses in Japan and Switzerland and to compare with similar data from other countries.
Data for Japan was collected between 1982 and 2009, and 467 cases with MPS were identified. The combined birth prevalence was 1.53 per 100,000 live births. The highest birth prevalence was 0.84 for MPS II, accounting for 55% of all MPS. MPS I, III, and IV accounted for 15, 16, and 10%, respectively. MPS VI and VII were more rare and accounted for 1.7 and 1.3%, respectively.
A retrospective epidemiological data collection was performed in Switzerland between 1975 and 2008 (34 years), and 41 living MPS patients were identified. The combined birth prevalence was 1.56 per 100,000 live births. The highest birth prevalence was 0.46 for MPS II, accounting for 29% of all MPS. MPS I, III, and IV accounted for 12, 24, and 24%, respectively. As seen in the Japanese population, MPS VI and VII were more rare and accounted for 7.3 and 2.4%, respectively.
The high birth prevalence of MPS II in Japan was comparable to that seen in other East Asian countries where this MPS accounted for approximately 50% of all forms of MPS. Birth prevalence was also similar in some European countries (Germany, Northern Ireland, Portugal and the Netherlands) although the prevalence of other forms of MPS is also reported to be higher in these countries. Birth prevalence of MPS II in Switzerland and other European countries is comparatively lower. The birth prevalence of MPS III and IV in Switzerland is higher than in Japan but comparable to that in most other European countries. Moreover, the birth prevalence of MPS VI and VII was very low in both Switzerland and Japan. Overall, the frequency of MPS varies for each population due to differences in ethnic backgrounds and/or founder effects that affect the birth prevalence of each type of MPS, as seen for other rare genetic diseases. Methods for identification of MPS patients are not uniform across all countries, and consequently, if patients are not identified, recorded prevalence rates will be aberrantly low.
Keywords: mucopolysaccharidoses, incidence, prevalence, epidemiology, Japan, Switzerland
2. Introduction
Mucopolysaccharidoses
The Mucopolysaccharidoses (MPS) are a group of inherited metabolic diseases caused by the deficiency of enzymes required to degrade glycosaminoglycans (GAGs) in the lysosome. GAGs are sulfated polysaccharides comprising repeating disaccharides, uronic acid (or galactose) and hexosamines including chondroitin sulfate (CS), dermatan sulfate (DS), heparan sulfate (HS), and keratan sulfate (KS). Hyaluronan is an exception in the GAG family because it is a non-sulfated polysaccharide. Lysosomal enzymes are crucial for stepwise degradation of GAGs to provide a normal function of tissues and extracellular matrix (ECM). The deficiency of one or more lysosomal enzyme(s) results in accumulation of undegraded GAGs causing cell, tissue, and organ dysfunction. Accumulated GAGs of various tissues and their ECM are secreted into the blood circulation and then excreted in urine. MPS are autosomal recessive disorders except for MPS II that is X-linked.
Types of MPS
Seven types of MPS are categorized based on lack or defect in one of eleven specific lysosomal enzymes and are described as MPS I through MPS IX (excluding MPS V and VIII, which are no longer used). Clinical features vary with the type of MPS and clinical severity of the disease.
MPS I (Hurler syndrome, Hurler/Scheie syndrome, Scheie syndrome)
MPS I affected individuals have traditionally been classified as having one of three MPS I syndromes (Hurler syndrome, Hurler-Scheie syndrome, or Scheie syndrome), but no easily measurable biochemical differences have been identified [1] and the clinical findings overlap. Affected individuals are best described as having either severe or attenuated MPS I, a distinction that influences therapeutic options. The greatest variability is observed in individuals with attenuated MPS I. MPS I with either severe or attenuated form has limited activity of the enzyme α-L-iduronidase (IDUA) that breaks down DS and HS. These GAGs remain stored in cells causing progressive damage of various tissues including bone and, in severe cases, brain. MPS I presents with a wide range of symptoms. Typical manifestations include coarse face, corneal clouding, developmental delay, mental retardation, growth retardation, contractures of the joints, kyphoscoliosis, dysostosis multiplex, hearing loss, thickening of the heart valves, hepatosplenomegaly, and umbilical and inguinal hernias. Scheie syndrome, the milder form of the disease, is characterized by a coarse face and stocky physique with normal intelligence while Hurler syndrome displays neurological symptoms such as dyslexia, thermanesthesia, and cognitive degradation. Patients with Hurler syndrome develop initial symptoms like hernias, hepatomegaly, kyphosis and developmental delay within a year and die within a decade if untreated, while Scheie syndrome patients can live more than 50 years [2].
MPS II (Hunter syndrome)
Like MPS I, MPS II also has both mild and severe forms. Their clinical symptoms are very different. The severe form has features similar to Hurler syndrome, except for the lack of corneal clouding and slower progression of somatic and central nervous system involvement. The mild form is analogous to Hurler/Scheie or Scheie syndrome, with a longer life span, a slower progression of somatic deterioration, and retention of intelligence. Both forms arise from the deficiency of iduronate-2-sulfatase (I2S) that catalyzes DS and HS degradation. MPS II is inherited in an X-linked manner, resulting in mostly males being affected. However, on rare occasions, heterozygous females manifest findings of MPS II. This is thought to result from skewed inactivation of the normal paternally inherited X chromosome and expression of the maternally inherited mutated IDS allele [3, 4].
Clinical features of MPS II usually become apparent by two to four years of age. Abnormalities may include developmental delay, mental retardation, coarsening of facial features including thickening of lips, tongue, and nostrils, dysostosis multiplex, progressive growth delay resulting in short stature, and joint stiffness associated with restriction of movements. Affected children may also have an abnormally large head (macrocephaly), a short neck, broad chest, delayed tooth eruption, hearing loss, hepatosplenomegaly, and inguinal and umbilical hernias. The life expectancy also differs in the two forms; patients with the mild form reach adulthood, while patients affected by the severe form usually die within the first two decades of life.
MPS III (Sanfilippo syndrome)
MPS III is characterized by four types; MPS IIIA, IIIB, IIIC, and IIID due to lack of heparan-N-sulfatase (SGHS), α-N-acetylglucosaminidase (NAGLU), α-glucosaminidase acetyltransferase (HGSNAT), and N-acetylglucosamine 6-sulfatase (GNS), respectively. A defect in any of the four enzymes compromises the degradation of HS. Phenotypic variation exists among MPS III patients but to a lesser degree than in the other MPS, possibly because very mild forms of MPS III may not be diagnosed. MPS III is characterized by severe central nervous system (CNS) degeneration and progressive developmental delay and mental retardation. It is noteworthy that MPS I, II, III and MPS VII share partial or incomplete degradation of HS that results in CNS involvement.
MPS IV (Morquio syndrome)
MPS IV is caused by the deficiency of any of two distinct enzymes, N-acetylgalactosamine-6-sulfate sulfatase (GALNS) and β-galactosidase (GLB1) resulting in MPS IVA and IVB. Both chondroitin-6-sulfate (C6S) and KS accumulate in MPS IVA whereas only KS accumulates in MPS IVB. Accumulation of KS (and C6S) in MPS IV directly results in impairment of cartilage and bone development leading to systemic skeletal dysplasia. MPS IV has minimal impact on cognitive function in these patients, distinguishing this from other forms of MPS. Clinical features include coarsening of facies, short neck and trunk dwarfism, fine corneal clouding, skeletal dysplasia, small teeth with thin enamel and frequent caries formation, atlantoaxial subluxation, cervical spinal cord compression, pectus carinatum, kyphoscoliosis, bilateral genu valgum, pes planus, hyperlaxity of joints, waddling gait with a tendency to fall, and hearing loss. In addition, patients with a severe form of MPS IVA have easy fatigue, sleep apnea, short breath with restrictive lung, tracheal obstruction, and recurrent ear and respiratory infection. The clinical features of MPS IVB are usually milder than those associated with MPS IVA.
MPS VI (Maroteaux-Lamy syndrome)
MPS VI is caused by the deficiency of N-acetylgalactosamine-4-sulfatse enzyme that degrades C4S and DS. This form of MPS varies greatly among affected individuals. Some affected individuals only experience a few mild symptoms while others develop a more severe form of the disorder. Symptoms include coarse facial features, umbilical hernia, pectus carinatum, joint contractures, corneal clouding, and hepatosplenomegaly. Skeletal malformations and heart disease may occur in individuals with this form of MPS. In most cases, intelligence is normal.
MPS VII (Sly syndrome)
MPS VII is characterized by a deficiency of β-D-glucuronidase enzyme that causes accumulation of C4S, C6S, DS, and HS in tissues. MPS VII patients have moderate mental retardation that does not progress with the growth of the patients. Skeletal abnormalities are often present. Hernias, corneal clouding, hydrocephalus, short stature, heart disease, and coarse facial features have also been reported. Newborns with a severe form may experience abnormal accumulation of body fluids in various tissues (hydrops fetalis).
MPS IX (Natowicz syndrome)
MPS IX is an extremely rare form of MPS due to a deficiency of the enzyme hyaluronidase, which is needed to breakdown the hyaluronan (hyaluronic acid). This form of MPS was first described in 1996. In 2011, Imundo et al. described 3 siblings of Middle Eastern origin with MPS IX [5]. The putatively causative mutations were found in the HYAL1 gene, encoding one of three hyaluronidases [6]. Symptoms may include short stature, cysts, frequent ear infections, cleft palate, and the development of soft-tissue masses.
3. Birth prevalence of MPS
In this article, we use the term birth prevalence, which is calculated by the number of MPS cases during a period divided by the total number of live births during the same period and expressed as cases per 100,000 live births [7, 8]. In the literature, the term incidence is often used, and incidence rate at birth is equivalent to birth prevalence. The first study on the frequency of MPS was done by Lowry and Renwick in British Columbia, in 1971 [9], which was later updated between 1990 and 2000 [10, 11].
The birth prevalence of MPS has been extensively reported in European countries, and there have been several reports from Asian countries including Taiwan, China, South Korea, and India [7, 8, 12–20]. Newborn screens for MPS are becoming available to enable early treatment options and consequently to improve their quality of life. In this article, we present the first data on the birth prevalence of MPS in Japan and Switzerland and compare these new data with results published for other countries to show the range of birth prevalence worldwide.
3.1. Birth Prevalence of MPS in Japan
We investigated the birth prevalence of MPS patients in Japan between 1982 and 2009. Diagnosis of MPS was made in a high-risk population (suspected to be MPS) as described below.
Method
Screening of Urinary GAG
The screening of urinary GAG occurred across the country for 18 years from 1982 to 1999 and tests were performed at Gifu University. A urine specimen was sent to Gifu University when the patient was suspected as MPS clinically by the local physician. From 2000 to 2003 no data were obtained. In 2003, the urinary analysis was performed by Special Reference Laboratories, Inc (SRL), and interpreted by Dr. Orii (Department of Pediatrics at Gifu University, School of Medicine). The uronic acid quantitative test was carried out using an acid carbazole method as described [21].
Results
Number and type of MPS patients (1982–1999)
In total, 331 cases were diagnosed for all MPS types. The relative occurrence of the different MPSs identified during this period were; MPS I (14.8%, n = 49), MPS II (53%, n = 176), MPS III A (8.5%, n = 28), MPS III B (8.2%, n = 27), MPS III C (3.3%, n = 11), MPS IVA (9%, n = 30), MPS VI (1.2%, n = 4) and MPS VII (1.8%, n = 6).
Number and type of MPS patients (2003–2009)
136 patients were diagnosed in the seven years between 2003 to 2009. Relative occurrence of MPS I (16.2%, n = 22), MPS II (58.1%, n = 79), MPS III A (3.0%, n = 4), MPS III B (8.1%, n = 11), MPS IVA (11.0%, n = 15), MPS IVB (0.7%, n = 1), MPS VI (2.9%, n = 4) was similar to that measured between 1982 and 1999.
The combined birth prevalence, 1.53 per 100,000, was calculated by the number of MPS cases diagnosed, divided by the total number of live births during the two time periods between 1982 and 2009. One case of MPS I was found for every 430,000 births, 1 out of 119,000 for MPS II, 1 out of 385,000 for all MPS III (1 out of 1,000,000 for MPS IIIA, 1 out of 834,000 for MPS IIIB, 1 out of 2.5 million for MPS IIIC),1 out of 667,000 for MPS IVA, 1 out of 33 million for MPS IVB, 1 out of 3.3 million for MPS VI, and 1 out of 5 million for MPS VII. The birth prevalence of MPS I, II, III, IV, VI, or VII was 0.23, 0.84, 0.26, 0.15, 0.03, or 0.02 per 100,000 live births, respectively. MPS II accounted for more than half of all MPS. MPS I, MPS III, and MPS IV were 15, 16, and 10%, respectively. MPS VI and VII accounted for only 1.7 and 1.3%, respectively, as shown in Fig. 1.
Figure 1. Incidence of MPS (%).
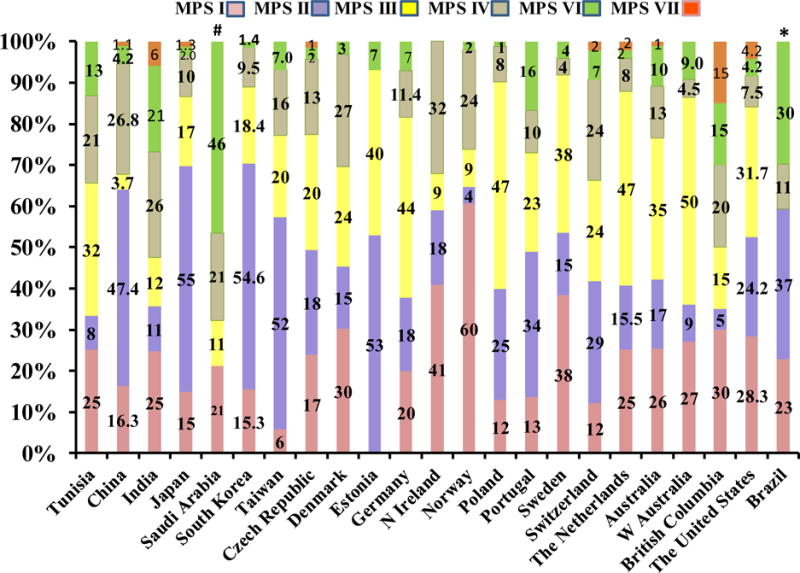
*Brazilian report did not include data for MPS III and MPS VII.
#Saudi Arabia has no MPS II data.
3.2. Birth Prevalence of MPS in Switzerland
Materials and subjects
The Swiss working group for Lysosomal Disorder (SALS) established a national registry for patients with lysosomal disorders in 2009. This electronically based registry is technically supported by a non-university institution, and all the data are saved on an external and secure server. Informed consent of the patients or their parents, was obtained in accordance with requirements of the Local Ethics Committees of the referring centers. The study was approved by the Local Ethics Committees. The retrospective study was carried out to identify all diagnosed MPS patients in Switzerland between 1975 and 2008. Patients enrolled were diagnosed either by measurement of reduced enzyme activity in leukocytes or fibroblasts and/or by molecular analysis. Abnormal urinary quantitative or qualitative GAGs alone were not satisfactory for a final diagnosis but were performed in all cases. Diagnosed patients are all followed in dedicated university centers (Bern, Lausanne, and Zurich) and were thus identified through these centers. Data collection was performed by a masters student at three centers. Data collection was based on the medical records.
The frequency of different subtypes of MPS was determined. The age of patients at the onset of symptoms and diagnosis was used to calculate the pre-diagnostic interval. The registry was used to determine the birth prevalence of the MPS patients.
Results
Switzerland has an average 85,000 births/year. In total, 51 patients with MPS were identified between 1975 and 2008 (34 years). 10 patients who died prior to creation of the registry are not included in the study. Of the remaining 41 patients, one MPS III and one MPS VI died during the study. A total of five patients had MPS I (female 1; male 4); 12 MPS II (male), 10 MPS III (female 5; male 5), 10 MPS IV (female 9; male 1), 3 MPS VI (female), and one MPS VII (male). The average age of onset of symptoms was 8 months for MPS I, 40 months for MPS II, 21 months for MPS III, 18 months for MPS IV, and 31 months for MPS VI, respectively. The average age at diagnosis was 15 (MPS I), 56 (MPS II), 54 (MPS III), 38 (MPS IV), and 69 (MPS VI) months, respectively. The total live birth during this period was 2,621,036. The combined birth prevalence for diagnosed MPS was 1.56 per 100,000 live births. This could be as high as 1.95 if the 10 MPS patients not included in the registry were born after 1975. MPS II had the highest birth prevalence of 0.46 per 100,000 live births. Birth prevalence of MPS I, III, IV, VI, and VII was 0.19, 0.38, 0.38, 0.11, and 0.038, respectively. MPS II comprised 29% of all MPS, and MPS III and MPS IV each accounted for 24%. MPS I accounted for 12%. MPS VII showed the lowest incidence in the Swiss MPS population as shown in Fig. 1.
3.3. Other countries
Method
We have extensively searched the literature for incidence and birth prevalence of MPS in different parts of the world and summarize the data in this section. The criteria for the diagnosis of MPS patients was made by clinical manifestation and clinical and molecular tests. The general diagnosis was made by elevated urinary GAGs and/or enzyme assay in serum, leukocytes, and/or fibroblasts.
3.3.1. Birth Prevalence of MPS in Saudi Arabia
In Saudi Arabia, Saudi Aramco Medical Services Organization (SAMSO) provided comprehensive free health care for its employees and their dependents. Data from 1983 to 2008 (26 years) was collected from SAMSO. The total number of live births during the study period was 165,130, and 248 cases of various metabolic diseases were diagnosed. 28 of these cases were MPSs [22]. The birth prevalence of MPS I and MPS IV was each calculated to be 3.62 per 100,000 live births, and each accounted 21% of all MPS. MPS VI had the highest birth prevalence at 7.85 per 100,000 live births, comprising 46% of all MPS cases diagnosed. The birth prevalence of MPS III was comparatively low (1.8) and accounted for 11% as shown in Fig. 1. No case of MPS II and VII was detected during this period.
3.3.2. Birth Prevalence of MPS in Taiwan
The diagnosis of MPS in Taiwan was made during a period of 21 years between 1984 and 2004. The data of live births were obtained from Department of Statistics, Ministry of the Interior, Taiwan. A total of 6,377,299 live births and 3,323,160 male live births were recorded during the period of study. 130 MPS cases were diagnosed, giving a combined birth prevalence of 2.04 per 100,000 live births [15]. The diagnosis of all patients was confirmed by two-dimensional electrophoresis of urinary GAGs and/or enzyme assay in serum, leukocytes, and/or fibroblasts. MPS II had the highest calculated birth prevalence of 1.07 per 100,000 (2.05 per 100,000 male live births), comprising 52% of all MPS cases diagnosed. Overall, the birth prevalence of all MPS was low and reported as 0.11, 0.39, 0.33, and 0.14 per 100,000 live births for MPS I, MPS III, MPS IV, and MPS VI, respectively. The percentage of MPS I, III, IV, and VI were 6, 20, 16, and 7%, respectively as shown in Fig. 1. In Taiwan, no case of MPS VII was reported.
3.3.3. Birth Prevalence of MPS in South Korea
In South Korea, 147 MPS patients were identified between 1994 and 2013 [23]. The combined birth prevalence of all MPS was calculated to be 1.35 per 100,000 live births. The birth prevalence of MPS II was 0.74 per 100,000 live births. The birth prevalence of MPS I, III, IV, and VI was 0.21, 0.25, 0.13, and 0.019, respectively. The most common subtype of MPS was MPS II (54.6%), followed by MPS III (18.4%). MPS I and IV account for 15.3 and 9.5%, respectively. MPS VI was very low (1.4%), and no MPS VII was identified during this time [23].
3.3.4. Relative incidence of MPSs in China
In China, a LSD study was carried out between 2006 and 2012. A total of 376 cases out of 1331 suspected patients were diagnosed for 17 different LSDs [24]. Patients in this study were from 21 provinces and municipalities in China, with 85.8% (n = 322) from East China. A small proportion of patients were from Northern China (n = 2), Northeast China (n = 3), Southwest China (n = 8), Northwest China (n = 9), Southern China (n = 11) and Central China (n = 21). MPSs represented 50.5% of the LSDs. Within the MPS group, MPS II represented nearly half of all MPS cases diagnosed (47.4%), followed by MPS IVA (26.8%), I (16.3%), VI (4.2%), IIIA/B (3.7%), and VII (1.1%) [24]. Only one patient was diagnosed with MPS IVB. Total numbers of births from the areas of China in these studies were not recorded, so birth prevalence cannot be calculated.
3.3.5. Relative incidence of MPSs in India
Various LSD determinations were performed by Sheth et al. in an Indian population [25]. 1,110 children referred from various Indian states and two neighboring countries (Sri Lanka and Afghanistan) from January 2002 to December 2012. It comprises of 938 (84.50 %) children from Western part of India, 121 (10.9 %) from Southern, 30 (2.7 %) from Northern, 1 (0.09 %) each from Eastern and central parts, 18 (1.62 %) from Sri Lanka, and 1 (0.09 %) from Afghanistan. The majority of these LSDs were glycolipid storage disorders (48%). MPS accounted for 22% of these LSDs [25, 26]. A total of 85 MPS cases were identified. MPS IV was the most common comprising 26%, followed by MPS I with 25%. MPS II, III, and VI accounted for 11, 12, and 21%, respectively. MPS VII was the lowest among all MPS and accounted for 6%.
3.3.6. Birth Prevalence of MPS in Tunisia
In Tunisia, MPS patients were diagnosed based on urinary GAG and/or appropriate enzyme assay during the period of 1988 to 2005. Of the 132 suspected MPS, only 96 cases were confirmed as MPS [27]. The birth prevalence was calculated including only 76 patients and the total number of live births (3,309,091) during this time period. The combined birth prevalence of all MPS was 2.27 per 100,000 live births. The birth prevalence of MPS III was the highest among all MPS, 0.7, comprising 32% of all MPS cases diagnosed. The birth prevalence of MPS I, II, IV, and VI was 0.63, 0.29 (male live births), 0.45, and 0.30, respectively, which accounted for 25, 8, 21, and 13% of all MPS, as shown in Fig. 1. No case for MPS VII was reported.
3.3.7. Birth Prevalence of MPS in Australia
In Australia, 188 MPS patients were diagnosed over a period of 17 years (1980–1996). The combined birth prevalence of all MPS was 4.46 per 100,000 live births [17]. The birth prevalence of MPS II, MPS IVA, and MPS VI was 0.74, 0.59, and 0.43 per 100,000 live births, respectively, which accounted for 17, 13, and 10%, respectively as shown in Fig. 1. The birth prevalence of MPS III was the highest, 1.51 per 100,000 live births, comprising 35% of all MPS. The birth prevalence of MPS I (1.14) was slightly lower than MPS III and accounted for 26% of all MPS. The birth prevalence of MPS VII was 0.047 per 100,000 live births and accounted for 1.1%. An epidemiological study of MPS was carried out by Nelson et al. in Western Australia during 1969 and 1996. Only 22 MPS cases were diagnosed with a combined birth prevalence of 3.43 per 100,000 live births [19]. The birth prevalence of MPS III was very high 1.71 per 100,000 live births that accounted for 50% of all MPS. The birth prevalence of MPS I was 0.94 and accounted 27%. The birth prevalence of MPS II and MPS VI was both 0.31 (MPS II 0.61 for male live births), each accounting for 9% of MPS. The birth prevalence of MPS IVA (0.16) was comparatively lower than all other MPS, accounting for 4.5 of all MPS, as shown in Fig. 1. In Australia, no case of MPS VII has been reported.
3.3.8. Birth Prevalence of MPS in British Columbia, Canada
Lowry and Renwick [9] studied on the frequency of MPS in British Columbia, Canada in 1971. Lowry et al. and Applegarth et al. [10, 11] updated the study. A total of 20 MPS cases were diagnosed over a period of 28 years between 1969 and 1996 [9–11]. The combined birth prevalence of all MPS was 1.94 per 100,000 live births. The birth prevalence of MPS I was the highest (0.58) among all MPS comprising 30%. The birth prevalence of each MPS II, III, IV, VI, and VII was 0.10 (0.19 per 100,000 male live births), 0.29, 0.39, 0.29, and 0.29 per 100,000 live births, respectively, accounting for 5, 15, 20, 15, and 15% of all MPS, as shown in Fig. 1.
3.3.9. Birth Prevalence of MPS in Brazil
The Brazilian Health System database reported 56,587,867 live births between 1994 and 2012 (19 years). A total of 600 cases of MPS were diagnosed in this period (138 cases with MPS I, 220 with MPS II, 65 with MPS IVA, and 177 with MPS VI). This report [28] did not provide data for MPS III, MPS VII, and MPS IX. The combined birth prevalence was 1.04 per 100,000 live births. The highest birth prevalence was associated with MPS II which was 0.38 (0.75 per 100,000 male live births) that accounted for 37% of all MPS cases (not including MPS III, VII, and IX). The birth prevalence of MPS I, IVA, and VI was 0.24, 0.11, and 0.31 per 100,000 live births, respectively that accounted for 23, 11, and 30% of all reported MPS, as shown in Fig. 1.
3.3.10. Birth Prevalence of MPS in the Czech Republic
In the Czech Republic, LDS cases were diagnosed at the Institute of Inherited Metabolic Disorders in Prague between 1975 and 2008 (34 years). During this period, there were 4,261,897 live births. Out of 478 LDS patients, 119 patients were identified as MPS. The initial diagnosis was based on demonstration of accumulation of substrates, confirmed by the deficiency of relevant enzymes. The combined birth prevalence of all MPS was 3.72 per 100,000 live births [8]. The birth prevalence of MPS III was the highest, 0.91 per 100,000 live births that accounted for 20% of all MPS. The birth prevalence of MPS I and MPS IV was almost the same (0.72 and 0.73, respectively), which was slightly lower in the case of MPS II (0.43 or 0.83 male live births). The birth prevalence of unspecified MPS and multiple sulfatase deficiency (MSD) was 0.6 and 0.26 per 100,000 live births, respectively. The birth prevalence of MPS VI and VII was very low (0.05 and 0.02 per 100,000 live births). The percentages of MPS I, II, and IV were 17, 18, and 13%, respectively. MPS VI and VII had a very low incidence as shown in Fig. 1.
3.3.11. Birth Prevalence of MPS in Denmark, Norway, and Sweden
In Denmark and Sweden, data was collected over a period of 30 years between 1975 and 2004; and in Norway data was collected over 26 years between 1979 and 2004 [16].
Denmark
In Denmark, diagnosis of MPS was performed at the Kennedy Institute in Glostrup and the Department of Clinical Genetics at Rigshospitale, Copenhagen. 33 MPS cases were diagnosed with a combined birth prevalence of 1.77 per 100,000 live births [16]. The birth prevalence of MPS I, II, III, IV, and VI was 0.54, 0.27, 0.43, 0.48, and 0.05, respectively, accounting for 30, 15, 24, 27 and 3% of all MPS. No case of MPS VII was reported.
Norway
The diagnosis of MPS was carried out at the Department of Clinical Chemistry and Neurochemistry in Molndal, and more recently at the section for Genetics at Rikshospitalet, Oslo [16]. 45 MPS cases were diagnosed with a combined birth prevalence of 3.08 per 100,000 live births. The birth prevalence of MPS I was very high, 1.85, accounting for 60% of all MPS, followed by MPS IV (0.76) comprising 24%. The birth prevalence of MPS II (4%), III (9%), and VI (2%) was 0.13, 0.27, and 0.07 per 100,000 live births, respectively [16].
Sweden
Diagnosis of MPS was done by two laboratories, Center for Inborn Errors of Metabolism, Karolinska University Hospital, Huddinge, Stockholm, and Department of Clinical Chemistry and Neurochemistry in Molndal, Sahlgrenska University Hospital Gothenburg. Fifty-two cases of MPS were diagnosed with a combined birth prevalence of 1.75 per 100,000 live births. The birth prevalence of MPS I and III was both 0.67, each comprising 38% of all MPS. The birth prevalence of MPS II was 0.27 that accounted 15%. The birth prevalence of both MPS IV and VI was low (0.07) comprising only 4% each. No case of MPS VII was found [16].
3.3.12. Birth Prevalence of MPS in Estonia
In Estonia, diagnosis of MPS was carried out between 1985 and 2006 (22 years) [14]. The data was collected from Children’s Hospital in Tallinn and the Department of Genetics of Tartu University Hospital. Selective screening of MPS was done by toluidine blue spot test [29] followed by quantitative analysis of GAGs in urine. When urinary GAG levels were elevated, enzyme analysis was performed to confirm the MPS. 15 MPS cases were identified, and a total of 370,298 live births were recorded during the study period, to give a combined birth prevalence of 4.05 per 100,000 live births [14]. MPS II had the highest birth prevalence of 2.16 (4.2 per 100,000 male live births), followed by MPS IIIA with 1.62 per 100,000 live births. The birth prevalence of MPS VI was 0.27 per 100,000 live births. Among all MPS cases, MPS II contributed to 53% of all MPS. MPS III and MPS VI comprised 40 and 7%, respectively as shown in Fig. 1. In Estonia, no case of MPS I, IV, and VII was identified.
3.3.13. Birth Prevalence of MPS in Germany
A retrospective epidemiological survey study of MPS was carried out in Germany to estimate cumulative incidences of different types of MPS during a period of 16 years between 1980 and 1995 [12]. According to data obtained from the German Bureau of Statistics, there were 13,410,924 live births during this period. 474 cases of MPS were identified, giving a combined birth prevalence of 3.51 per 100,000 live births. The diagnosis of all cases were confirmed by enzyme assay in serum, leukocytes and/or fibroblasts [12]. The birth prevalence for MPS I was 0.69 per 100,000 live births. The birth prevalence for MPS II was 0.64 per 100,000 live births (1.3 per 100,000 male live births). The birth prevalence for MPS III was very high, at 1.57 per 100,000 live births. Birth prevalence for MPS IVA and MPS VI was lower, at 0.38 and 0.23 per 100,000 live births, respectively. MPS III accounted for 44% of all MPS. MPS I, II, IV and VI accounted for 20, 18, 11, and 7%, respectively, as shown in Fig. 1. Interestingly, the births of Turkish origin in this study (617,013) accounted for a relatively high proportion of patients with MPS IIIB (33%), IVA (22%), and VI (52%) even though they accounted for less than 5% of total births. No case of MPS VII was detected in Germany.
3.3.14. Birth Prevalence of MPS in the Netherlands
In the Netherlands, 331 MPS cases were diagnosed based on enzyme assay during a period of 27 years between 1970 and 1996 with the combined birth prevalence of 4.5 per 100,000 live births [7]. During this period, the number of live births for different MPS was determined in detail [7]. MPS I had a birth prevalence of 1.19 per 100,000 live births. The birth prevalence of all types of MPS III was 1.89 per 100,000 live births while the birth prevalence of MPS II was 0.67 per 100,000 live births (1.3 per 100,000 male live births). The birth prevalence of MPS IVA, IVB, VI, and VII was 0.22, 0.14, 0.15, and 0.24, respectively. The combined MPS III accounted for 47%, followed by MPS I and MPS II that accounted for 25 and 15.5%, respectively. MPS IV accounted for 8%; however, both VI and VII comprised 2% as shown in Fig. 1.
3.3.15. Birth Prevalence of MPS in Northern Ireland
An epidemiological study of MPS was carried out in Northern Ireland over a period of 28 years between 1958 and 1985, and in total, 34 MPS cases were reported. The diagnosis was confirmed by appropriate enzyme assay [18]. There were 7 suspected MPS I cases that had died during the ascertainment period. All children had typical clinical and radiological findings. They were also positive with urinary GAGs, and α-L-iduronidase enzyme activity showed intermediate values. There were also 4 MPS II cases that died during the ascertainment period before definitive enzymatic analysis [18]. During this period, 839,517 total live births and 432,849 male live births were registered. The combined birth prevalence was 4.0 per 100,000 live births. MPS I had the highest birth prevalence of 1.66 per 100,000 live births followed by MPS IVA with 1.3 per 100,000 live births. The birth prevalence of MPS II and MPS III was 0.71 (1.39 male live births) and 0.36 per 100,000 live births, respectively. Among all MPS cases, MPS I accounted for the highest proportion (41%), followed by MPS IVA, II, and III, which were 32, 18, and 9%, respectively, as shown in Fig. 1. No case for MPS VI and VII was identified.
3.3.16. Birth Prevalence of MPS in Poland
In Poland, a retrospective epidemiological survey study of MPS was implemented to estimate birth prevalence. The live births registered in the Polish Bureau of statistics over a period of 4 decades were 21,686,890 from 1970 until 2010. A total of 392 cases were diagnosed with MPS to give a combined birth prevalence of 1.80 per 100,000 live births [13]. Electrophoresis method was used for the initial diagnosis of MPS which was based on the demonstration of accumulated GAGs in the body fluids. Confirmation of various MPS was made by the deficiency of relevant enzymes. In Poland, MPS III had the highest birth prevalence of 0.86 per 100,000 live births, followed by MPS II, I, and IV with 0.46, 0.22, and 0.14 per 100,000 live births, respectively. The birth prevalence of unspecified MPS was 0.11 per 100,000 live births. The birth prevalence of MPS VI was very low (0.0132) as compared to other MPSs. Among all MPS cases, MPS III accounted for 48%. MPS I, II, IV, and VI accounted 12, 25, 8, and 1%, respectively as shown in Fig. 1. No case for MPS VII was reported in Poland.
3.3.17. Birth Prevalence of MPS in Portugal
In Portugal, 62 MPS cases were identified over a period of 20 years from 1982 until 2001 [20]. The combined birth prevalence was 4.8 per 100,000 live births. The diagnosis was determined by urinary GAGs which were confirmed by enzyme activity in the blood sample. The birth prevalence of MPS I was 1.33 per 100,000 live births and accounted 13%. The birth prevalence of MPS II, III, IVA, and VI was estimated as 1.09, 0.84, 0.6, and 0.42 per 100,000 live births, respectively. The birth prevalence of MSD was 0.48 per 100,000 live births. MPS II comprised 34% of all MPS followed by MPS III (23%), VI (16%), and IVA (10%) as shown in Fig. 1.
3.3.18. Birth Prevalence of MPS I in the United States
In the United States, four States; Illinois, Missouri, Kentucky and Pennsylvania have implemented newborn screening for MPS I which is based on measurement of IDUA enzyme activity level in dried blood spot (DBS) specimens (http://www.babysfirsttest.org). Illinois and Missouri have published data from their screening programs. In addition, a University of Washington group reported an assessment of MS/MS LSD-multiplex screening methods on anonymous DBS, with follow-up genetic testing on these same DBS samples [30, 31]. Published reports of population-based pilot newborn screening for MPS I with diagnostic confirmation have come from programs in Italy and Taiwan [32, 33].
State of Illinois
According to Illinois NBS program contacts, from November 2014 through December 18, 2014, 17,300 newborns were screened [31]. Of the 17,300, 17 newborns were called out (0.1%) and repeated in triplicate before reporting. Referral results were as follows: None of these were confirmed as positive for MPS I; 15 were false positives, and 2 had not yet been confirmed.
State of Missouri
In Missouri as of 2014, 174,636 DBS had been screened for MPS I [31]. The screen yielded 70 hits. Out of 70, they confirmed one severe MPS I; 58 were false positives, 9 had not yet been confirmed, and 2 samples were lost to follow-up. NBS results showed the birth prevalence of MPS I (of all types), 1.1 per 100,000 live births, in Illinois and Missouri [31].
University of Washington Study
Scott et al. [30] evaluated MS/MS multiplex screening procedures for three LSDs using anonymous DBS from the Washington State newborn screening program. For MPS I disease, a cutoff of IDUA activity ≤ 1.15 μmol/h/l, corresponding to ≤32% of the mean, was used. Of the 106,526 samples, 9 screened positive for low IDUA activity. Based on these findings, overall birth prevalence of infants who “may eventually develop clinical symptoms of MPS I” was 1/35,700 (95% CI: 1/43,000–1/11,100). However, the phenotype of these hits is not known and is likely an over estimate of prevalence.
Puckett et al. [34] described the incidence and birth prevalence of MPS in the United States using the database from the National MPS Society between 1995 and 2005. Population information was obtained from the National U.S. Census Bureau. The combined birth prevalence was calculated to be 1.2 per 100,000 live births. Individual birth prevalence of MPS I, II, III, IV, VI, or VII, was 0.34, 0.29, 0.38, 0.09, 0.05, or 0.05 per 100,000 live births, respectively. MPS III had the highest incidence, followed by MPS I, II, and IV accounting for 31.7, 28.3, 24.2, and 7.5%, respectively. MPS VI and VII was 4.2% each as shown in Fig. 1.
The overall birth prevalence and period of study is summarized in Table 1. A demographic distribution of the combined birth prevalence of in different countries around the world is shown in Fig. 2A.
Table 1.
MPS cases and prevalence in different countries by continent.
| Country | Study period | Total MPS Cases | MPS I | MPS II | MPS III all type | MPS IV A &B | MPS VI | MPS VII | Conbined Prevalence | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| Africa | ||||||||||
| Tunisia | 1970–2005 | 96 | 0.63 | 0.29 | 0.7 | 0.45 | 0.3 | N/A | 2.27 | Turkia et al. [2009] |
| Asia | ||||||||||
| Japan | 1982–2009 | 467 | 0.23 | 0.84 | 0.26 | 0.15 | 0.03 | 0.02 | 1.53 | Present Study* |
| Saudi Arabia | 1983–2008 | 28 | 3.62 | N/A | 1.8 | 3.62 | 7.85 | N/A | 16.9 | Moammar et al. [2010] |
| South Korea | 1994–2013 | 147 | 0.21 | 0.74 | 0.25 | 0.13 | 0.019 | N/A | 1.35 | Cho et al. [2014] |
| Taiwan | 1984–2004 | 130 | 0.11 | 1.07 | 0.39 | 0.33 | 0.14 | N/A | 2.04 | Lin et al. [2009] |
| Australia | ||||||||||
| Australia | 1980–1996 | 188 | 1.14 | 0.74 | 1.51 | 0.59 | 0.43 | 0.047 | 4.46 | Meikle et al. [1999] |
| W Australia | 1969–1996 | 18 | 0.94 | 0.31 | 1.71 | 0.16 | 0.31 | N/A | 3.43 | Nelson et al. [2003] |
| Europe | ||||||||||
| Czech Republic | 1975–2008 | 119 | 0.72 | 0.43 | 0.91 | 0.73 | 0.05 | 0.02 | 3.72 | Poupetova et al. [2010] |
| Denmark | 1975–2004 | 33 | 0.54 | 0.27 | 0.43 | 0.48 | 0.05 | N/A | 1.77 | Malm et al. [2008] |
| Estonia | 1985–2006 | 15 | N/A | 2.16 | 1.62 | N/A | 0.27 | N/A | 4.05 | Krabbi et al. [2012] |
| Germany | 1980–1995 | 474 | 0.69 | 0.64 | 1.57 | 0.38 | 0.23 | N/A | 3.51 | Baehner et al. [2005] |
| Northern Ireland | 1958–1985 | 34 | 1.66 | 0.71 | 0.36 | 1.3 | N/A | N/A | 4 | Nelson et al. [1997] |
| Norway | 1979–2004 | 45 | 1.85 | 0.13 | 0.27 | 0.76 | 0.07 | N/A | 3.08 | Malm et al. [2008] |
| Poland | 1970–2010 | 392 | 0.22 | 0.46 | 0.86 | 0.14 | 0.0132 | N/A | 1.8 | Zurecka et al. [2015] |
| Portugal | 1982–2001 | 353 | 1.33 | 1.09 | 0.84 | 0.6 | 0.42 | N/A | 4.8 | Pinto et al. [2004] |
| Sweden | 1975–2004 | 52 | 0.67 | 0.27 | 0.67 | 0.07 | 0.07 | N/A | 1.75 | Malm et al. [2008] |
| Switzerland | 1975–2008 | 41 | 0.19 | 0.46 | 0.38 | 0.38 | 0.11 | 0.038 | 1.56 | Present study* |
| The Netherlands | 1970–1996 | 331 | 1.19 | 0.67 | 1.89 | 0.36 | 0.15 | 0.24 | 4.5 | Poorthuis et al. [1999] |
| North America | ||||||||||
| British Columbia | 1969–1996 | 20 | 0.58 | 0.1 | 0.29 | 0.39 | 0.29 | 0.29 | 1.94 | Applegrath et al. [2000] |
| The United States | 1995–2005 | N/A | 0.34 | 0.29 | 0.38 | 0.09 | 0.05 | 0.05 | 1.2 | Yana Puckett et al. [2017] |
| South America | ||||||||||
| Brazil | 1994–2012 | 1069 | 0.24 | 0.38 | N/A | 0.11 | 0.31 | N/A | 1.04 | Federhen et al. [abstract, 2016] |
Figure 2.
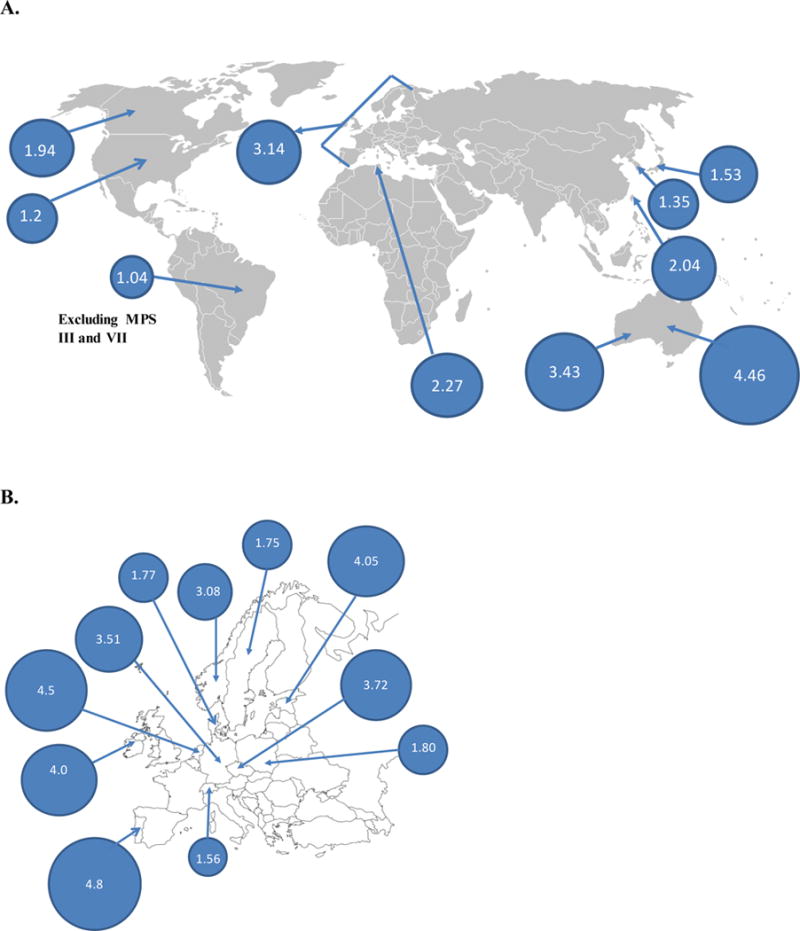
Demographic distribution of the combined birth prevalence of MPS.
A. The combined birth prevalence of Europe is 3.14; the birth prevalence of individual countries is shown in Fig. 2B. The birth prevalence of Japan, South Korea, and Taiwan is 1.53, 1.35, and 2.04, respectively. The birth prevalence of Western Australia is 3.43; overall, Australian birth prevalence is shown as 4.46. The birth prevalence of Tunisia is 2.27. The birth prevalence of British Columbia, the United States, and Brazil is 1.94, 1.2, and 1.04, respectively.
B. Demographic distribution of the combined birth prevalence of MPS in Europe.
4. Molecular Genetics
Method
The data in this section was obtained from the literature and mutation has been updated from the HGMD data base, which is described below.
MPS disorders are distributed worldwide; however, there are regional differences in their distribution of overall MPS or a specific type of MPS.
4.1. MPS I Mutations
MPS I is caused by mutations in the gene encoding IDUA, which is located on chromosome 4p16.3 [35, 36]. The birth prevalence of MPS I is higher in Northern European countries such as Norway (accounts for 60% of all MPS), Demark, Sweden [16], and Northern Ireland [18]. Frequency is also high in Germany [12] and the Netherlands [7].
As of October of 2016, there were 224 mutations identified in HGMD database (http://www.hgmd.cf.ac.uk/ac/gene.php?gene=IDUA); missense/nonsense mutations are most common (58.5 %) with 131 different mutations, followed by splice site mutations (16.1%), deletions (13.8%), and insertions (7.1%) [37].
The two most common IDUA mutations are p.W402X and p.Q70X. The frequency of the p.W402X mutation in MPS I patients is estimated to be 4% in Russia [38], 11% in Italy [39, 40], and 20% in Brazil [41, 42]. p.W402X, accounts for 38.8% of mutant alleles in Spain and 24% in Poland [43]. The frequency of this mutation in the United States is 39%, [44].
The p.Q70X mutation is also common in Europe, accounting for 35% of mutant alleles [45]. Bertola et al. [43] reported that in a cohort of 102 European patients with MPS I, mutation p.Q70X accounted 39% IDUA alleles in Poland but was lower in Italy (14.9%), Spain (8.3%), and Turkey (7.1%). The United States the frequency of this mutation in MPS I patients is 30% [44]. A genotype/phenotype relationship of these two common mutations has been reported [43].
By contrast, c.704ins5 and p.R89Q are the most common in Japanese MPS I patients and p.A79V and p.L346R are most commonly found in Chinese MPS I patients [46–48]. In 2016, Kwak et al. reported the p.L346R and c.704ins5 mutations as the most commonly found mutation in Korean patients, which were different to those found in Caucasian patients [45, 46].
The overlap in common mutations in the Asian populations indicates early mutations have spred between these populations. The higher incidence of MPS I in most European countries indicates that mutations found in Caucasian populations are more common than those found in the Asian populations.
4.2. MPS II Mutations
MPS II is associated with a mutation in the IDS gene located on chromosome Xq28. The IDS gene comprises 9 exons that encode the active IDS enzyme with 550 amino acids. Almost half of the patients with MPS in South Korea and Taiwan have MPS II (Hunter syndrome) [15, 23], and the same is true for other Asian countries including Japan (current study) and China [24]. There are a total of 584 mutations reported for IDS in the HGMD database (http://www.hgmd.cf.ac.uk/ac/gene.php?gene=IDS). Missense/nonsense mutations, have a total of 296 mutations (50.7 %), followed by deletions (106;18.2%), splice site mutations (54; 9.2%), and insertions (46; 7.9%) [37].
In most cases, the severe phenotype of MPS II is associated with gross alterations of the structure of the IDS gene, such as large deletions and recombination events involving IDS-IDS2, whereas a nucleotide change by missense mutation or that in minimal mutations by deletion, insertion, or even both may exhibit attenuated clinical presentations.
The higher birth prevalence of MPS II in East Asian populations is possibly due to the higher allele frequency of the p.R468 mutations in the IDS gene. Five patients in a group of 43 Japanese individuals with MPS II (11.6%) had p.R468 mutations [49]. Three had p.R468Q, and the other two had p.R468W and p.R468L, respectively [49]. In Taiwanese patients, p.R468 mutations accounted for nearly half of all mutations (6/14; 42.9%) [50]. These were equally divided into p.R468W and p.R468Q mutations [50]. In addition, in a cohort of 38 Chinese MPS II patients, 13.2% had the p.R468 mutation [51]. R468 was mutated in 5 individuals, of whom three were p.R468W, one was p.R468Q, and one showed the combination of p.R468W with p.P423S. In contrast, only 6% of mutant alleles in MPS II patients in South America had R468 mutations [52]. It is noteworthy that the mutated codon for R468 is considered as “hot spot” for IDS gene mutation and different mutations have been identified in distinct populations [49–51, 53–56]. Kosuga et al. found 8 recombination events involving IDS-IDS2 in the Japanese population [57]. IDS-IDS2 recombination mutations were also observed most frequently in Korean patients with a severe phenotype [23].
4.3. MPS III Mutations
MPS III is characterized by four subtypes based on a deficiency of four different enzymes that are described in types of MPS section above. The relative frequency of MPS III (IIIA; 20–30% and IIIB; 10–14%) is higher in European countries including Estonia [14], Germany [12], Poland [13], Sweden [16], and the Netherlands [7]. Frequency is also high in Australia [19]. MPS IIIB is the most common subtype in Greece [58, 59]. MPS IIIC is common in European countries (Germany, The Netherlands, Czech Republic, and Portugal etc.) but less common than MPS IIIA. MPS IIID is the rarest among all four groups of MPS III and frequency is similar in all regions.
MPS IIIA has the most severe phenotype amongst the MPS III groups as it has an early onset, rapid symptoms progression and short survival. The gene locus for human SGSH is on chromosome 17q25.3 [60]. The human SGSH gene has an approximate length of 11 kb with 8 exons [61]. As of October of 2016, 142 mutations in the SGSH gene have been described including missense/nonsense mutations (109 different mutations, 78.8%), small deletions (17; 12%), insertions (9; 6.3%), and splicing mutations (2; 1.4%) (Human Gene Mutation Database-HGMD; http://www.hgmd.cf.ac.uk/ac/gene.php?gene=SGSH). MPS IIIA is the most common subtype in Northern Europe [7, 12, 18, 58, 59, 62] with p.R245H as a common mutation. In the Netherlands, the incidence of this mutation in 45 unrelated MPS IIIA patients was present in 51 alleles, representing 56.7% of the total mutant alleles [62]. Therefore, this common mutation correlates with the higher frequency of MPS IIIA in Northern European countries. In Australia, a common mutation, p.R245H, in SGSH alleles accounted for 31% of MPS IIIA patients [63].
MPS IIIB is caused by a defect in the NAGLU gene. The gene was isolated, characterized, and localized to chromosome 17q21.3 [64]. This gene spans 83 kb, contains 6 exons and encodes a protein of 743 amino acids, which plays a critical role in the cleavage of heparin glycosilic bounds. As of October of 2016, 156 different mutations in the NAGLU gene have been identified, including missense/nonsense mutations (107; 68.6%), small deletions (23; 14.7%), small insertions (13; 8.3%), and splicing substitutions (5; 3.2%) (http://www.hgmd.cf.ac.uk/ac/gene.php?gene=NAGLU). MPS IIIB is the most common subtype in Greece accounting for 81% of all MPS III cases diagnosed (26 of 32 cases). Three mutations (p.Y140C, p.H414R, and p.R626X) were found at high frequency and accounted for approximately 70% of the studied alleles [65]. Mutation analysis in MPS IIIB revealed a broad allelic heterogeneity that reflects the wide spectrum of clinical phenotypes reported for MPS IIIB patients. The p.R565P mutation is common in Japanese MPS III patients. Weber et al. reported p.R643C and p.R297X as common mutations, each accounted for around 20% of MPS IIIB alleles in Dutch patients. Mutation p.R643C is associated with an attenuated form of the disease [66]. Mutations p.R297X, p.P521L, p.R565W, and p.R626X each accounted for 6% in Australian MPS IIIB population and conferred a severe phenotype [66]. A common mutation in Portuguese MPS IIIB patients is p.R234C (32% of the mutated alleles) [67] which is a founder mutation in the Portuguese population.
MPS IIIC is a severe disorder, caused by mutations in the HGSNAT gene and is characterized by progressive neurological deterioration, with retinal degeneration as a prominent feature. This gene is localized to chromosome 8p11.1 is 62.4 kb long, and contains 18 exons coding for a protein of 635 amino acids [68, 69]. As of October of 2016, 66 mutations in the HGSNAT gene have been reported including, missense/nonsense mutations (a total of 37, 56%), splicing site mutations (14, 21.2%), deletions (5, 7.8%) and insertions (5, 7.8%). The deduced HGSNAT amino acid sequence predicts 11 transmembrane domains with the N-terminus being inside the lysosome and the C-terminus being in the cytosol [69]. Ruijter et al. have reported two common mutations p.R344C and p.S518F in Dutch MPS IIIC patients that accounted for 22.0% and 29.3%, respectively [70]. Mutation p.R344C was also found in families from France, Germany, Singapore, and the UK.
MPS IIID was first reported in 1980 by Kresse et al. in a patient from East India [71]. Since then, only 19 MPS IIID patients have been reported [72–82]. As of October of 2016, 24 mutations in the GNS gene have been reported; missense/nonsense mutations (a total of 7 mutations, 29.2%), deletions (5, 20.8%), insertions (4, 16.7%), and splicing site mutations (3, 12.5%) (http://www.hgmd.cf.ac.uk/ac/gene.php?gene=GNS). MPS IIID is the rarest subtype of MPS III in the Netherlands [7]. The GNS gene was identified by Robertson et al. [83]. It consists of 14 exons [84] dispersed over approximately 43 kb and is located on chromosome 12. The predicted protein has 552 amino acids including a leader peptide of 36 amino acids, a sulfatase domain (45–492, Swiss-Prot database, entry P15586), and contains 13 potential N-glycosylation sites. In 2003, Beesley et al. and Mok et al. [72, 84] identified the first 2 disease-causing mutations in GNS. In 2007, another five mutations were reported [73, 77]. In 2010, Valstar et al. reported 12 new patients with MPS IIID and 15 novel GNS mutations in 16 patients [85].
4.4. MPS IV Mutations
MPS IVA is associated with a mutation in GALNS gene coding for GALNS enzyme. The GALNS gene is located on chromosome arm 16q24.3 [86, 87] and spans approximately 50 kb containing 13 introns and 14 exons. A single splice product, 1566-bp GALNS mRNA, codes for a 522-amino acid precursor. After the cleavage of a 21-amino acid N-terminal signal peptide and glycosylation, it becomes 40- and 15-kDa subunits of the mature active enzyme [88, 89]. Several different common mutations for MPS IVA represents different region irrespective of its incidence. Tomatsu et al. summarized overall 148 unique mutations until 2005 [90]. As of October of 2016, 328 mutations in the GALNS gene have been reported (http://www.hgmd.cf.ac.uk/ac/gene.php?gene=GALNS); missense/nonsense mutations account for a total of 243 kinds (74.1%), deletions (32; 9.8%), splicing site mutations (32; 9.8%), and insertions (5; 1.5%). Several mutations are common; the most prevalent recurrent mutations in the GALNS gene are c.1156C4T (p.R386C), c.901G4T (p.G301C), c.337A4T (p.I113F), c.1A4G (p.M1?), c.757C4T (p.R253W), c.871G4A (p.A291 T), c.935C4G (p.T312S), and c.1171A4G (p.M391 V), accounting for 8.9%, 6.8%, 5.7%, 2.3%, 2.1%, 1.8%, 1.8%, 1.8%, and 1.8%, respectively [91–97].
The p.I113F and p.T312S mutations are specific to British/Irish populations and account for 18% and 14% of all British/Irish mutations, respectively [96, 98]. These two founder mutations are associated with a three to six-fold increase in the incidence of MPS IVA in Northern Ireland compared to other ethnic populations. Both mutations were probably introduced to Australia by Irish migrants during the 19th century. Over 90% of p.G301C mutations were found in Colombian MPS IVA patients with the same haplotype, and this mutation accounted for around 70% of mutant alleles in Colombians, suggesting a founder mutation [91]. The p.R386C, p.A291T, and p.M391V mutations have been detected randomly in various ethnic populations. The most prevalent c.1156C>T transitional mutation at CpG dinucleotide (p.R386C) was found in 34 alleles from 25 patients of American (Caucasian), Argentine, Brazilian, British, Chilean, Colombian, German, Italian, Japanese, Mexican, Polish, and Turkish origin [90, 93, 94, 97, 99–101].
The p.M1? (Mutation occurs in initiation codon where A is changed to G. As a result, no functional protein is predicted) mutation is the most common mutation in Italian MPS IVA patients, accounting for 26.7% of the mutant alleles [94]. The p.G340D common mutation was found in the Chinese MPS IVA patients, accounting for 16.7% of the total number of mutant alleles [47]. Pajares S, et al. described mutations in GALNS gene in 16 Spanish MPS IV A patients. Mutations, p.G301C, p.R386C, c.423_566del and p.Y254C, accounted for 60% of the alleles [102]. Common mutations, p.D288G and p.R386C, in GALNS were reported in Tunisia by Chkioua et al. [103].
Thus, different common mutations for MPS IVA are representing different region irrespective of its incidence.
MPS IVB is caused by mutations in the β-galactosidase (GLB1) gene that leads to accumulation of ganglioside GM1 and keratan sulfate. If accumulation of ganglioside GM1 predominates, patients are diagnosed with GM1 gangliosidosis while accumulation of keratan sulfate results in a MPS IV B diagnosis. Paschke et al. performed mutation analyses in 17 juvenile and adult patients from various European regions with a deficiency in beta-galactosidase and skeletal abnormalities [104]. A two-base substitution (851-852TG>CT; p.W273L) was present in 14 of the 15 Morquio B cases, indicating a strong founder effect. Mutations have also been reported in specific populations in a few small studies [105–107]. In Spain, a p.R59H mutation was more prevalent than others, accounting for 19 of the 68 alleles (27.9%) [108]. Mutations c.588_591insT and p.D441N were present in four alleles each (5.9%). Mutations p.R201H, p.K346N, and p.R590C were each found in 3 alleles (4.4%), while the remaining 47.1% mutations found once or twice. In 2012, Lei et al. reported GLB1 activities and mutation analysis of three patients from mainland of China, one with Morquio B syndrome and two with GM1 gangliosidosis [109]. The three novel mutations, p.Y485C, p.Y270F, and p.L337P, are thought to be disease-causing mutations. As of October of 2016, 210 mutations in the GLB1 gene have been reported (http://www.hgmd.cf.ac.uk/ac/gene.php?gene=GLB1); missense/nonsense mutations have a total of 160 mutations (76.2%), deletions (8.1%), splicing site mutations (7.6%), and insertions (5.7%).
4.5. MPS VI Mutations
MPS VI is caused by mutations in N-acetylgalactosamine-4-sulfatase (4S; or ARSB), which degrades C4S and DS. As of October of 2016, 192 mutations were found in the ARSB gene (http://www.hgmd.cf.ac.uk/ac/gene.php?gene=ARSB); missense/nonsense mutations have a total of 143 mutations (74.5%), deletions (12.5%), splicing site mutations (5.7%), and insertions (3.1%). In 2001, Litjens and Hopwood reported 45 clinically relevant mutations in the human 4S gene [110]. Missense mutations were the largest group, with 31 identified mutations. Nonsense mutations and small insertions or deletions comprise the remainder, with seven mutations each. In 2003 and 2005, Petry et al. described a common mutation, 1533del23, among Brazilian MPS VI patients found in 6/26 alleles (23.1%), which also occurs in Portuguese MPS VI patients, although the frequency is unknown [111–116]. In 2007, Karageorgos et al. reported a cohort of 105 MPS VI patients from 15 countries, but primarily from Australia, Europe, North America, and South America that represented more than 10% of the world’s MPS VI patients [113]. The missense mutations, p.R315Q and p.S384N, were frequent mutations appearing in 11.4% and 12.4% of patients, respectively. The p.R315Q and p.S384N were detected in 5 patients on the same allele. Three of which came from Portugal, and two have been detected on the same allele from Brazil [113]. The presence of both mutations on the same allele has a founder effect on the Portuguese patients.
4.6. MPS VII Mutations
MPS VII is caused by a mutation in the β-D-glucuronidase enzyme (GUSB) responsible for degradation of CS, DS, and HS. The GUSB gene is located on chromosome 7. GUSB mRNA encodes a 651-amino acid residue precursor polypeptide. After cleavage of a 22-amino acid N-terminal signal peptide and glycosylation, a 78-kDa monomer is transported to lysosomes and becomes the mature active enzyme [117, 118]. GUSB contains three domains, a jelly roll-like domain containing the lysosomal targeting residues, a TIM barrel domain (active site), and an immunoglobulin region constant domain [119, 120]. 49 unique mutations have been observed in the patients suffering from Sly syndrome of which 36 were missense mutations, two splice site alterations, five deletions, and six nonsense mutations [121]. A total of 63 mutations are currently listed for GUSB gene in the Human Gene Mutation Database (HGMD) (http://www.hgmd.cf.ac.uk/ac/gene.php?gene=GUSB); 52 are missense/nonsense mutations (82.5%), 4 deletions (6.3%), and 5 splicing site mutations (7.9%). Missense mutations such as p.L176F, p.P415L, p.P408S, p.A619V, p.R216W, p.R382C, and p.R477W were frequent [122, 123], while p.L176F was the most prevalent in the patients [124, 125]. The heterogeneity in GUSB gene mutations contributes to the extensive clinical variability among patients with MPS VII [121, 126–128].
4.7. MPS IX Mutations
Triggs-Raine et al. [6] showed mutation in HYAL1, a member of a tandemly distributed multigene family, encoding disparate hyaluronidase activities. There were two mutations in the HYAL1 alleles of one patient, one being a 1412G > A mutation that introduces a nonconservative amino acid substitution (p.E268K) in a putative active site residue and another was a complex intragenic rearrangement, 1361del37ins14, that resulted in a premature termination codon. It had previously been thought that lysosomal hyaluronidase was a single enzyme and that a genetic defect had not been identified because its importance in embryonic morphogenesis made its deficiency lethal in early development [129]. However, Triggs-Raine et al. [6] showed that there are two hyaluronidase genes, and a third was recently discovered. Each gene has markedly different tissue expression patterns, consistent with differing roles of each gene in HA metabolism. These duplicate genes provide an explanation for the unexpectedly mild phenotype of mucopolysaccharidosis IX and predict the existence of other hyaluronidase deficiency disorders. Imundo et al. described another single consanguineous family with three affected children with MPS IX, and thus, only two patients have been detected with MPS IX to date [5]. They found c.104delT deletion in HYAL1, resulting in a premature termination codon, p.V35AfsX25. No mutation has been found in HYAL2 and HYALA3.
5. Discussion
In this study, we have explored the birth prevalence of MPS in two countries, Japan and Switzerland and compared our results with the known birth prevalence in other countries.
We have demonstrated that 1) the birth prevalence rate of all MPS is similar in Japan (1.53) and Switzerland (1.56 per 100,000 live births), 2) the incidence of MPS II in Japan is as high as that in other East Asian countries, 3) the incidence and birth prevalence of MPS varies in each country and region, and 4) the high incidence and/or birth prevalence of some MPS is reflected by founder mutations.
The birth prevalence of MPS in Japan and Switzerland was lower, compared to most European countries including the Czech Republic (3.72), Estonia (4.05), Germany (3.51), Northern Ireland (4.0), Norway (3.08), Portugal (4.8), and the Netherlands (4.5) [7, 8, 12, 14, 16, 18, 20]. However, the birth prevalence of MPS in Japan was comparable to central European countries such as Denmark (1.77), Poland (1.80), and Sweden (1.75) [13, 16]. Some other Asian or African countries including Taiwan (2.04) and Tunisia (2.27) have a similar birth prevalence [15, 27]. The birth prevalence of MPS in British Columbia is also similar (1.94) to that in these countries [10]. The limitation in countries having a small number of MPS including British Columbia, Denmark, Estonia, Northern Ireland, Norway Sweden, and Switzerland is due to a small number of population. Therefore the data may not be very significant.
In MPS, there are unique common mutations in different MPS genes. Some common mutations in a particular MPS directly correspond to the high incidence of that particular MPS in a certain region or ethnic background; however, these mutations are not equally distributed among different regions. The birth prevalence of MPS often correlates with specific mutations that can predict the phenotype.
The birth prevalence of MPS I in Switzerland is low (0.19) as compared to that in Japan (0.23). This finding is incompatible with the fact that the incidence of MPS I is higher in European countries such as Norway (account for 60%), Demark (30%), Sweden (38%) [16], and Northern Ireland (41%) due to two common mutations, p.W402X and p.Q70X, in the IDUA gene [18], and moderate-high in Germany (19.5%) [12] and the Netherlands (25%) [7]. The frequency of p.W402X and p.Q70X mutations varies in different countries. The p.W402X mutation was the most common, with 38.8 % in Spain [43], and p.Q70X was the most common, with 39% and 62 %, respectively, in Poland and Scandinavia [43, 45]. Additionally, p.W402X and p.Q70X accounted for 39% and 30%, respectively, in the United States, showing the highest frequency [44].
Norway has the highest birth prevalence of MPS I with 1.85 per 100,000 live births. Other European countries including Northern Ireland (1.66), Portugal (1.33), and the Netherlands (1.19) have comparable birth prevalence. In Australia, the birth prevalence of MPS I is also high (1.14) comparable to European countries. In addition, British Columbia (0.58) and Tunisia (0.63) have a moderately high birth prevalence of MPS I comparable to other European countries such as the Czech Republic (0.72), Denmark (0.54), Germany (0.69), and Sweden (0.67). Moore et al. have reported an exclusive MPS I study based on the severity of disease and found 1.07/100,000 live births in England and Wales that are consistent with other European countries [130]. The newborn screening (NBS) for MPS I, is in progress in some US states suggests an overall birth prevalence of 1.1 per 100,000 live births (data from Missouri and Illinois [31]). However, recent data [34] described the birth prevalence of MPS in the United States as 1.2 per 100,000 live births. MPS I had the birth prevalence of 0.34/100,000 live births similar to Brazil and Asian countries as described by Federhen et al. [28].
In contrast to the incidence of MPS I, it is of great interest that in East Asian countries such as China [24], Japan (current study), South Korea [23], and Taiwan [15], the incidence of MPS II accounts for about 50% of all MPS. The birth prevalence of MPS II in most European countries is between 0.4 and 0.99 per 100,000 live births but notably very high in Estonia (2.16) and Portugal (1.09). The birth prevalence of MPS II in Japan (0.84) is similar to that in Taiwan (1.07) and other East Asian Countries. The birth prevalence of MPS II in Brazil is lower (0.38), similar to Czech Republic, Denmark, Norway, Sweden, Switzerland, and Tunisia. In general, East Asian countries including China, Japan, and Taiwan have a high incidence of MPS II that could be due to common mutation R468 in IDS gene [24, 49–51]. In contrast, in South Korea, IDS-IDS2 recombination mutations were most frequently found in Korean patients with a severe phenotype [23]. Another common mutation included p.G374G splicing mutation causing an attenuated phenotype [23].
The incidence of MPS III is also very high in European countries including Estonia [14], Germany [12], Poland [13], Sweden [16], and the Netherlands [7] and also Australia [19]. The incidence of MPS III (IIIA; 20–30% and IIIB; 10–14%) is higher in Germany, the Netherlands, and Western Australia than other MPS subtypes [7, 12, 19].
The birth prevalence of MPS III in Switzerland is 0.38, but the birth prevalence in Japan is even lower with 0.26. Most European countries have a high birth prevalence of MPS III (all types) per 100,000 live births that include the Czech Republic (0.91), Estonia (1.62), Germany (1.57), Poland (0.86), Portugal (0.84), Sweden (0.67), and the Netherlands (1.89). Australia also has high birth prevalence (1.51) of MPS III comparable to European countries. MPS IIIA is the most common subtype in Northern Europe [7, 12, 18, 58, 59, 62]. However, MPS IIIB is the most common subtype in Greece [65]. In addition, MPS IIID is the more common in the Netherlands, compared to other countries [7]. The similar birth prevalence of MPS III, 0.7 per 100,000 births, was also found in Tunisia. Heron et al. [131] carried out a study in France for MPS III patients and showed the incidence between 1990 and 2006 (period of 17 years), compared with Greece and United Kingdom. The birth prevalence of MPS III was 0.68 per 100,000 live births in France; however, in the UK, diagnosis of MPS III was evaluated in children born between 1990 and 2006, which was 1.16 comparable to other European countries listed above. In Greece, the birth prevalence was 0.97 per 100,000 live births for MPS III. The incidences of MPS III are higher in Germany, the Netherlands, and Western Australia with the common p.R245H mutation in MPS IIIA gene. In contrast, the incidence of MPS IIIB in Japan is only 8%, less than of Western countries, although the p.R565P mutation is present as a common mutation.
The birth prevalence of MPS IV is also variable in different countries; the birth prevalence in Switzerland is 0.38, but low in Japan (0.15). The birth prevalence of MPS IV was high in the Czech Republic (0.73), Northern Ireland (1.3), Norway (0.76), and Portugal (0.6).
The birth prevalence of MPS VI in Switzerland was low (0.11), comprising only 7.3% of all MPS and comparable to most of the European countries. However, the birth prevalence of MPS VI in Japan was very low (0.03). Brazil has a relatively high incidence of MPS VI (18.48% of all MPS) [132]. Moreover, Brazil had a higher birth prevalence of MPS VI (0.31) than most European countries but similar to Estonia, Tunisia, and West Australia [28]. There is a common mutation, 1533del23, among Brazilian MPS VI patients found in 23.1% of alleles, which also occurs in Portuguese MPS VI patients [111–116] that could reflect the high incidence of Brazilian MPS VI.
Jurecka et al. have reported MPS VI cases between 1983 and 2011 in Kazakhstan and Russia and compared with central and eastern European countries [133]. A high incidence of the p.R152W mutation was observed both in the whole series (42%) as well as in Russian patients (43%). The incidence rate ranged from 0.0363 to 0.64 per 100,000 live births in Poland and Lithuania, respectively.
MPS VII is one of the rarest MPS, along with MPS III D and MPS IX (which is the rarest one), with a very low birth prevalence. The prevalence of MPS is also influenced by the detection method in each country; in some cases with MPS III and IV, DMB-based spectrophotometry can give rise to false negatives, and therefore, it is important to consider other methods such as enzyme activity in serum, leukocytes, and/or fibroblasts for the confirmatory diagnosis.
To obtain accurate data on birth prevalence of MPS, it is essential that all patients are identified and that the total numbers of births in the population that gave rise to these patients is determined correctly. For example, MPS II patients are missing from the Brazilian data and as these can account for up to 50% of all MPS types, the overall birth prevalence in this country is likely to be under-estimated. Birth prevalence is highest in Europe and Australia; this may in part be due to availability of comprehensive medical records in these countries. Nevertheless, our data show that birth prevalence of MPS in Switzerland is low when compared to prevalence in other European countries. Our data from the Japanese population supports data from other East Asian countries that indicate that birth prevalence in Asian countries is low compared to European countries. Comprehensive population screening (e.g. newborn screening) for MPS would help determine true birth prevalence.
In conclusion, the overall birth prevalence of MPS (from 1.04 to 4.8/100,000) varies depending on the country and region or ethnic background. Some common mutations in specific regions directly contribute to the birth prevalence of each specific MPS.
This information is important as specific treatments are available (or in clinical development) for most MPS types, making early diagnosis important to properly manage each case and positively modify the natural history of these debilitating diseases.
Acknowledgments
This work was supported by grants from the Austrian MPS Society and International Morquio Organization (Carol Ann Foundation). This work was also supported by Japanese MPS Family Society. R.W.M. and S.T. were supported by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of NIH under grant number P30GM114736. S.T. was supported by the National Institutes of Health grant R01HD065767. The content of the article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or other sponsors. Editorial assistance to the manuscript was provided by Michelle Stofa at Nemours/Alfred I. duPont Hospital for Children. We thank C. Kuehni, who supervised the Master thesis of F. Wiesbauer: A register of lysosomal storage dieseases in Switzerland, piloted by the example of the Mucopolysaccharidoses, Master thesis 2015 at the Institute of Social and Preventive Medicine, University of Bern. We thank Alabus who technically supports the SALS registry that is funded by co-sponsoring from Actelion, Genzyme, and Biomarin, Switzerland.
Abbreviation
- MPS
mucopolysaccharidoses
- GAG
glycosaminoglycans
- CS
chondroitin sulfate
- DS
dermatan sulfate
- HS
heparan sulfate
- KS
keratan sulfate
- ECM
extracellular matrix
- IDUA
α-L-iduronidase
- I2S
iduronate-2-sulfatse
- SGHS
heparan-N-sulfatase
- NAGLU
α-N-acetylglucosaminidase
- HGSNAT
α-glucosaminidase acetyltransferase
- GNS
N-acetylglucosamine 6-sulfatase
- GALNS
N-acetylgalactosamine-6-sulfate sulfatase
- GLB
1β-galactosidase
- G4S
N-acetylgalactosamine-4-sulfatse
- GUSB
β-D-glucuronidase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest:
All the authors contributed to the original article and had no conflict of interest with any other party. Shaukat A. Khan, Hira Peracha, Alfred Wiesbauer, Tadao Orii, Roberto Giugliani, Matthias Gautschi, Marianne Rohrbach, Diana Ballhausen and Shunji Tomatsu declare that they have no conflict of interests.
Contributions to the project:
Shaukat A. Khan is an expert in molecular biology and biochemistry. He has over 10 years of experience in MPS field from basic science to the development of therapy. He has contributed to the concept and planning of the article, collection of previous articles and data, and reporting of the work described.
Hira Peracha is the primary author of this article and expert in biology. She has contributed to the planning, data analysis, and reporting of the work described.
Alfred Wiesbauer is the primary author of this article. He has a child affected with MPS IVA. He entered the data of the Swiss LSD patients in a database (Swiss registry for LSD patients) and analyzed them in the context of a master thesis in public health. He has thus contributed to the data collection, and reporting of the work described.
Marianne Rohrbach is a pediatrician and geneticist specialized in LSD. She has 14 years’ experience in LSD and works at the University Children’s Hospital in Zürich, one of the three Swiss centers for inborn errors of metabolism, where she diagnosed MPS patients and collected data. She is a mentor of Alfred Wiesbauer and contributed to the concept, planning, data analysis and reporting of the Swiss registry for LSD patients.
Diana Ballhausen is a pediatrician specialized in LSD. She first worked during 6 years in Zurich and now since 11 years in Lausanne where she diagnosed and followed a couple of the Swiss MPS patients. She is also responsible for the molecular diagnosis of MPS patients in Switzerland.
Matthias Gautschi is a pediatrician specialized in LSD and works since a couple of years at University Children’s Hospital in Bern where he diagnosed MPS patients and collected and analyzed the data of his patients.
Robert W. Mason has contributed to data analysis, and reporting of the work described.
Roberto Giugliani has diagnosed a large number of MPS patients in Brazil and contributed to data analysis, and reporting of the work described.
Tadao Orii is a medical doctor with 50 years of clinical and research experiences in MPS. He diagnosed over 500 MPS patients in his career in Japan. He published over 150 articles and chapter books in this field. He has contributed to the planning, data analysis, and reporting of the work described.
Shunji Tomatsu is a Principal Investigator and has 30 years of clinical and research experience in MPS, publishing over 150 articles in this field. He has contributed to the concept of the project, planning, analysis of data, and reporting of the work described in the article.
References
- 1.Muenzer J. The mucopolysaccharidoses: a heterogeneous group of disorders with variable pediatric presentations. The Journal of pediatrics. 2004;144:S27–34. doi: 10.1016/j.jpeds.2004.01.052. [DOI] [PubMed] [Google Scholar]
- 2.Neufeld E MJ. The Metabolic and Molecular Bases of Inherited Disease. In: C.B.A.S.W.V.D. Scriver, editor. The mucopolysaccharidoses. McGraw-Hill; New York: 2001. pp. 3421–3452. [Google Scholar]
- 3.Guillen-Navarro E, Domingo-Jimenez MR, Alcalde-Martin C, Cancho-Candela R, Couce ML, Galan-Gomez E, Alonso-Luengo O. Clinical manifestations in female carriers of mucopolysaccharidosis type II: a Spanish cross-sectional study. Orphanet journal of rare diseases. 2013;8:92. doi: 10.1186/1750-1172-8-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jurecka A, Krumina Z, Zuber Z, Rozdzynska-Swiatkowska A, Kloska A, Czartoryska B, Tylki-Szymanska A. Mucopolysaccharidosis type II in females and response to enzyme replacement therapy. American journal of medical genetics Part A. 2012;158A:450–454. doi: 10.1002/ajmg.a.34415. [DOI] [PubMed] [Google Scholar]
- 5.Imundo L, Leduc CA, Guha S, Brown M, Perino G, Gushulak L, Triggs-Raine B, Chung WK. A complete deficiency of Hyaluronoglucosaminidase 1 (HYAL1) presenting as familial juvenile idiopathic arthritis. Journal of inherited metabolic disease. 2011;34:1013–1022. doi: 10.1007/s10545-011-9343-3. [DOI] [PubMed] [Google Scholar]
- 6.Triggs-Raine B, Salo TJ, Zhang H, Wicklow BA, Natowicz MR. Mutations in HYAL1, a member of a tandemly distributed multigene family encoding disparate hyaluronidase activities, cause a newly described lysosomal disorder, mucopolysaccharidosis IX. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:6296–6300. doi: 10.1073/pnas.96.11.6296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Poorthuis BJ, Wevers RA, Kleijer WJ, Groener JE, de Jong JG, van Weely S, Niezen-Koning KE, van Diggelen OP. The frequency of lysosomal storage diseases in The Netherlands. Human genetics. 1999;105:151–156. doi: 10.1007/s004399900075. [DOI] [PubMed] [Google Scholar]
- 8.Poupetova H, Ledvinova J, Berna L, Dvorakova L, Kozich V, Elleder M. The birth prevalence of lysosomal storage disorders in the Czech Republic: comparison with data in different populations. Journal of inherited metabolic disease. 2010;33:387–396. doi: 10.1007/s10545-010-9093-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lowry RB, Renwick DH. Relative frequency of the Hurler and Hunter syndromes. The New England journal of medicine. 1971;284:221–222. doi: 10.1056/NEJM197101282840425. [DOI] [PubMed] [Google Scholar]
- 10.Applegarth DA, Toone JR, Lowry RB. Incidence of inborn errors of metabolism in British Columbia, 1969–1996. Pediatrics. 2000;105:e10. doi: 10.1542/peds.105.1.e10. [DOI] [PubMed] [Google Scholar]
- 11.Lowry RB, Applegarth DA, Toone JR, MacDonald E, Thunem NY. An update on the frequency of mucopolysaccharide syndromes in British Columbia. Human genetics. 1990;85:389–390. doi: 10.1007/BF00206770. [DOI] [PubMed] [Google Scholar]
- 12.Baehner F, Schmiedeskamp C, Krummenauer F, Miebach E, Bajbouj M, Whybra C, Kohlschutter A, Kampmann C, Beck M. Cumulative incidence rates of the mucopolysaccharidoses in Germany. Journal of inherited metabolic disease. 2005;28:1011–1017. doi: 10.1007/s10545-005-0112-z. [DOI] [PubMed] [Google Scholar]
- 13.Jurecka A, Lugowska A, Golda A, Czartoryska B, Tylki-Szymanska A. Prevalence rates of mucopolysaccharidoses in Poland. Journal of applied genetics. 2015;56:205–210. doi: 10.1007/s13353-014-0262-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krabbi K, Joost K, Zordania R, Talvik I, Rein R, Huijmans JG, Verheijen FV, Ounap K. The live-birth prevalence of mucopolysaccharidoses in Estonia. Genetic testing and molecular biomarkers. 2012;16:846–849. doi: 10.1089/gtmb.2011.0307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin HY, Lin SP, Chuang CK, Niu DM, Chen MR, Tsai FJ, Chao MC, Chiu PC, Lin SJ, Tsai LP, Hwu WL, Lin JL. Incidence of the mucopolysaccharidoses in Taiwan, 1984–2004. American journal of medical genetics Part A. 2009;149A:960–964. doi: 10.1002/ajmg.a.32781. [DOI] [PubMed] [Google Scholar]
- 16.Malm G, Lund AM, Mansson JE, Heiberg A. Mucopolysaccharidoses in the Scandinavian countries: incidence and prevalence. Acta paediatrica. 2008;97:1577–1581. doi: 10.1111/j.1651-2227.2008.00965.x. [DOI] [PubMed] [Google Scholar]
- 17.Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. Jama. 1999;281:249–254. doi: 10.1001/jama.281.3.249. [DOI] [PubMed] [Google Scholar]
- 18.Nelson J. Incidence of the mucopolysaccharidoses in Northern Ireland. Human genetics. 1997;101:355–358. doi: 10.1007/s004390050641. [DOI] [PubMed] [Google Scholar]
- 19.Nelson J, Crowhurst J, Carey B, Greed L. Incidence of the mucopolysaccharidoses in Western Australia. American journal of medical genetics Part A. 2003;123A:310–313. doi: 10.1002/ajmg.a.20314. [DOI] [PubMed] [Google Scholar]
- 20.Pinto R, Caseiro C, Lemos M, Lopes L, Fontes A, Ribeiro H, Pinto E, Silva E, Rocha S, Marcao A, Ribeiro I, Lacerda L, Ribeiro G, Amaral O, Sa Miranda MC. Prevalence of lysosomal storage diseases in Portugal. European journal of human genetics : EJHG. 2004;12:87–92. doi: 10.1038/sj.ejhg.5201044. [DOI] [PubMed] [Google Scholar]
- 21.Hopwood JJ, Harrison JR. High-resolution electrophoresis of urinary glycosaminoglycans: an improved screening test for the mucopolysaccharidoses. Analytical biochemistry. 1982;119:120–127. doi: 10.1016/0003-2697(82)90674-1. [DOI] [PubMed] [Google Scholar]
- 22.Moammar H, Cheriyan G, Mathew R, Al-Sannaa N. Incidence and patterns of inborn errors of metabolism in the Eastern Province of Saudi Arabia, 1983–2008. Ann Saudi Med. 2010;30:271–277. doi: 10.4103/0256-4947.65254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cho SY, Sohn YB, Jin DK. An overview of Korean patients with mucopolysaccharidosis and collaboration through the Asia Pacific MPS Network. Intractable & rare diseases research. 2014;3:79–86. doi: 10.5582/irdr.2014.01013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen X, Qiu W, Ye J, Han L, Gu X, Zhang H. Demographic characteristics and distribution of lysosomal storage disorder subtypes in Eastern China. Journal of human genetics. 2016;61:345–349. doi: 10.1038/jhg.2015.155. [DOI] [PubMed] [Google Scholar]
- 25.Sheth J, Mistri M, Sheth F, Shah R, Bavdekar A, Godbole K, Nanavaty N, Datar C, Kamate M, Oza N, Ankleshwaria C, Mehta S, Jackson M. Burden of lysosomal storage disorders in India: experience of 387 affected children from a single diagnostic facility. JIMD reports. 2014;12:51–63. doi: 10.1007/8904_2013_244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Verma PK, Ranganath P, Dalal AB, Phadke SR. Spectrum of Lysosomal storage disorders at a medical genetics center in northern India. Indian pediatrics. 2012;49:799–804. doi: 10.1007/s13312-012-0192-4. [DOI] [PubMed] [Google Scholar]
- 27.Ben Turkia H, Tebib N, Azzouz H, Abdelmoula MS, Ben Chehida A, Chemli J, Monastiri K, Chaabouni M, Sanhagi H, Zouari B, Kaabachi N, Ben Dridi MF. Incidence of mucopolysaccharidoses in Tunisia La. Tunisie medicale. 2009;87:782–785. [PubMed] [Google Scholar]
- 28.Federhen A, Burin M, Leistner-Segal S, Matte U, Tirelli K, Facchin A, Pasqualim G, Bender F, Rafaelli C, Giugliani R. Minimal Estimated Incidence of MPS I, II, IV-A and VI in Brazil and Comparision to the rest of the world. In: Giugliani R, editor. 14th International Symposium on MPS and Related Diseases. Bonn, Germany: 2016. pp. 1–82. [Google Scholar]
- 29.Berry HK. Screening for mucopolysaccharide disorders with the Berry spot test. Clinical biochemistry. 1987;20:365–371. doi: 10.1016/s0009-9120(87)80088-7. [DOI] [PubMed] [Google Scholar]
- 30.Scott CR, Elliott S, Buroker N, Thomas LI, Keutzer J, Glass M, Gelb MH, Turecek F. Identification of infants at risk for developing Fabry, Pompe, or mucopolysaccharidosis-I from newborn blood spots by tandem mass spectrometry. The Journal of pediatrics. 2013;163:498–503. doi: 10.1016/j.jpeds.2013.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kemper AR. A Systematic Review of Evidence Report of Final Findings Final Version 1.1. Duke University; 2015. Newborn Screening for Mucopolysaccharidosis Type 1 (MPS I) pp. 3–60. [Google Scholar]
- 32.Lin SP, Lin HY, Wang TJ, Chang CY, Lin CH, Huang SF, Tsai CC, Liu HL, Keutzer J, Chuang CK. A pilot newborn screening program for Mucopolysaccharidosis type I in Taiwan. Orphanet journal of rare diseases. 2013;8:147. doi: 10.1186/1750-1172-8-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paciotti S, Persichetti E, Pagliardini S, Deganuto M, Rosano C, Balducci C, Codini M, Filocamo M, Menghini AR, Pagliardini V, Pasqui S, Bembi B, Dardis A, Beccari T. First pilot newborn screening for four lysosomal storage diseases in an Italian region: identification and analysis of a putative causative mutation in the GBA gene. Clinica chimica acta; international journal of clinical chemistry. 2012;413:1827–1831. doi: 10.1016/j.cca.2012.07.011. [DOI] [PubMed] [Google Scholar]
- 34.Pucketta Y, Buib E, Zelicoffc A, Montano A. Epidemiology of mucopolysaccharidoses (MPS) in the United States: challenges and opportunities. MGM. 2017;120:S111. doi: 10.1186/s13023-021-01880-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Scott HS, Ashton LJ, Eyre HJ, Baker E, Brooks DA, Callen DF, Sutherland GR, Morris CP, Hopwood JJ. Chromosomal localization of the human alpha-L-iduronidase gene (IDUA) to 4p16.3. American journal of human genetics. 1990;47:802–807. [PMC free article] [PubMed] [Google Scholar]
- 36.Uttarilli A, Ranganath P, Matta D, Md Nurul Jain J, Prasad CK, Babu AS, Girisha KM, Verma IC, Phadke SR, Mandal K, Puri RD, Aggarwal S, Danda S, H SV, Kapoor S, Bhat M, Gowrishankar K, Hasan AQ, Nair M, Nampoothiri S, Dalal A. Identification and Characterization of 20 Novel Pathogenic Variants in 60 unrelated Indian patients with Mucopolysaccharidoses (MPS) type I and type II. Clinical genetics. 2016 doi: 10.1111/cge.12795. [DOI] [PubMed] [Google Scholar]
- 37.Cooper D, Ball E, Stenson P, Phillips AD, Evans K, Heywood S, Hayden M, Mort M, Hussain M. The Human Gene Mutation Database. Cardiff University; UK: 2015. [Google Scholar]
- 38.Voskoboeva EY, Krasnopolskaya XD, Mirenburg TV, Weber B, Hopwood JJ. Molecular genetics of mucopolysaccharidosis type I: mutation analysis among the patients of the former Soviet Union. Molecular genetics and metabolism. 1998;65:174–180. doi: 10.1006/mgme.1998.2745. [DOI] [PubMed] [Google Scholar]
- 39.Gatti R, DiNatale P, Villani GR, Filocamo M, Muller V, Guo XH, Nelson PV, Scott HS, Hopwood JJ. Mutations among Italian mucopolysaccharidosis type I patients. Journal of inherited metabolic disease. 1997;20:803–806. doi: 10.1023/a:1005323918923. [DOI] [PubMed] [Google Scholar]
- 40.Venturi N, Rovelli A, Parini R, Menni F, Brambillasca F, Bertagnolio F, Uziel G, Gatti R, Filocamo M, Donati MA, Biondi A, Goldwurm S. Molecular analysis of 30 mucopolysaccharidosis type I patients: evaluation of the mutational spectrum in Italian population and identification of 13 novel mutations. Human mutation. 2002;20:231. doi: 10.1002/humu.9051. [DOI] [PubMed] [Google Scholar]
- 41.Matte U, Yogalingam G, Brooks D, Leistner S, Schwartz I, Lima L, Norato DY, Brum JM, Beesley C, Winchester B, Giugliani R, Hopwood JJ. Identification and characterization of 13 new mutations in mucopolysaccharidosis type I patients. Molecular genetics and metabolism. 2003;78:37–43. doi: 10.1016/s1096-7192(02)00200-7. [DOI] [PubMed] [Google Scholar]
- 42.Matte U, Leistner S, Lima L, Schwartz I, Giugliani R. Unique frequency of known mutations in Brazilian MPS I patients. American journal of medical genetics. 2000;90:108–109. doi: 10.1002/(sici)1096-8628(20000117)90:2<108::aid-ajmg3>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 43.Bertola F, Filocamo M, Casati G, Mort M, Rosano C, Tylki-Szymanska A, Tuysuz B, Gabrielli O, Grossi S, Scarpa M, Parenti G, Antuzzi D, Dalmau J, Di Rocco M, Dionisi Vici C, Okur I, Rosell J, Rovelli A, Furlan F, Rigoldi M, Biondi A, Cooper DN, Parini R. IDUA mutational profiling of a cohort of 102 European patients with mucopolysaccharidosis type I: identification and characterization of 35 novel alpha-L-iduronidase (IDUA) alleles. Human mutation. 2011;32:E2189–2210. doi: 10.1002/humu.21479. [DOI] [PubMed] [Google Scholar]
- 44.Li P, Wood T, Thompson JN. Diversity of mutations and distribution of single nucleotide polymorphic alleles in the human alpha-L-iduronidase (IDUA) gene. Genetics in medicine : official journal of the American College of Medical Genetics. 2002;4:420–426. doi: 10.1097/00125817-200211000-00004. [DOI] [PubMed] [Google Scholar]
- 45.Bunge S, Kleijer WJ, Steglich C, Beck M, Zuther C, Morris CP, Schwinger E, Hopwood JJ, Scott HS, Gal A. Mucopolysaccharidosis type I: identification of 8 novel mutations and determination of the frequency of the two common alpha-L-iduronidase mutations (W402X and Q70X) among European patients. Human molecular genetics. 1994;3:861–866. doi: 10.1093/hmg/3.6.861. [DOI] [PubMed] [Google Scholar]
- 46.Kwak MJ, Huh R, Kim J, Park HD, Cho SY, Jin DK. Report of 5 novel mutations of the alpha-L-iduronidase gene and comparison of Korean mutations in relation with those of Japan or China in patients with mucopolysaccharidosis I. BMC medical genetics. 2016;17:58. doi: 10.1186/s12881-016-0319-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang X, Zhang W, Shi H, Qiu Z, Meng Y, Yao F, Wei M. Mucopolysaccharidosis I mutations in Chinese patients: identification of 27 novel mutations and 6 cases involving prenatal diagnosis. Clinical genetics. 2012;81:443–452. doi: 10.1111/j.1399-0004.2011.01680.x. [DOI] [PubMed] [Google Scholar]
- 48.Yamagishi A, Tomatsu S, Fukuda S, Uchiyama A, Shimozawa N, Suzuki Y, Kondo N, Sukegawa K, Orii T. Mucopolysaccharidosis type I: identification of common mutations that cause Hurler and Scheie syndromes in Japanese populations. Human mutation. 1996;7:23–29. doi: 10.1002/(SICI)1098-1004(1996)7:1<23::AID-HUMU3>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 49.Isogai K, Sukegawa K, Tomatsu S, Fukao T, Song XQ, Yamada Y, Fukuda S, Orii T, Kondo N. Mutation analysis in the iduronate-2-sulphatase gene in 43 Japanese patients with mucopolysaccharidosis type II (Hunter disease) Journal of inherited metabolic disease. 1998;21:60–70. doi: 10.1023/a:1005363414792. [DOI] [PubMed] [Google Scholar]
- 50.Lin SP, Chang JH, Lee-Chen GJ, Lin DS, Lin HY, Chuang CK. Detection of Hunter syndrome (mucopolysaccharidosis type II) in Taiwanese: biochemical and linkage studies of the iduronate-2-sulfatase gene defects in MPS II patients and carriers. Clinica chimica acta; international journal of clinical chemistry. 2006;369:29–34. doi: 10.1016/j.cca.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 51.Zhang H, Li J, Zhang X, Wang Y, Qiu W, Ye J, Han L, Gao X, Gu X. Analysis of the IDS gene in 38 patients with Hunter syndrome: the c.879G>A (p.Gln293Gln) synonymous variation in a female create exonic splicing. PloS one. 2011;6:e22951. doi: 10.1371/journal.pone.0022951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brusius-Facchin AC, Schwartz IV, Zimmer C, Ribeiro MG, Acosta AX, Horovitz D, Monlleo IL, Fontes MI, Fett-Conte A, Sobrinho RP, Duarte AR, Boy R, Mabe P, Ascurra M, de Michelena M, Tylee KL, Besley GT, Garreton MC, Giugliani R, Leistner-Segal S. Mucopolysaccharidosis type II: identification of 30 novel mutations among Latin American patients. Molecular genetics and metabolism. 2014;111:133–138. doi: 10.1016/j.ymgme.2013.08.011. [DOI] [PubMed] [Google Scholar]
- 53.Goldenfum SL, Young E, Michelakakis H, Tsagarakis S, Winchester B. Mutation analysis in 20 patients with Hunter disease. Human mutation. 1996;7:76–78. doi: 10.1002/(SICI)1098-1004(1996)7:1<76::AID-HUMU14>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 54.Rathmann M, Bunge S, Beck M, Kresse H, Tylki-Szymanska A, Gal A. Mucopolysaccharidosis type II (Hunter syndrome): mutation “hot spots” in the iduronate-2-sulfatase gene. American journal of human genetics. 1996;59:1202–1209. [PMC free article] [PubMed] [Google Scholar]
- 55.Sukegawa K, Tomatsu S, Fukao T, Iwata H, Song XQ, Yamada Y, Fukuda S, Isogai K, Orii T. Mucopolysaccharidosis type II (Hunter disease): identification and characterization of eight point mutations in the iduronate-2-sulfatase gene in Japanese patients. Human mutation. 1995;6:136–143. doi: 10.1002/humu.1380060206. [DOI] [PubMed] [Google Scholar]
- 56.Whitley CB, Anderson RA, Aronovich EL, Crotty PL, Anyane-Yeboa K, Russo D, Warburton D. Caveat to genotype-phenotype correlation in mucopolysaccharidosis type II: discordant clinical severity of R468W and R468Q mutations of the iduronate-2-sulfatase gene. Human mutation. 1993;2:235–237. doi: 10.1002/humu.1380020313. [DOI] [PubMed] [Google Scholar]
- 57.Kosuga M, Mashima R, Hirakiyama A, Fuji N, Kumagai T, Seo JH, Nikaido M, Saito S, Ohno K, Sakuraba H, Okuyama T. Molecular diagnosis of 65 families with mucopolysaccharidosis type II (Hunter syndrome) characterized by 16 novel mutations in the IDS gene: Genetic, pathological, and structural studies on iduronate-2-sulfatase. Molecular genetics and metabolism. 2016;118:190–197. doi: 10.1016/j.ymgme.2016.05.003. [DOI] [PubMed] [Google Scholar]
- 58.Beratis NG, Sklower SL, Wilbur L, Matalon R. Sanfilippo disease in Greece. Clinical genetics. 1986;29:129–132. doi: 10.1111/j.1399-0004.1986.tb01235.x. [DOI] [PubMed] [Google Scholar]
- 59.Michelakakis H, Dimitriou E, Tsagaraki S, Giouroukos S, Schulpis K, Bartsocas CS. Lysosomal storage diseases in Greece. Genetic counseling. 1995;6:43–47. [PubMed] [Google Scholar]
- 60.Scott HS, Blanch L, Guo XH, Freeman C, Orsborn A, Baker E, Sutherland GR, Morris CP, Hopwood JJ. Cloning of the sulphamidase gene and identification of mutations in Sanfilippo A syndrome. Nature genetics. 1995;11:465–467. doi: 10.1038/ng1295-465. [DOI] [PubMed] [Google Scholar]
- 61.Karageorgos LE, Guo XH, Blanch L, Weber B, Anson DS, Scott HS, Hopwood JJ. Structure and sequence of the human sulphamidase gene. DNA research : an international journal for rapid publication of reports on genes and genomes. 1996;3:269–271. doi: 10.1093/dnares/3.4.269. [DOI] [PubMed] [Google Scholar]
- 62.Weber B, van de Kamp JJ, Kleijer WJ, Guo XH, Blanch L, van Diggelen OP, Wevers R, Poorthuis BJ, Hopwood JJ. Identification of a common mutation (R245H) Sanfilippo A patients from The Netherlands Journal of inherited metabolic disease. 1998;21:416–422. doi: 10.1023/a:1005362826552. [DOI] [PubMed] [Google Scholar]
- 63.Weber B, Guo XH, Wraith JE, Cooper A, Kleijer WJ, Bunge S, Hopwood JJ. Novel mutations in Sanfilippo A syndrome: implications for enzyme function. Human molecular genetics. 1997;6:1573–1579. doi: 10.1093/hmg/6.9.1573. [DOI] [PubMed] [Google Scholar]
- 64.Zhao HG, Li HH, Bach G, Schmidtchen A, Neufeld EF. The molecular basis of Sanfilippo syndrome type B. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:6101–6105. doi: 10.1073/pnas.93.12.6101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Beesley C, Moraitou M, Winchester B, Schulpis K, Dimitriou E, Michelakakis H. Sanfilippo B syndrome: molecular defects in Greek patients. Clinical genetics. 2004;65:143–149. doi: 10.1111/j.0009-9163.2004.00210.x. [DOI] [PubMed] [Google Scholar]
- 66.Weber B, Guo XH, Kleijer WJ, van de Kamp JJ, Poorthuis BJ, Hopwood JJ. Sanfilippo type B syndrome (mucopolysaccharidosis III B): allelic heterogeneity corresponds to the wide spectrum of clinical phenotypes. European journal of human genetics, EJHG. 1999;7:34–44. doi: 10.1038/sj.ejhg.5200242. [DOI] [PubMed] [Google Scholar]
- 67.Mangas M, Nogueira C, Prata MJ, Lacerda L, Coll MJ, Soares G, Ribeiro G, Amaral O, Ferreira C, Alves C, Coutinho MF, Alves S. Molecular analysis of mucopolysaccharidosis type IIIB in Portugal: evidence of a single origin for a common mutation (R234C) in the Iberian. Peninsula Clinical genetics. 2008;73:251–256. doi: 10.1111/j.1399-0004.2007.00951.x. [DOI] [PubMed] [Google Scholar]
- 68.Fan X, Zhang H, Zhang S, Bagshaw RD, Tropak MB, Callahan JW, Mahuran DJ. Identification of the gene encoding the enzyme deficient in mucopolysaccharidosis IIIC (Sanfilippo disease type C) American journal of human genetics. 2006;79:738–744. doi: 10.1086/508068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hrebicek M, Mrazova L, Seyrantepe V, Durand S, Roslin NM, Noskova L, Hartmannova H, Ivanek R, Cizkova A, Poupetova H, Sikora J, Urinovska J, Stranecky V, Zeman J, Lepage P, Roquis D, Verner A, Ausseil J, Beesley CE, Maire I, Poorthuis BJ, van de Kamp J, van Diggelen OP, Wevers RA, Hudson TJ, Fujiwara TM, Majewski J, Morgan K, Kmoch S, Pshezhetsky AV. Mutations in TMEM76* cause mucopolysaccharidosis IIIC (Sanfilippo C syndrome) American journal of human genetics. 2006;79:807–819. doi: 10.1086/508294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ruijter GJ, Valstar MJ, van de Kamp JM, van der Helm RM, Durand S, van Diggelen OP, Wevers RA, Poorthuis BJ, Pshezhetsky AV, Wijburg FA. Clinical and genetic spectrum of Sanfilippo type C (MPS IIIC) disease in The Netherlands. Molecular genetics and metabolism. 2008;93:104–111. doi: 10.1016/j.ymgme.2007.09.011. [DOI] [PubMed] [Google Scholar]
- 71.Kresse H, Paschke E, von Figura K, Gilberg W, Fuchs W. Sanfilippo disease type D: deficiency of N-acetylglucosamine-6-sulfate sulfatase required for heparan sulfate degradation. Proceedings of the National Academy of Sciences of the United States of America. 1980;77:6822–6826. doi: 10.1073/pnas.77.11.6822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Beesley CE, Burke D, Jackson M, Vellodi A, Winchester BG, Young EP. Sanfilippo syndrome type D: identification of the first mutation in the N-acetylglucosamine-6-sulphatase gene. Journal of medical genetics. 2003;40:192–194. doi: 10.1136/jmg.40.3.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Beesley CE, Concolino D, Filocamo M, Winchester BG, Strisciuglio P. Identification and characterisation of an 8.7 kb deletion and a novel nonsense mutation in two Italian families with Sanfilippo syndrome type D (mucopolysaccharidosis IIID) Molecular genetics and metabolism. 2007;90:77–80. doi: 10.1016/j.ymgme.2006.07.014. [DOI] [PubMed] [Google Scholar]
- 74.Coppa GV, Giorgi PL, Felici L, Gabrielli O, Donti E, Bernasconi S, Kresse H, Paschke E, Mastropaolo C. Clinical heterogeneity in Sanfilippo disease (mucopolysaccharidosis III) type D: presentation of two new cases. European journal of pediatrics. 1983;140:130–133. doi: 10.1007/BF00441662. [DOI] [PubMed] [Google Scholar]
- 75.del Canho H, van den Bergh FA, Duran M, Hennekam RC, Groniger AM, Poorthuis BJ. Type D Sanfilippo disease in an 8-year-old boy; a rare cause of mental retardation. Nederlands tijdschrift voor geneeskunde. 1993;137:969–972. [PubMed] [Google Scholar]
- 76.Gatti R, Borrone C, Durand P, De Virgilis S, Sanna G, Cao A, von Figura K, Kresse H, Paschke E. Sanfilippo type D disease: clinical findings in two patients with a new variant of mucopolysaccharidosis III. European journal of pediatrics. 1982;138:168–171. doi: 10.1007/BF00441147. [DOI] [PubMed] [Google Scholar]
- 77.Jansen AC, Cao H, Kaplan P, Silver K, Leonard G, De Meirleir L, Lissens W, Liebaers I, Veilleux M, Andermann F, Hegele RA, Andermann E. Sanfilippo syndrome type D: natural history and identification of 3 novel mutations in the GNS. Gene Archives of neurology. 2007;64:1629–1634. doi: 10.1001/archneur.64.11.1629. [DOI] [PubMed] [Google Scholar]
- 78.Jones MZ, Alroy J, Rutledge JC, Taylor JW, Alvord EC, Jr, Toone J, Applegarth D, Hopwood JJ, Skutelsky E, Ianelli C, Thorley-Lawson D, Mitchell-Herpolsheimer C, Arias A, Sharp P, Evans W, Sillence D, Cavanagh KT. Human mucopolysaccharidosis IIID: clinical, biochemical, morphological and immunohistochemical characteristics. Journal of neuropathology and experimental neurology. 1997;56:1158–1167. [PubMed] [Google Scholar]
- 79.Kaplan P, Wolfe LS. Sanfilippo syndrome type D. The Journal of pediatrics. 1987;110:267–271. doi: 10.1016/s0022-3476(87)80171-3. [DOI] [PubMed] [Google Scholar]
- 80.Ozand PT, Thompson JN, Gascon GG, Sarvepalli SB, Rahbeeni Z, Nester MJ, Brismar J. Sanfilippo type D presenting with acquired language disorder but without features of mucopolysaccharidosis. Journal of child neurology. 1994;9:408–411. doi: 10.1177/088307389400900415. [DOI] [PubMed] [Google Scholar]
- 81.Siciliano L, Fiumara A, Pavone L, Freeman C, Robertson D, Morris CP, Hopwood JJ, Di Natale P, Musumeci S, Horwitz AL. Sanfilippo syndrome type D in two adolescent sisters. Journal of medical genetics. 1991;28:402–405. doi: 10.1136/jmg.28.6.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tylki-Szymanska A, Czartoryska B, Gorska D, Piesiewicz-Grzonkowska E. Type III D mucopolysaccharidosis (Sanfilippo D): clinical course and symptoms. Acta paediatrica Japonica; Overseas edition. 1998;40:492–494. doi: 10.1111/j.1442-200x.1998.tb01977.x. [DOI] [PubMed] [Google Scholar]
- 83.Robertson DA, Callen DF, Baker EG, Morris CP, Hopwood JJ. Chromosomal localization of the gene for human glucosamine-6-sulphatase to 12q14. Human genetics. 1988;79:175–178. doi: 10.1007/BF00280560. [DOI] [PubMed] [Google Scholar]
- 84.Mok A, Cao H, Hegele RA. Genomic basis of mucopolysaccharidosis type IIID (MIM 252940) revealed by sequencing of GNS encoding N-acetylglucosamine-6-sulfatase. Genomics. 2003;81:1–5. doi: 10.1016/s0888-7543(02)00014-9. [DOI] [PubMed] [Google Scholar]
- 85.Valstar MJ, Bertoli-Avella AM, Wessels MW, Ruijter GJ, de Graaf B, Olmer R, Elfferich P, Neijs S, Kariminejad R, Suheyl Ezgu F, Tokatli A, Czartoryska B, Bosschaart AN, van den Bos-Terpstra F, Puissant H, Burger F, Omran H, Eckert D, Filocamo M, Simeonov E, Willems PJ, Wevers RA, Niermeijer MF, Halley DJ, Poorthuis BJ, van Diggelen OP. Mucopolysaccharidosis type IIID: 12 new patients and 15 novel mutations. Human mutation. 2010;31:E1348–1360. doi: 10.1002/humu.21234. [DOI] [PubMed] [Google Scholar]
- 86.Baker E, Guo XH, Orsborn AM, Sutherland GR, Callen DF, Hopwood JJ, Morris CP. The morquio A syndrome (mucopolysaccharidosis IVA) gene maps to 16q24.3. American journal of human genetics. 1993;52:96–98. [PMC free article] [PubMed] [Google Scholar]
- 87.Masuno M, Tomatsu S, Nakashima Y, Hori T, Fukuda S, Masue M, Sukegawa K, Orii T. Mucopolysaccharidosis IV A: assignment of the human N-acetylgalactosamine-6-sulfate sulfatase (GALNS) gene to chromosome 16q24. Genomics. 1993;16:777–778. doi: 10.1006/geno.1993.1266. [DOI] [PubMed] [Google Scholar]
- 88.Masue M, Sukegawa K, Orii T, Hashimoto T. N-acetylgalactosamine-6-sulfate sulfatase in human placenta: purification and characteristics. Journal of biochemistry. 1991;110:965–970. doi: 10.1093/oxfordjournals.jbchem.a123697. [DOI] [PubMed] [Google Scholar]
- 89.Tomatsu S, Fukuda S, Masue M, Sukegawa K, Fukao T, Yamagishi A, Hori T, Iwata H, Ogawa T, Nakashima Y, et al. Morquio disease: isolation, characterization and expression of full-length cDNA for human N-acetylgalactosamine-6-sulfate sulfatase. Biochemical and biophysical research communications. 1991;181:677–683. doi: 10.1016/0006-291x(91)91244-7. [DOI] [PubMed] [Google Scholar]
- 90.Tomatsu S, Montano AM, Nishioka T, Gutierrez MA, Pena OM, Tranda Firescu GG, Lopez P, Yamaguchi S, Noguchi A, Orii T. Mutation and polymorphism spectrum of the GALNS gene in mucopolysaccharidosis IVA (Morquio A) Human mutation. 2005;26:500–512. doi: 10.1002/humu.20257. [DOI] [PubMed] [Google Scholar]
- 91.Kato Z, Fukuda S, Tomatsu S, Vega H, Yasunaga T, Yamagishi A, Yamada N, Valencia A, Barrera LA, Sukegawa K, Orii T, Kondo N. A novel common missense mutation G301C in the N-acetylgalactosamine-6-sulfate sulfatase gene in mucopolysaccharidosis IVA. Human genetics. 1997;101:97–101. doi: 10.1007/s004390050594. [DOI] [PubMed] [Google Scholar]
- 92.Montano AM, Kaitila I, Sukegawa K, Tomatsu S, Kato Z, Nakamura H, Fukuda S, Orii T, Kondo N. Mucopolysaccharidosis IVA: characterization of a common mutation found in Finnish patients with attenuated phenotype. Human genetics. 2003;113:162–169. doi: 10.1007/s00439-003-0959-8. [DOI] [PubMed] [Google Scholar]
- 93.Tomatsu S, Dieter T, Schwartz IV, Sarmient P, Giugliani R, Barrera LA, Guelbert N, Kremer R, Repetto GM, Gutierrez MA, Nishioka T, Serrato OP, Montano AM, Yamaguchi S, Noguchi A. Identification of a common mutation in mucopolysaccharidosis IVA: correlation among genotype, phenotype, and keratan sulfate. Journal of human genetics. 2004;49:490–494. doi: 10.1007/s10038-004-0178-8. [DOI] [PubMed] [Google Scholar]
- 94.Tomatsu S, Filocamo M, Orii KO, Sly WS, Gutierrez MA, Nishioka T, Serrato OP, Di Natale P, Montano AM, Yamaguchi S, Kondo N, Orii T, Noguchi A. Mucopolysaccharidosis IVA (Morquio A): identification of novel common mutations in the N-acetylgalactosamine-6-sulfate sulfatase (GALNS) gene in Italian patients. Human mutation. 2004;24:187–188. doi: 10.1002/humu.9265. [DOI] [PubMed] [Google Scholar]
- 95.Tomatsu S, Fukuda S, Cooper A, Wraith JE, Rezvi GM, Yamagishi A, Yamada N, Kato Z, Isogai K, Sukegawa K, et al. Mucopolysaccharidosis type IVA: identification of six novel mutations among non-Japanese patients. Human molecular genetics. 1995;4:741–743. doi: 10.1093/hmg/4.4.741. [DOI] [PubMed] [Google Scholar]
- 96.Yamada N, Fukuda S, Tomatsu S, Muller V, Hopwood JJ, Nelson J, Kato Z, Yamagishi A, Sukegawa K, Kondo N, Orii T. Molecular heterogeneity in mucopolysaccharidosis IVA in Australia and Northern Ireland: nine novel mutations including T312S, a common allele that confers a mild phenotype. Human mutation. 1998;11:202–208. doi: 10.1002/(SICI)1098-1004(1998)11:3<202::AID-HUMU4>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 97.Tomatsu S, Nishioka T, Montano AM, Gutierrez MA, Pena OS, Orii KO, Sly WS, Yamaguchi S, Orii T, Paschke E, Kircher SG, Noguchi A. Mucopolysaccharidosis IVA: identification of mutations and methylation study in GALNS gene. Journal of medical genetics. 2004;41:e98. doi: 10.1136/jmg.2003.018010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tomatsu S, Fukuda S, Cooper A, Wraith JE, Rezvi GM, Yamagishi A, Yamada N, Kato Z, Isogai K, Sukegawa K, et al. Mucopolysaccharidosis IVA: identification of a common missense mutation I113F in the N-Acetylgalactosamine-6-sulfate sulfatase gene. American journal of human genetics. 1995;57:556–563. [PMC free article] [PubMed] [Google Scholar]
- 99.Bunge S, Kleijer WJ, Tylki-Szymanska A, Steglich C, Beck M, Tomatsu S, Fukuda S, Poorthuis BJ, Czartoryska B, Orii T, Gal A. Identification of 31 novel mutations in the N-acetylgalactosamine-6-sulfatase gene reveals excessive allelic heterogeneity among patients with Morquio A syndrome. Human mutation. 1997;10:223–232. doi: 10.1002/(SICI)1098-1004(1997)10:3<223::AID-HUMU8>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 100.Fukuda S, Tomatsu S, Masuno M, Ogawa T, Yamagishi A, Rezvi GM, Sukegawa K, Shimozawa N, Suzuki Y, Kondo N, Imaizumi K, Kuroki Y, Okabe T, Orii T. Mucopolysaccharidosis IVA: submicroscopic deletion of 16q24.3 and a novel R386C mutation of N-acetylgalactosamine-6-sulfate sulfatase gene in a classical Morquio disease. Human mutation. 1996;7:123–134. doi: 10.1002/(SICI)1098-1004(1996)7:2<123::AID-HUMU6>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 101.Tomatsu S, Fukuda S, Cooper A, Wraith JE, Ferreira P, Di Natale P, Tortora P, Fujimoto A, Kato Z, Yamada N, Isogai K, Yamagishi A, Sukegawa K, Suzuki Y, Shimozawa N, Kondo N, Sly WS, Orii T. Fourteen novel mucopolysaccharidosis IVA producing mutations in GALNS gene. Human mutation. 1997;10:368–375. doi: 10.1002/(SICI)1098-1004(1997)10:5<368::AID-HUMU6>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 102.Pajares S, Alcalde C, Couce ML, Del Toro M, Gonzalez-Meneses A, Guillen E, Pineda M, Pintos G, Gort L, Coll MJ. Molecular analysis of mucopolysaccharidosis IVA (Morquio A) in Spain. Molecular genetics and metabolism. 2012;106:196–201. doi: 10.1016/j.ymgme.2012.03.006. [DOI] [PubMed] [Google Scholar]
- 103.Chkioua L, Khedhiri S, Hafsi H, Grissa O, Ben Turkia H, Miled A, Laradi S, Froissart R, Alif N. Molecular analysis in a GALNS study cohort of 15 Tunisian patients: description of a novel mutation. Diagnostic pathology. 2016;11:51. doi: 10.1186/s13000-016-0498-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Paschke E, Milos I, Kreimer-Erlacher H, Hoefler G, Beck M, Hoeltzenbein M, Kleijer W, Levade T, Michelakakis H, Radeva B. Mutation analyses in 17 patients with deficiency in acid beta-galactosidase: three novel point mutations and high correlation of mutation W273L with Morquio disease type B. Human genetics. 2001;109:159–166. doi: 10.1007/s004390100570. [DOI] [PubMed] [Google Scholar]
- 105.Caciotti A, Bardelli T, Cunningham J, D’Azzo A, Zammarchi E, Morrone A. Modulating action of the new polymorphism L436F detected in the GLB1 gene of a type-II GM1 gangliosidosis patient. Human genetics. 2003;113:44–50. doi: 10.1007/s00439-003-0930-8. [DOI] [PubMed] [Google Scholar]
- 106.Morrone A, Bardelli T, Donati MA, Giorgi M, Di Rocco M, Gatti R, Parini R, Ricci R, Taddeucci G, D’Azzo A, Zammarchi E. beta-galactosidase gene mutations affecting the lysosomal enzyme and the elastin-binding protein in GM1-gangliosidosis patients with cardiac involvement. Human mutation. 2000;15:354–366. doi: 10.1002/(SICI)1098-1004(200004)15:4<354::AID-HUMU8>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 107.Silva CM, Severini MH, Sopelsa A, Coelho JC, Zaha A, d’Azzo A, Giugliani R. Six novel beta-galactosidase gene mutations in Brazilian patients with GM1-gangliosidosis. Human mutation. 1999;13:401–409. doi: 10.1002/(SICI)1098-1004(1999)13:5<401::AID-HUMU9>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 108.Santamaria R, Chabas A, Coll MJ, Miranda CS, Vilageliu L, Grinberg D. Twenty-one novel mutations in the GLB1 gene identified in a large group of GM1-gangliosidosis and Morquio B patients: possible common origin for the prevalent p.R59H mutation among gypsies. Human mutation. 2006;27:1060. doi: 10.1002/humu.9451. [DOI] [PubMed] [Google Scholar]
- 109.Lei HL, Ye J, Qiu WJ, Zhang HW, Han LS, Wang Y, Gu XF. Beta-galactosidase deficiencies and novel GLB1 mutations in three Chinese patients with Morquio B disease or GM1 gangliosidosis. World journal of pediatrics : WJP. 2012;8:359–362. doi: 10.1007/s12519-012-0382-0. [DOI] [PubMed] [Google Scholar]
- 110.Litjens T, Hopwood JJ. Mucopolysaccharidosis type VI: Structural and clinical implications of mutations in N-acetylgalactosamine-4-sulfatase. Human mutation. 2001;18:282–295. doi: 10.1002/humu.1190. [DOI] [PubMed] [Google Scholar]
- 111.Petry MF, Dieter T, Burin M, Giugliani R, Leistner S. Identification of a novel mutation in the ARSB gene that is frequent among Brazilian MPSVI patients. Genetic testing. 2003;7:347–349. doi: 10.1089/109065703322783743. [DOI] [PubMed] [Google Scholar]
- 112.Petry MF, Nonemacher K, Sebben JC, Schwartz IV, Azevedo AC, Burin MG, de Rezende AR, Kim CA, Giugliani R, Leistner-Segal S. Mucopolysaccharidosis type VI: Identification of novel mutations on the arylsulphatase B gene in South American patients. Journal of inherited metabolic disease. 2005;28:1027–1034. doi: 10.1007/s10545-005-0020-2. [DOI] [PubMed] [Google Scholar]
- 113.Karageorgos L, Brooks DA, Pollard A, Melville EL, Hein LK, Clements PR, Ketteridge D, Swiedler SJ, Beck M, Giugliani R, Harmatz P, Wraith JE, Guffon N, Leao Teles E, Sa Miranda MC, Hopwood JJ. Mutational analysis of 105 mucopolysaccharidosis type VI patients. Human mutation. 2007;28:897–903. doi: 10.1002/humu.20534. [DOI] [PubMed] [Google Scholar]
- 114.Garrido E, Chabas A, Coll MJ, Blanco M, Dominguez C, Grinberg D, Vilageliu L, Cormand B. Identification of the molecular defects in Spanish and Argentinian mucopolysaccharidosis VI (Maroteaux-Lamy syndrome) patients, including 9 novel mutations. Molecular genetics and metabolism. 2007;92:122–130. doi: 10.1016/j.ymgme.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 115.Giugliani R, Federhen A, Rojas MV, Vieira T, Artigalas O, Pinto LL, Azevedo AC, Acosta A, Bonfim C, Lourenco CM, Kim CA, Horovitz D, Bonfim D, Norato D, Marinho D, Palhares D, Santos ES, Ribeiro E, Valadares E, Guarany F, de Lucca GR, Pimentel H, de Souza IN, Correa J, Sr, Fraga JC, Goes JE, Cabral JM, Simionato J, Llerena J, Jr, Jardim L, Giuliani L, da Silva LC, Santos ML, Moreira MA, Kerstenetzky M, Ribeiro M, Ruas N, Barrios P, Aranda P, Honjo R, Boy R, Costa R, Souza C, Alcantara FF, Avilla SG, Fagondes S, Martins AM. Mucopolysaccharidosis I, II, and VI: Brief review and guidelines for treatment. Genetics and molecular biology. 2010;33:589–604. doi: 10.1590/S1415-47572010005000093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Giugliani R, Harmatz P, Wraith JE. Management guidelines for mucopolysaccharidosis VI. Pediatrics. 2007;120:405–418. doi: 10.1542/peds.2006-2184. [DOI] [PubMed] [Google Scholar]
- 117.Brot FE, Bell CE, Jr, Sly WS. Purification and properties of beta-glucuronidase from human placenta. Biochemistry. 1978;17:385–391. doi: 10.1021/bi00596a001. [DOI] [PubMed] [Google Scholar]
- 118.Oshima A, Kyle JW, Miller RD, Hoffmann JW, Powell PP, Grubb JH, Sly WS, Tropak M, Guise KS, Gravel RA. Cloning, sequencing, and expression of cDNA for human beta-glucuronidase. Proceedings of the National Academy of Sciences of the United States of America. 1987;84:685–689. doi: 10.1073/pnas.84.3.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hassan MI, Waheed A, Grubb JH, Klei HE, Korolev S, Sly WS. High resolution crystal structure of human beta-glucuronidase reveals structural basis of lysosome targeting. PloS one. 2013;8:e79687. doi: 10.1371/journal.pone.0079687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Jain S, Drendel WB, Chen ZW, Mathews FS, Sly WS, Grubb JH. Structure of human beta-glucuronidase reveals candidate lysosomal targeting and active-site motifs. Nature structural biology. 1996;3:375–381. doi: 10.1038/nsb0496-375. [DOI] [PubMed] [Google Scholar]
- 121.Tomatsu S, Montano AM, Dung VC, Grubb JH, Sly WS. Mutations and polymorphisms in GUSB gene in mucopolysaccharidosis VII (Sly Syndrome) Human mutation. 2009;30:511–519. doi: 10.1002/humu.20828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Tomatsu S, Fukuda S, Sukegawa K, Ikedo Y, Yamada S, Yamada Y, Sasaki T, Okamoto H, Kuwahara T, Yamaguchi S, et al. Mucopolysaccharidosis type VII: characterization of mutations and molecular heterogeneity. American journal of human genetics. 1991;48:89–96. [PMC free article] [PubMed] [Google Scholar]
- 123.Tomatsu S, Sukegawa K, Ikedo Y, Fukuda S, Yamada Y, Sasaki T, Okamoto H, Kuwabara T, Orii T. Molecular basis of mucopolysaccharidosis type VII: replacement of Ala619 in beta-glucuronidase with. Val Gene. 1990;89:283–287. doi: 10.1016/0378-1119(90)90019-n. [DOI] [PubMed] [Google Scholar]
- 124.Vervoort R, Islam MR, Sly WS, Zabot MT, Kleijer WJ, Chabas A, Fensom A, Young EP, Liebaers I, Lissens W. Molecular analysis of patients with beta-glucuronidase deficiency presenting as hydrops fetalis or as early mucopolysaccharidosis VII. American journal of human genetics. 1996;58:457–471. [PMC free article] [PubMed] [Google Scholar]
- 125.Wu BM, Tomatsu S, Fukuda S, Sukegawa K, Orii T, Sly WS. Overexpression rescues the mutant phenotype of L176F mutation causing beta-glucuronidase deficiency mucopolysaccharidosis in two Mennonite siblings. The Journal of biological chemistry. 1994;269:23681–23688. [PubMed] [Google Scholar]
- 126.Lacombe D, Germain DP. Genetic aspects of mucopolysaccharidoses. Archives de pediatrie : organe officiel de la Societe francaise de pediatrie. 2014;21(Suppl 1):S22–26. doi: 10.1016/S0929-693X(14)72255-9. [DOI] [PubMed] [Google Scholar]
- 127.Wang P, Sorenson J, Strickland S, Mingus C, Haskins ME, Giger U. Mucopolysaccharidosis VII in a Cat Caused by 2 Adjacent Missense Mutations in the GUSB Gene. Journal of veterinary internal medicine/American College of Veterinary Internal Medicine. 2015;29:1022–1028. doi: 10.1111/jvim.13569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Khan FI, Shahbaaz M, Bisetty K, Waheed A, Sly WS, Ahmad F, Hassan MI. Large scale analysis of the mutational landscape in beta-glucuronidase: A major player of mucopolysaccharidosis type VII. Gene. 2016;576:36–44. doi: 10.1016/j.gene.2015.09.062. [DOI] [PubMed] [Google Scholar]
- 129.Roden L, Campbell P, Fraser JR, Laurent TC, Pertoft H, Thompson JN. Enzymic pathways of hyaluronan catabolism Ciba Foundation symposium. 1989;143:60–76. doi: 10.1002/9780470513774.ch5. discussion 76–86, 281–285. [DOI] [PubMed] [Google Scholar]
- 130.Moore D, Connock MJ, Wraith E, Lavery C. The prevalence of and survival in Mucopolysaccharidosis I: Hurler, Hurler-Scheie and Scheie syndromes in the UK. Orphanet journal of rare diseases. 2008;3:24. doi: 10.1186/1750-1172-3-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Heron B, Mikaeloff Y, Froissart R, Caridade G, Maire I, Caillaud C, Levade T, Chabrol B, Feillet F, Ogier H, Valayannopoulos V, Michelakakis H, Zafeiriou D, Lavery L, Wraith E, Danos O, Heard JM, Tardieu M. Incidence and natural history of mucopolysaccharidosis type III in France and comparison with United Kingdom and Greece. American journal of medical genetics Part A. 2011;155A:58–68. doi: 10.1002/ajmg.a.33779. [DOI] [PubMed] [Google Scholar]
- 132.Coelho JC, Wajner M, Burin MG, Vargas CR, Giugliani R. Selective screening of 10,000 high-risk Brazilian patients for the detection of inborn errors of metabolism. European journal of pediatrics. 1997;156:650–654. doi: 10.1007/s004310050685. [DOI] [PubMed] [Google Scholar]
- 133.Jurecka A, Zakharova E, Cimbalistiene L, Gusina N, Malinova V, Rozdzynska-Swiatkowska A, Golda A, Kulpanovich A, Kaldenovna Abdilova G, Voskoboeva E, Tylki-Szymanska A. Mucopolysaccharidosis type VI in Russia, Kazakhstan, and Central and Eastern Europe. Pediatrics international : official journal of the Japan Pediatric Society. 2014;56:520–525. doi: 10.1111/ped.12281. [DOI] [PubMed] [Google Scholar]