

Abstract
Antiviral transcriptional responses and regulated cell death are critical components of the host response to virus infection. However, in contrast to the signaling pathways that promote antiviral transcription, those that initiate cell death following virus infection remain less understood. Several recent studies identified pattern recognition receptors (PRRs) of the mammalian innate immune system that activate cell death pathways. These same receptors also have established roles in the induction of antiviral gene expression. In this review, we discuss the mechanisms by which PRRs can serve dual roles as initiators of inflammatory gene expression and inducers of apoptosis and necroptosis following virus infection.
Initiation of Cell Death upon Virus Infection
Viruses are obligate intracellular pathogens that require living host cells to propagate. Consequently, hosts have evolved mechanisms to recognize viral infection and inhibit virus replication. The antiviral response initiated by virus infection is multifaceted and consists of the induction of an antiviral transcriptional program, including expression of interferons (IFNs), cytokines, chemokines, and the activation of cell death pathways (apoptosis, necroptosis, and pyroptosis) [1, 2]. Individually, these responses provide distinct benefits to the host following virus infection. IFN can control virus replication by promoting expression of IFN-stimulated genes (ISGs), which directly antagonize the virus lifecycle [3]. This response limits virus replication in the infected cell and promotes an antiviral state in adjacent uninfected cells. In contrast, programmed cell death eliminates the infected-cell, eradicating the replicative niche that facilitates virus production [1]. In addition, expression of cytokines and the activation of inflammatory cell death pathways contribute to the recruitment of immune cells to the site of infection and promote adaptive immunity [4]. Therefore, the combined activities of antiviral transcription and cell death are critical to control viral infection and establish prolonged immunity.
The host-encoded proteins that initiate antiviral transcription are collectively known as pattern recognition receptors (PRRs). Of these proteins, the Toll-like receptors (TLRs), the RIG-I-like receptors (RLRs), and cGMP-AMP (cGAMP) synthase (cGAS) have attracted the most attention. These proteins function as receptors to detect viral nucleic acids in the form of incoming genomic material or replication intermediates. TLRs are membrane-bound receptors that survey the extracellular space or endosomes for viral nucleic acids [2]. The endosomal TLRs -3, -7/-8, and -9 detect double-stranded viral RNA (vRNA), single-stranded vRNA, and unmethylated CpG-rich viral DNA (vDNA), respectively [5-8]. Cytosolic vRNA is detected by the RLRs, RIG-I and MDA5 [9-11]. Upon recognition of their ligands, RIG-I and MDA5 engage the adaptor protein MAVS, which provides a scaffold to activate the NF-κB and IRF3 transcription factors (Sun et al 2006). In contrast, the genomes of cytosolic DNA viruses can be sensed by cGAS, an enzyme that produces the second messenger cGAMP [12, 13]. cGAMP binds and activates the endoplasmic reticulum (ER)-localized stimulator of IFN genes (STING) protein, which coordinates activation of the IFN-inducing transcription factor IRF3 [13, 14].
In addition to inducing inflammatory and antiviral transcriptional responses, recent studies have demonstrated that several of the PRRs described above also facilitate programmed cell death. This observation is particularly evident in apoptosis and necroptosis. In contrast, PRRs involved in pyroptosis are not known to engage signaling cascades resulting in transcription factor activation. In this review, we highlight recent advances in the mechanistic understanding of how apoptosis and necroptosis are initiated following virus infection, and the consequences of these responses for virus replication and host immunity. Due to space constraints, we will not discuss the role of pyroptosis in viral immunity, but we point readers to a recent review by Lupfer and colleagues [15].
Apoptosis
Apoptosis is a non-inflammatory type of programmed cell death characterized by morphological changes, including cell shrinkage, nuclear condensation, and plasma membrane blebbing [16]. These outcomes are the consequence of degradation of numerous cellular proteins by intracellular enzymes, most notably caspases. Apoptotic caspase activity is induced by cell-extrinsic and -intrinsic mechanisms, and the biochemical signaling events that regulate these pathways are well-characterized [17]. The extrinsic apoptotic pathway is induced by signals originating in the extracellular space. This pathway is initiated by recognition of cognate ligands by specific cell surface receptors, including tumor necrosis factor receptor (TNFR) and Fas-ligand (FASL) receptor [18]. Activation of these receptors induces formation of a protein complex consisting of death receptor adaptor proteins TNFR-associated death domain (TRADD) and/or Fas-associated death domain (FADD), which activate the intracellular protease caspase-8 [18]. In turn, caspase-8 cleaves the effector caspases-3 and -7, which mediate apoptotic cell death by cleavage of many cellular proteins, including poly (ADP-ribose) polymerase (PARP-1) and inhibitor of caspase-activated DNase (ICAD) [19].
In contrast, the intrinsic apoptotic pathway is activated in response to intracellular stresses, including DNA damage, nutrient deprivation, and endoplasmic reticulum (ER) stress, and requires mitochondrial outer membrane permeabilization (MOMP) [17]. MOMP is induced by the proteins Bax or Bak, which in the absence of pro-apoptotic stimuli are sequestered in an inactive state in the cytoplasm by anti-apoptotic members of the Bcl-2 family of proteins. In response to apoptotic stimuli, Bax/Bak translocate to the mitochondria where they disrupt outer membrane integrity through a poorly understood mechanism. MOMP allows for release of the intramembrane mitochondrial component cytochrome C, which facilitates oligomerization of apoptotic protease-activating factor 1 (APAF-1) and formation of the heptameric complex known as the apoptosome [20]. Caspase-9 is then recruited to the apoptosome where it is activated and cleaves caspase-3 and -7 resulting in cell death [17].
Cross talk occurs between the cell-intrinsic and -extrinsic apoptosis pathways in a cell-type dependent manner. Consequently, it is now recognized that two types of cells exist, in terms of the mechanisms used to promote apoptosis. These cell types are distinguished by their differential dependence on mitochondrial factors by extrinsic signals to induce cell death. In type I cells, MOMP is not required to activate caspase-3 and -7 downstream of TNF or FASL [17]. In contrast, type II cells require MOMP to cleave these caspases. In type II cells, death receptor activated caspase-8 cleaves Bcl-2 homology 3-interacting domain death agonist (Bid), which results in accumulation of truncated Bid (tBid) [17]. tBID associates with the mitochondrial membrane and activates Bax to initiate the MOMP signaling pathway [21].
Apoptotic cell death is important in the host response to viral infection, as demonstrated by the increased susceptibility of bax-/- mice to several viral infections [22, 23]. Viral infection can induce the extrinsic apoptotic pathway through PRR-dependent expression of death receptor ligands, including TNFα. In addition to regulating extrinsic apoptotic signals, recent evidence suggests PRRs activate apoptosis through intrinsic transcription-independent mechanisms. The RLR signaling pathway was implicated in cell death by the identification of MAVS as a regulator of apoptosis following infection by Sendai virus (SeV), vesicular stomatitis virus (VSV) or encephalomyocarditis virus (ECMV) [22, 24]. Apoptosis in primary murine embryonic fibroblasts (MEFs) and HT1080 cells mediated by RNA virus infection or transfection of a synthetic RNA ligand (poly[I:C]) that mimics double-stranded vRNA requires RIG-I and IRF3 [22, 25, 26]. Based on these observations, the pathway was subsequently named the RLR-induced IRF3-mediated pathway of apoptosis (RIPA) [22]. The DNA binding domain of IRF3 is not necessary to mediate apoptosis following RIG-I activation in MEFs [25], and overexpression of the alternatively translated form of human MAVS known as miniMAVS, which is incapable of inducing antiviral transcriptional responses, is sufficient to induce apoptosis in HEK293T cells [27], indicating that the transcriptional activity of IRF3 is not required to promote RIPA. Instead, linear ubiquitination of IRF3 appears to induce its association with Bax, and promote mitochondrial translocation of the IRF3/Bax complex, resulting in cytochrome C release and activation of caspase-9 [23, 25]. The receptor proximal events that promote this non-transcriptional activity of IRF3 are unclear, but it is possible that MAVS and/or miniMAVS (in human cells) coordinate the poly-ubiquitination of IRF3 by the linear ubiquitin chain complex (LUBAC). In vivo, an IRF3-mutant incapable of inducing transcription is protective following SeV infection [23]. Therefore, more work is necessary to define the relative roles IRF3-mediated transcriptional and non-transcriptional responses play in host protection.
PRR-dependent activation of apoptosis has recently been extended to the detection of viral DNA following adenovirus (AdV) and herpes simplex virus-1 (HSV-1) infection. In certain cell types (e.g., HEK293T and Epstein-Barr virus-positive burkitt lymphoma cell lines), RIG-I-dependent detection of DNA viruses occurs through the production of RNA transcripts by RNA polymerase III [28, 29]. RNA polymerase III activity is required for AdV induced apoptosis in HT1080 cells, suggesting AdV can induce apoptosis via a RNA intermediate [22] (Figure 1). In contrast, HSV-1 induced PARP-1 cleavage in primary human fibroblasts is dependent on the cGAS/STING pathway [30] (Figure 1). The cellular components downstream of cGAS/STING that facilitate PARP-1 cleavage have not been determined. However, similar to the RIG-I/MAVS signaling pathway, activated STING engages IRF3 to promote antiviral transcription [14]. Therefore, the apoptotic response to HSV-1 DNA may involve the mitochondrial IRF3/Bax signaling pathway initiated by RIPA.
Figure 1. Viral infections activate distinct and overlapping signaling pathways to initiate apoptosis and necroptosis.
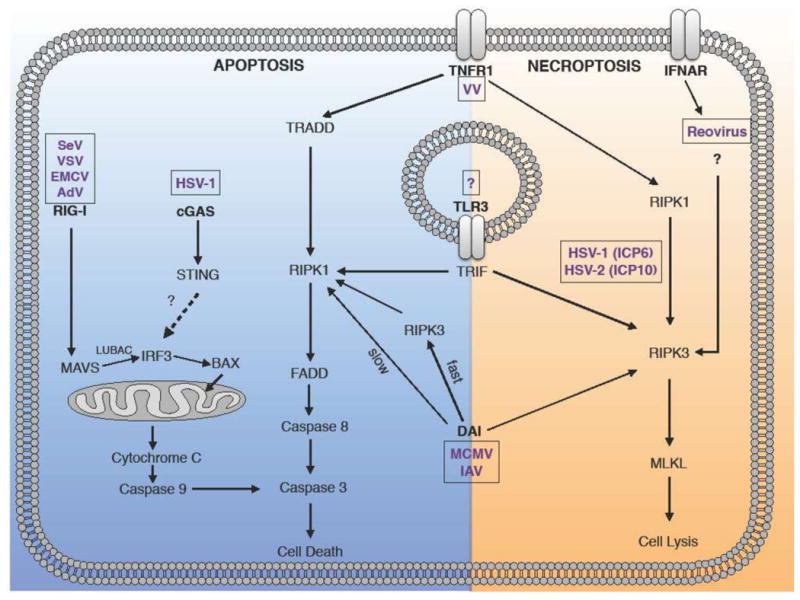
(Apoptosis) SeV, VSV, EMCV, and AdV activate a RIG-I-dependent signaling cascade resulting in cleavage of caspase-3 and cell death. In contrast, HSV-1 initiates apoptosis through a parallel pathway involving cGAS and STING. TNFR, TLR3/TRIF, and DAI signal through distinct signaling complexes that activate caspase-8. The initial DAI-dependent response requires RIPK3, but RIPK3-independent apoptosis is observed at later times post-receptor engagement. (Necroptosis). TNFR and TLR3/TRIF initiate necroptosis through RIPK1/RIPK3 and RIPK3, respectively. MCMV and IAV activate DAI-dependent necroptosis via RIPK3. Reovirus is sensed by an unknown IFN-inducible factor, which activates RIPK3. HSV-1 ICP6 and HSV-2 ICP10 proteins induce necroptosis by binding to RIPK1/RIPK3. SeV (Sendai virus), VSV (vesicular stomatitis virus), ECMV (encephalomyocarditis virus) HSV-1 (herpes simplex virus 1), MCMV (murine cytomegalovirus), IV (influenza virus), Reovirus (respiratory enteric orphan virus), VV (vaccinia virus).
The activation of apoptosis by distinct cytosolic PRRs that sense viral infection suggests this response might be a general feature downstream of receptors that signal through IRF3. However, the ability of TLRs that utilize IRF3 to initiate apoptosis following virus infection remains poorly characterized. All TLRs have the potential to induce the extrinsic apoptotic pathway, as all members of this receptor family induce TNFa expression [31]. In addition to this common ability to induce TNFα, a subset of TLRs activate TNFα-independent apoptosis via a pathway that engages several classic extrinsic apoptosis signaling components. These TLRs (TLR3 and TLR4) are unique in their abilities to engage the cytosolic adaptor TRIF, which seeds the formation of a multi protein-complex known as the ripoptosome [32]. The ripoptosome consists of TRIF, FADD, FLICE-like inhibitor protein (cFLIP), receptor-interacting protein (RIP) kinase 1 (RIPK1), and caspase-8 [32-34] (Figure 1). Assembly of the ripoptosome correlates with this ability of TLR4 and TLR3 to induce an extrinsic-like apoptosis pathway, as this complex promotes caspase-8 activation [32, 34]
The regulation of TLR-induced apoptosis is still unclear, as TRIF signaling downstream of TLR3 and TLR4 does not always lead to apoptosis. For example, activation of TLR3 by poly (I:C) induces apoptosis in a cell type-dependent manner [25, 35, 36]. In certain human cancer cell lines and primary human keratinocytes, treatment with poly (I:C) induces apoptosis via a TLR3-dependent mechanism, but similar treatments of mouse keratinocytes induce cytokine expression but not cell death [35-37]. While the context-dependent mechanisms that influence TLR-induced apoptosis remain undefined, genetic analysis has established a critical role of TLR3 in host defense following several viral infections [36, 38, 39]. Interestingly, whereas the LUBAC is required for the RLR-mediated induction of apoptosis, this complex negatively regulates death pathways induced by TLR3 [36]. These results suggest that the signaling pathway that regulates TLR3-mediated apoptosis differs from that induced by cytosolic PRRs. This may reflect an intrinsic difference between endosomal and cytosolic PRRs in their ability activate IRF3-mediated apoptosis, even though critical signaling components (e.g., IRF3 and LUBAC) are shared between these pathways.
In vivo studies demonstrate that apoptosis plays a critical role in host defense against viral infections [22, 23, 40]. However, while apoptosis can suppress virus replication by eliminating infected cells, it is a poor inducer of host immunity due to the non-inflammatory nature of the cell death. In contrast to apoptosis, an alternative form of cell death known as necroptosis (discussed below) can eliminate infected cells and initiate a strong pro-inflammatory response critical for host immunity.
Necroptosis
Necrosis is an inflammatory type of cell death characterized by cell swelling, loss of plasma membrane permeability, and release of cytosolic contents into the extracellular space [16]. As this cell death process is caspase-independent, it was initially thought to be an accidental (non-regulated) form of cell death [16]. Work in recent years has changed this view, and a subtype of necrosis known as necroptosis is now recognized as a tightly regulated process that is induced by inflammatory stimuli and encounters with pathogens [41]. Unlike the immune-suppressive nature of apoptosis, necroptosis an highly inflammatory process. Necroptosis mediates release of intracellular danger associated molecular patterns (DAMPs), including interleukin 1 alpha, HMGB1, uric acid, ATP and DNA [42], resulting in the recruitment of pro-inflammatory cell types to sites of infection [4, 43]. Release of DAMPs during necroptosis is mediated by mixed lineage kinase domain-like protein (MLKL), the executioner of necroptosis [44, 45]. Activated (i.e., phosphorylated) MLKL forms a homotrimeric complex that translocates to the plasma membrane where it forms a pore and induces cell lysis [46, 47]. Unlike apoptosis, necroptosis is not dependent on caspase activity. Rather, necroptosis is inhibited by pro-apoptotic caspase-8 [48, 49]. As caspase-8 inhibition is a common mechanism employed by viruses to prevent apoptosis [50], the ability of caspase-8 inhibited cells to induce necroptosis provides an important backup mechanism of restricting immune-evasive viral replication. Caspase-8 activity therefore differentiates between apoptotic cell death and necroptosis, with the latter being induced during immune-evasive virus infection.
Membrane receptor initiated necroptosis
Similar to apoptosis, necroptosis can be initiated following viral infection by external and internal stimuli, including activators of cell death receptors, viral nucleic acids, and direct sensing of viral proteins. TLRs that sense viral nucleic acids (TLR3, TLR7, TLR9) induce necroptosis upon ligand binding and caspase-8 inhibition, but these receptors elicit this response via different mechanisms. For example, similar to apoptosis, only TLRs that rely on TRIF (i.e., TLR3) promote necroptosis through an intrinsic mechanism [51, 52]. TLRs that signal through the adaptor MyD88 induce necroptosis indirectly through the expression of TNFα [52]. While TNFR signaling is normally associated with apoptosis, this receptor can also induce necroptosis under conditions of caspase-8 inhibition. When caspase-8 is inhibited, TNFR signaling induces the formation of a protein complex known as the necrosome, which includes the RIPK family members, RIPK1 [49] and RIPK3 [53-55] (Figure 1). RIPK1 associates with RIPK3 via a shared RIP homotypic interaction motif (RHIM) domain [56]. This interaction promotes RIPK3-mediated phosphorylation of RIPK1, which then phosphorylates RIPK3. Activated RIPK3 recruits and phosphorylates MLKL, resulting in cell lysis. Like TNFR, TLR3 has the potential to activate apoptosis and necroptosis (Figure 1). However, in contrast to TNFR-mediated necroptosis, RIPK1 is not required for TLR3-mediated necroptosis [52]. Instead, a RHIM domain in the TLR3 adaptor protein TRIF directly interacts with RIPK3 to facilitate MLKL activation (Figure 1).
The importance of TNFR- and TLR3-mediated apoptosis or necroptosis following virus infection in vivo remains undefined. The strongest argument for an antiviral role of TNFR-mediated necroptosis derives from studies of vaccinia virus (VV). VV encodes the B13R protein, which blocks caspase-8 activity and sensitizes infected cells to TNFα-mediated necroptosis in vitro [57]. In addition, tnfr2-/- and ripk3-/- mice are more susceptible to VV-mediated lethality [53, 58, 59], and tnfr1-/- mice have impaired cutaneous immune responses to infection [58]. Furthermore, these studies noted reduced inflammatory responses to VV infection in the absence of these signaling components, suggesting an important role for necroptosis in the antiviral response to VV infection. However, TNFR and RIPK3 have additional necroptosis-independent activities [60-63], and the contribution of these activities to VV antiviral immunity in vivo have not been ruled out. Future studies investigating the susceptibility of mlkl-/- and tnf1-/-/mlkl-/- mice to VV infection will clarify the role of TNFR-mediated necroptosis in antiviral immunity.
Tlr3-/- mice infected with various viruses have enhanced viral loads and more pronounced disease following infection, and TLR3-mediated viral control has been attributed to the activation of an antiviral transcriptional response [64]. However, the contribution of TLR3-mediated necroptosis in mediating these effects has not been determined. Interestingly, no viral infection has been shown to induce TLR3-dependent necroptosis in vitro, and elucidation of the necroptotic signaling pathway initiated by TLR3 has been performed solely through use of synthetic RNAs that mimic viral nucleic acids [51, 52]. Therefore, it will be important to ascertain whether TLR3 mediates necroptosis during a bona fide virus infection and whether this response contributes to antiviral immunity in vivo.
Cytosolic receptor initiated necroptosis
Viral nucleic acids can initiate cytosolic PRR-dependent necroptosis in response to DNA and RNA virus infection. Under these conditions, necroptosis is dependent on DNA dependent activator of IRFs (DAI), a protein encoded by the zbp1 gene. Originally, described as a sensor of vDNA upstream of IFN and cytokine expression [65], DAI was later demonstrated to play a critical role in promoting necroptosis in MEFs following infection with murine cytomegalovirus (MCMV) [66] (Figure1). In contrast to TNFR-induced necroptosis, DAI-induced necroptosis following MCMV infection requires RIPK3, but not RIPK1 [66, 67]. The antiviral roles of DAI or RIPK3 following MCMV infection in vitro and in vivo were only revealed with a virus expressing a mutant viral inhibitor of RIP activation (vIRA) protein, which blocks the interaction between DAI and RIPK3 [66]. vIRA mutant viruses replicate poorly in wild-type (wt) mice and MEFs [68, 69], but this replication defect is reversed in the absence of DAI [66]. Interestingly, the human CMV (HCMV) also blocks necroptosis in human fibroblasts, but at a different stage of this cell death pathway and independently of the HCMV homologue of MCMV vIRA (UL45) [70]. HCMV inhibits TNFR-induced necroptosis downstream of MLKL phosphorylation via an undefined mechanism that requires viral gene expression [71].
Similar to MCMV, influenza A virus (IAV) infection induces cell death in MEFs and human lung epithelial cells via a DAI/RIPK3-dependent signaling axis [72, 73] (Figure 1). However, mlkl-/- MEFs are as susceptible as wt MEFs for IAV-induced cell death, suggesting a necroptosis-independent role for DAI following IAV infection [72, 74]. Genetic analysis revealed that in the absence of MLKL, IAV-infected MEFs undergo DAI-dependent apoptosis, and that cell survival can only be rescued by simultaneous deletion of mlkl and the apoptosis regulator fadd [72, 74]. These results indicate a role for apoptosis and necroptosis in DAI-induced cell death. DAI-induced apoptosis appears to differ from that induced by TNFR in that RIPK1 expression, but not its kinase activity, is required for cell death [74]. Furthermore, the requirement for RIPK3 in this process is kinetic; i.e., RIPK3 facilitates a faster IAV-induced apoptotic death, but is not required at later time points post-infection [72] (Figure1). Mice expressing a catalytically inactive RIPK3 die in a RIPK1 and caspase-8 dependent manner [75] and, similarly, RIPK3 regulated apoptosis following IAV infection occurs independent of RIPK3 kinase activity [72], arguing for a dual role for RIPK3 in virus-induced necroptosis and apoptosis [75]. DAI-induced apoptosis is likely initiated in MCMV-infected cells, but is functionally blocked by expression of the viral inhibitor of caspase-8-induced apoptosis (vICA) [76].
Unlike the clear role for DAI in controlling MCMV infection in vivo [66], the functional consequences of DAI activity and cell death following IAV infection remain controversial. In one study, DAI was required to protect mice against IAV lethality [72], and consistent with this observation, IAV replication was enhanced in zbp1-/- mice. In contrast, another study observed decreased susceptibility of zbp1-/- mice to IAV lethality, although they also observed enhanced virus replication [73]. The reasons for these inter-study differences studies are not understood.
In vivo studies examining the role of cell death components in antiviral immunity have recently become more difficult to interpret with the identification of cell death-independent activities of RIPK1 and RIPK3. In contrast to IAV-infected MEFs, IAV-infected murine bone marrow-derived macrophages (BMDM) do not die but rather promote ifnβ transcription and translation in a RIPK1 or RIPK3-dependent manner, respectively [63]. In BMDM, RIPK3 negatively regulates an interaction between RIPK1 and MAVS resulting in decreased ifnβ transcription, while simultaneously promoting PKR phosphorylation and ifnβ mRNA stability. Consequently, ripk3-/- BMDM secrete less IFNβ, which may contribute to the increased susceptibility of ripk3-/- mice to IAV infection[63]. RIPK3 has also been shown to promote cell death-independent neuroinflammation following subcutaneous West Nile virus (WNV) infection [62]. WNV-infected neurons from ripk3-/- mice show reduced ccl2 and cxcl10 expression, which correlates with a reduced accumulation of infiltrating leukocytes in the brains of infected-mice [62]. Therefore, cell-type dependent differences in RIPK3 activity may govern whether a RIPK3-dependent phenotype is regulated by cell death or cytokine expression. It is therefore important to determine whether the executioners of apoptosis or necroptosis (i.e., caspase-8 or MLKL) are involved in phenotypes ascribed to RIPK3 in vivo.
Despite the initial observation that DAI is a sensor of viral DNA (vDNA) [65], vRNA can be immunoprecipitated with DAI following IAV infection, indicating that DAI may also sense vRNA. Presumably, the ligand recognized by DAI following MCMV infection is vDNA, but its ability to bind MCMV DNA has not been demonstrated. This raises the question of whether DAI senses vRNA and vDNA or whether MCMV infection produces a RNA ligand recognized by DAI. As mentioned previously, viral DNAs can act as templates to produce immunostimulatory RNAs for RIG-I dependent responses [28, 29]. Therefore, an analogous system for the detection of MCMV by DAI may exist.
In addition to detecting vRNA, DAI is proposed to detect IAV nucleoprotein (NP) and polymerase subunit PB1 [73]. NP and PB1 bind to the C-terminus of DAI, which was previously identified as the critical region for DAI binding to TBK1 [65]. However, whether these proteins regulate necroptosis remains undetermined. Alternatively, the ability of PB1 and NP to associate with DAI may reflect an uncharacterized evasion strategy, and further investigation of these interactions is therefore warranted. The ability of viral proteins, rather than viral nucleic acids, to initiate necroptosis was first demonstrated in response to HSV infection. HSV-1 and HSV-2 induce necroptosis in murine cells, and the viral RHIM domain containing proteins ICP6 (HSV-1) and ICP10 (HSV-2) mediate this response [77, 78]. Homodimers of ICP6 and ICP10 associate with RIPK1/RIPK3 via RHIM:RHIM protein interactions and induce MLKL-dependent necroptosis. Ripk3-/- mice are more susceptible to wt, but not ICP6-deficient, HSV-1 infection [77], indicating this signaling pathway plays an important antiviral role in vivo. However, in contrast to observations made in murine cells, HSV does not induce necroptosis in human cells. In HT-29 cells ICP6 and ICP10 inhibit TNFα-induced necroptosis by competing with RIPK3 for binding to the RIPK1 RHIM domain [77, 79]. Simultaneously, these viral proteins bind to and inactivate caspase-8, indicating HSV can directly antagonize apoptosis and necroptosis through a single viral protein. Furthermore, these observations suggest that necroptosis may represent a species-specific barrier to HSV infections [80].
Additional sensors that initiate necroptosis following virus infection likely exist, as infection of L929 cells with the T3D strain of respiratory enteric orphan virus (reovirus) induces caspase-independent cell death [52] (Figure 1). Cell death following T3D infection is dependent on de novo synthesis of viral dsRNA and requires RIPK1, but is independent of the known initiators of necroptosis, TNFR, DAI or TLR3 [81]. In addition, this response requires production of IFN and signaling through the type I IFN receptor, suggesting an IFN stimulated gene(s) might sense or regulate reovirus-mediated cell death [81]. Further identification and characterization of this response remains an important area of future study.
Concluding Remarks
Cell death plays an important role in the host response to viral infection, and we are now beginning to understand the diversity of intracellular sensors and signaling components required to initiate these responses. Interestingly, PRRs and signaling proteins predominantly described to function in antiviral transcriptional cascades have additional activities in initiating cell death pathways. Conversely, cellular components initially described as regulators of cell death are increasingly observed to be important contributors to signaling cascades culminating in antiviral transcription. In addition, it is now clear that there is substantial overlap between the apoptotic and necroptotic signaling pathways, beginning at the earliest stages of receptor utilization. A common theme emerging for PRRs that initiate apoptosis and necroptosis is their reliance on proteins that contain RHIM domains, and their ability to initiate signaling pathways regulated by caspase-8. In contrast, the cytosolic sensor RIG-I (and perhaps cGAS), which activates apoptosis independent of caspase-8, does not directly induce necroptosis following virus infection. Despite the substantial increase in our knowledge of how cells initiate cell death following virus infection, several fundamental questions remain (see below; Outstanding Questions), including a comprehensive understanding of how different viruses induce cell death and the roles of these death pathways in host defense in vivo. Furthermore, utilization of the same receptors and signaling proteins for antiviral transcription and cell death raises the question of how these responses are temporally regulated. It is unclear whether PRR-dependent gene expression or cell death is dominant in the infected cell, or even whether both responses can occur simultaneously. Therefore, future studies will provide additional important information with regards to the interplay between PRR-induced transcription and cell death and their individual roles in regulating viral infection.
Trends Box.
RIG-I induces apoptosis via a transcription-independent activity of IRF3.
TLRs utilizing TRIF can activate apoptosis and necroptosis through the formation of different signaling complexes.
DAI recognizes RNA and DNA virus infection to induce apoptosis and necroptosis
An undefined sensor initiates RIPK3-mediated necroptosis following reovirus infection.
Outstanding Questions.
What is the spectrum of viruses that induce the cytosolic PRR-dependent apoptosis and necroptosis pathways?
Does RIPA contribute to the antiviral activity of IRF3 for all RNA viruses that initiate RIG-I signaling?
How does the cGAS/STING pathway induce apoptosis? Does this play a role in cGAS-mediated control of virus infection in vivo?
How does DAI detect RNA and DNA viruses?
What mediates the selective nature of DAI in detecting specific RNA virus infections?
Are their examples of viruses that activate apoptosis or necroptosis through TLR3?
What is the identity of the IFN-inducible factor that initiates necroptosis following reovirus infection? Are there additional cytosolic or membrane-bound sensors that initiate cell death?
Does necroptosis (i.e., MLKL activation) account for the in vivo phenotypes of virus infected ripk1-/- and ripk3-/- mice?
Do additional activities of RIPK1 and RIPK3 contribute to antiviral resistance?
Does antiviral transcription or cell death dominate when utilizing the same PRR-dependent signaling pathway?
Acknowledgments
We apologize to our colleagues whose work we could not reference due to space limitations. This work is supported by NIH grants R01AI093589 and R01AI116550 to J.C.K. M.H.O. is supported by a NIAID T32 training grant (T32AI007512).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Upton JW, Chan FK. Staying alive: cell death in antiviral immunity. Mol Cell. 2014;54(2):273–80. doi: 10.1016/j.molcel.2014.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chow J, et al. PRRs are watching you: Localization of innate sensing and signaling regulators. Virology. 2015;479-480:104–9. doi: 10.1016/j.virol.2015.02.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schoggins JW. Interferon-stimulated genes: roles in viral pathogenesis. Curr Opin Virol. 2014;6:40–6. doi: 10.1016/j.coviro.2014.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garlanda C, et al. The interleukin-1 family: back to the future. Immunity. 2013;39(6):1003–18. doi: 10.1016/j.immuni.2013.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alexopoulou L, et al. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413(6857):732–8. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 6.Diebold SS, et al. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 2004;303(5663):1529–31. doi: 10.1126/science.1093616. [DOI] [PubMed] [Google Scholar]
- 7.Heil F, et al. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science. 2004;303(5663):1526–9. doi: 10.1126/science.1093620. [DOI] [PubMed] [Google Scholar]
- 8.Hemmi H, et al. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408(6813):740–5. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 9.Yoneyama M, et al. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol. 2004;5(7):730–7. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- 10.Gitlin L, et al. Essential role of mda-5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. Proc Natl Acad Sci U S A. 2006;103(22):8459–64. doi: 10.1073/pnas.0603082103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kato H, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441(7089):101–5. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- 12.Sun L, et al. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339(6121):786–91. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu J, et al. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science. 2013;339(6121):826–30. doi: 10.1126/science.1229963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455(7213):674–8. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lupfer C, et al. Inflammasome control of viral infection. Curr Opin Virol. 2015;12:38–46. doi: 10.1016/j.coviro.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Galluzzi L, et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012;19(1):107–20. doi: 10.1038/cdd.2011.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tait SW, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol. 2010;11(9):621–32. doi: 10.1038/nrm2952. [DOI] [PubMed] [Google Scholar]
- 18.Wilson NS, et al. Death receptor signal transducers: nodes of coordination in immune signaling networks. Nat Immunol. 2009;10(4):348–55. doi: 10.1038/ni.1714. [DOI] [PubMed] [Google Scholar]
- 19.Fischer U, et al. Many cuts to ruin: a comprehensive update of caspase substrates. Cell Death Differ. 2003;10(1):76–100. doi: 10.1038/sj.cdd.4401160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li P, et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91(4):479–89. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 21.Lovell JF, et al. Membrane binding by tBid initiates an ordered series of events culminating in membrane permeabilization by Bax. Cell. 2008;135(6):1074–84. doi: 10.1016/j.cell.2008.11.010. [DOI] [PubMed] [Google Scholar]
- 22.Chattopadhyay S, et al. The IRF-3/Bax-mediated apoptotic pathway, activated by viral cytoplasmic RNA and DNA, inhibits virus replication. J Virol. 2011;85(8):3708–16. doi: 10.1128/JVI.02133-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chattopadhyay S, et al. Ubiquitination of the Transcription Factor IRF-3 Activates RIPA, the Apoptotic Pathway that Protects Mice from Viral Pathogenesis. Immunity. 2016;44(5):1151–61. doi: 10.1016/j.immuni.2016.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lei Y, et al. MAVS-mediated apoptosis and its inhibition by viral proteins. PLoS One. 2009;4(5):e5466. doi: 10.1371/journal.pone.0005466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chattopadhyay S, et al. Viral apoptosis is induced by IRF-3-mediated activation of Bax. EMBO J. 2010;29(10):1762–73. doi: 10.1038/emboj.2010.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.White CL, et al. Phosphatidylinositol 3-kinase signaling delays sendai virus-induced apoptosis by preventing XIAP degradation. J Virol. 2011;85(10):5224–7. doi: 10.1128/JVI.00053-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brubaker SW, et al. A bicistronic MAVS transcript highlights a class of truncated variants in antiviral immunity. Cell. 2014;156(4):800–11. doi: 10.1016/j.cell.2014.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chiu YH, et al. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell. 2009;138(3):576–91. doi: 10.1016/j.cell.2009.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ablasser A, et al. RIG-I-dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III-transcribed RNA intermediate. Nat Immunol. 2009;10(10):1065–72. doi: 10.1038/ni.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Diner BA, et al. Viral DNA Sensors IFI16 and Cyclic GMP-AMP Synthase Possess Distinct Functions in Regulating Viral Gene Expression, Immune Defenses, and Apoptotic Responses during Herpesvirus Infection. MBio. 2016;7 doi: 10.1128/mBio.01553-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brubaker SW, et al. Innate immune pattern recognition: a cell biological perspective. Annu Rev Immunol. 2015;33:257–90. doi: 10.1146/annurev-immunol-032414-112240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tenev T, et al. The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol Cell. 2011;43(3):432–48. doi: 10.1016/j.molcel.2011.06.006. [DOI] [PubMed] [Google Scholar]
- 33.Estornes Y, et al. dsRNA induces apoptosis through an atypical death complex associating TLR3 to caspase-8. Cell Death Differ. 2012;19(9):1482–94. doi: 10.1038/cdd.2012.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Feoktistova M, et al. cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol Cell. 2011;43(3):449–63. doi: 10.1016/j.molcel.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Salaun B, et al. TLR3 can directly trigger apoptosis in human cancer cells. J Immunol. 2006;176(8):4894–901. doi: 10.4049/jimmunol.176.8.4894. [DOI] [PubMed] [Google Scholar]
- 36.Zinngrebe J, et al. --LUBAC deficiency perturbs TLR3 signaling to cause immunodeficiency and autoinflammation. J Exp Med. 2016;213(12):2671–2689. doi: 10.1084/jem.20160041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grimstad O, et al. TLR3 mediates release of IL-1beta and cell death in keratinocytes in a caspase-4 dependent manner. J Dermatol Sci. 2013;72(1):45–53. doi: 10.1016/j.jdermsci.2013.05.006. [DOI] [PubMed] [Google Scholar]
- 38.Totura AL, et al. Toll-Like Receptor 3 Signaling via TRIF Contributes to a Protective Innate Immune Response to Severe Acute Respiratory Syndrome Coronavirus Infection. MBio. 2015;6(3):e00638–15. doi: 10.1128/mBio.00638-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang SY, et al. TLR3 deficiency in patients with herpes simplex encephalitis. Science. 2007;317(5844):1522–7. doi: 10.1126/science.1139522. [DOI] [PubMed] [Google Scholar]
- 40.Kerr DA, et al. BCL-2 and BAX protect adult mice from lethal Sindbis virus infection but do not protect spinal cord motor neurons or prevent paralysis. J Virol. 2002;76(20):10393–400. doi: 10.1128/JVI.76.20.10393-10400.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vandenabeele P, et al. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11(10):700–14. doi: 10.1038/nrm2970. [DOI] [PubMed] [Google Scholar]
- 42.Kaczmarek A, et al. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity. 2013;38(2):209–23. doi: 10.1016/j.immuni.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 43.Kim B, et al. The Interleukin-1alpha Precursor is Biologically Active and is Likely a Key Alarmin in the IL-1 Family of Cytokines. Front Immunol. 2013;4:391. doi: 10.3389/fimmu.2013.00391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhao J, et al. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc Natl Acad Sci U S A. 2012;109(14):5322–7. doi: 10.1073/pnas.1200012109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sun L, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148(1-2):213–27. doi: 10.1016/j.cell.2011.11.031. [DOI] [PubMed] [Google Scholar]
- 46.Cai Z, et al. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol. 2014;16(1):55–65. doi: 10.1038/ncb2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang H, et al. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell. 2014;54(1):133–46. doi: 10.1016/j.molcel.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 48.Degterev A, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1(2):112–9. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- 49.Holler N, et al. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol. 2000;1(6):489–95. doi: 10.1038/82732. [DOI] [PubMed] [Google Scholar]
- 50.Mocarski ES, et al. Necroptosis: The Trojan horse in cell autonomous antiviral host defense. Virology. 2015:479–480. 160–6. doi: 10.1016/j.virol.2015.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.He S, et al. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc Natl Acad Sci U S A. 2011;108(50):20054–9. doi: 10.1073/pnas.1116302108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kaiser WJ, et al. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J Biol Chem. 2013;288(43):31268–79. doi: 10.1074/jbc.M113.462341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cho YS, et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137(6):1112–23. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.He S, et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137(6):1100–11. doi: 10.1016/j.cell.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 55.Zhang DW, et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325(5938):332–6. doi: 10.1126/science.1172308. [DOI] [PubMed] [Google Scholar]
- 56.Sun X, et al. Identification of a novel homotypic interaction motif required for the phosphorylation of receptor-interacting protein (RIP) by RIP3. J Biol Chem. 2002;277(11):9505–11. doi: 10.1074/jbc.M109488200. [DOI] [PubMed] [Google Scholar]
- 57.Li M, Beg AA. Induction of necrotic-like cell death by tumor necrosis factor alpha and caspase inhibitors: novel mechanism for killing virus-infected cells. J Virol. 2000;74(16):7470–7. doi: 10.1128/jvi.74.16.7470-7477.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tian T, et al. Disruption of TNF-alpha/TNFR1 function in resident skin cells impairs host immune response against cutaneous vaccinia virus infection. J Invest Dermatol. 2012;132(5):1425–34. doi: 10.1038/jid.2011.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chan FK, et al. A role for tumor necrosis factor receptor-2 and receptor-interacting protein in programmed necrosis and antiviral responses. J Biol Chem. 2003;278(51):51613–21. doi: 10.1074/jbc.M305633200. [DOI] [PubMed] [Google Scholar]
- 60.Brenner D, et al. Regulation of tumour necrosis factor signalling: live or let die. Nat Rev Immunol. 2015;15(6):362–74. doi: 10.1038/nri3834. [DOI] [PubMed] [Google Scholar]
- 61.Alvarez-Diaz S, et al. The Pseudokinase MLKL and the Kinase RIPK3 Have Distinct Roles in Autoimmune Disease Caused by Loss of Death-Receptor-Induced Apoptosis. Immunity. 2016;45(3):513–26. doi: 10.1016/j.immuni.2016.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Daniels BP, et al. RIPK3 Restricts Viral Pathogenesis via Cell Death-Independent Neuroinflammation. Cell. 2017;169(2):301–313 e11. doi: 10.1016/j.cell.2017.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Downey J, et al. RIPK3 interacts with MAVS to regulate type I IFN-mediated immunity to Influenza A virus infection. PLoS Pathog. 2017;13(4):e1006326. doi: 10.1371/journal.ppat.1006326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang SY, et al. TLR3 immunity to infection in mice and humans. Curr Opin Immunol. 2013;25(1):19–33. doi: 10.1016/j.coi.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Takaoka A, et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature. 2007;448(7152):501–5. doi: 10.1038/nature06013. [DOI] [PubMed] [Google Scholar]
- 66.Upton JW, et al. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe. 2012;11(3):290–7. doi: 10.1016/j.chom.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Upton JW, et al. Virus inhibition of RIP3-dependent necrosis. Cell Host Microbe. 2010;7(4):302–13. doi: 10.1016/j.chom.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Brune W, et al. A ribonucleotide reductase homolog of cytomegalovirus and endothelial cell tropism. Science. 2001;291(5502):303–5. doi: 10.1126/science.291.5502.303. [DOI] [PubMed] [Google Scholar]
- 69.Lembo D, et al. The ribonucleotide reductase R1 homolog of murine cytomegalovirus is not a functional enzyme subunit but is required for pathogenesis. J Virol. 2004;78(8):4278–88. doi: 10.1128/JVI.78.8.4278-4288.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Patrone M, et al. The human cytomegalovirus UL45 gene product is a late, virion-associated protein and influences virus growth at low multiplicities of infection. J Gen Virol. 2003;84(Pt 12):3359–70. doi: 10.1099/vir.0.19452-0. [DOI] [PubMed] [Google Scholar]
- 71.Omoto S, et al. Suppression of RIP3-dependent necroptosis by human cytomegalovirus. J Biol Chem. 2015;290(18):11635–48. doi: 10.1074/jbc.M115.646042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Thapa RJ, et al. DAI Senses Influenza A Virus Genomic RNA and Activates RIPK3-Dependent Cell Death. Cell Host Microbe. 2016;20(5):674–681. doi: 10.1016/j.chom.2016.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kuriakose T, et al. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci Immunol. 2016;1(2):aag2045. doi: 10.1126/sciimmunol.aag2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nogusa S, et al. RIPK3 Activates Parallel Pathways of MLKL-Driven Necroptosis and FADD-Mediated Apoptosis to Protect against Influenza A Virus. Cell Host Microbe. 2016;20(1):13–24. doi: 10.1016/j.chom.2016.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Newton K, et al. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science. 2014;343(6177):1357–60. doi: 10.1126/science.1249361. [DOI] [PubMed] [Google Scholar]
- 76.McCormick AL, et al. Differential function and expression of the viral inhibitor of caspase 8-induced apoptosis (vICA) and the viral mitochondria-localized inhibitor of apoptosis (vMIA) cell death suppressors conserved in primate and rodent cytomegaloviruses. Virology. 2003;316(2):221–33. doi: 10.1016/j.virol.2003.07.003. [DOI] [PubMed] [Google Scholar]
- 77.Huang Z, et al. RIP1/RIP3 binding to HSV-1 ICP6 initiates necroptosis to restrict virus propagation in mice. Cell Host Microbe. 2015;17(2):229–42. doi: 10.1016/j.chom.2015.01.002. [DOI] [PubMed] [Google Scholar]
- 78.Wang X, et al. Direct activation of RIP3/MLKL-dependent necrosis by herpes simplex virus 1 (HSV-1) protein ICP6 triggers host antiviral defense. Proc Natl Acad Sci U S A. 2014;111(43):15438–43. doi: 10.1073/pnas.1412767111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Guo H, et al. Herpes simplex virus suppresses necroptosis in human cells. Cell Host Microbe. 2015;17(2):243–51. doi: 10.1016/j.chom.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Guo H, et al. Manipulation of apoptosis and necroptosis signaling by herpesviruses. Med Microbiol Immunol. 2015;204(3):439–48. doi: 10.1007/s00430-015-0410-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Berger AK, et al. Viral RNA at Two Stages of Reovirus Infection Is Required for the Induction of Necroptosis. J Virol. 2017;91(6):e02404–16. doi: 10.1128/JVI.02404-16. [DOI] [PMC free article] [PubMed] [Google Scholar]