

Abstract
Background
Acute Chest Syndrome (ACS) is one of the leading causes of death among children with Sickle Cell Disease (SCD). Disruption of microvascular integrity is critical to the pathophysiology of ACS, but the factors governing its phenotypic variability are incompletely understood. Because circulating exosomes have been implicated in vascular dysfunction in various diseases, we hypothesized that exosomes induce endothelial dysfunction in patients who experience ACS.
Procedure
Cross-sectional cohort study including 33 outpatients with SCD (without new health-related complaints or recent transfusions) and a cohort of control patients. Exosomes were isolated from platelet-free plasma.
Results
ImageStream showed that exosome counts were greatly increased in patients with SCD compared with controls, but there were few differences in the concentrations of exosomes between patients who had experienced ACS (ACS(+)) and those who had not (ACS(−)). Exosomes were added to human microvascular endothelial cells, and the exosomal effects on monolayer integrity was determined using Electric Cell-substrate Impedance Sensing (ECIS). Exosomes from SCD patients without ACS differed minimally from control patients; however, exosomes from ACS(+) decreased endothelial cell resistance compared to ACS(−), (Relative resistance: ACS(+): 0.981±0.055 vs. ACS(−): 1.124±0.042; p = 0.006). Treatment of endothelial cultures with exosomes from ACS(−) patients increased endothelial Nitric Oxide Synthase (eNOS) mRNA expression, while ACS(+)-derived exosomes were not able to increase eNOS expression above that of controls.
Conclusions
These findings demonstrate that patients with SCD have circulating exosomes that produce differential effects that may contribute to the pathophysiology of ACS and may serve as risk-related biomarkers.
Introduction
Sickle Cell Disease (SCD), caused by a single amino acid substitution in the β-globin polypeptide, is the third commonest cause of death in childhood in African-Americans. Pathognomonic features of SCD include hemolysis and recurrent vaso-occlusive crises (VOC); these result from hemoglobin polymerization and erythrocyte sickling, culminating in endothelial injury, inflammation, and tissue hypoxia.1,2 Acute Chest Syndrome (ACS), is a syndrome of lung damage caused by vaso-occlusion, infection, or fat embolism. ACS is one of the leading causes of death from SCD in childhood.3 There are wide variations in the risk of developing ACS (and other complications) among individuals with SCD. Since the mechanisms accounting for this phenotypic variability are incompletely understood, improved knowledge could lead to better prevention and treatment of ACS.1,4
ACS results from a combination of aberrant multi-cellular interactions, many of which involve the endothelium, including adhesion, ischemia-reperfusion injury, infection and inflammation2,4. In patients with SCD, the endothelium is damaged by activated platelets, cell free hemoglobin and heme, causing alterations that include decreased bioavailability of nitric oxide and increased inflammation5. Even in the absence of red blood cells, plasma from patients with SCD elicits unique (and potentially pathologic) changes in endothelial gene expression compared with non-SCD patients.6
Recent evidence has shown that extracellular vesicles in plasma are paracrine effectors of endothelial function, suggesting their possible importance in the pathophysiology of ACS.7 Extracellular vesicles (ECVs) with a diameter between 100 and 1000 nm are present in the circulation and have been shown to increase in SCD and impact disease pathogenesis.8 Only rather limited studies have been conducted to examine the involvement of smallest ECVs, exosomes, in sickle cell pathophysiology. Exosomes are ECVs composed of bilayered proteolipids that contain selectively packaged, tissue-specific proteins, mRNAs, microRNAs, lipids, and occasionally DNA.9 Exosomes are present in normal plasma, and their abundance is increased in patients with various pathologies, such as cancer and inflammatory disorders.9 Exosomes can selectively bind to endothelium and enter the cells via transcytosis to deliver RNAs encoding inflammatory cytokines, regulatory microRNAs, and/or specific proteins that in turn modulate endothelial cell function, induce inflammation, alter gene expression, and enhance cellular migration, injury or death.9 We recently showed that extracellular vesicles from SCD patients contain different miRNAs based on general SCD severity.10 However, exosomes have not been evaluated as potential modulators of SCD phenotypic variance.
Accordingly, we hypothesized that circulating exosomes from patients with SCD who experienced 1 or more episodes of ACS (ACS(+)) would induce increased dysfunction of naïve endothelium in vitro as compared to exosomes from SCD patients who never experienced an ACS episode (ACS(−)). We evaluated this hypothesis in a convenience cohort of pediatric patients with SCD at the University of Chicago and LaRabida Children’s Hospital.
Materials and Methods
Patient Characteristics
33 patients with SCD at the University of Chicago Comer or LaRabida Children’s Hospital were prospectively enrolled, with parents providing informed consent, and assent being obtained in subjects ≥9 years of age. Controls (n=6) were recruited from the general pediatric clinic, were of African-American ancestry, had a BMI <85th percentile, did not to carry a diagnosis of asthma or inflammatory disorder, and were having blood drawn for screening. All protocols were approved by the IRB (protocol # 14-0466 and 15-0263) and were conducted in accordance with the guidelines set by the Declaration of Helsinki. Patients were excluded if less than 2 years of age, currently in an aplastic crisis, had a previous stroke, or were receiving chronic transfusion therapy. All patients were in a steady-state of disease (free of infection, new pain, or transfusion for the 4 weeks prior to their participation). Clinical and demographic characteristics of SCD patients and controls are shown in Table 1 and supplementary Table 1, respectively. After clinically necessary blood work, 6 mL of blood were drawn in EDTA, centrifuged at 2000×g at 4°C for 20 minutes and platelet-free plasma was separated and frozen at −80°C until further use.
Table 1. Clinical characteristics patients with SCD without ACS and with ACS.
Patient characteristics, demographics and hematologic values
| No ACS (n =10) Median (25th, 75th) or frequency (%) |
ACS (n=23) Median (25th, 75th) or frequency (%) |
p-value | |
|---|---|---|---|
| Age, median (range), y | 14 (3–20) | 15 (8–23) | 0.18 |
| Sex, n (%) | 0.19 | ||
| Male | 8 (80) | 18 (67) | |
| Female | 2 (20) | 9 (33) | |
| Clinical Characteristics | |||
|
| |||
| Hemoglobin Genotype | 0.06 | ||
| SS | 5 | 19 | |
| SC | 4 | 3 | |
| Sb+ | 1 | 0 | |
| S-O-Arab | 0 | 1 | |
| Rate of ACS (# / year) | 0 | 0.22 (0.07, 0.3) | 0.001 |
| Absolute # ACS | 0 | 2.87 (1,4) | 0.003 |
| Rate of Pain | 0.08 (0, 0.02) | 0.40 (0, 0.62) | 0.03 |
| Absolute # Pain | 0–12 | 0–26 | 0.08 |
| Hydroxyurea | 4 (40) | 14 (60) | 0.16 |
| Asthma, # (%) | 0 (0) | 16 (70) | 0.0002 |
| OSA (%) | 3 (30) | 9 (39) | 0.28 |
| Splenectomy (%) | 1 (10) | 2 (9) | 0.46 |
| Cholecystectomy | 0 (0) | 7 (30) | 0.06 |
| Hematologic Values | |||
|
| |||
| White blood cell count (×10*3/uL) | 12.05 (9.05, 11.43) | 12.13 (9.00, 15.20) | 0.97 |
| Hemoglobin (g/dL) | 8.23 (7.53, 8.90) | 9.12 (7.83, 10.30) | 0.18 |
| MCV (fL) | 80.56 (76.00, 85.48) | 90.05 (81.98, 96.8) | 0.006 |
| Absolute retic (K/uL) | 274.4 (201.0, 303.0) | 247.3 (178.3, 331.3) | 0.47 |
| Platelet (×10*3/uL) | 425.4 (328.5, 474.5) | 364.1 (3018, 450.8) | 0.15 |
| Bilirubin (mg/dL) | 2.78 (2.25, 3.33) | 3.83 (2.00, 4.00) | 0.46 |
Results are expressed as median (25 %ile, 75 %ile). P-values: overall significance between groups. MCV: mean corpuscular volume.
Event Characteristics
ACS was defined as a new infiltrate on chest x-ray accompanied by fever, supplemental oxygen requirement, tachypnea, wheezing, cough or chest pain. Admissions for VOC were defined by use of parenteral opioids to treat pain without the presence of another etiology. The rate of VOC or ACS was calculated by dividing the total number of events by the number of patient-years at the time of the blood draw. Asthma was defined by either a documented ICD9 code for asthma (493.0 – 493.2, 493.8, and 493.9) or EMR documentation of a long-acting controller asthma medication. Splenectomy and cholecystectomy were identified by procedure codes.
Exosome Isolation
Exosomes were isolated from plasma using the Total Exosome Isolation kit from Life Technologies (Carlsbad, CA) per manufacturer’s guidelines. All downstream experiments were performed with exosomes at a 1:100 dilution based on sensitivity curves determined previously.10 Detailed procedure in supplemental methods.
Exosome Quantification and Cellular Origin
The ImageStream (ISXMkII) image cytometer (Millipore/Amnis, Seattle, WA) combines the immunofluorescent sensitivity and capacity of a traditional flow cytometer with bright-field high resolution microscopy and image analysis. Briefly, 100 μL of exosomes were stained with appropriately titrated antibodies (BioLegend, San Diego, CA) as follows: CD14 Alexa 488, CD309 PE, CD133 PECy7, CD34 BV421, CD31 BV605, CD42b APC, CD45 APCCy7, CD235a PerCPCy5.5. Exosome size verification and matching was accomplished using 500 size appropriate beads of each color, and an auto-compensation routine was performed using the IDEAS 6.0 data analysis software (Millipore/Amnis, Seattle, WA). Detailed procedures are in the Supplemental Methods.
Electric Cell-substrate Impedance Sensing (ECIS)
Endothelial monolayer barrier integrity was measured using the ECIS system (Applied Biophysics; http://www.biophysics.com/products-ecisz0.php) which monitors the electrical impedance across small 250-micrometer diameter electrodes used as substrates for cell growth. Baseline measurements were established for each array using culture medium (300 μl/well). HMVEC cells were seeded (3 × 105 cells/well) onto an 8W10E array and grown to confluence with media containing only 2% FBS. Once confluent, exosomes were added in duplicate wells, and trans-endothelial electrical resistance was monitored continuously for up to 36 hrs.
eNOS expression
Exosomes were added to HMVEC cells that had been grown to confluent after seeding at 3×105. RNA was isolated with Qiagen RNeasy mini kit (Qiagen, Crawley, UK). DNase-treated mRNA was reverse-transcribed using the high-capacity cDNA reverse transcription kit (Applied Biosystems, Warrington, UK). Expression of eNOS was determined by real-time quantitative PCR using assay-on-demand primer sets were analyzed using ABI7500 SDS version 2 (Applied Biosystems). The relative expression of eNOS was normalized to GAPDH using the ΔCT method and expressed as the relative quantity(RTL).
Results
Clinical Characteristics
Thirty-three patients with SCD completed the study. The demographic, hematologic and clinical characteristics of ACS(−) and ACS(+) patients are shown in Table 1. Most variables, including the number of patients receiving hydroxyurea did not differ between the two groups. The presence of asthma (p =0.0002), frequency of pain (p = 0.03) and MCV values (ACS(−) 80.56± 2.156 vs. ACS(+) 90.05±2.28; p = 0.006) were significantly higher in ACS(+) patients (Table 1).
Exosomes in SCD
Plasma exosomes were isolated from the 33 patients in the outpatient clinic, while at baseline. We quantified the exosomes originating from erythrocytes, lymphocytes, hematopoietic progenitor cells, endothelial cells and platelets by ImageStream. Two example ImageStream plots are shown in Figure 1A. The total number of plasma exosomes was significantly higher in SCD patients as compared to controls.
Figure 1. Total exosome count is increased in SCD compared to control with minimal changes between ACS(−) and ACS(+) patients.
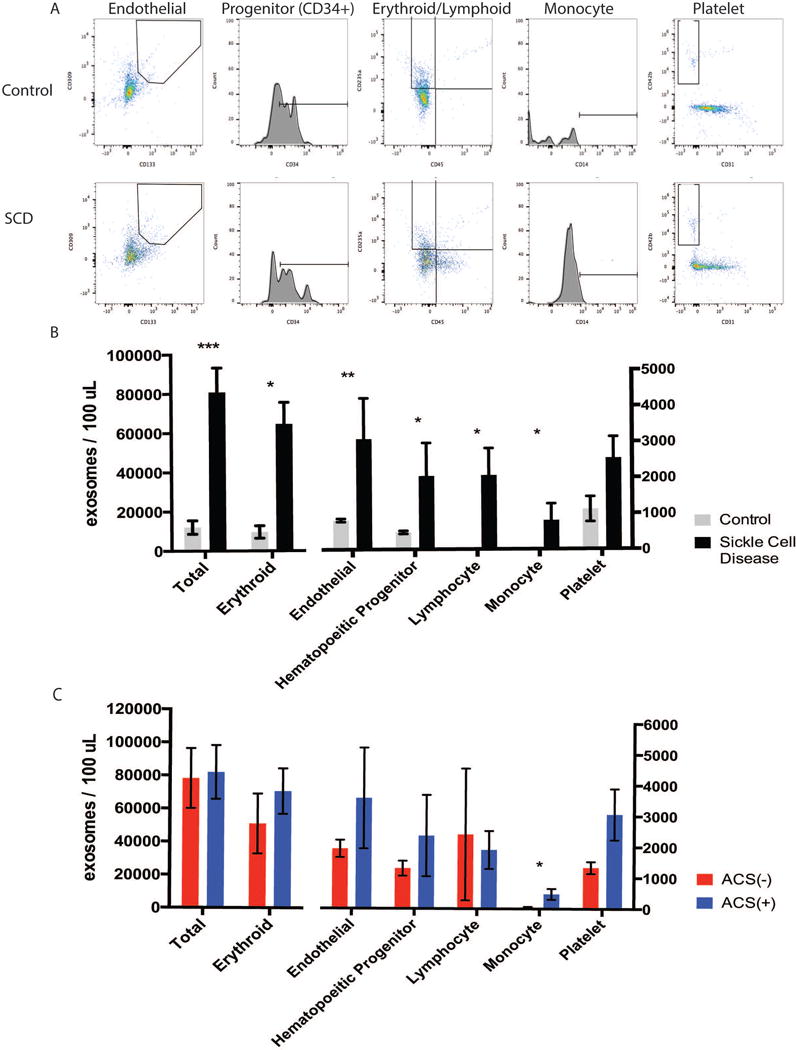
A. Representative images of flow cytometry results obtained using Image Stream X that identify and quantify the relative proportion of exosomes derived from endothelial cells, hematologic progenitor cells, monocytes, platelets and erythrocytes in a control patient and a patient with SCD. B. Summary of numbers of exosomes from each cellular origin across all patients with SCD compared with control patients. C. Number of exosomes from each cellular origin in patients with SCD that did not experience ACS (ACS-) versus those with a history of (ACS+) episodes. *p<0.05, ** p<0.005, and *** p<0.0005.
Similarly, the numbers of exosomes derived from each cell type in patients with SCD was increased significantly (Fig 1B). Although there was significant variability between patients, erythrocyte-derived exosomes exhibited the highest increase, with an overall 3-fold increase from control to SCD (Fig 1B; controls: 9,661 ± 3,195 /100 uL vs. SCD: 31,338 ± 5,323 /100 uL, p<0.007). Endothelial cell-derived exosomes also revealed marked differences between SCD and controls (Fig 1B; controls: 814± 45/100 uLvs. SCD 1,967±255 /100 uL, p < 0.0001). Platelet exosomes were the only subtype where an increase was observed but did not reach significance. (Fig 1 B; controls 1,116±350 vs. SCD 2,702±670, p=0.0535)
In contrast to the marked increase of exosome numbers observed between SCD patients and controls, we observed only minor differences in the exosome numbers between SCD patients with or without ACS. No significant differences were detected in the abundant exosomes from erythroid, hematopoietic progenitor, or lymphocyte origins between ACS(+) and ACS(−)(Fig 1C). In contrast, there was a significant increase in monocyte-derived exosomes between ACS(−) and ACS(+) which correspond to a small fraction of the total exosome population (Fig 1C; monocyte exosomes ACS(−): 45.89 ± 22.41 /100 uL vs. ACS(+): 477.4 ± 173.7 /100 uL, p=0.0218). Platelet-derived exosomes showed a trend towards an increase.(Fig 1 C; platelet exosomes ACS(−): 1,336 ± 192.2 /100 uL, SCD: 3,064 ± 828.7 /100 uL, p=0.0533).
Endothelial impedance changes after exposure to exosomes
To evaluate potential functional differences between the exosomes from ACS(−) and ACS(+) patients in a qualitative manner, we treated human microvascular endothelial cell monolayers with exosomes from each patient, and monitored the effects on the integrity of the monolayers using an endothelial impedance assay. In Fig 2A. representative curves from one patient in each clinical group (Control, ACS(−) and ACS(+), respectively) are presented. Exosomes from all sources caused an initial transient severe decrease in impedance that recovered to above baseline within a few hours. Then, control and ACS (−) exosome treated cultures showed a gradual increase of impedance that likely corresponds to cell proliferation. In contrast, cultures treated with ACS (+) exosomes showed a severe decline in impedance starting about 24 hours after application of exosomes.
Figure 2. Plasma-derived exosomes from patients with ACS disrupt endothelial monolayer function to a greater degree than those without ACS.
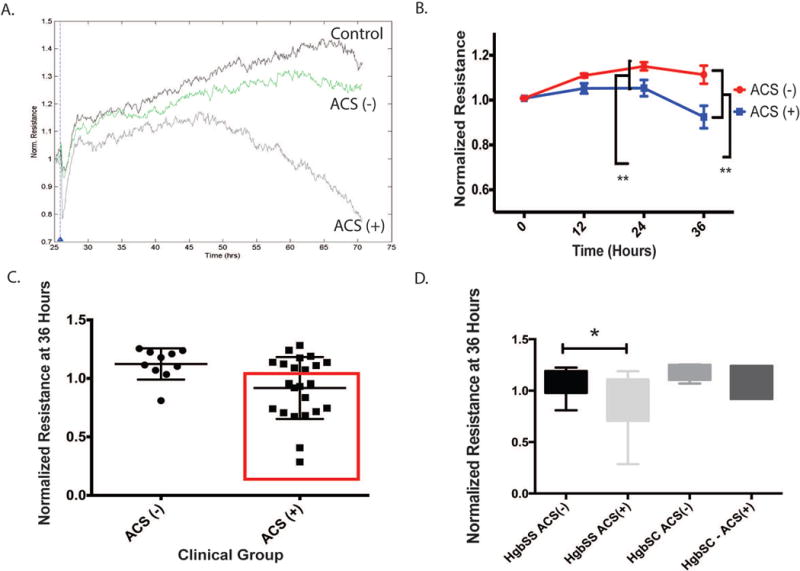
Impedance measurements were acquired after exposing an endothelial monolayer to exosomes from each patient. The assay was repeated twice. A. Representative time course recordings of impedance measurements following addition of exosomes from a control, ACS(−) or ACS(+) patient to endothelial monolayers B. Graph shows the average resistance (normalized to time = 0) from all patients with or without ACS. Data are presented as mean ± S.E.M. C. Graph shows the mean normalized resistance for each patient (■) at 36 hours after exosome addition and error bars represent standard error of the mean., (ACS(+):0.981±0.055 vs. 1.124±0.042 relative resistance; p = 0.006). Significant variability existed in the resistance pattern of the ACS(+) – derived exosomes, with a sub-group indicated in the red box as having greater endothelial damage (ACS(E+)). D. We separated exosomes based on patient genotype and found that the pattern of decreased resistance in ACS(+) –derived is present in HgbSS/O-Arab patients more so than HgbSC patients. At 36 hours, statistically significant decreases in endothelial cell monolayer resistance were apparent after exposure to ACS(+) –derived exosomes in the patients with HgbSS and S-O-Arab, as measured by ECIS. In contrast HgbSC-derived exosomes did not induce a difference whether or not they were from ACS(−) or ACS(+) patients. (HgbSS/S-O-Arab ACS(−): 1.077 ± 0.06 vs. HgbSS/S-O-Arab ACS(+): 0.8703 ± 0.06 relative resistance; p = 0.0263 and HgbSC ACS(−): 1.193 ± 0.04 vs. HgbSC ACS(+): 1.099 ± 0.06 relative resistance; p = 0.2383)
We repeated the assay twice in samples from all patients, averaging the results for all samples tested at 0, 12, 24 and 36 hours after addition of exosomes (Figure 2B). ACS(+)-derived plasma exosomes elicited significantly more disruption of endothelial integrity when compared to ACS(−) exosomes (Figure 2B). Differences in impedance were already apparent at 24 hours, and continued to evolve thereafter(at 36 hours ACS(−): 1.124±0.042 vs. ACS(+): 0.981±0.055, p = 0.006).
There was increased variance in the effect of exosomes on naïve endothelium within the ACS(+) compared to the ACS(−) – derived exosomes (F-test; p = 0.032). To explore potential clinical and hematological differences within the ACS(+)-derived exosomes we compared those exosomes that did not cause endothelial damage (ACS(E−); n = 10) and those who did (ACS(E+)); Figure 2C; red square; n = 13). ACS(E+) compared with ACS(E−) patients did not exhibit differences in ACS rates, absolute # of ACS, pain frequency, the presence of asthma, hematologic parameters, hydroxyurea use, gender, time since last transfusion or genotype between the two groups. However, at 36 hours, a pattern emerged that distinguished HgbSS and S-O-Arab as a “severe” group whose ACS(+)-derived exosomes lead to endothelial disruption as opposed to those from HgbSC (Fig 2D: “Severe” ACS (−): 1.08 ± 0.06 vs. ACS(+): 0.89 ± 0.06, p = 0.043; HgbSC ACS (−): 1.19 ± 0.04 vs. ACS(+): 1.1 ± 0.09, p = 0.44). The “severe” genotype group demonstrated increased disruption of endothelium monolayers by exosomes from ACS(+) patients. In contrast, there was no difference in exosome induced endothelial activity in HgbSC patients regardless of their history of ACS.
Effects of exosomes on eNOS gene expression
Since no tissue-specific differences were detected in exosomes derived from patients with SCD, we evaluated which aspect of endothelial integrity might be regulated by exosomes. Knowing that exosomes have previously been shown to impact ischemia-reperfusion injury via PISKT/eNOS, we tested eNOS expression 24 hours after endothelial cells were exposed to exosomes.11 After exposure to exosomes, naïve endothelial cells displayed divergent changes in the expression of eNOS (Figure 3A). We restricted our analysis to patients with HgbSS and HgbS-O-Arab since they demonstrated a significant differential based on ACS status in endothelial monolayer impedance testing(Figure 2 D). We additionally restricted the analysis to children older than 10 in order to better age-match the groups. Exosomes from controls yielded no significant changes in eNOS expression when compared to basal expression levels in untreated endothelial cells. Exosomes from HgbSS ACS(−) patients led to a ~50% increase in eNOS expression (controls;1.01± 0.00 versus ACS(−); 1.61 ± 0.06; p = 0.01). However, ACS(+)-derived exosomes failed to induce any significant changes in eNOS expression (ACS(+): 0.99 ± 0.05; p < 0.0001 vs. ACS(−)). Of note, patients with ACS whose plasma exosomes did not induce disruption of the monolayer in the ECIS experiments also manifested improved ability to induce eNOS expression (Figure 3A; ACS(E−); 1.15 ± 0.06 versus ACS(E+); 0.90 ± 0.05; p = 0.0127). eNOS expression partially correlates with normalized resistance at 36 hours (Fig 3B; r2 = 0.2052).
Figure 3. eNOS, a known endothelial protective gene, is differentially expressed based on SCD/ACS disease status.
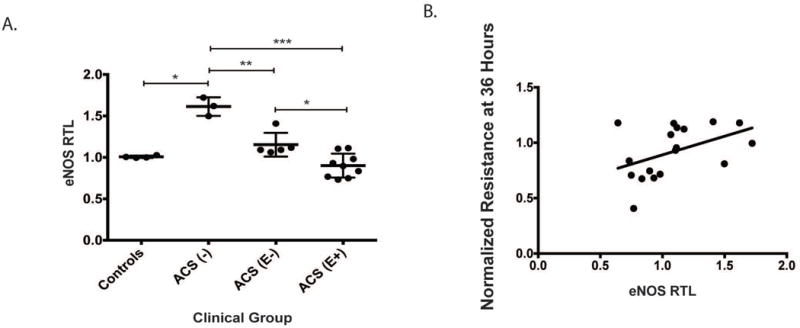
eNOS expression was evaluated in endothelial cells 24 hours after exposure to exosomes from patients with HgbSS, >10 years of age. A. Exosomes from ACS(−) patients elicited a 50% increase in eNOS mRNA compared to controls, while exosomes from ACS(+) patients do not induce increased eNOS expression. Within the ACS(+) group, those that exhibited more endothelial damage on ECIS also induced lower eNOS responses in naïve endothelium. B. eNOS expression partially correlates with normalized resistance at 36 hours. (r2 = 0.2052)
Discussion
In the present pilot study, we have shown that the plasma from patients with SCD contains a much higher concentration of exosomes than that of control patients. While microparticles are known to be produced at higher rates in SCD, limited knowledge exists about exosomes in SCD.8 The exosomes identified in this study derive from a variety of hematopoietic and endothelial cell sources, but most prominently from erythrocytes. The exosomes from SCD patients in our study exert variable effects on endothelial integrity in vitro depending on the ACS history of the patient. ACS(+)-derived exosomes produce a delayed, but sustained disruption of the integrity of the endothelial monolayer in vitro. In contrast, ACS(−)-derived exosomes induce increased eNOS expression (a potentially protective response) in the endothelium.
Quantification of exosomes based on their cell-origin revealed increases in exosomes from multiple sources in SCD compared to controls. Platelet-derived exosomes were not significantly different from controls, despite the known importance of platelet activation in SCD and the typical increase of exosomes produced by activated platelets.12 Among the exosomes from SCD patients there were few significant differences in cellular origin based on history of ACS. Notably, there were increased monocyte-derived exosomes from the ACS(+) patients compared to the ACS(−) patients. Although they made up a small fraction of the total exosomes, they are intriguing because monocytes are capable of targeting specific tissues.
SCD is a disease of baseline inflammation and endothelial damage making it rare to find markers of disease at baseline that are not already altered. We observed two corollary functional differences, in trans-endothelial resistance and eNOS induction, that derive from differences between the baseline ACS(+) and ACS(−) exosomes in vitro. Trans-endothelial resistance decreased after exposure to ACS(+)-derived but not ACS(−)-derived exosomes. When applied to the endothelium the ACS(+)-derived exosomes exerted on initial insult, which recovered to baseline then followed by a slow decline in endothelial resistance in vitro. The relatively delayed and long time course of changes that we have observed suggests that there might be specific components of the exosomes that cause transcriptional changes associated with differing clinical phenotypes. Identification of critical exosome contents may provide both future therapeutic targets and serve as disease phenotype biomarkers13. Exosomes contain multiple elements that are potentially biologically active (miRNA, proteins, and lipids)9,14–16. In subsequent studies, it will be important to identify exosome contents that underlie the preponderant biological differences identified in our experiments.
In addition to detecting a difference between ACS(+) and ACS(−)-derived exosomes on the endothelium, we found another interesting aspect of the ACS(+)-derived exosomes. There was a population that appeared not to have any effect on the endothelium, mirroring the ACS(−)-derived exosomes and a group that was clearly harmful to the endothelium. We defined this group as ACS(E+), having ACS and disrupting the endothelium. Few clinical differences existed between ACS(E−) and ACS(E+) patient groups. However, when the exosomes were separated based on genotype, HgbSC-derived exosomes did do not differ in the disruption of endothelial resistance based on ACS status. Thus, further analysis of exosomes in SCD may elucidate some of the pathophysiologic differences between HgbSS and HgbSC disease. Additional studies might also enable us to describe the contributions of genotype, gender, concurrent asthma and other clinical complications of SCD on exosome-related vascular damage.
Tissue hypoxia is a central component of SCD pathophysiology. Occlusion occurs in a regular, but intermittent pattern, exposing vessels to the dual effects of hypoxia and re-oxygenation mimicking ischemia-reperfusion injury. Exosomes isolated from cultured cells treated with hypoxia contain altered miRNA profiles and produce barrier protection of endothelial cells in vitro17. Exosomes have been previously shown to demonstrate protective effects after ischemia-reperfusion injury and carry non-target effects of ionizing radiation, but never both18–21. In our study, exosomes from patients with SCD afforded both protective and damaging effects, diverging based on the severity of disease and genotype; exosomes from ACS(−) patients promoted continued growth of the endothelial monolayer and from ACS(+) patients, exosomes caused endothelial damage. We speculate that there may be differing miRNAs responsible for these differential effects.
To test our hypothesis that exosomes in the setting of SCD induced ischemia-reperfusion injury act via PISKT/eNOS, we tested eNOS expression 24 hours after endothelial cells were exposed to exosomes. ENOS is a critical regulator of nitric oxide and consequently of endothelial integrity. It contributes to leukocyte adhesion, vascular tone, and platelet aggregation – all of which are critical in the pathogenesis of SCD.22 In murine models of whole body hypoxic preconditioning, eNOS is selectively increased (as opposed to inducible or neural NOS), and induces anti-inflammatory protection.23 Interestingly, when that protective mechanism is not available, such as in eNOS knock-out mice, pulmonary ischemia results in severe pulmonary edema.24 In humans, at least one eNOS polymorphism is associated with an increased risk of ACS in females with SCD.25 In our cohort of patients with HgbSS and older than 10 years of age, we observed a 50% increase in eNOS expression in naïve endothelium induced by a subset of ACS(−) exosomes. This suggests that a subset of patients with SCD generate exosomes that protect them from endothelial dysfunction, by inducing a large increase in eNOS and potentially through additional pathways. In contrast, ACS(+) exosomes did not induce eNOS compared with ACS(−)-derived exosomes potentially suggesting that they could not induce a protective effect in endothelial cells. Interestingly, within the group of patients that were ACS (+) and then stratified based on their impedance ACS(E−)-derived exosomes induced greater eNOS than ACS(E+)-derived exosomes. eNOS expression was lower after exposure to ACS(+)-derived exosomes overall and within the ACS(+) group lower eNOS induction correlated with exosomes from the ACS(E+) group. A small correlation was detected between endothelial cell impedance and eNOS, suggesting there are additional pathways contributing to disruption of the monolayer (Fig 3B).
Although this exploratory study investigated only a relatively small number of patients originating from a single center, significant differences were detected. These results should stimulate design and execution of future larger studies with a priori estimates of statistical power. As with many studies of SCD, the rather prominent phenotypic variance in clinical course analysis is both challenging and intriguing. The importance of a second potential limitation, the contribution of hydroxyurea therapy to the findings, remains unclear; although hydroxyurea use was similar in ACS(+) and ACS(−) groups, the MCV values were only elevated in patients with previous ACS episodes.
Our data provide a novel insight into a method of intercellular communication in SCD that can be harnessed to improve outcomes. While further work needs to be done, with a large multi-institutional cohort, there is potential to utilize exosomes to predict which patients may develop ACS and to tailor therapy. It is clear that exosomes in patients with SCD have differential effects based on their disease state. With a better understanding of patient-specific and treatment-specific effects we could potentially increase production of protective exosomes. Our next steps include evaluating all the pathways that are regulated by exosomes in SCD and evaluating how we can clinically modulate those pathways.
Supplementary Material
Acknowledgments
The authors would like to thank the Chicago Sickle Cell Disease Research Group (CSCDRG) for the significant support, intellectual input and assistance with sample collection.
Financial support: Dr Lapping-Carr’s research reported in this publication was supported by the National Institutes of Health under Award Numbers 5K12HL119995 and UL1TR000430. Dr Cunningham received funding from the Donald N. Pritzker Professorship in Pediatrics. Dr. Gozal was supported by the Herbert T. Abelson Endowed Chair in Pediatrics
References
- 1.Rees DC, Williams TN, Gladwin MT. Sickle-cell disease. The Lancet. 2010;376(9757):2018–2031. doi: 10.1016/S0140-6736(10)61029-X. [DOI] [PubMed] [Google Scholar]
- 2.Hoppe CC. Inflammatory mediators of endothelial injury in sickle cell disease. Hematol Oncol Clin North Am. 2014;28(2):265–286. doi: 10.1016/j.hoc.2013.11.006. [DOI] [PubMed] [Google Scholar]
- 3.Miller AC, Gladwin MT. Pulmonary Complications of Sickle Cell Disease. Am J Respir Crit Care Med. 2012;185(11):1154–1165. doi: 10.1164/rccm.201111-2082CI. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hebbel RP, Vercellotti GM, Nath KA. A systems biology consideration of the vasculopathy of sickle cell anemia: the need for multi-modality chemo-prophylaxis. Cardiovasc Hematol Disord Drug Targets. 2009;9(4):271. doi: 10.2174/1871529x10909040271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kassim AA, DeBaun MR. Sickle cell disease, vasculopathy, and therapeutics. Annu Rev Med. 2013;64:451–466. doi: 10.1146/annurev-med-120611-143127. [DOI] [PubMed] [Google Scholar]
- 6.Klings ES, Safaya S, Adewoye AH, Odhiambo A, Frampton G, Lenburg M, Gerry N, Sebastiani P, Steinberg MH, Farber HW. Differential gene expression in pulmonary artery endothelial cells exposed to sickle cell plasma. Physiol Genomics. 2005;21(3):293–298. doi: 10.1152/physiolgenomics.00246.2004. [DOI] [PubMed] [Google Scholar]
- 7.Salomon C, Ryan J, Sobrevia L, Kobayashi M, Ashman K, Mitchell M, Rice GE. Exosomal Signaling during Hypoxia Mediates Microvascular Endothelial Cell Migration and Vasculogenesis. PLoS One. 2013;8(7):e68451. doi: 10.1371/journal.pone.0068451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hebbel RP, Key NS. Microparticles in sickle cell anaemia: promise and pitfalls. Br J Haematol. 2016:n/a–n/a. doi: 10.1111/bjh.14112. [DOI] [PubMed] [Google Scholar]
- 9.Pant S, Hilton H, Burczynski ME. The multifaceted exosome: Biogenesis, role in normal and aberrant cellular function, and frontiers for pharmacological and biomarker opportunities. Biochem Pharmacol. 2012;83(11):1484–1494. doi: 10.1016/j.bcp.2011.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Khalyfa AKA, Akbaroiy M, Connes P, Romana M, Lapping-Carr G, Zhang C, Andrade J, Gozal D. Extracellular Microvesicles MicroRNAs in Children with Sickle Cell Anemia with Divergent Clinical Phenotypes. Br J Haematol. doi: 10.1111/bjh.14104. Accepted for publication. [DOI] [PubMed] [Google Scholar]
- 11.Ju R, Zhuang ZW, Zhang J, Lanahan AA, Kyriakides T, Sessa WC, Simons M. Angiopoietin-2 secretion by endothelial cell Exosomes Regulation by the phosphatidylinositol 3-kinase (Pi3k)/akt/endothelial nitric oxide synthase (enos) and syndecan-4/syntenin pathways. J Biol Chem. 2014;289(1):510–519. doi: 10.1074/jbc.M113.506899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heijnen HF, Schiel AE, Fijnheer R, Geuze HJ, Sixma JJ. Activated Platelets Release Two Types of Membrane Vesicles: Microvesicles by Surface Shedding and Exosomes Derived From Exocytosis of Multivesicular Bodies and α-Granules. Blood. 1999;94(11):3791–3799. [PubMed] [Google Scholar]
- 13.Fais S, O’Driscoll L, Borras FE, Buzas E, Camussi G, Cappello F, Carvalho J, Cordeiro da Silva A, Del Portillo H, El Andaloussi S, Ficko Trček T, Furlan R, Hendrix A, Gursel I, Kralj-Iglic V, Kaeffer B, Kosanovic M, Lekka ME, Lipps G, Logozzi M, Marcilla A, Sammar M, Llorente A, Nazarenko I, Oliveira C, Pocsfalvi G, Rajendran L, Raposo G, Rohde E, Siljander P, van Niel G, Vasconcelos MH, Yáñez-Mó M, Yliperttula ML, Zarovni N, Zavec AB, Giebel B. Evidence-Based Clinical Use of Nanoscale Extracellular Vesicles in Nanomedicine. ACS Nano. 2016 doi: 10.1021/acsnano.5b08015. [DOI] [PubMed] [Google Scholar]
- 14.Boon RA, Vickers KC. Intercellular Transport of MicroRNAs. Arterioscler Thromb Vasc Biol. 2013;33(2):186–192. doi: 10.1161/ATVBAHA.112.300139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Choi D-S, Kim D-K, Kim Y-K, Gho YS. Proteomics, transcriptomics and lipidomics of exosomes and ectosomes. PROTEOMICS. 2013;13(10–11):1554–1571. doi: 10.1002/pmic.201200329. [DOI] [PubMed] [Google Scholar]
- 16.Valadi H, Ekström K, Bossios A, Sjöstrand M, Lee JJ, Lötvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9(6):654–659. doi: 10.1038/ncb1596. [DOI] [PubMed] [Google Scholar]
- 17.Gray WD, French KM, Ghosh-Choudhary S, Maxwell JT, Brown ME, Platt MO, Searles CD, Davis ME. Identification of therapeutic covariant microRNA clusters in hypoxia-treated cardiac progenitor cell exosomes using systems biology. Circ Res. 2015;116(2):255–263. doi: 10.1161/CIRCRESAHA.116.304360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Doeppner TR, Herz J, Görgens A, Schlechter J, Ludwig A-K, Radtke S, de Miroschedji K, Horn PA, Giebel B, Hermann DM. Extracellular vesicles improve post-stroke neuroregeneration and prevent postischemic immunosuppression. Stem cells translational medicine. 2015;4(10):1131–1143. doi: 10.5966/sctm.2015-0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nojima H, Freeman CM, Schuster RM, Japtok L, Kleuser B, Edwards MJ, Gulbins E, Lentsch AB. Hepatocyte exosomes mediate liver repair and regeneration via sphingosine-1-phosphate. J Hepatol. 2016;64(1):60–68. doi: 10.1016/j.jhep.2015.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li L, Jin S, Zhang Y. Ischemic preconditioning potentiates the protective effect of mesenchymal stem cells on endotoxin-induced acute lung injury in mice through secretion of exosome. Int J Clin Exp Med. 2015;8(3):3825. [PMC free article] [PubMed] [Google Scholar]
- 21.Al-Mayah A, Bright S, Chapman K, Irons S, Luo P, Carter D, Goodwin E, Kadhim M. The non-targeted effects of radiation are perpetuated by exosomes. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis. 2015;772:38–45. doi: 10.1016/j.mrfmmm.2014.12.007. [DOI] [PubMed] [Google Scholar]
- 22.Oliveira-Paula GH, Lacchini R, Tanus-Santos JE. Endothelial nitric oxide synthase: From biochemistry and gene structure to clinical implications of NOS3 polymorphisms. Gene. 2016;575(2):584–599. doi: 10.1016/j.gene.2015.09.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaminski A, Kasch C, Zhang L, Kumar S, Sponholz C, Choi Y-H, Ma N, Liebold A, Ladilov Y, Steinhoff G. Endothelial nitric oxide synthase mediates protective effects of hypoxic preconditioning in lungs. Respir Physiol Neurobiol. 2007;155(3):280–285. doi: 10.1016/j.resp.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 24.Kaminski A, Pohl CB, Sponholz C, Ma N, Stamm C, Vollmar B, Steinhoff G. Up-regulation of endothelial nitric oxide synthase inhibits pulmonary leukocyte migration following lung ischemia-reperfusion in mice. The American journal of pathology. 2004;164(6):2241–2249. doi: 10.1016/S0002-9440(10)63780-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sharan K, Surrey S, Ballas S, Borowski M, Devoto M, Wang KF, Sandler E, Keller M. Association of T-786C eNOS gene polymorphism with increased susceptibility to acute chest syndrome in females with sickle cell disease. Br J Haematol. 2004;124(2):240–243. doi: 10.1046/j.1365-2141.2003.04762.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.