

Abstract
The ubiquitous plasticizer, diethylhexyl phthalate (DEHP), is a known endocrine disruptor. However, DEHP exposure effects are not well understood. Changes in industrial and agricultural practices have resulted in increased prevalence of DEHP exposure and has coincided with the heightened occurrence of metabolic syndrome and obesity. DEHP and its metabolites are detected in the umbilical cord blood of newborns; however, the prenatal and perinatal effects of DEHP exposure have not been intensively studied. Previously, we discovered that phosphorylation (p) of proliferating cell nuclear antigen (PCNA) at tyrosine 114 (Y114) is required for adipogenesis and diet-induced obesity in mice. Here, we show the unique ability of DEHP to induce p-Y114 in PCNA in vitro. We also show that while DEHP promotes adipogenesis of wild type (WT) murine embryonic fibroblasts, mutation of Y114 to phenylalanine (Y114F) in PCNA blocked adipocyte differentiation. Given the induction of p-Y114 in PCNA by DEHP and the relationship to obesity, WT and Y114F PCNA mice were exposed to DEHP during gestation or lactation, followed by high fat diet feeding. Paradoxically, in utero exposure of Y114F PCNA females to DEHP led to a significant increase in body mass and was associated with augmented expression of PPARγ, a critical regulator of obesity, compared to WT controls. In utero exposure of WT mice to DEHP led to insulin sensitivity while Y114F mutation ablated this phenotype, indicating that PCNA is an important regulator of early DEHP exposure and ensuing metabolic phenotypes.
Keywords: phthalate exposure, proliferating cell nuclear antigen, PCNA, obesity, prenatal exposure
Graphical abstract

INTRODUCTION
For several decades, diethylhexyl phthalate (DEHP) has been used as an industrial plasticizer in the production of polyvinylchloride among other universal products; DEHP is present within plumbing, medical instruments, food products, and more. The release of DEHP into the environment, via manufacturing and leaching from DEHP-containing products, is well documented, posing an environmental risk which is compounded by low environmental degradation rate of the chemical (Wams 1987). As a result, large populations of industrialized nations are exposed to DEHP daily (Snyder, Westerhoff et al. 2003, Casals-Casas, Feige et al. 2008). There is increasing evidence supporting that endocrine disorders correlate with phthalates (including DEHP) owing to their activity as endocrine disruptors. The ubiquitous nature of DEHP poses complex challenges for expecting mothers, as DEHP and its metabolites (most notably the monoester MEHP) have been detected in the umbilical cord blood of newborns in a multitude of studies (Latini, De Felice et al. 2003, Jurewicz and Hanke 2011, Huang, Li et al. 2014, Darbre 2017, Minatoya, Araki et al. 2017); however, the effects of the introduction of DEHP during gestation and shortly after birth are not well understood.
A gap in knowledge exists with regards to both the mechanism of action and extent of influence of phthalates in human disease. Modern calorie-rich diets are tightly linked to the pandemic of metabolic disorders including obesity and diabetes, which may be compounded in the presence of environmental toxins. Indeed, increased prevalence of metabolic diseases has been found to correlate with changes in agricultural and industrial practices which are known to introduce endocrine disruptors into the environment. A subset of endocrine disruptors leads to metabolic deregulation, manifested by increased adipose deposition and disorder of glucose metabolism (Casals-Casas and Desvergne 2011, Heindel, Newbold et al. 2015). However, much of the current knowledge about in utero and postnatal exposure to endocrine disruptors have been focused on development, particularly of reproductive systems (Barakat, Lin et al. 2017, Cardoso, Alves et al. 2017). The potential impact of interaction between endocrine disruptors and diet on global metabolic programming remains understudied.
Endocrine disruptors are thought to work through binding to nuclear receptors with similar mechanisms as hormones (Snyder, Westerhoff et al. 2003, Henley and Korach 2006, Casals-Casas and Desvergne 2011). With respect to DEHP, the nuclear receptor known as peroxisome proliferator-activated receptor gamma (PPARγ) has been identified to be a specific molecular target in which MEHP, the monoester metabolite of DEHP, reportedly functions as an agonist in in vitro assays (Feige, Gelman et al. 2007, Casals-Casas, Feige et al. 2008). PPARγ’s function lies at the crossroads of both metabolic syndrome and obesity; this protein is required for the deposition of adipose tissues and is known to be the molecular target of insulin- sensitizing compounds thiazolidinediones (TZDs), which are used clinically to treat type 2 diabetes (Spiegelman 1998, Feige, Gelman et al. 2007).
Previously, our laboratory has shown that phosphorylation (p) of proliferating cell nuclear antigen (PCNA) at tyrosine 114 (Y114) plays a significant role in adipocyte maturation and subsequent clonal expansion in mice (Lo, Ho et al. 2013). When placed on a high-fat diet (HFD), mice defective for Y114 phosphorylation on PCNA exhibit a less obese phenotype compared to the wild type mice over a prolonged time course, establishing a role of Y114 phosphorylation (p-Y114) of PCNA to HFD-induced obesity. As the functional consequences of PCNA phosphorylation with respect to metabolic endocrine regulation are not well understood, particularly in the context of exposure to endocrine disruptors, we reasoned that our mouse model was suitable to study the effects of early exposure to DEHP in the context of a calorie-rich diet. In this study, we conducted analyses for the in vivo mechanism of DEHP exposure and subsequent obesity and metabolic morbidity. Using in utero and postnatal (lactational) DEHP exposure models with dosing congruent to that of human exposure (Kavlock, Boekelheide et al. 2002, Hao, Cheng et al. 2012), we characterized the effects on the offspring of DEHP exposed mothers during this sensitive developmental window with a focus on obesity and metabolic outcome.
MATERIALS AND METHODS
Chemicals and Antibodies
Antibodies for analysis included: anti-PCNA (Santa Cruz), anti-PPARγ (Cell Signaling Technologies), anti-actin C4 antibody (Abcam), and anti-Phospho Y114 PCNA generated via rabbits immunized with a KLH-conjugated peptide (synthetic sequence CNQEKVSD-pY-EMKLMD; Yenzym) and subsequent isolation of serum and extraction via a phosphopeptide affinity matrix and clean-up with an affinity matrix conjugated to unphosphorylated peptides (Lo, Ho et al. 2013). Diethylhexyl phthalate (DEHP), benzylbutyl phthalate (BBP), diisononyl phthalate (DNP) and Oil Red O were purchased from Sigma.
In Vitro Adipogenesis
Mouse embryonic fibroblasts (MEFs) used for in vitro studies were previously described (Lo, Ho et al. 2013). After plating MEFs and allowing for attachment, media with or without 0.5 μM DEHP was utilized for the totality of the adipogenesis protocol as previously described (Tang, Otto et al. 2003). Briefly, confluent MEFs are provided Dulbecco’s Modification of Eagle Medium with 10% FBS, 1 μg/ml insulin, 1 μM dexamethasone, and 0.5 mM 3-isobutyl-1-methylxanthine for two days, then DMEM with 10% Fetal Bovine Serum and 1 μg/ml insulin for two days followed by culturing in DMEM with 10% FBS. Visualization with 0.1% Oil Red O staining occurs 8 days after the initiation of differentiation. Adipogenesis statistics were performed using a Student’s t-test.
Mice
Wild type (WT/WT) mice and mice with a point mutation of PCNA at residue 114 which changes tyrosine to phenylalanine (PCNAY114F/Y114F)(Lo, Ho et al. 2013) in a FVB background were maintained per approved University of Cincinnati Institutional Animal Care and Use Committee (IACUC) protocols. Mice genotypes were confirmed as previously described (Lo, Ho et al. 2013). Exposure in both gestational and lactation exposed mice occurred through oral gavage of the pregnant/lactating mouse of either corn oil (vehicle), 0.05 mg/kg/day DEHP, or 500 mg/kg/day DEHP. Offspring were weaned onto high fat diet (45 kcal% fat, Research Diets, Inc.) at 21 days of age. Body mass monitored weekly until study mice were euthanized at 22 weeks of age. Body weight curve statistics were performed using either a Student’s t-test of data points at individual time points (Figure 3BC) or two-way ANOVA analyses of data points at individual time points (Figure 2BC).
Figure 3. Obesity phenotypes in WT and PCNAY114F/Y114F male and female mice.

(A) Representative images of hematoxylin and eosin stained sections of visceral fat pads isolated from WT and PCNAY114F/Y114F male and female mice with vehicle or DEHP exposure during gestation and/or lactation following 19 weeks of HFD feeding. Body mass of male (B) and female (C) WT and PCNAY114F/Y114F mice at low and high concentrations of DEHP exposed through gestation and lactation. For male mice: sample sizes of WT: lactation: control n=7, low dose n=8, high dose n=4; gestation: control n=6, low dose n=6, high dose n=7; PCNAY114F/Y114F lactation: control n=6, low dose n=7, high dose n=5; PCNAY114F/Y114F gestation: control n=4, low dose n=6, high dose n=7. For female mice: sample sizes of WT/WT mice: lactation: control n=4, low dose n=9, high dose n=6; gestation: control n=5, low dose n=9, high dose n=6; PCNAY114F/Y114F lactation: control n=8, low dose n=8, high dose n=6; PCNAY114F/Y114F gestation: control n=5, low dose n=8, high dose n=6. (D) Visceral and inguinal fat pad mass of WT and PCNAY114F/Y114F female mice at a high concentration of DEHP exposed through gestation and lactation. For both visceral and inguinal fat pads: sample sizes of WT mice: gestation n=6, lactation n=6; PCNAY114F/Y114F mice: gestation n=6, lactation n=6. *, p<0.05; **, p<0.001; ***, p<0.0001 with statistical analysis performed using a Student’s t-test. Body weight statistics performed using Student’s t-test of data points at individual time points.
Figure 2. DEHP exposure at low concentration (50 ug/kg/day) or high concentration (500 mg/kg/day) during gestation or through lactation has no overt impact on body mass weight in WT FVB mice.
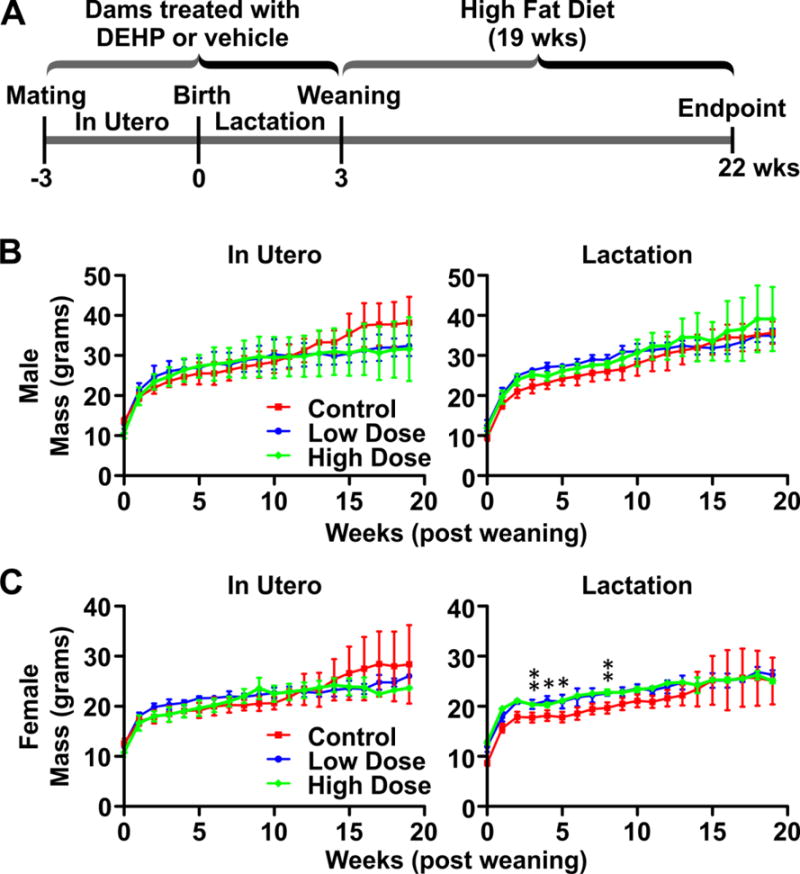
(A) Schematic of DEHP exposure and high fat diet (HFD) feeding. Dams were subjected to oral gavage with either corn oil (vehicle control) or DEHP. Treatments of the dams initiated at the first sign of a vaginal plug (for the In Utero group) or immediately after birth (for the Lactation group). Treatment of the dams ceased at weaning for both groups. At 3 weeks of age, all offspring of DEHP or control exposed dams were weaned onto a HFD. After 19 weeks of HFD feeding, offspring were euthanized. The body weight of the offspring was measured temporally. Body mass curves of wild type (WT) male (B) and female (C) offspring were generated and compared across control, low, and high concentrations of DEHP with HFD. (Males: lactation: control n=7, low dose n=8, high dose n=4; gestation: control n=6, low dose n=6, high dose n=7) (Females: lactation: control n=4, low dose n=9, high dose n=6; gestation: control n=5, low dose n=9, high dose n=6). *, p<0.05; **, p<0.01; Body weight statistics performed using ANOVA analyses of data points at individual time points.
Tissue Examination
Visceral and inguinal fat pads were dissected and weighed at the point of euthanasia. Samples were homogenized in RIPA lysis buffer (10 mM Tris-HCl, 1 mM EDTA, 0.5 mM EGTA, 1% Triton-X 100, 0.1% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 150 mM NaCl, 1 mM PMSF, Aprotinin 2 μg/mL, sodium fluoride 5 mM, sodium orthovanadate 1 mM) for analysis via immunoblot, or were fixed in 10% formalin, embedded in paraffin, and stained with hematoxylin and eosin for histological examination of fat pad morphology.
Glucose Tolerance and Insulin Tolerance Testing
Glucose tolerance testing (GTT) was performed using 1g/kg intra-peritoneal glucose injection (Andrikopoulos, Blair et al. 2008, Stuart, Brown et al. 2015); glucose levels were measured using Accu-Chek Aviva Plus meter and test strips (Roche) from blood collected via tail nicks. GTT statistics were performed using two-way ANOVA analyses of data at individual time points. Insulin tolerance testing (ITT) was performed using 0.5 U/kg as per published protocols (Feige, Gelman et al. 2007, Stuart, Brown et al. 2015). GTT and ITT were performed at 12 and 13 weeks of HFD feeding, respectively. ITT statistics were performed using a Fisher’s exact test.
RESULTS
DEHP treatment induces phosphorylation of PCNA at Y114 and promotes adipocyte differentiation in vitro
Utilizing mouse embryonic fibroblasts (MEF) derived from wild type (WT) mice cultured ex vivo, we sought to examine the effect of phthalates on PCNA protein levels and status of phosphorylation at the Y114 residue. MEFs were exposed to benzylbutyl phthalate (BBP), diisononyl phthalate (DNP), and diethylhexyl phthalate (DEHP) after which whole cell lysates were prepared to be examined p-Y114 by Western blot analysis. As shown in Figure 1A, DEHP strongly induced p-Y114 PCNA whereas BBP and DNP showed no discernable induction over endogenous p-Y114 PCNA levels observed in the vehicle control group. Thus, DEHP was selected to be further studied in a dose course of p-Y114 increasing concentrations of DEHP and cell lysates were examined for p-Y114 PCNA by Western blot analysis (Figure 1B). The result shows that DEHP induces p-Y114 of PCNA in a dose-dependent manner.
Figure 1. Adipogenesis induced via DEHP requires p-Y114 PCNA.
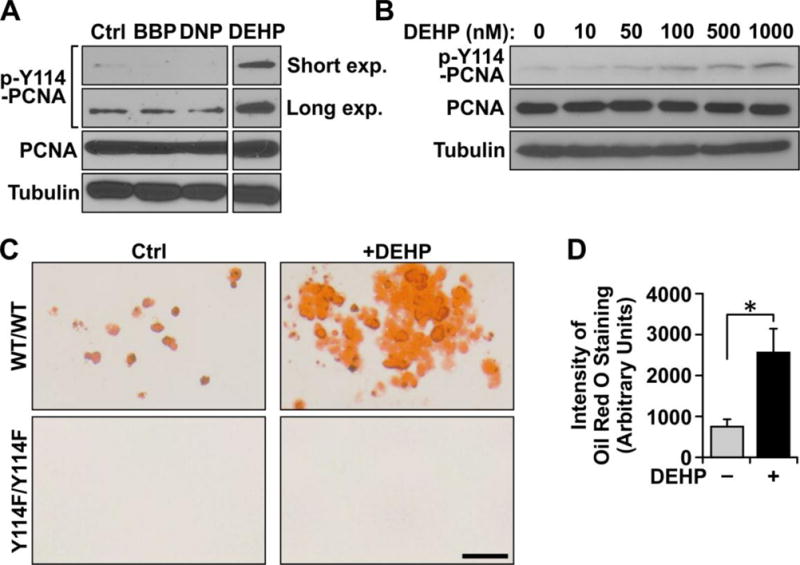
(A) Immunoblot of phosphorylated (p-Y114) PCNA and total PCNA in response to phthalates BBP, DNP, and DEHP each at 1 μM for 24 hours. (B) Immunoblot of p-Y114 PCNA and total PCNA after DEHP administration at increasing concentrations. (C) Oil Red O staining to visualize adipogenesis of PCNA WT and PCNAY114F/Y114F mutant MEFs in the presence or absence of DEHP and (D) quantitation of staining of WT cells measuring intensity of Oil Red O staining. The staining intensity was averaged from three experimental replicates; error bars depict standard error). *, p<0.05 with statistical analysis performed using a Student’s t-test.
DEHP has been shown to induce adipogenesis in vitro (Hurst and Waxman 2003, Bility, Thompson et al. 2004, Feige, Gelman et al. 2007, Hao, Cheng et al. 2012). Previously, we demonstrated that p-Y114 PCNA induction is required for in vitro adipogenesis, and therefore sought to understand whether DEHP-induced adipogenesis is through the function of p-Y114 PCNA during adipocyte differentiation. To test this linkage, we compared WT MEFs to MEFs isolated from mice containing a tyrosine to phenylalanine substitution at residue 114 in PCNA (PCNAY114F/Y114F) in an in vitro adipogenic assay. Basal levels of adipogenesis are detected in the WT control group as visualized by Oil Red O staining (Figure 1C). An approximately three-fold induction of adipocyte differentiation potential was evident when WT MEFs were exposed to DEHP during differentiation relative to vehicle exposed group (roughly 800 arbitrary units in the vehicle exposed group versus roughly 2500 arbitrary units in the DEHP exposed group) (Figure 1D). In contrast, no adipogenesis was evident in PCNAY114F/Y114F MEFs either under control conditions or when DEHP was introduced. These results suggest that DEHP exposure is able to enhance in vitro adipogenesis in the presence of p-Y114 PCNA, but is not sufficient to overcome the adipogenic defect of PCNAY114F/Y114F MEFs.
In utero and perinatal exposure to DEHP
To investigate the in vivo ramifications of DEHP exposure and PCNA phosphorylation, an in vivo exposure scheme was developed in mice to model the effects of in utero and perinatal exposure to DEHP utilizing daily oral gavage of dams with vehicle (corn oil), or DEHP at high (DEHPHigh 500 mg/kg) or low (DEHPLow 0.05 mg/kg) concentration (Figure 2A). For in utero exposure, dams were subjected to DEHP or vehicle treatment via daily oral gavage at the first sign of a vaginal plug following mating. Treatment of the dams continued until resulting offspring were weaned at 3 weeks of age. For lactation exposure, treatment of the mothers via oral gavage initiated concurrently with the birth of the offspring and lasted until weaning. All offspring were subsequently weaned onto a high fat diet (HFD; 45% kcal% fat) to mimic the calorie-rich diet prominent in the western countries (Troiano, Briefel et al. 2000). During HFD feeding, body mass was measured weekly for 19 weeks. Interestingly, body mass curves from wild type (WT) mice were not statistical different following exposure to vehicle, low dose of DEHP, or high dose of DEHP and was irrespective of gender (Figure 2B, 2C). These results suggest that in utero and perinatal DEHP exposure of WT mice does not have an impact on weight gain of offspring following HFD feeding.
Relative to WT FVB mice, our previous published study identified a decreased diet-induced body weight gain in PCNAY114F/Y114F FVB mice after long-term (23 weeks and longer) HFD feeding. Prior to 23 weeks of HFD feeding, the body mass of PCNAY114F/Y114F mice was found to be similar to that of WT (Lo, Ho et al. 2013). Taking this information into account, the endpoint of this study was designed such that any body mass alteration could be solely attributable to DEHP exposure and not genotype dependent. Thus, PCNAY114F/Y114F mice and their corresponding WT controls were exposed to DEHP as outlined in Figure 2A and were examined following 19 weeks of HFD feeding. As depicted in Figure 3A, no overt morphological changes were observed in histological analysis of individual visceral fat pads in response to DEHP exposure, regardless of Y114 phosphorylation of PCNA. Importantly, several differences were observed in body mass measurements when comparing WT with PCNAY114F/Y114F mice under specific exposure conditions (Figure 3B, 3C). For males exposed to vehicle during lactation, there was a slight but statistically significant decrease in body mass of PCNAY114F/Y114F mice compared to that of WT male mice during HFD feeding. A similar trend of decreased body mass in PCNAY114F/Y114F males relative to that of WT was also observed in mice exposed to vehicle in utero, although this decrease was not statistically significant. Interestingly, this relative decrease of body mass dissipated after DEHPLow or DEHPHigh exposure. These data provide evidence suggesting that early exposure of PCNAY114F/Y114F males to DEHP allows for a slight weight gain in response to HFD feeding compared to DEHP exposed WT males.
Female mice did not show a noticeable difference with respect to body mass in vehicle exposed groups in either in utero or lactation exposure windows. However, after DEHPHigh exposure in utero, PCNAY114F/Y114F mice showed increased body mass at both early and late time points relative to WT mice. To extend this observation, visceral and inguinal fat pads were collected from female mice at the end of the study period and weighed. Statistically significant increases in fat pad mass of female PCNAY114F/Y114F mice were observed compared to their WT counterparts at high doses of DEHP in both in utero as well as in the lactation exposure groups (Figure 3D). Taken together, these data suggest that early exposure to DEHP leads to a p-Y114 PCNA dependent effect, wherein the inability to phosphorylate this site leads to enhanced body mass in response to HFD feeding.
p-Y114 PCNA-dependent metabolic consequences of DEHP gestation exposure
In addition to overt obesity phenotypes, the role of p-Y114 PCNA in regulating molecular metabolism in vivo, particularly with respect to glucose metabolism, was examined. Given the similarities of the mice to high and low DEHP exposure and the slightly more pronounced weight gain observed in female PCNAY114F/Y114F mice during gestational exposure, we chose to focus further studies on female mice in response to high DEHP exposure groups. To examine glucose metabolism, an intraperitoneal bolus injection of glucose was given after 18 hours of fasting; blood glucose levels were subsequently followed until returning to the baseline. As shown in Figure 4A, no difference in glucose metabolism, irrespective of ability to phosphorylate Y114 on PCNA or exposure to DEHP (Figure 4A), was found. A similar experiment was performed to evaluate insulin sensitivity. WT and PCNAY114F/Y114F female mice fed HFD were equally tolerant of insulin treatment in the absence of DEHP exposure (Figure 4B). However, in sharp contrast to the comparable abilities of DEHP-exposed female WT and PCNAY114F/Y114F mice to respond to glucose, dramatic differences to insulin were observed between genotypes in response to DEHP. Shortly after intraperitoneal injection of insulin, all DEHP-exposed WT mice became lethargic and all but one mouse entered hypoglycemic shock necessitating experimental termination (Figure 4B). All PCNAY114F/Y114F mice, however, were insulin tolerant (Figure 4B) remaining active and returned to normal blood glucose levels similar to non DEHP-exposed mice (data not shown). These studies suggest that early maternal DEHP exposure dramatically impairs the ability of WT offspring to respond to insulin under HFD conditions and that the mechanism associated with this effect is due to p-Y114 PCNA.
Figure 4. Female mice exposed to DEHP in utero exhibit insulin sensitivity in a p-Y114 PCNA dependent manner.

(A) Glucose tolerance testing of WT and PCNAY114F/Y114F mice in the presence and absence of DEHP exposure at a high concentration during gestation. Data are not statistically significant using ANOVA analyses of data at individual time points using 5% significance threshold. (B) Frequency of mice showing tolerance or sensitivity to Insulin. Sample sizes of WT mice: control n=5, DEHP n=6; PCNAY114F/Y114F mice: control n=6, DEHP n=6. *, p<0.05 with statistical analysis performed using a Fisher’s exact test. (C) Representative PPARy and Actin Immunoblots of cell lysates of visceral fat pad tissue from WT and PCNAY114F/Y114F female mice exposed to vehicle or DEHP during gestation (D) and densitometric quantitation of PPARy expression normalized to actin. *, p<0.05 with statistical analysis performed using ANOVA analyses.
To examine molecular consequences accompanying the p-Y114 PCNA-dependent insulin sensitive phenotype following DEHP exposure, protein levels of PPARγ were examined in visceral fat pads. In vehicle exposed mice, a significantly higher level of PPARγ was observed in WT compared to PCNAY114F/Y114F animals despite no discernable effects on glucose metabolism or insulin sensitivity. Maternal DEHP exposure lead to an increase in expression of PPARγ in PCNAY114F/Y114F mice while levels of PPARγ expression in WT mice slightly decreased compared to vehicle treated controls (Figure 4C, 4D).
DISCUSSION
Endocrine function is crucial for regulating the metabolism of food taken in as well as in maintaining the equilibrium between energy use and storage. As a lack of exercise and a calorie-rich diet cannot fully explain the high occurrence of obesity and metabolic syndrome prevalent in the US population, we sought to examine ubiquitous environmental factors, specifically endocrine disruptors, for their ability to exacerbate weight gain and metabolic phenotypes. In particular, exposure to phthalates, a common chemical found in plasticizers, has been increasingly associated with obesity (Hurst and Waxman 2003, Snyder, Westerhoff et al. 2003, Bility, Thompson et al. 2004, Casals-Casas and Desvergne 2011, Janesick and Blumberg 2011, Heindel, Newbold et al. 2015, Barakat, Lin et al. 2017, Cardoso, Alves et al. 2017), and recent studies suggest that phthalate exposure during the fetal period may increase this risk (Hatch, Nelson et al. 2008). The underlying mechanism associated with the early effect of phthalate exposure on obesity has not been well established and is a topic of intensive investigation. Our prior work identified proliferating cell nuclear antigen (PCNA) as a novel regulator of adipogenesis and we recently discovered that phosphorylation of PCNA at tyrosine residue 114 (Y114) of PCNA is necessary to augment the development of obesity in response to a calorie-rich diet (Lo, Ho et al. 2013). Given the emerging evidence relating phthalates and PCNA to obesity, we sought to investigate whether a metabolic connection existed between phthalate exposure and Y114 phosphorylation of PCNA in response to a high fat diet.
To study the link between endocrine disruption and PCNA, the induction of p-Y114 PCNA was examined in response to a variety of structurally similar phthalate esters. Remarkably, our data show that p-Y114 PCNA stimulation occurs selectively in response to DEHP, with no p-Y114 induction observed in response to the other phthalates BBP and DNP. Intriguingly, in the context of endocrine disruption of the estrogen receptor, BBP showed strong estrogenic activity, followed by DNP, while DEHP entirely lacked estrogenic activity (Harris, Henttu et al. 1997). Thus, despite the similarity in structure of phthalate esters, side chain variations lead to drastic differences in molecular and cellular responses. The selectivity of DEHP to induce p-Y114 PCNA is further supported by reports which link DEHP to induction of adipogenesis (Hao, Cheng et al. 2012). Additionally, DEHP has reported functional links with PPARγ (Hurst and Waxman 2003, Casals-Casas, Feige et al. 2008), whereas no links exist for DNP and BBP, suggesting that DEHP is an appropriate phthalate for this study over DNP and BBP. Our data show that DEHP exposure enhances adipogenesis of WT MEFs but is not sufficient to induce adipogenesis in MEFs without p-Y114 PCNA. These data suggest a novel role for p-Y114 PCNA as a molecular mechanism which may, at least in part, regulate the response to DEHP exposure. The in vitro study in Figure 1 more closely resembles a direct exposure model, which is not directly employed in the in vivo studies. Thus, the ability of DEHP to induce p-Y114 PCNA, as well as the lack therein when exposed to DNP or BBP may only be significant in direct exposure experiments. These data do not preclude involvement of p-Y114 PCNA in DNP or BBP exposure during developmental windows, however the induction in response to DEHP suggests functional ties between p-Y114 PCNA and DEHP, which led to the use of DEHP over other phthalates for our in vivo studies.
To more closely model exposure to DEHP in a physiological setting (Ward, Peters et al. 1998, Kloting, Hesselbarth et al. 2015, Lv, Cheng et al. 2016), we examined the effects of endocrine disruption in critical developmental windows susceptible to external cues. Specifically, the lurking phenotype of DEHP exposure during gestation and lactation with subsequent HFD was examined. Early exposure to high or low levels of DEHP, at either developmental time point, did not affect weight gain in WT FVB mice as judged by similar body mass measurements over the course of 19 weeks of HFD feeding between vehicle and DEHP exposed groups (Figure 2B, 2C). In PCNAY114F/Y114F mice, body mass increases over control mice were observed to varying extents in both male and female mice. Inherent body weight gains were also observed in control male mice compared to the PCNAY114F/Y114F male mice following exposure to corn oil vehicle during lactation; the mechanism associated with this change is not known and will require further characterization. Most significant was the body weight increase identified in female PCNAY114F/Y114F mice over control mice exposed to high DEHP levels during gestation. This body mass elevation was consistent with statistically significant increases in visceral and inguinal fat pad mass observed in female PCNAY114F/Y114F animals (Figure 3D). Taken together, these data suggest that PCNA phosphorylation at Y114 limits the accumulation of fat in adipose tissue in response to early DEHP exposure and a high-calorie diet may play a role in systemic regulation of metabolism controlling the distribution of body mass. Our studies, however, cannot distinguish if p-Y114 PCNA is required transiently during development leading to a pre-disposition to metabolic dysregulation, or if p-Y114 PCNA is required into adulthood after being exposed to DEHP in utero. Nonetheless, these data suggest that exposure to DEHP in this brief developmental window in the absence of Y114 PCNA phosphorylation may lead to enhanced obesity-related morbidity. It is worth noting that this study was limited to mice placed on HFD in order to mimic the energy-rich diet prevalent in the United States. Future studies including mice on normal chow would be useful to understand the involvement of diet on the effects observed here.
Most remarkable, however, was the insulin sensitive phenotype discovered in the WT female mice exposed to a high concentration of DEHP during gestation. In the absence of p-Y114 PCNA, the heightened insulin sensitivity was abolished (Figure 4B). Interestingly, this insulin sensitive phenotype was only observed in WT mice following DEHP exposure as the vehicle exposed mice of both WT and PCNAY114F/Y114F were able to tolerant insulin injection; thus, this sensitivity is dependent on early DEHP exposure and is p-Y114 PCNA-dependent. We consider the phenotype to be insulin sensitization of WT mice rather than insulin resistance of PCNAY114F/Y114F because both WT and PCNAY114F/Y114F mice not exposed to DEHP were tolerant of the same dose of insulin. In contrast to the reported insulin sensitization activity of PPARγ (Miles, Barak et al. 2000, Feige, Gelman et al. 2007), insulin sensitization of the WT mice after in utero DEHP exposure was correlated with a decrease in expression of PPARγ. While this finding is difficult to interpret, it is not unprecedented as a prior study with HFD-fed mice with a single allele of PPARγ exhibited an insulin sensitivity relative to mice with two alleles (Kubota, Terauchi et al. 1999). Within the context of direct exposure to DEHP, a previous study by others showed that mice directly exposed to DEHP beginning at 11 weeks of age exhibited insulin resistance (Kloting, Hesselbarth et al. 2015). This study also reported the similar effect of losing PPARγ expression after DEHP exposure. Reconciling these data with our current work, early developmental DEHP exposure may induce metabolic alterations that are distinct from exposure occurring later in life, and as such the role of PPARγ in this process may be more complex than previously assumed. Additionally, the role of p-Y114 PCNA in this context has not previously been examined. Though the insulin sensitivity phenotype is novel, experiments were performed on mice that were exposed to a high concentration of DEHP. It is not yet known if a lower exposure concentration would be sufficient to induce this phenotype and further experiments are warranted.
Metabolic research over the past couple of decades has identified adipose tissue to play an important role as an endocrine regulator with great influence over obesity and metabolic syndrome. Through secretion of hormonal regulators, such as leptin, TNFα, IL-6, MCP-1, PAI-1, ASP, resistin, and adiponectin, modulation and regulation of metabolic functions of the pancreas, liver, and muscles are coordinated and balanced in normal tissue (Troiano, Briefel et al. 2000, Tang, Otto et al. 2003, Andrikopoulos, Blair et al. 2008, Stuart, Brown et al. 2015). Human obesity-related diseases, including metabolic syndrome, may be a consequence of unbalanced regulation in adipose tissue, such as observed in the insulin resistance phenotype of A-ZIP/F-1 mice (mutation inducing a severe form of lipoatrophic diabetes) which is reversed via surgical implantation of adipose tissue grafts (Gavrilova, Marcus-Samuels et al. 2000, Kim, Gavrilova et al. 2000). The use of genetic murine models paired with surgical implantation of fat pads from DEHP treated mice would be a useful system to further study adipose tissue as a potential mediator of the effects of early DEHP exposure on endocrine functions. Further, future studies with use of fat pads from WT and PCNAY114F/Y114F mice implanted in A-ZIP/F-1 mice would provide insight into the role of p-Y114 PCNA in coordinating these processes with and without DEHP exposure.
Chemicals present in daily life, such as DEHP, may be sufficient to perturb endocrine functions of adipose tissue, with obesity and metabolic syndrome being potential consequences. However current knowledge is still rudimentary with respect to the impact that such chemicals have on human health. In this report, we demonstrated a novel molecular mechanism governing adverse health effects after exposure to an endocrine disruptor and anticipate that this information will lay the scientific foundation for the development of new biomarkers and targets in obesity related phenotypes.
Acknowledgments
We thank Glenn Doerman for graphic editing. Funding for this research was supported by the National Institutes of Health Research Grant ES023942 (SEW, S-CW).
Footnotes
CONFLICT OF INTEREST
The authors declare no competing financial interest.
References
- Andrikopoulos S, Blair AR, Deluca N, Fam BC, Proietto J. Evaluating the glucose tolerance test in mice. Am J Physiol Endocrinol Metab. 2008;295(6):E1323–1332. doi: 10.1152/ajpendo.90617.2008. [DOI] [PubMed] [Google Scholar]
- Barakat R, Lin PP, Rattan S, Brehm E, Canisso IF, Abosalum ME, Flaws JA, Hess R, Ko C. Prenatal Exposure to DEHP Induces Premature Reproductive Senescence in Male Mice. Toxicol Sci. 2017;156(1):96–108. doi: 10.1093/toxsci/kfw248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bility MT, Thompson JT, McKee RH, David RM, Butala JH, Vanden Heuvel JP, Peters JM. Activation of mouse and human peroxisome proliferator-activated receptors (PPARs) by phthalate monoesters. Toxicological Sciences. 2004;82(1):170–182. doi: 10.1093/toxsci/kfh253. [DOI] [PubMed] [Google Scholar]
- Cardoso AM, Alves MG, Mathur PP, Oliveira PF, Cavaco JE, Rato L. Obesogens and male fertility. Obes Rev. 2017;18(1):109–125. doi: 10.1111/obr.12469. [DOI] [PubMed] [Google Scholar]
- Casals-Casas C, Desvergne B. Endocrine disruptors: from endocrine to metabolic disruption. Annu Rev Physiol. 2011;73:135–162. doi: 10.1146/annurev-physiol-012110-142200. [DOI] [PubMed] [Google Scholar]
- Casals-Casas C, Feige JN, Desvergne B. Interference of pollutants with PPARs: endocrine disruption meets metabolism. Int J Obes (Lond) 2008;32(Suppl 6):S53–61. doi: 10.1038/ijo.2008.207. [DOI] [PubMed] [Google Scholar]
- Darbre PD. Endocrine Disruptors and Obesity. Curr Obes Rep. 2017;6(1):18–27. doi: 10.1007/s13679-017-0240-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feige JN, Gelman L, Rossi D, Zoete V, Metivier R, Tudor C, Anghel SI, Grosdidier A, Lathion C, Engelborghs Y, Michielin O, Wahli W, Desvergne B. The endocrine disruptor monoethyl-hexyl-phthalate is a selective peroxisome proliferator-activated receptor gamma modulator that promotes adipogenesis. J Biol Chem. 2007;282(26):19152–19166. doi: 10.1074/jbc.M702724200. [DOI] [PubMed] [Google Scholar]
- Gavrilova O, Marcus-Samuels B, Graham D, Kim JK, Shulman GI, Castle AL, Vinson C, Eckhaus M, Reitman ML. Surgical implantation of adipose tissue reverses diabetes in lipoatrophic mice. J Clin Invest. 2000;105(3):271–278. doi: 10.1172/JCI7901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao C, Cheng X, Xia H, Ma X. The endocrine disruptor mono-(2-ethylhexyl) phthalate promotes adipocyte differentiation and induces obesity in mice. Biosci Rep. 2012;32(6):619–629. doi: 10.1042/BSR20120042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris CA, Henttu P, Parker MG, Sumpter JP. The estrogenic activity of phthalate esters in vitro. Environ Health Perspect. 1997;105(8):802–811. doi: 10.1289/ehp.97105802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatch EE, Nelson JW, Qureshi MM, Weinberg J, Moore LL, Singer M, Webster TF. Association of urinary phthalate metabolite concentrations with body mass index and waist circumference: a cross-sectional study of NHANES data, 1999–2002. Environ Health. 2008;7:27. doi: 10.1186/1476-069X-7-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heindel JJ, Newbold R, Schug TT. Endocrine disruptors and obesity. Nature reviews Endocrinology. 2015;11(11):653. doi: 10.1038/nrendo.2015.163. [DOI] [PubMed] [Google Scholar]
- Henley DV, Korach KS. Endocrine-disrupting chemicals use distinct mechanisms of action to modulate endocrine system function. Endocrinology. 2006;147(6):s25–s32. doi: 10.1210/en.2005-1117. [DOI] [PubMed] [Google Scholar]
- Huang Y, Li J, Garcia JM, Lin H, Wang Y, Yan P, Wang L, Tan Y, Luo J, Qiu Z, Chen JA, Shu W. Phthalate levels in cord blood are associated with preterm delivery and fetal growth parameters in Chinese women. PLoS One. 2014;9(2):e87430. doi: 10.1371/journal.pone.0087430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst CH, Waxman DJ. Activation of PPARα and PPARγ by environmental phthalate monoesters. Toxicological Sciences. 2003;74(2):297–308. doi: 10.1093/toxsci/kfg145. [DOI] [PubMed] [Google Scholar]
- Janesick A, Blumberg B. Endocrine disrupting chemicals and the developmental programming of adipogenesis and obesity. Birth Defects Research Part C: Embryo Today: Reviews. 2011;93(1):34–50. doi: 10.1002/bdrc.20197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurewicz J, Hanke W. Exposure to phthalates: reproductive outcome and children health. A review of epidemiological studies. Int J Occup Med Environ Health. 2011;24(2):115–141. doi: 10.2478/s13382-011-0022-2. [DOI] [PubMed] [Google Scholar]
- Kavlock R, Boekelheide K, Chapin R, Cunningham M, Faustman E, Foster P, Golub M, Henderson R, Hinberg I, Little R, Seed J, Shea K, Tabacova S, Tyl R, Williams P, Zacharewski T. NTP Center for the Evaluation of Risks to Human Reproduction: phthalates expert panel report on the reproductive and developmental toxicity of butyl benzyl phthalate. Reprod Toxicol. 2002;16(5):453–487. doi: 10.1016/s0890-6238(02)00029-1. [DOI] [PubMed] [Google Scholar]
- Kim JK, Gavrilova O, Chen Y, Reitman ML, Shulman GI. Mechanism of insulin resistance in A-ZIP/F-1 fatless mice. J Biol Chem. 2000;275(12):8456–8460. doi: 10.1074/jbc.275.12.8456. [DOI] [PubMed] [Google Scholar]
- Kloting N, Hesselbarth N, Gericke M, Kunath A, Biemann R, Chakaroun R, Kosacka J, Kovacs P, Kern M, Stumvoll M, Fischer B, Rolle-Kampczyk U, Feltens R, Otto W, Wissenbach DK, von Bergen M, Bluher M. Di-(2-Ethylhexyl)-Phthalate (DEHP) Causes Impaired Adipocyte Function and Alters Serum Metabolites. PLoS One. 2015;10(12):e0143190. doi: 10.1371/journal.pone.0143190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota N, Terauchi Y, Miki H, Tamemoto H, Yamauchi T, Komeda K, Satoh S, Nakano R, Ishii C, Sugiyama T, Eto K, Tsubamoto Y, Okuno A, Murakami K, Sekihara H, Hasegawa G, Naito M, Toyoshima Y, Tanaka S, Shiota K, Kitamura T, Fujita T, Ezaki O, Aizawa S, Kadowaki T, et al. PPAR gamma mediates high-fat diet-induced adipocyte hypertrophy and insulin resistance. Mol Cell. 1999;4(4):597–609. doi: 10.1016/s1097-2765(00)80210-5. [DOI] [PubMed] [Google Scholar]
- Latini G, De Felice C, Presta G, Del Vecchio A, Paris I, Ruggieri F, Mazzeo P. Exposure to Di(2-ethylhexyl)phthalate in humans during pregnancy. A preliminary report. Biol Neonate. 2003;83(1):22–24. doi: 10.1159/000067012. [DOI] [PubMed] [Google Scholar]
- Lo YH, Ho PC, Chen MS, Hugo E, Ben-Jonathan N, Wang SC. Phosphorylation at tyrosine 114 of Proliferating Cell Nuclear Antigen (PCNA) is required for adipogenesis in response to high fat diet. Biochem Biophys Res Commun. 2013;430(1):43–48. doi: 10.1016/j.bbrc.2012.11.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv Z, Cheng J, Huang S, Zhang Y, Wu S, Qiu Y, Geng Y, Zhang Q, Huang G, Ma Q, Xie X, Zhou S, Wu T, Ke Y. DEHP induces obesity and hypothyroidism through both central and peripheral pathways in C3H/He mice. Obesity (Silver Spring) 2016;24(2):368–378. doi: 10.1002/oby.21359. [DOI] [PubMed] [Google Scholar]
- Miles PD, Barak Y, He W, Evans RM, Olefsky JM. Improved insulin-sensitivity in mice heterozygous for PPAR-gamma deficiency. J Clin Invest. 2000;105(3):287–292. doi: 10.1172/JCI8538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minatoya M, Araki A, Miyashita C, Sasaki S, Goto Y, Nakajima T, Kishi R. Prenatal di-2-ethylhexyl phthalate exposure and cord blood adipokine levels and birth size: The Hokkaido study on environment and children’s health. Sci Total Environ. 2017;579:606–611. doi: 10.1016/j.scitotenv.2016.11.051. [DOI] [PubMed] [Google Scholar]
- Snyder SA, Westerhoff P, Yoon Y, Sedlak DL. Pharmaceuticals, personal care products, and endocrine disruptors in water: implications for the water industry. Environmental Engineering Science. 2003;20(5):449–469. [Google Scholar]
- Spiegelman BM. PPAR-gamma: adipogenic regulator and thiazolidinedione receptor. Diabetes. 1998;47(4):507–514. doi: 10.2337/diabetes.47.4.507. [DOI] [PubMed] [Google Scholar]
- Stuart WD, Brown NE, Paluch AM, Waltz SE. Loss of Ron receptor signaling leads to reduced obesity, diabetic phenotypes and hepatic steatosis in response to high-fat diet in mice. Am J Physiol Endocrinol Metab. 2015;308(7):E562–572. doi: 10.1152/ajpendo.00467.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang QQ, Otto TC, Lane MD. CCAAT/enhancer-binding protein beta is required for mitotic clonal expansion during adipogenesis. Proc Natl Acad Sci U S A. 2003;100(3):850–855. doi: 10.1073/pnas.0337434100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troiano RP, Briefel RR, Carroll MD, Bialostosky K. Energy and fat intakes of children and adolescents in the united states: data from the national health and nutrition examination surveys. Am J Clin Nutr. 2000;72(5 Suppl):1343S–1353S. doi: 10.1093/ajcn/72.5.1343s. [DOI] [PubMed] [Google Scholar]
- Wams TJ. Diethylhexylphthalate as an environmental contaminant–a review. Sci Total Environ. 1987;66:1–16. doi: 10.1016/0048-9697(87)90072-6. [DOI] [PubMed] [Google Scholar]
- Ward JM, Peters JM, Perella CM, Gonzalez FJ. Receptor and nonreceptor-mediated organ-specific toxicity of di(2-ethylhexyl)phthalate (DEHP) in peroxisome proliferator-activated receptor alpha-null mice. Toxicol Pathol. 1998;26(2):240–246. doi: 10.1177/019262339802600208. [DOI] [PubMed] [Google Scholar]