

Abstract
Human cytomegalovirus (HCMV) encodes a viral cyclin-dependent kinase (v-CDK), the UL97 protein. UL97 phosphorylates Rb, p107 and p130, thereby inactivating all three retinoblastoma (Rb) family members. Rb proteins function through regulating the activity of transcription factors to which they bind. Therefore, we examined whether the UL97-mediated regulation of the Rb tumor suppressors also extended to their binding partners. We observed that UL97 phosphorylates LIN52, a component of p107- and p130-assembled transcriptionally repressive DREAM complexes that control transcription during the G0/G1 phases, and the Rb-associated E2F3 protein that activates transcription through G1 and S phases. Intriguingly, we also identified FoxM1B, a transcriptional regulator during the S and G2 phases, as a UL97 substrate. This survey extends the influence of UL97 beyond simply the Rb proteins themselves to their binding partners, as well as past the G1/S transition into later stages of the cell cycle.
Keywords: human cytomegalovirus, viral CDK, UL97, cell cycle, Rb pathway, E2F3, DREAM complex, LIN52, FoxM1
1. Introduction
Human cytomegalovirus (HCMV) is a member of the beta-herpesvirus family that causes disease in neonates and adults with suppressed or impaired immune function (Britt, 2008). HCMV infection may also play a role in proliferative diseases such as atherosclerosis (Melnick et al., 1995), restenosis (Speir et al., 1994), and cancer, most notably glioblastoma multiforme brain tumors (Dziurzynski et al., 2012; Liu et al., 2017; Mitchell et al., 2015; Ranganathan et al., 2012; Soroceanu and Cobbs, 2011). Examining how HCMV modulates cellular proliferation will increase our understanding of the cell cycle pathways that control oncogenesis, and may lead to novel insights into potential roles for HCMV in human malignancies.
Progression through each phase of the cell cycle, and the transition from one phase to the next, is controlled in large part by cyclin-dependent kinase (CDK) mediated phosphorylation of target proteins to alter their localization, function, or stability (Suryadinata et al., 2010). HCMV, like the other beta- and gamma-herpesviruses (Kuny et al., 2010), encodes its own (viral) v-CDK, the UL97 protein. Like the cellular CDKs, UL97 phosphorylates the retinoblastoma family of tumor suppressors, Rb, p107, and p130 (Hume et al., 2008; Iwahori et al., 2017; Prichard et al., 2008). Rb proteins form transcriptionally repressive complexes that arrest cell cycle progression. Phosphorylation on specific serine and threonine residues followed by a proline, the consensus motif targeted by cellular CDKs and UL97, disrupts these complexes, and thereby inactivates the Rb proteins (Iwahori et al., 2015; Rubin, 2013). Rb proteins can also be inactivated without phosphorylation when bound by virally encoded oncoproteins such as Adenovirus E1A, SV40 T antigen, and Papillomavirus E7 (Felsani et al., 2006; Helt and Galloway, 2003; Lee and Cho, 2002). These viral oncoproteins use an LXCXE motif (Singh et al., 2005) to associate with a specific binding cleft found in all Rb family proteins, disrupt their assembled complexes, and inactivate their transcriptional repression and tumor suppressor function.
In G0 and the early G1 phase of the cell cycle, p130 associates with E2F4 and the MuvB core assembly (LIN9, LIN37, LIN52, LIN54 and RBBP4) to form the transcriptionally repressive DREAM complex (DP, p130, E2F4 and MuvB complex). Later in the S phase, p107 nucleates a similar DREAM complex. The MuvB component LIN52 binds directly to p107 and p130 through an LXCXE-like motif. Phosphorylation of a nearby serine residue (Ser28) on LIN52 by the DYRK1A kinase is required for binding to p107 and p130 (Guiley et al., 2015; Litovchick et al., 2011). Progression through the G1 and S phases is controlled by Rb in complex with E2F1, E2F2, E2F3a, E2F3b and E2F4 (Dyson, 1998). E2F3a and 3b represent different transcriptional products of the same gene. Individual promoters drive the expression of separate transcripts with unique first exons that splice to a common second exon (Adams et al., 2000; Leone et al., 2000). In S and G2 phases, the MuvB core associates with B-Myb and FoxM1 to form the MMB-FoxM1 complex that regulates the transcription of genes required for mitosis (Sadasivam et al., 2012).
UL97 utilizes an LXCXE motif (termed L1) to target the Rb family members for phosphorylation and inactivation, disrupting Rb-E2F1, Rb-E2F2, Rb-E2F3a, and Rb-E2F3b complexes (Iwahori et al., 2015; Iwahori et al., 2017). Interestingly, while UL97 inactivates both p130 and p107 through phosphorylation, it does not disrupt their complexes with E2F4 (Iwahori et al., 2017). Furthermore, a substitution mutant disrupting the L1 LXCXE motif of UL97 (UL97-L1m) retains the ability to disrupt Rb-E2F complexes but fails to reverse Rb-mediated transcriptional repression (Iwahori et al., 2015). Therefore, while complex disruption is important, other means to regulate these complexes must exist.
To reveal additional levels of regulation within the Rb pathway, we examined UL97-mediated phosphorylation of other members of these transcriptionally regulating complexes. We identified the DREAM complex member LIN52, the Rb-associated E2F3, and the MMB-associated FoxM1 as UL97 substrates. Our study demonstrates that UL97 phosphorylates factors that act at multiple cell cycle stages, suggesting that UL97 influences cell cycle progression by targeting more than just the Rb tumor suppressors.
2. Results
2.1. UL97 directs the phosphorylation of DREAM complex member LIN52
The DREAM complex component LIN52 has 6 identified phosphorylation sites, Ser7, Ser28, Ser45, Ser48, Ser52, and Ser53 (PhosphoSitePlus: http://www.phosphosite.org/homeAction.action). LIN52 is phosphorylated by DYRK1A on Ser28 (Litovchick et al., 2011) to promote p130 binding, DREAM complex assembly, and cellular quiescence. It is the only phosphorylation event of the MuvB components of the DREAM complex with a defined phenotype. In our experiments, ectopic expression was utilized because we had difficulty detecting the endogenous protein with commercially available antibodies.
Ectopically expressed FLAG-tagged LIN52 migrated as a single band upon Western blot analysis of transiently transfected U-2 OS cell lysates, but was converted to multiple slower migrating species by co-expression of UL97 (Fig. 1A). Co-transfection of UL97-L1 motif mutant, Cyclin D2/CDK4, Cyclin E1/CDK2, or Cyclin A2/CDK2 also generated slower migrating forms of LIN52, whereas neither Cyclin B1/CDK1 nor a kinase dead derivative of UL97 efficiently induced slower migrating forms of LIN52. The slower migration of the kinase-initiated forms of LIN52 was magnified by phosphate-affinity (Phos-tag) SDS-PAGE, but eliminated by lysate incubation with lambda protein phosphatase (Fig. 1A). We conclude UL97 as well as the G1/S cellular CDKs direct LIN52 phosphorylation.
Figure 1. UL97 directs LIN52 phosphorylation at multiple sites.

(A) U-2 OS cells were transfected with expression plasmids for Flag-tagged LIN52 (Flag-LIN52) together with either an empty vector (EV), plasmids expressing HA-tagged UL97 (97), a UL97-L1 motif mutant (L1), a UL97 kinase-deficient mutant (KD), cyclin A2 (A2)/CDK2 (K2), cyclin E1 (E1)/CDK2, cyclin D2 (D2)/CDK4 (K4) or cyclin B1 (B1)/CDK1 (K1). Lysates harvested 48 hours after transfection were harvested and incubated with (+) or without (−) lambda protein phosphatase (l-PPase) prior to analysis by western blot with the indicated antibodies. The phosphorylation-dependent band shifts of LIN52 were detected in phosphate affinity (Phos-tag) gels. (B, C) Transfections were performed as in panel A except Flag-LIN52 alleles carrying single or multiple serine to alanine exchanges were also included.
Both UL97 (Oberstein et al., 2015) and the cellular CDKs (Ubersax and Ferrell, 2007) demonstrate preferential phosphorylation of serine or threonine residues followed by a proline. LIN52 has four residues that match this consensus, Ser7, Ser28, Ser48, and Ser53. All are demonstrated phosphorylation sites (PhosphoSitePlus) (Litovchick et al., 2011). Mutation of any one of these potential phosphorylation sites to a non-phosphorylatable alanine residue resulted in altered UL97-induced migration patterns, most easily discernable during phosphate affinity (Phos-tag) SDS-PAGE (Fig. 1B). Such altered migration is indicative of a change in phosphorylation, therefore we conclude transfected UL97 phosphorylates LIN52 on Ser7, Ser28, Ser48, and Ser53. Combining the non-phosphorylatable mutant residues in pairs (S7A/S28A; S28A/S48A; S28A/S53A) or in a triple mutant (S28A/S48A/S53A) further reduced phosphorylation (Fig. 1C). A quadruple mutant (S7A/S28A/S48A/S53A) showed a dramatic reduction in phosphorylation, but did retain a slower migrating band whose presence depended upon the kinase activity of UL97 (Fig. 1C). Of the two known phosphorylation sites remaining in LIN52 (Ser45 and Ser52), we suspect this remaining band represents phosphorylation at Ser52 because it is precisely adjacent to the proline-directed Ser53 site, and because Ser45 is not a proline-directed site.
LIN52 was also phosphorylated in HCMV infected cells in a UL97-dependent manner. Ectopically expressed FLAG-tagged LIN52 was visualized as a slowly migrating band upon phosphate-affinity (Phos-tag) SDS-PAGE and Western blot analysis of HCMV infected HFF cell lysates (Fig. 2A). The UL97 small molecule inhibitor Maribavir (MBV) (Chou, 2008) altered the HCMV-induced slower migration of LIN52 (Fig 2A), indicating a role for UL97 in LIN52 phosphorylation during HCMV infection. S7A or S48A mutants showed identical phosphorylation patterns as wild type LIN52 (Fig. 2A), indicating these residues are unlikely to be phosphorylated by UL97 during HCMV infection. However, both S28A and S53A mutants showed altered migration patterns compared to wild type LIN52 (Fig. 2A). The migration of the S28A mutant was further altered by MBV but not by Harmine (Fig. 2A and 2B), an inhibitor of the DYRK1A kinase known to phosphorylate Ser28. However, Harmine enforced another previously described (Chen et al., 2013) biological outcome in these cells, elevating the level of cyclin D1. Thus, Ser53 appears to be a target of UL97. The migration of the S53A mutant was not noticeably altered by MBV (Fig. 2A and 2B), Harmine (Fig. 2B), or a combined treatment with both inhibitors (Fig. 2B). Thus, we are, at this time, unsure of which kinase (DYRK1A, UL97 or potentially cellular CDKs) is responsible for phosphorylating Ser28 during HCMV infection, and all may participate. Phosphorylation site combination mutants of LIN52 in which Ser28 and Ser53 were both substituted with alanine were not detectably phosphorylated upon HCMV infection (Fig. 2A). We conclude that Ser28 and Ser53 are the major phosphorylation sites during HCMV infection and UL97 contributes their phosphorylation.
Figure 2. LIN52 is phosphorylated on Ser28 and Ser53 during HCMV infection.

(A) HFFs were transfected with the indicated alanine substitution alleles of Flag-tagged LIN52. At 18 hr post-transfection the cultures were serum-starved. After 48 hr cells were infected with HCMV at an MOI of 3. At 2 hr post-infection cultures were treated with 10 μM maribavir (MBV) or DMSO as a vehicle. Whole cell lysates were prepared at 48 hr post-infection and analyzed by western blot with the indicated antibodies. The phosphorylation-dependent band shifts of LIN52 were detected in phosphate affinity (Phos-tag) gels. (B) In the upper panel, HCMV-infected cells were treated and analyzed as described in panel A except including DYRK1A inhibitor Harmine. Where indicated (+), Harmine was added at 5 μM. In the lower panel, mock-infected serum-starved cells were stimulated by 15% FBS for 22 hr with or without 5 μM Harmine. Lysates were analyzed by western blot with the indicated antibodies. (C) U-2 OS cells were transfected with expression plasmids for the indicated alleles of Flag-tagged LIN52 together with expression plasmids for the indicated alleles of UL97. Lysates harvested 48 hr after transfection were subjected to immunoprecipitation with the Flag antibody. Input lysates and immunoprecipitates (IP) were analyzed by western blot with the indicated antibodies. C, mouse IgG control.
Despite UL97-mediated p130 phosphorylation, p130 still binds to MuvB components in UL97-transfected or HCMV infected cells (Iwahori et al., 2017). This binding appears to require Ser28 phosphorylation as the LIN52 S28A mutant failed to interact with DREAM components p107, p130, or E2F4 (Fig. 2C). Ser53 phosphorylation does not seem to be required for this interaction as LIN52 S53A associated with all three proteins (Fig. 2C). The DREAM complex is also found assembled in cells transfected with kinase-deficient UL97 (Fig. 2C) presumably because of prior phosphorylation of Ser28 by DYRK1A. We conclude that phosphorylation of Ser28, as previously reported (Litovchick et al., 2011) but not Ser53 is required for DREAM complex formation.
2.2. UL97 directs the phosphorylation of the E2F family member E2F3
E2F3 has two isoforms designated as E2F3a and E2F3b. Human E2F3a has 10 identified phosphorylation sites (PhosphoSitePlus). E2F3a is phosphorylated on Ser124 by Chk1 in response to DNA damage (Gong et al., 2016; Martinez et al., 2010). This phosphorylation stabilizes E2F3a and increases its transactivation potential. It is the only phosphorylation event on E2F3 with a defined phenotype. Ser124 is not present in E2F3b.
E2F3a (465 amino acids, predicted molecular weight 49 kDa) and E2F3b (340 amino acids, predicted molecular weight 38 kDa) can be distinguished on Western blots by comparison to the migration of the proteins expressed from individual epitope-tagged cDNA constructs (Fig. 3A). Both E2F3a and E2F3b exhibited a level of phosphorylation that was increased upon HCMV infection (Fig. 3B). Phosphatase treatment of mock-infected lysates caused a down-shift in electrophoretic mobility, and the HCMV infection-induced additional slower migrating forms also collapsed upon phosphatase treatment. Note the slowest migrating band revealed by the E2F3-specific antibody (Fig. 3A and Fig. 3B) is insensitive to phosphatase treatment (Fig. 3B) and therefore either represents a form of E2F3 not modified by phosphorylation or non-specifically reacting protein. We conclude that HCMV infection induces the phosphorylation of both E2F3a and E2F3b.
FIGURE 3. UL97 directs E2F3b phosphorylation during HCMV infection.
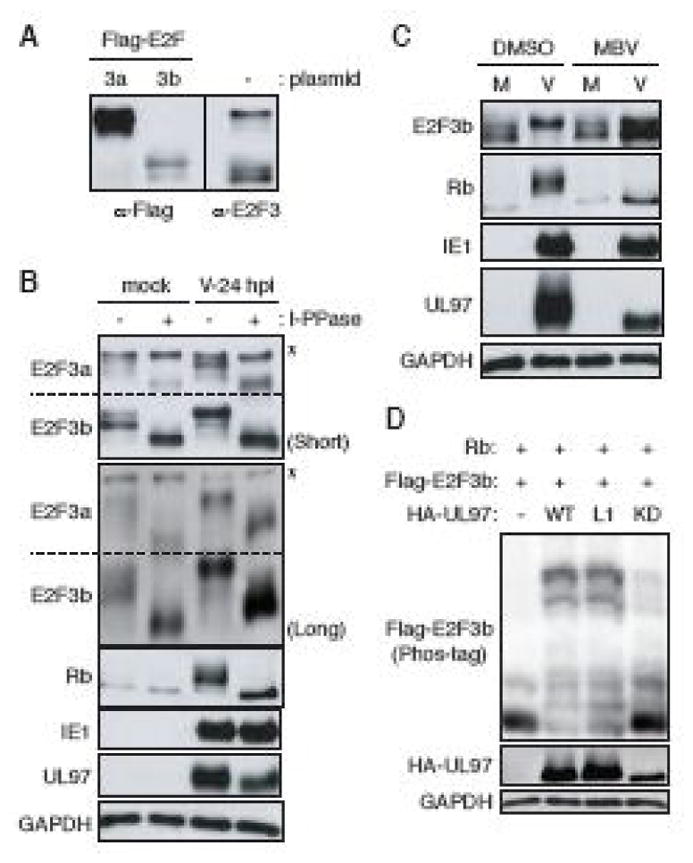
(A) HFFs were transfected with either an empty vector (−) or expression vectors for Flag-tagged E2F3a (3a) or E2F3b (3b). At 48 hr post-transfection, cells were harvested and analyzed by western blot with Flag or E2F3 antibodies. (B) Serum-starved human fibroblasts (HFFs) were mock-infected or infected with wild-type HCMV (V) at an MOI of 3. At 24 hr post-infection (hpi) protein lysates were harvested and incubated with (+) or without (−) lambda protein phosphatase (l-PPase) prior to analysis by western blot with the indicated antibodies. Short: short-term electrophoresis in 10% gel. Long: long-term electrophoresis in 7.5% gel. *: non-phosphorylated or non-specific band. (C) Serum-starved HFFs were infected with HCMV as in panel B except maribavir (MBV) or DMSO was added as indicated. Lysates were analyzed by western blot with the indicated antibodies. M, mock infection. (D) Saos-2 cells were transfected with both expression plasmids for Flag-tagged E2F3b and Rb together with either an empty vector (−) or one expressing HA-tagged wild-type (WT), L1 motif mutant (L1) or a kinase deficient (KD) UL97. Lysates harvested 48 hr after transfection were analyzed by western blot with the indicated antibodies. The phosphorylation-dependent band shifts of E2F3b were detected in phosphate affinity (Phos-tag) gels.
The only known phosphorylation site unique to E2F3a is Ser124; it is not followed by a proline residue and is a known site for Chk1. For these reasons, we suspect Ser124 is unlikely to be phosphorylated by UL97. Of the remaining known phosphorylation sites, all are conserved between E2F3a and E2F3b (Table 1). Furthermore, E2F3b steady state levels appear higher than those of E2F3a upon HCMV infection (Fig. 3B). Therefore, our examination of UL97-mediated phosphorylation of E2F3 proceeded only with E2F3b. Maribavir altered the HCMV-induced slower migration of E2F3b (Fig 3C), as well as the known UL97 substrate Rb, indicating a role for UL97 in E2F3b phosphorylation during HCMV infection. Ectopically expressed FLAG-tagged E2F3b migrated as a single major band upon phosphate-affinity (Phos-tag) SDS-PAGE and Western blot analysis of transiently transfected Saos-2 cell lysates, but was converted to multiple slower migrating species by co-expression of UL97 (Fig. 3D). Co-transfection of UL97-L1 motif mutant, but not a kinase dead derivative of UL97 also generated slower migrating forms of E2F3b. We conclude that UL97 influences the phosphorylation state of E2F3 during transient transfections and HCMV infections.
Table 1.
Potential phosphorylation residues on E2F3
| E2F3aa | E2F3bb | S/T-Pc | |
|---|---|---|---|
| Ser | 124 | - | − |
| Thr | 152 | 27 | + |
| Ser | 163 | 38 | + |
| Ser | 166 | 41 | + |
| Thr | 169 | 44 | + |
| Ser | 172 | 47 | + |
| Ser | 174 | 49 | − |
| Ser | 235 | 110 | − |
| Ser | 267 | 142 | − |
| Tyr | 349 | 224 | − |
Residue numbers on E2F3a.
Residue numbers on E2F3b.
Serine/threonine is followed by proline (+) or not (−).
Of the 10 known E2F3 phosphorylation sites, 5 are proline directed. In E2F3b, these sites are Thr27, Ser38, Ser41, Thr44, and Ser47 (Table 1). Interestingly, these sites abut the DNA binding domain of E2F3 (Leone et al., 2000). Mutation of Thr27, Ser41, Thr44, or Ser47 to a non-phosphorylatable alanine residue (T27A, S41A, T44A or S47A) did not noticeably affect UL97-induced migration patterns during phosphate affinity (Phos-tag) SDS-PAGE of lysates from transiently transfected Saos-2 cells (Fig. 4A). However, the slowly migrating E2F3b band elicited by UL97 was not present in cells expressing the S38A mutant (Fig. 4A). Compared to ectopically expressed wild type, phosphorylation of ectopically expressed E2F3b-S38A was also diminished in HFF cells transiently transfected with UL97 (Fig. 4B) or infected with HCMV (Fig. 4C). Furthermore, UL97 phosphorylated wild type E2F3b in vitro, but when E2F3b-S38A was used as the substrate, phosphorylation was significantly reduced (Fig. 4D and 4E). We conclude that Ser38 is the major E2F3b phosphorylation site for UL97 during HCMV infection.
FIGURE 4. UL97 likely phosphorylates E2F3b on Ser38.

(A) Saos-2 cells were transfected with expression plasmids for Flag-tagged alleles of wild-type E2F3b or E2F3b in which the indicated Ser or Thr residues are replaced with alanines (27, T27A; 38, S38A; 41, S41A; 44, T44A; 47, S47A; DA, T27- and S38-double alanine mutant) together with either an empty vector (−) or one expressing HA-tagged wild-type (WT) UL97 or a kinase deficient (KD) UL97. Lysates harvested 48 hr after transfection were analyzed by western blot with the indicated antibodies. The phosphorylation-dependent band shifts of transiently-expressed E2F3b were detected in phosphate affinity (phos-tag) gels. (B) HFFs were transfected with Flag-tagged alleles of wild-type E2F3b or E2F3b-S38A and analyzed as in panel A. (C) HFFs were transfected with Flag-tagged alleles of wild-type E2F3b or E2F3b-S38A and then subjected to serum starvation. After 48 hr cells were infected with HCMV at an MOI of 2 and harvested after 27 hrs and analyzed as in panel A. (D) GST-tagged wild-type E2F3b and E2F3b-S38A were purified from bacteria. Wild-type (WT) or kinase-deficient (KD) UL97 were incubated with the GST-tagged substrates in the presence of [γ-32P]ATP. After these in vitro kinase reactions, the samples were resolved by SDS-PAGE and visualized by autoradiography or subjected to western blot with the indicated antibodies. (E) The 32P incorporation into E2F3b in panel D was measured using a Typhoon 9200 (Amersham Biosciences) and ImageQuant 5.2 software and then normalized to protein amounts of E2F3b measured by LI-COR. Values are presented relative to the value in wild-type E2F3b phosphorylation by wild-type UL97 (set at 100%). Error bars denote the standard deviation. Statistical analysis utilized a two-tailed unpaired Student’s t test. ** P < 0.01.
In addition to the v-CDK UL97, the cellular CDKs also phosphorylate E2F3b. In serum starvation and release experiments, E2F3b was converted to a slower migrating, phosphorylated form with kinetics similar to the known CDK substrate, Rb (Fig. 5A). Cyclin E/CDK2 phosphorylated wild type E2F3b in vitro (Fig. 5B). When E2F3b-S38A was used as the substrate, Cyclin E/CDK2-mediated phosphorylation was modestly reduced (Fig. 5B and 5C), indicating that one or more residues in addition to Ser38 are likely to be CDK target sites. Ectopically expressed Cyclin A2/CDK2 phosphorylated ectopically expressed E2F3b in transiently transfected Saos-2 cells (Fig. 5D). Mutation of Ser41, Thr44, or Ser47 to a non-phosphorylatable alanine residue did not noticeably affect Cyclin A2/CDK2 phosphorylation of E2F3b (Fig. 5D). However, the slowest migrating E2F3b band elicited by Cyclin A2/CDK2 was not present in cells expressing either the T27A or the S38A mutant (Fig. 5D). Similarly, Cyclin E1/CDK2- and Cyclin D2/CDK4-mediated phosphorylation of E2F3b occurred on both Thr27 and Ser38 (Fig. 5E). None of these Cyclin/CDK pairs were able to appreciably phosphorylate an E2F3b mutant with alanine replacements at both Thr27 and Ser38 (Fig. 5E). We conclude the cellular CDKs phosphorylate E2F3b on Thr27 and Ser38.
FIGURE 5. Cellular cyclin/CDK complexes likely phosphorylate E2F3b on Thr27 and Ser38.

(A) HFFs were serum-starved for 48 hrs and then stimulated with 15% serum. Serum-stimulated HFFs were harvested at the indicated hrs and analyzed by western blot with the indicated antibodies. The percentage of the slower-migrating form of E2F3b was quantified using NIH ImageJ software and is listed under each lane. (B) An in vitro kinase reaction was conducted and analyzed as in Fig. 4D except using purified cyclin E/CDK2 complex. (C) The 32P incorporation into E2F3b in panel B was measured as in Fig. 4E. Values are presented relative to the value of wild-type E2F3b phosphorylation by cyclin E/CDK2 (set at 100%). Error bars denote the standard deviation. Statistical analysis utilized a two-tailed unpaired Student’s t test. ** P < 0.01. (D) Saos-2 cells were transfected with expression plasmids for Flag-tagged alleles of wild-type (WT) E2F3b or E2F3b in which the indicated Ser or Thr residues are replaced with alanines (27, T27A; 38, S38A; 41, S41A; 44, T44A; 47, S47A) together with either an empty vector (−) or expression plasmids for cyclin A2 and CDK2. Lysates harvested 48 hr after transfection were analyzed by western blot with the indicated antibodies. (E) Saos-2 cells were transfected with Flag-tagged alleles of wild-type E2F3b or E2F3b containing double alanine (DA) mutations of Thr-27 and Ser-38 as in panel D except in the presence of expression plasmids of cyclin E1 and CDK2 or cyclin D2 and CDK4.
In a luciferase assay measuring activation of the E2F-responsive E2F-1 promoter, we detected no differences between wild type E2F3b and the nonphosphorylatable T27A/S38A double mutant, nor the phosphomimetic mutants T27E, S38D, or the double T27E/S38D mutant (Fig. 6A). Despite our inability to demonstrate a phenotype for E2F3b phosphorylation at Ser38, this modification may play a role in human cancers. The amino acid corresponding to Arg162 of E2F3a and Arg37 of E2F3b has been found substituted by glutamine in endometrial and stomach cancers (Catalogue of Somatic Mutations in Cancer; ID: COSM1076417). In transfected cells, we discovered the R37Q mutation in E2F3b inhibited phosphorylation of the protein by both UL97 and Cyclin E1/CDK2 (Fig. 6B). Therefore, were this substitution mutation to play a phenotypic role in human cancers, the contribution may be functionally mediated by impaired phosphorylation.
FIGURE 6. Cancer associated R37Q mutation prevents E2F3b phosphorylation by UL97 or cellular cyclin E/CDK2.

(A) Saos-2 cells were transfected with a luciferase reporter driven by the E2F1 promoter together with either an empty vector (−) or different amounts (0.1, 0.2, or 0.5 μg/35-cm2 area) of expression plasmids for DP1 or the indicated allele of Flag-tagged E2F3b. WT, wild-type; DA, T27A/S38A; 27E, T27E; 38D, S38D; ED, T27E/S38D. Lysates harvested 48 hr after transfection were analyzed for luciferase activity (top) and protein expression with the indicated antibodies (bottom). Luciferase activity was normalized to total protein concentration and is presented relative to the activity of the reporter without E2F3b or DP1 (set at 1). Error bars denote the standard deviation of more than 3 replicates. Statistical analysis utilized a two-tailed unpaired Student’s t test. n.s., not significant. (B) Saos-2 cells were transfected with expression plasmids for Flag-tagged alleles of wild-type (WT) E2F3b or E2F3b with single amino acid mutation (RQ, R37Q; 27, T27A; 38, S38A) and either an empty vector (−) or one expressing HA-tagged wild-type UL97 (97) or cyclin E1/CDK2 (E/2). Lysates harvested 48 hr after transfection were analyzed by western blot with the indicated antibodies. The phosphorylation-dependent band shifts of E2F3b were detected in phosphate affinity (phos-tag) gels.
2.3. UL97 directs the phosphorylation of MMB-associated FoxM1
The MMB-interacting protein, FoxM1 has 30 identified phosphorylation sites (PhosphoSitePlus). FoxM1 is phosphorylated by Cyclin D1/CDK4, Cyclin D3/CDK6 (Anders et al., 2011), CDK1 (Sullivan et al., 2012), and Cyclin A/CDK2 complexes (Laoukili et al., 2008) on a series of residues within the carboxy terminal transcriptional activation domain, including Thr600. This hyper phosphorylation, including specific phosphorylation of Thr600, increases the ability of FoxM1 to activate transcription. We observed that ectopically expressed UL97 also phosphorylated ectopically expressed FoxM1 on Thr600 (Fig. 7A). UL97-mediated FoxM1 phosphorylation required UL97 to be an active kinase but did not require the L1 LXCXE motif of UL97 (Fig. 7A). Ectopically expressed FoxM1 was also phosphorylated on Thr600 during HCMV infection in a UL97-dependent manner (Fig. 7B). We conclude that UL97 directs the phosphorylation of FoxM1 on Thr600.
Figure 7. UL97 directs FoxM1 phosphorylation on Thr600.
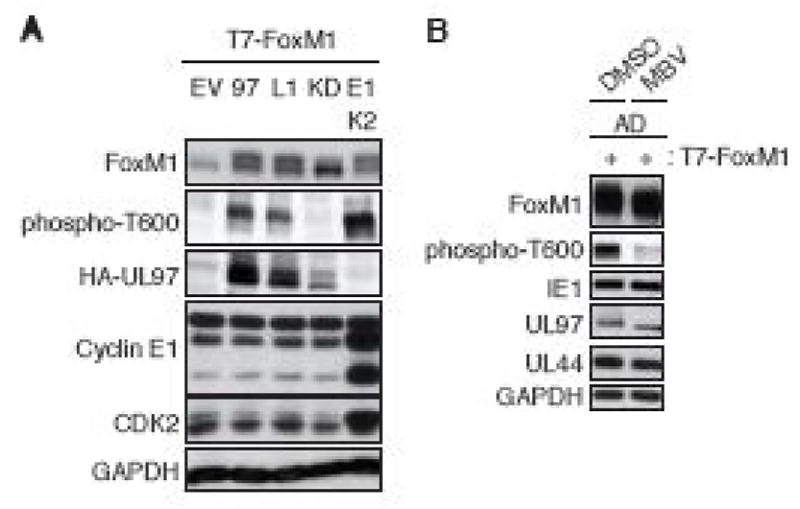
(A) Saos-2 cells were transfected with expression plasmids for T7-tagged FoxM1 together with either an empty vector (EV), HA-tagged wild-type UL97 (97), the indicated allele of UL97, or cyclin E1/CDK2. Lysates harvested 48 hr after transfection were analyzed by western blot with the indicated antibodies. (B) NHDFs were transfected with the CMV promoter-driven T7-tagged FoxM1. At 18 hr post-transfection the transfected NHDFs were serum-starved. After 48 hr cells were infected with HCMV at an MOI of 3. At 2 hr post-infection infected cells were treated with 10 μM maribavir (MBV) or DMSO as a vehicle. Whole cell lysates were prepared at 48 hr post-infection and analyzed by western blot with the indicated antibodies.
3. Discussion
UL97 phosphorylates all three retinoblastoma family proteins, Rb, p107 and p130 reversing their ability to repress E2F-dependent transcription (Iwahori et al., 2015; Iwahori et al., 2017). This UL97-mediated phosphorylation disrupts Rb-E2F complexes (Iwahori et al., 2015) but not p107-E2F or p130-E2F complexes (Iwahori et al., 2017). A mutation in the first LXCXE motif (L1) in UL97 renders the mutant protein unable to inactivate Rb, though it still disrupts Rb-E2F complexes (Iwahori et al., 2015). How UL97 inactivates p107 and p130 without complex disruption, and how the L1 LXCXE motif of UL97 contributes to Rb inactivation remains to be explored, and highlights the complex and incompletely understood nature of how the Rb family proteins regulate cell cycle progression. Here we identify novel substrates of UL97 that extend its regulatory reach beyond the Rb proteins themselves, and beyond the G1/S transition. We found LIN52, E2F3, and FoxM1 are all phosphorylated by UL97. Interestingly, none of these phosphorylation events required the L1 LXCXE motif, indicating that additional layers of regulation exist.
UL97 phosphorylated LIN52 on Ser53 and possibly Ser28 during HCMV infection, and on additional sites (Ser7, Ser48, and an unidentified site likely to be Ser52) when ectopically expressed. The significance of phosphorylation at Ser53 is not known, and we have not tested whether or not cellular CDKs can phosphorylate this site. However, Ser53, unlike Ser28 does not regulate LIN52 binding to p130 (Fig. 2B). Phosphorylation at Ser28 appears to be mediated by DYRK1A, UL97 and potentially cellular CDKs, implying a need to maintain an assembled DREAM complex in HCMV infected cells.
E2F proteins are not only regulated by interaction with Rb family members but also by the post-translational modifications of phosphorylation, ubiquitination, methylation, sumoylation, and acetylation (Black and Azizkhan-Clifford, 1999; Munro et al., 2012). These modifications can affect interactions with Rb family members, nuclear export, stability, and DNA binding activity. Here we detected UL97-mediated phosphorylation of E2F3b on Ser38, and cellular CDK-mediated phosphorylation on Thr27 and Ser38. We could detect no effect of mutation of these residues to either non-phosphorylatables or phospho-mimetics using a standard E2F transcriptional activation assay. Assays more tailored to specific E2F3a or E2F3b functions, when developed, could yield different results. The possibility for phosphorylation-dependent regulation of E2F3 activity is made more appealing by our discovery that a cancer-associated mutation in E2F3 precludes cellular CDK-mediated phosphorylation (Fig. 6B). Whether this point mutation or its effect on E2F3 phosphorylation is a driving event in human cancers remains to be determined. Furthermore, while the functional significance of E2F3b phosphorylation remains unclear, we do note that UL97 targets only one of the two sites phosphorylated by cellular CDKs, implying that modification of these two sites may have independent functional significance.
The ability of FoxM1 to activate the transcription of genes required for progression through the G2/M phases of the cell cycle is strengthened by CDK-dependent phosphorylation on a series of residues including Thr600 (Laoukili et al., 2008; Sullivan et al., 2012). Here, we detect UL97-medited phosphorylation of FoxM1 on Thr600, implying that UL97 may activate this transcription factor. HCMV productive (lytic) infection of fibroblasts arrests their cell cycle progression at an aberrant state most resembling the G1/S border. Perhaps UL97 induces the expression of FoxM1-regulated genes with augmenting functions for HCMV replication that would otherwise not accumulate due to the viral-induced cell cycle arrest. Furthermore, this phosphorylation event may be pro-proliferative in other cell types where the cell cycle effects of HCMV infection are less studied.
In summary, we have identified three new cell cycle substrates for the v-CDK UL97 of human cytomegalovirus (Fig. 8). These novel substrates widen the footprint of UL97’s influence on the cell cycle, expanding it past the Rb tumor suppressors themselves and to phases beyond G1 and S.
Figure 8. UL97 phosphorylates multiple Rb pathway proteins throughout the cell cycle.

UL97 phosphorylates DREAM complex members (LIN52, p130, p107), Rb complex members (Rb, E2F3), and an MMB-interacting protein (FoxM1), each of which regulate transcription at different stages of the cell cycle.
4. Materials and methods
4.1. Cells
Saos-2 (containing truncated Rb), U-2 OS, primary human foreskin fibroblasts (HFF), and primary normal human dermal fibroblasts (NHDFs) (clonetics) were grown and maintained at 37°C in Dulbecco’s modified Eagle’s medium (DMEM, Sigma-Aldrich) supplemented with 10% fetal bovine serum (FBS, Sigma-Aldrich), 100 U/ml penicillin, 100 μg/ml streptomycin, and 0.292 mg/ml glutamine (PSG, Sigma-Aldrich). All infections were performed under serum-starved conditions. For serum starvation, sub-confluent fibroblasts were incubated in DMEM with 0.1% FBS for 48 hours. For serum stimulation, serum-starved fibroblasts were incubated with 15% serum. Saos-2 and U-2 OS cells were transfected using TransIT-2020 (Mirus), and HFF and NHDF cells were transfected using nucleofector kits for human dermal fibroblast (NHDF) and an Amaxa Nucleofector II (Lonza).
4.2. Virus and inhibitors
The wild-type HCMV strain was AD169. Maribavir (Acme Bioscience, A4028; 10 μM) or Harmine (Abcam, 5 μM) was added at 2 hr after infection.
4.3. Plasmids
Expression plasmids have been described: pCGN-HA UL97wt and pCGN-HA UL97kd, (Kuny et al., 2010); Rc/cyclin A2 and Rc/cyclin E1 (Hinds et al., 1992); Rc/cyclin D2 (Baker et al., 2005); CMV-CDK4 (van den Heuvel and Harlow, 1993); LIN52-V5-pEF6 (Guiley et al., 2015); pCMV-T7-FoxM1B (Chen et al., 2009); pCMV-DP1 (Helin et al., 1993); pE2F1-Luc(-242) (Johnson et al., 1994); pCMV-Rb (Qin et al., 1992). pCDK2-HA, pUHD-CDK1-WT-HA, p-cyclin B1-mCherry, pCMV-Neo-Bam-E2F3a and pCMV-Neo-Bam-E2F3b were purchased from Addgene (#1884, #27652, #26063, #37970 and #37975) (Gavet and Pines, 2010; Lees et al., 1993; van den Heuvel and Harlow, 1993). pCMV-Flag-E2F3a, pCMV-Flag-E2F3b and pCMV-FLAG-LIN52 were constructed by inserting PCR products amplified from pCMV-Neo-Bam-E2F3a, pCMV-Neo-Bam-E2F3b or LIN52-V5-pEF6 into the HindIII and XbaI sites of p3xFlag-CMV-10 (Sigma-Aldrich) using an In-Fusion HD cloning kit (Clontech). pGEX2T-E2F3b was constructed by inserting PCR products amplified from pCMV-Neo-Bam-E2F3b into the BamHI and EcoRI sites of pGEX-2T (GE Healthcare) using an In-Fusion HD cloning kit. Expression plasmids generated during this study by site-directed mutagenesis techniques are based on p3xFlag-CMV-10 or pGEX-2T and contain these substitutions: E2F3b-T27A, S38A, S41A, T44A, S47A, T27A/S38A, T27E, S38D, T27E/S38D and R37Q, and p3xFlag-CMV-10-based LIN52-S7A, S28A, S48A, S53A, S7/28A, S28/48A, S28/53A, S28/48/53A and S7/28/48/53A. All alleles generated by PCR have been confirmed by sequencing. The sequences of primers for mutagenesis are available upon request.
4.4. Antibodies
Primary antibodies were purchased from Abcam (Cyclin A2, catalog# ab32498; DYRK1A, ab54944), Ambion (GAPDH, AM4300), BD Transduction Laboratories (CDK1, 610038; CDK2, 610146), Bethyl laboratories (LIN9, A300-BL2981; LIN54, A303-799A), Cell Signaling (FoxM1, 5436; FoxM1-pT600, 14655; GST, 2624; Rb, 9309), Covance (HA, MMS-101P), Virusys (UL44, CA006-100), Santa Cruz (CDK4, sc-23896; cyclin B1, sc-245; cyclin D1, sc-718; cyclin D2, sc-181; cyclin E, sc-247; E2F3, sc-878; E2F4, sc-866; p107, sc-318; p130, sc-317), and Sigma-Aldrich (α-tubulin, T9026; Flag-M2, F1804). Antibodies against HCMV IE1 (1B12) and UL97 have been previously described (He et al., 1997; Zhu et al., 1995). Mouse TrueBlot Ultra (eBioscience, 18-8817) and rabbit TrueBlot (18-8816) were used as secondary antibodies for Western blotting of immunoprecipitation samples.
4.5. In vivo phosphorylation assays
To detect E2F3b or FoxM1 phosphorylation, Saos-2 cells were transfected with wild-type and mutated E2F3b or wild-type FoxM1B expression plasmid DNA together with wild-type and kinase-deficient HCMV UL97 or cellular Cyclin/CDKs. To detect LIN52 phosphorylation, transfections were performed as for E2F3b phosphorylation except using U-2 OS cells and wild-type and mutated LIN52 expression plasmid. At 48 hours post-transfection, the transfected cells were harvested and subjected to Western blotting.
4.6. Luciferase assay
U-2 OS cells (2.5×105/6-well) were transfected with expression plasmids for alleles of Flag-tagged E2F3b (0.1, 0.2, and 0.5 μg) together with CMV-DP1 (same amount as E2F3b) and pE2F1-Luc(-242) (0.02 μg) using TransIT-2020. Luciferase assays were performed as described previously (Iwahori et al., 2015).
4.7. Protein purification and in vitro phosphorylation assay
GST-tagged wild-type E2F3b and S38A mutant proteins were purified using Glutathione Sepharose 4B (GE Healthcare) from pGEX2T-based wild-type E2F3b and S38A mutant-transformed bacteria as previously described (Iwahori et al., 2007). Wild-type and kinase-deficient UL97 were purified as previously described (Iwahori et al., 2017). Cyclin E/CDK2 was purchased from Millipore. In vitro kinase reactions with E2F3b were performed at 37°C for 30 min in the kinase reaction buffer (50 mM Tris, pH8; 5 mM β-glycerophosphate, 10 mM MgCl2, 2 mM DTT, 0.05 mM cold ATP, 2 μCi [γ-32P]ATP). The reactions were incubated at 37°C for 30 min, stopped with SDS-PAGE loading buffer, and boiled for 10 min. Half of the samples were run on a 7.5% SDS-PAGE gel, dried and exposed to film for autoradiography. The other half of the samples were subjected to Western blotting.
4.8. Westerns
E2F3b or FoxM1-transfected Saos-2 cells, HCMV-infected fibroblasts or LIN52-transfected U-2 OS cells were lysed in an SDS solution (1% SDS, 2% β-mercaptoethanol) by boiling for 10 minutes followed by vortexing as previously described (Hume et al., 2008). Equal amounts of proteins were separated by 7.5%, 10%, or 12% SDS-PAGE and transferred onto nitrocellulose membranes. The phosphate-affinity (Phos-tag, NARD institute) electrophoresis was performed following the manufacturer’s instructions. Lambda protein phosphatase (l-PPase) reactions were performed as described previously (Iwahori et al., 2007). Protein bands were quantified with the Odyssey Fc Imager and the Image Studio version 2.1.10 software (LI-COR).
Highlights.
HCMV v-CDK UL97 phosphorylates multiple cell cycle substrates
DREAM complex assembly likely driven in part by UL97-mediated phosphorylation
Cancer relevant E2F3 mutation impairs phosphorylation by UL97 and cellular CDKs
G2 substrate FoxM1 targeted by UL97
Acknowledgments
We thank our lab managers Phil Balandyk and Diccon Fiore for expert technical assistance, the members of our lab for helpful discussion, and these investigators for providing research materials: Don Coen, Jim DeCapprio, Greg Enders, Ed Harlow, Philip Hinds, Jacqueline Lees, Larisa Litovchick, Joe Nevins, Jonathon Pines, Pradip Raychaudhuri, and Sander van den Heuvel. SI was supported by a Japan Herpesvirus Infections Forum scholarship award in herpesvirus infection research. This work was supported by grants from the NIH to RFK (R01-AI080675) and Paul Lambert (P01-CA022443).
Footnotes
Conflict of interest
The authors declare that they have no conflicts of interest with the contents of this article.
Author Contributions
SI and RFK designed the experiments and wrote the paper. SI performed the experiments.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams MR, Sears R, Nuckolls F, Leone G, Nevins JR. Complex transcriptional regulatory mechanisms control expression of the E2F3 locus. Mol Cell Biol. 2000;20:3633–3639. doi: 10.1128/mcb.20.10.3633-3639.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders L, Ke N, Hydbring P, Choi YJ, Widlund HR, Chick JM, Zhai H, Vidal M, Gygi SP, Braun P, Sicinski P. A systematic screen for CDK4/6 substrates links FOXM1 phosphorylation to senescence suppression in cancer cells. Cancer Cell. 2011;20:620–634. doi: 10.1016/j.ccr.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker GL, Landis MW, Hinds PW. Multiple functions of D-type cyclins can antagonize pRb-mediated suppression of proliferation. Cell Cycle. 2005;4:330–338. [PubMed] [Google Scholar]
- Black AR, Azizkhan-Clifford J. Regulation of E2F: a family of transcription factors involved in proliferation control. Gene. 1999;237:281–302. doi: 10.1016/s0378-1119(99)00305-4. [DOI] [PubMed] [Google Scholar]
- Britt W. Manifestations of human cytomegalovirus infection: proposed mechanisms of acute and chronic disease. Curr Top Microbiol Immunol. 2008;325:417–470. doi: 10.1007/978-3-540-77349-8_23. [DOI] [PubMed] [Google Scholar]
- Chen JY, Lin JR, Tsai FC, Meyer T. Dosage of Dyrk1a shifts cells within a p21-cyclin D1 signaling map to control the decision to enter the cell cycle. Mol Cell. 2013;52:87–100. doi: 10.1016/j.molcel.2013.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YJ, Dominguez-Brauer C, Wang Z, Asara JM, Costa RH, Tyner AL, Lau LF, Raychaudhuri P. A conserved phosphorylation site within the forkhead domain of FoxM1B is required for its activation by cyclin-CDK1. J Biol Chem. 2009;284:30695–30707. doi: 10.1074/jbc.M109.007997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou S. Cytomegalovirus UL97 mutations in the era of ganciclovir and maribavir. Rev Med Virol. 2008;18:233–246. doi: 10.1002/rmv.574. [DOI] [PubMed] [Google Scholar]
- Dyson N. The regulation of E2F by pRB-family proteins. Genes Dev. 1998;12:2245–2262. doi: 10.1101/gad.12.15.2245. [DOI] [PubMed] [Google Scholar]
- Dziurzynski K, Chang SM, Heimberger AB, Kalejta RF, McGregor Dallas SR, Smit M, Soroceanu L, Cobbs CS. Consensus on the role of human cytomegalovirus in glioblastoma. Neuro Oncol. 2012;14:246–255. doi: 10.1093/neuonc/nor227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsani A, Mileo AM, Paggi MG. Retinoblastoma family proteins as key targets of the small DNA virus oncoproteins. Oncogene. 2006;25:5277–5285. doi: 10.1038/sj.onc.1209621. [DOI] [PubMed] [Google Scholar]
- Gavet O, Pines J. Progressive activation of CyclinB1-Cdk1 coordinates entry to mitosis. Dev Cell. 2010;18:533–543. doi: 10.1016/j.devcel.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong C, Liu H, Song R, Zhong T, Lou M, Wang T, Qi H, Shen J, Zhu L, Shao J. ATR-CHK1-E2F3 signaling transactivates human ribonucleotide reductase small subunit M2 for DNA repair induced by the chemical carcinogen MNNG. Biochim Biophys Acta. 2016;1859:612–626. doi: 10.1016/j.bbagrm.2016.02.012. [DOI] [PubMed] [Google Scholar]
- Guiley KZ, Liban TJ, Felthousen JG, Ramanan P, Litovchick L, Rubin SM. Structural mechanisms of DREAM complex assembly and regulation. Genes Dev. 2015;29:961–974. doi: 10.1101/gad.257568.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Z, He YS, Kim Y, Chu L, Ohmstede C, Biron KK, Coen DM. The human cytomegalovirus UL97 protein is a protein kinase that autophosphorylates on serines and threonines. J Virol. 1997;71:405–411. doi: 10.1128/jvi.71.1.405-411.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helin K, Wu CL, Fattaey AR, Lees JA, Dynlacht BD, Ngwu C, Harlow E. Heterodimerization of the transcription factors E2F-1 and DP-1 leads to cooperative trans-activation. Genes Dev. 1993;7:1850–1861. doi: 10.1101/gad.7.10.1850. [DOI] [PubMed] [Google Scholar]
- Helt AM, Galloway DA. Mechanisms by which DNA tumor virus oncoproteins target the Rb family of pocket proteins. Carcinogenesis. 2003;24:159–169. doi: 10.1093/carcin/24.2.159. [DOI] [PubMed] [Google Scholar]
- Hinds PW, Mittnacht S, Dulic V, Arnold A, Reed SI, Weinberg RA. Regulation of retinoblastoma protein functions by ectopic expression of human cyclins. Cell. 1992;70:993–1006. doi: 10.1016/0092-8674(92)90249-c. [DOI] [PubMed] [Google Scholar]
- Hume AJ, Finkel JS, Kamil JP, Coen DM, Culbertson MR, Kalejta RF. Phosphorylation of retinoblastoma protein by viral protein with cyclin-dependent kinase function. Science. 2008;320:797–799. doi: 10.1126/science.1152095. [DOI] [PubMed] [Google Scholar]
- Iwahori S, Hakki M, Chou S, Kalejta RF. Molecular determinants for the inactivation of the retinoblastoma tumor suppressor by the viral cyclin-dependent kinase UL97. J Biol Chem. 2015;290:19666–19680. doi: 10.1074/jbc.M115.660043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwahori S, Shirata N, Kawaguchi Y, Weller SK, Sato Y, Kudoh A, Nakayama S, Isomura H, Tsurumi T. Enhanced phosphorylation of transcription factor Sp1 in response to herpes simplex virus type 1 infection is dependent on the ataxia telangiectasia-mutated protein. J Virol. 2007;81:9653–9664. doi: 10.1128/JVI.00568-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwahori S, Umana AC, VanDeusen HR, Kalejta RF. Human cytomegalovirus-encoded viral cyclin-dependent kinase (v-CDK) UL97 phosphorylates and inactivates the retinoblastoma protein-related p107 and p130 proteins. J Biol Chem. 2017;292:6583–6599. doi: 10.1074/jbc.M116.773150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DG, Ohtani K, Nevins JR. Autoregulatory control of E2F1 expression in response to positive and negative regulators of cell cycle progression. Genes Dev. 1994;8:1514–1525. doi: 10.1101/gad.8.13.1514. [DOI] [PubMed] [Google Scholar]
- Kuny CV, Chinchilla K, Culbertson MR, Kalejta RF. Cyclin-dependent kinase-like function is shared by the beta- and gamma- subset of the conserved herpesvirus protein kinases. PLoS Pathog. 2010;6:e1001092. doi: 10.1371/journal.ppat.1001092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laoukili J, Alvarez M, Meijer LA, Stahl M, Mohammed S, Kleij L, Heck AJ, Medema RH. Activation of FoxM1 during G2 requires cyclin A/Cdk-dependent relief of autorepression by the FoxM1 N-terminal domain. Mol Cell Biol. 2008;28:3076–3087. doi: 10.1128/MCB.01710-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Cho Y. Interactions of SV40 large T antigen and other viral proteins with retinoblastoma tumour suppressor. Rev Med Virol. 2002;12:81–92. doi: 10.1002/rmv.340. [DOI] [PubMed] [Google Scholar]
- Lees JA, Saito M, Vidal M, Valentine M, Look T, Harlow E, Dyson N, Helin K. The retinoblastoma protein binds to a family of E2F transcription factors. Mol Cell Biol. 1993;13:7813–7825. doi: 10.1128/mcb.13.12.7813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leone G, Nuckolls F, Ishida S, Adams M, Sears R, Jakoi L, Miron A, Nevins JR. Identification of a novel E2F3 product suggests a mechanism for determining specificity of repression by Rb proteins. Mol Cell Biol. 2000;20:3626–3632. doi: 10.1128/mcb.20.10.3626-3632.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litovchick L, Florens LA, Swanson SK, Washburn MP, DeCaprio JA. DYRK1A protein kinase promotes quiescence and senescence through DREAM complex assembly. Genes Dev. 2011;25:801–813. doi: 10.1101/gad.2034211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Clark PA, Kuo JS, Kalejta RF. Human Cytomegalovirus-Infected Glioblastoma Cells Display Stem Cell-Like Phenotypes. mSphere. 2017:2. doi: 10.1128/mSphere.00137-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez LA, Goluszko E, Chen HZ, Leone G, Post S, Lozano G, Chen Z, Chauchereau A. E2F3 is a mediator of DNA damage-induced apoptosis. Mol Cell Biol. 2010;30:524–536. doi: 10.1128/MCB.00938-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melnick JL, Adam E, DeBakey ME. Cytomegalovirus and atherosclerosis. Bioessays. 1995;17:899–903. doi: 10.1002/bies.950171012. [DOI] [PubMed] [Google Scholar]
- Mitchell DA, Batich KA, Gunn MD, Huang MN, Sanchez-Perez L, Nair SK, Congdon KL, Reap EA, Archer GE, Desjardins A, Friedman AH, Friedman HS, Herndon JE, 2nd, Coan A, McLendon RE, Reardon DA, Vredenburgh JJ, Bigner DD, Sampson JH. Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature. 2015;519:366–369. doi: 10.1038/nature14320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro S, Carr SM, La Thangue NB. Diversity within the pRb pathway: is there a code of conduct? Oncogene. 2012;31:4343–4352. doi: 10.1038/onc.2011.603. [DOI] [PubMed] [Google Scholar]
- Oberstein A, Perlman DH, Shenk T, Terry LJ. Human cytomegalovirus pUL97 kinase induces global changes in the infected cell phosphoproteome. Proteomics. 2015;15:2006–2022. doi: 10.1002/pmic.201400607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prichard MN, Sztul E, Daily SL, Perry AL, Frederick SL, Gill RB, Hartline CB, Streblow DN, Varnum SM, Smith RD, Kern ER. Human cytomegalovirus UL97 kinase activity is required for the hyperphosphorylation of retinoblastoma protein and inhibits the formation of nuclear aggresomes. J Virol. 2008;82:5054–5067. doi: 10.1128/JVI.02174-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin XQ, Chittenden T, Livingston DM, Kaelin WG., Jr Identification of a growth suppression domain within the retinoblastoma gene product. Genes Dev. 1992;6:953–964. doi: 10.1101/gad.6.6.953. [DOI] [PubMed] [Google Scholar]
- Ranganathan P, Clark PA, Kuo JS, Salamat MS, Kalejta RF. Significant association of multiple human cytomegalovirus genomic Loci with glioblastoma multiforme samples. J Virol. 2012;86:854–864. doi: 10.1128/JVI.06097-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin SM. Deciphering the retinoblastoma protein phosphorylation code. Trends Biochem Sci. 2013;38:12–19. doi: 10.1016/j.tibs.2012.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadasivam S, Duan S, DeCaprio JA. The MuvB complex sequentially recruits B-Myb and FoxM1 to promote mitotic gene expression. Genes Dev. 2012;26:474–489. doi: 10.1101/gad.181933.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh M, Krajewski M, Mikolajka A, Holak TA. Molecular determinants for the complex formation between the retinoblastoma protein and LXCXE sequences. J Biol Chem. 2005;280:37868–37876. doi: 10.1074/jbc.M504877200. [DOI] [PubMed] [Google Scholar]
- Soroceanu L, Cobbs CS. Is HCMV a tumor promoter? Virus Res. 2011;157:193–203. doi: 10.1016/j.virusres.2010.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speir E, Modali R, Huang ES, Leon MB, Shawl F, Finkel T, Epstein SE. Potential role of human cytomegalovirus and p53 interaction in coronary restenosis. Science. 1994;265:391–394. doi: 10.1126/science.8023160. [DOI] [PubMed] [Google Scholar]
- Sullivan C, Liu Y, Shen J, Curtis A, Newman C, Hock JM, Li X. Novel interactions between FOXM1 and CDC25A regulate the cell cycle. PLoS One. 2012;7:e51277. doi: 10.1371/journal.pone.0051277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suryadinata R, Sadowski M, Sarcevic B. Control of cell cycle progression by phosphorylation of cyclin-dependent kinase (CDK) substrates. Biosci Rep. 2010;30:243–255. doi: 10.1042/BSR20090171. [DOI] [PubMed] [Google Scholar]
- Ubersax JA, Ferrell JE., Jr Mechanisms of specificity in protein phosphorylation. Nat Rev Mol Cell Biol. 2007;8:530–541. doi: 10.1038/nrm2203. [DOI] [PubMed] [Google Scholar]
- van den Heuvel S, Harlow E. Distinct roles for cyclin-dependent kinases in cell cycle control. Science. 1993;262:2050–2054. doi: 10.1126/science.8266103. [DOI] [PubMed] [Google Scholar]
- Zhu H, Shen Y, Shenk T. Human cytomegalovirus IE1 and IE2 proteins block apoptosis. J Virol. 1995;69:7960–7970. doi: 10.1128/jvi.69.12.7960-7970.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]