

Abstract
Little is known about how obesity affects the heart during sepsis and we sought to investigate the obesity-induced cardiac effects that occur during polymicrobial sepsis. Six-week old C57BL/6 mice were randomized to a high fat (HFD) (60% kcal fat) or normal diet (ND) (16% kcal fat). After 6 wks of feeding, mice were anesthetized with isoflurane and polymicrobial sepsis was induced by cecal ligation and puncture (CLP). Plasma and cardiac tissue were obtained for analysis. Echocardiography was performed on a separate cohort of mice at 0 and 18h after CLP. Following 6-wks of dietary intervention, plasma cardiac troponin I was elevated in obese mice at baseline compared to non-obese mice but troponin increased only in non-obese septic mice. IL-17a expression was 27-fold higher in obese septic mice versus non-obese septic mice. Cardiac phosphorylation of STAT3 at Ser727 was increased at baseline in obese mice and increased further only in obese septic mice. Phosphorylation of STAT3 at Tyr705 was similar in both groups at baseline and increased after sepsis. SOCS3, a downstream protein and negative regulator of STAT3, was elevated in obese mice at baseline compared to non-obese mice. After sepsis non-obese mice had an increase in SOCS3 expression that was not observed in obese mice. Taken together, we show that obesity affects cardiac function and leads to cardiac injury. Furthermore, myocardial injury in obese mice during sepsis may occur through alteration of the STAT3 pathway.
Keywords: sepsis, obesity, STAT3, cardiac inflammation
1. Introduction
Sepsis is a leading cause of death in patients in the intensive care unit (ICU), with up to 50% mortality [1, 2]. Sepsis involves a dysfunctional immune response which can lead to tissue injury and organ failure [3]. Co-morbidities, such as obesity, complicate sepsis by increasing hospitalization and readmissions [4]. Obesity is associated with chronic inflammation however in response to an acute insult such as trauma, obese human subjects have a depressed cytokine response compared to non-obese subjects [5]. Data from our laboratory also demonstrate a diminished cytokine response in obese mice after sepsis [6]. Additionally, obese mice have higher mortality after sepsis [6, 7].
Increased adiposity can alter the cardiac structure and have detrimental effects on the heart [8]. Mitra et al. showed that obese mice were more susceptible to doxorubicin-induced cardiotoxicity compared to normal weight controls [9]. However, in other studies obesity was cardio-protective and obese mice had decreased infarct size after myocardial infarction compared to non-obese mice [10]. Furthermore, studies in human subjects with obesity and heart disease show a lower mortality for obese patients, a concept termed the “obesity paradox” [11, 12]. Little is known about the obesity-induced effects on the heart that occur during sepsis.
Signal transducer and activator of transcription (STAT)-3 is an important acute phase reactant in sepsis. Leptin and interleukin (IL)-6 both activate the STAT3 signaling pathway and lead to phosphorylation at one of two sites, tyrosine 705 or serine 727. STAT3 is also important for the stress-induced response in the heart. Early activation of STAT3 is evident in the heart in response to β-adrenergic receptor agonist treatment and cardiac specific STAT3 deletion led to increased cardiac hypertrophy, necrosis, and apoptosis after chronic β-adrenergic stimulation [13]. Obesity also has effects on cardiac STAT3 expression, in that cardiac STAT3 expression was differentially regulated in Bama miniature pigs fed a long-term high-energy diet compared to controls [14]. The suppressor of the cytokine signaling family (SOCS3) acts as a negative regulator of STAT3 [15]. SOCS3 is increased in obesity, leading to down-regulation of phosphorylation of STAT3 [9]. These studies suggest that the STAT3 pathway is important in the heart during stress but is altered in obesity. Given the importance of STAT3 in the acute phase response to injury we sought to determine whether the STAT3 pathway is altered in the heart during sepsis and affected by obesity.
2. Materials and Methods
2.1 Mice and Diets
The investigation conformed to the Guide for the Care and Use of Laboratory Animals published by the U.S. National Institutes of Health [16] and was approved by the Institutional Animal Care and Use Committee at Cincinnati Children’s Hospital Medical Center. The experimental groups consisted of male C57BL/6 mice at six-weeks of age supplied from Charles River Laboratories International, Inc. (Wilmington, MA). The mice were housed in the animal facility at the Cincinnati Children’s Research Foundation (CCRF). Food and water were provided ad libitum. Animals were randomized to a high-fat diet (HFD) (TestDiet – 58Y1) (60% kcal provided by fat) or a normal diet (ND) (Formulab – 5008) (16% kcal provided by fat) for 6 weeks.
2.2. Body weight and body composition
Body weights were monitored once weekly throughout the diet phase of the study. To quantify fat mass, we analyzed body composition at the end of the 6-week diet phase using NMR imaging by EchoMRI body composition analyzers (EchoMRI, Houston, Tx).
2.3. Mouse model of cecal ligation and puncture (CLP)
Mice were fed a HFD or ND for 6 weeks and then underwent CLP to induce polymicrobial sepsis as previously described [17]. Mice were anesthetized with isoflurane. The abdomen was opened and the cecum exteriorized and ligated by a 6.0 ligature at its base. A double puncture technique was performed with a 22g needle and fecal material was expressed into the peritoneum. The abdominal incision was closed with liquid topical adhesive. Animals were fluid resuscitated with sterile saline (0.6 ml) injected subcutaneously and received imipenem (25mg/kg) for antimicrobial coverage. Plasma and whole heart tissue were collected at 6h after CLP for biochemical studies described below.
2.4. Troponin I
Plasma was obtained from mice at time of sacrifice. Plasma cardiac troponin I (cTnI) was quantified using the high sensitivity cardiac troponin-I ELISA kit (Life Diagnostics, West Chester, PA).
2.5. Measurement of myeloperoxidase activity
Myeloperoxidase activity was determined as an index of neutrophil accumulation as previously described [18]. Tissues were homogenized in a solution containing 0.5% hexa-decyl-trimethyl-ammonium bromide dissolved in 10 mM potassium phosphate buffer (pH7) and were centrifuged for 30 min at 4,000 × g at 4°C. An aliquot of the supernatant was allowed to react with a solution of tetra-methyl-benzidine (1.6 mM) and 0.1 mM hydrogen peroxide. The rate of change in absorbance was measured by spectrophotometry at 650 nm. Myeloperoxidase activity was defined as the quantity of enzyme degrading 1 μmol hydrogen peroxide per min at 37°C per 100 mg weight of tissue.
2.6. Echocardiographic studies
A separate cohort of mice were used for echocardiographic analysis. After the 6-week dietary intervention, mice were anesthetized with isoflurane and underwent CLP. All mice that underwent CLP survived to 18h and thus underwent echocardiography (n = 8 mice/group). Echocardiography was performed using a VisualSonics 2100 system (Toronto, Ontario, Canada) equipped with a 30-MHz transducer as previously described [19]. Heart rate, left ventricle internal dimensions, including end-diastolic and end-systolic dimensions, interventricular septal thickness in diastole and systole, and LV posterior wall thickness in diastole and systole were measured directly.
2.7. Gene expression analysis
RT-qPCR assays were performed according to the manufacture’s protocol using the Mouse Prime I-L6/STAT3 PCR Assay which profiles the expression of 96 IL-6/STAT3-related genes (BioRad catalog #10034344, Hercules, CA). Cardiac samples were homogenized in TRIzol (Invitrogen, Grand Island, NY). RNA was reversely transcribed using the RT2 First Strand Kit for cDNA synthesis according to the manufacture’s protocol (Qiagen, Valencia, CA). Cycle threshold was standardized between all samples. Each sample was normalized to two housekeeping genes (ΔCT). Normalized control samples (non-obese non-septic mice) were subtracted from normalized experimental samples (ΔΔCT). The non-obese non-septic group was set as the control group and non-obese septic, obese non-septic and obese septic groups were set as the experimental groups (n= 4 mice/group). The fold change in each tissue was calculated using the comparative CT method (2−ΔΔCT). Assay integrity was monitored by a genomic DNA control, 3 reverse-transcription controls and 3 positive PCR controls.
2.8. Subcellular fractionation and nuclear protein extraction
Heart tissue samples were homogenized in a buffer containing 0.32 M sucrose, 10 mM Tris-HCl, 1 mM ethylene glycol tetraacetic acid (EGTA), 2 mM ethylenediaminetetraacetic acid (EDTA), 5 mM NaN3, 10 mM β-mercaptoethanol, 50 mM NaF, 20 μM leupeptin, 0.15 μM pepstatin A, and 0.2 mM phenylmethylsulphonyl fluoride (PMSF), 1 mM sodium orthovanadate, 0.4 nM microcystin [20]. The homogenates were centrifuged (1,000 × g at 4°C, 10 min). The supernatant (cytosol + membrane extract) was collected and stored. The pellets were solubilized in Triton buffer (1% Triton X-100, 150 mM NaCl, 10 mM Tris-HCl (pH7.4), 1 mM EGTA, 1 mM EDTA, 0.2 mM sodium orthovanadate, 20 μM leupeptin A, and 0.2 mM PMSF). The lysates were centrifuged (15,000 × g, at 4°C, 30 min) and the supernatant (nuclear extract) collected. The amount of protein was quantified by Bradford assay.
2.9. Western blot analysis
Analysis was performed on nuclear extracts except where indicated. The Invitrogen NuPAGE gel electrophoresis system (Invitrogen) was used for all Western blotting. NuPAGE 10% Bis-Tris gels were used with NuPAGE MOPS buffer and Invitrogen Novex Mini-Cell, BioRad PowerPac 300. Membranes imaged using BioRad ChemiDoc XRS+ gel documentation system and analyzed using ImageLab v5.1 software (BioRad, Hercules, CA). The following antibodies were used to probe for proteins of interest: STAT3 (Cell Signaling Technologies, Danvers, MA), pSTAT3 (Tyrosine 705) (Cell Signaling Technologies), pSTAT3 (Serine 727) (Cell Signaling Technologies), SOCS3 (Santa Cruz Biotechnology, Inc., Dallas, Texas), gp130 (cytosol) (Cell Signaling Technologies), beta-Actin (nuclear and cytosol) (Santa Cruz Biotechnology, Inc., Dallas, Texas).
2.10. Statistical Analysis
Data were analyzed using Sigmaplot (Systat Systems, San Jose, CA). Values in the text and figures are expressed as mean and standard error of the mean (SEM) for parametric data and as median and interquartile range for nonparametric data. Data were analyzed by ANOVA and t-test for parametric data and Mann-Whitney test for non-parametric data. Echocardiographic data was analyzed with GraphPad InStat 3 for Macintosh using unpaired and paired t-tests. Significance was defined as P ≤ 0.05.
3. Results
3.1. High fat diet feeding leads to weight gain and increased fat mass
After six weeks of feeding on a diet with 60% kcal provided by fat (vs 16% in a normal diet) mice had significantly more total weight gain (11.7±1.9g vs 5.0±1.3g, p<0.001) and had significantly higher weight [36.3g (34.2–38.1g IQR) vs 27.8g (27.0–28.4g IQR)] than normal diet-fed mice. Fat mass gain by EchoMRI (Figure 1) was significantly higher in the HFD fed group (8.0±2.6g vs 0.6±0.5g, p<0.001). In addition, the HFD mice had less lean mass gain (2.7±0.7g vs 3.7±0.7g, p<0.001) and less total body water gain (2.6±0.8g vs 3.0±0.7g, p=0.03). Based on the higher total weight and increased fat mass, mice fed the high-fat diet are representative of human obesity.
Figure 1. EchoMRI to determine body composition of mice fed normal and high-fat diets for 6 weeks.
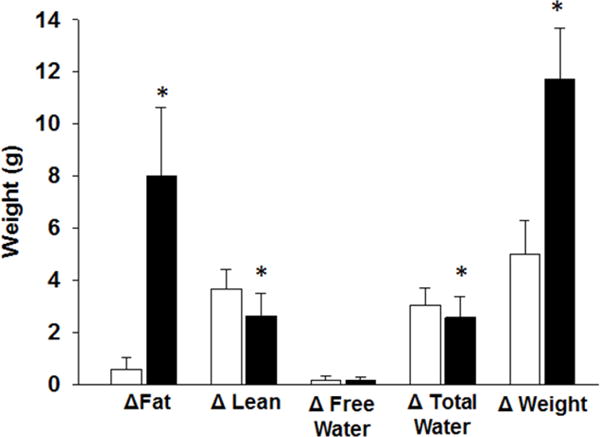
Changes in body fat, lean mass, free water, total water and total weight prior to and after dietary intervention. Values are means ± SEM. White bars=non-obese mice fed normal diet, Black bars=obese mice fed high-fat diet. n=12–13/group. *p<0.05 compared to normal diet.
3.2. Obesity affects cardiac dysfunction
Echocardiography was used to determine whether diet-induced obesity affected cardiac function before and after the induction of sepsis. Obese mice were compared to age- and gender-matched non-obese controls. All septic mice survived to 18h and underwent echocardiography. By echocardiography analysis, obese mice had lower systolic function as evidenced by lower fractional shortening (FS) compared to non-obese mice prior to sepsis (Table 1). Mice underwent CLP and survived to 18 hours post-CLP and thus underwent repeat echocardiography. After sepsis, non-obese mice had lower heart rates compared to baseline and obese mice. After sepsis, obese mice had improved cardiac function as evidenced by an increase in fractional shortening. Thus, although obese mice exhibit poor function at baseline, cardiac function increases in response to sepsis.
Table 1.
HR = heart rate; IVSd = diastolic interventricular septum thickness; LVIDs = systolic left ventricular internal dimension; LVIDd = diastolic left ventricular internal dimension; FS = left ventricular fractional shortening; LVM = left ventricular mass; LVPWd = diastolic left ventricular posterior wall thickness.
| Before CLP | After CLP | |||
|---|---|---|---|---|
| Non-obese | Obese | Non-obese | Obese | |
| HR (bpm) | 442 ± 85 | 437 ± 24 | 325 ± 46* | 430 ± 25# |
| IVSd (mm) | 0.85 ± 0.1 | 0.78 ± 0.02 | 1 ±0.16 | 0.86 ± 0.15 |
| LVIDs (mm) | 2.53 ± 0.35 | 3.17 ± 0.41# | 2.27 ± 0.85 | 2.54 ± 0.34* |
| LVIDd (mm) | 4 ± 0.41 | 4.20 ± 0.35 | 3.51 ± 0.76 | 3.68 ± 0.4* |
| FS (%) | 34 ±4 | 25 ± 5# | 37 ± 10 | 31 ± 4* |
| LVM (mg) | 96 ± 14.1 | 95.2 ± 14.8 | 91 ± 8 | 84 ± 7* |
| LVPWd (mm) | 0.74 ± 0.07 | 0.73 ± 0.1 | 0.88 ± 0.23 | 0.77 ± 0.07 |
HR = heart rate; IVSd = diastolic interventricular septum thickness; LVIDs = systolic left ventricular internal dimension; LVIDd = diastolic left ventricular internal dimension; F5 = left ventricular fractional shortening; LVM = left ventricular mass; LVPWd = diastolic left ventricular posterior wall thickness;.
p<0.05 versus non-obese by t-test,
p<0.05 versus before CLP by paired t-test. n=8 mice/group.
3.3 Cardiac injury and inflammation during sepsis
To assess myocardial injury at baseline and after sepsis we measured plasma cTnI in both obese and non-obese mice. At baseline, after 6 weeks of high-fat feeding, obese mice had elevated cTnI levels compared to non-obese mice [median 31 ng/ml (20–35 ng/ml IQR) vs 2 ng/ml (2–4 ng/ml IQR)] (Figure 2A). Following sepsis, cTnI increased significantly in non-obese mice [45 ng/ml (40–47 ng/ml IQR)] but was not altered in obese mice after sepsis compared to baseline (CLP 0h). Neutrophil infiltration of the cardiac tissue can be used as a marker of inflammation, and to that end we measured myeloperoxidase (MPO) activity in the myocardial tissue. After sepsis, MPO activity increased in both non-obese and obese mice and was significantly higher in obese septic mice (Figure 2B).
Figure 2. Plasma levels of troponin I and myeloperoxidase activity in obese and non-obese mice subjected to polymicrobial sepsis.
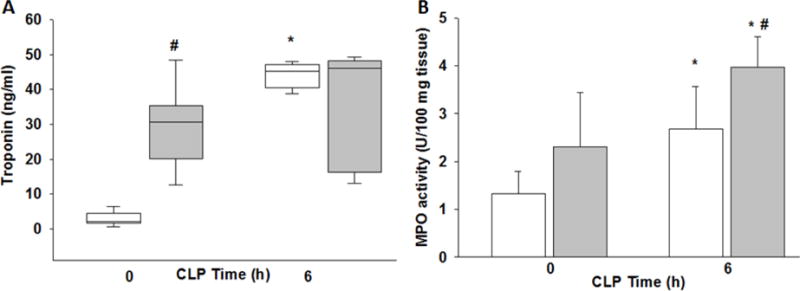
(A) Plasma cardiac troponin I and (B) cardiac neutrophil infiltration as determined by myeloperoxidase assay (MPO), white bars/boxes=non-obese mice, grey bars/boxes=obese mice. *p<0.05 vs. time 0, #p<0.05 vs. non-obese mice by 2-way ANOVA. n=4/group for MPO and n=6–8/group for troponin.
3.4. Cardiac gene expression in obese septic mice
To identify important regulators of sepsis that are affected by obesity we investigated differences in gene expression in obese and non-obese septic and non-septic mice. Gene expression was determined using the PrimePCR Assay to profile 90 select genes using the IL-6/STAT3 pathway (BioRad). In Figure 3A, center dots represent unchanged gene expression, boundary lines indicate 4-fold gene regulation cutoff between septic obese and septic non-obese samples. The red line indicates a four-fold upregulation and the green line indicates a four-fold downregulation. Examination of differentially expressed genes at 6h after CLP demonstrates that IL-17a was the most upregulated gene (26.5-fold increase) in septic obese mice compared to septic non-obese mice (Figure 3 A&B). IL-17a, Csf2 and IL-10 demonstrate differences that represent more than a doubling (log 2 changes of >1.0) and that were statistically significant at P≤0.05 (Figure 3).
Figure 3. Cardiac gene expression in obese and non-obese mice after sepsis.
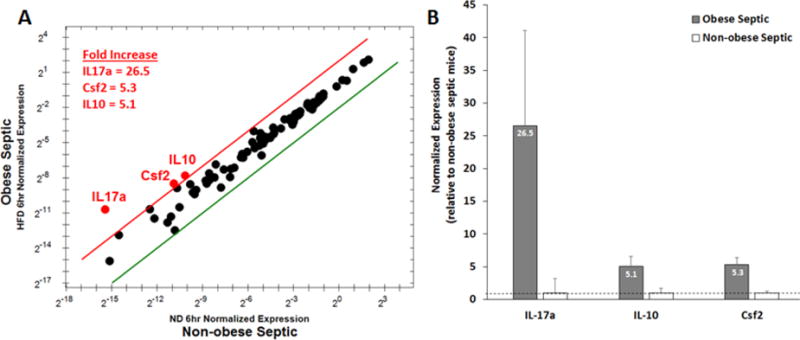
Gene expression profile of 90 genes related to IL-6/STAT3 signaling were evaluated at 6h after CLP in obese and non-obese mice. (A) Center dots represent unchanged gene expression. Boundary lines indicate 4-fold gene regulation cut-off between septic obese and septic non-obese samples (red line, 4-fold upregulation; green line 4-fold downregulation). IL-17a, Csf2 and IL-10 demonstrate differences that represent more than a doubling (log 2 changes of >1) and that were statistically significant at P≤0.05. (B) Relative expression of IL-17a, Csf2 and IL-10 in obese septic mice normalized to non-obese septic mice. Grey bar = obese septic mice, White bar = non-obese septic mice. Dashed line is placed at 1 for reference. n=4 mice/group.
3.5. Obesity alters phosphorylation of STAT3 in sepsis
Since IL-17 production is affected by STAT3 activity we sought to determine whether activation of STAT3 was differentially regulated in obese and non-obese mice during sepsis. Western blot expression revealed that phosphorylation of STAT3 at serine 727 was higher in nuclear cardiac extracts in obese mice at baseline (0.79 ± 0.15 relative units) compared to non-obese mice (0.3 ± 0.15 relative units, p<0.05) (Figure 4 A&C). After sepsis, phosphorylation of STAT3 at serine 727 increased in obese and non-obese mice compared to baseline and phosphorylation of STAT3 at serine 727 remained higher in obese mice compared to non-obese mice. Western blot analysis assessing nuclear phosphorylation of STAT3 at tyrosine 705 demonstrated similar protein expression in obese and non-obese mice at baseline. Phosphorylation of STAT3 at tyrosine 705 increased significantly in both non-obese and obese mice after sepsis (0.6±0.4 vs 11.1±1.7 relative units, p<0.01; 0.8±1.0 vs 11.6±6.1 relative units, p<0.01), but there was no dietary difference (Figure 4 B&D).
Figure 4. Phosphorylation of cardiac STAT3.
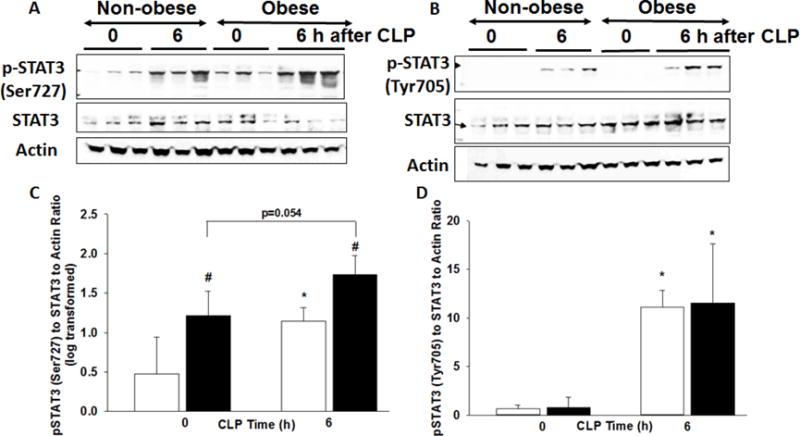
(A) changes in phosphorylation of STAT3 at serine 727 and (B) tyrosine 705, total STAT3 and actin expression in heart nuclear extracts by Western blot at 0 and 6h after CLP. Densitometric analysis of (C) pSTAT3 (Ser727) to STAT3 to Actin ratio log transformed and (D) pSTAT3 (Tyr705) to STAT3 to Actin ratio. *p<0.05 vs. time 0, #p<0.05 vs. non-obese mice by 2-way ANOVA. White bars=non-obese, Black bars=obese. n=3-4mice/group for densitometric analysis.
3.6. Obesity alters regulators of the STAT3 pathway in sepsis
SOCS3, a downstream negative regulator of STAT3, was increased in obese mice at baseline compared to non-obese mice (3.8±0.4 relative units vs 1±0.4 relative units, p<0.001) (Figure 5). After CLP, SOCS3 expression increased in non-obese mice to levels comparable to obese mice (3.7±0.7, p<0.001). But sepsis did not induce a change in SOCS3 expression in obese mice. Glycoprotein-130 (gp130) is a receptor subunit that binds many cytokines including IL-6 and IL-27. Total gp130 protein levels were significantly less in obese mice 6h after CLP due to the decreased variability of gp130 in non-obese mice (Figure 6).
Figure 5. Cardiac expression of SOCS3.
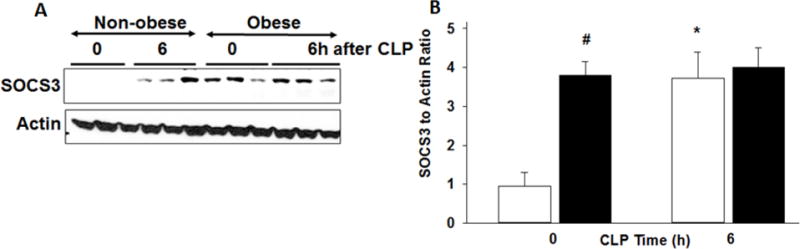
(A) Expression of SOCS3 and actin in heart nuclear extracts by Western blot analysis at 0 and 6h after CLP. (B) Densitometric analysis of SOCS to actin ratio. *p<0.05 vs. time 0, #p<0.05 vs non-obese group by 2-way ANOVA. White bars=non-obese, Black bars=obese. n=3–4mice/group for densitometric analysis.
Figure 6. Cardiac expression of gp130.
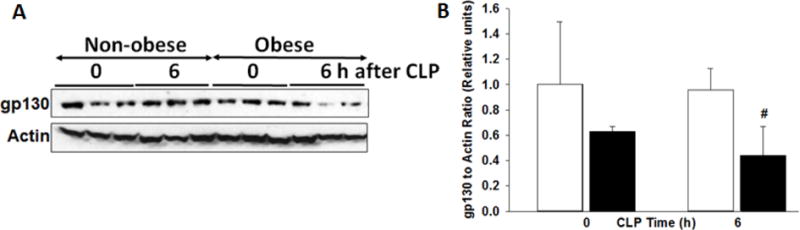
(A) Expression of gp130 and actin in heart cytosol extracts by Western blot analysis at 0 and 6h after CLP. (B) Densitometric analysis of gp130 to actin ratio. #p<0.05 vs non-obese group by 2-way ANOVA. White bars=non-obese, Black bars=obese. n=3–4mice/group for densitometric analysis.
4. Discussion
Our model of high fat feeding creates obese mice with increases in both total weight and fat mass. This replication of human obesity allows us to investigate the effects of obesity during an acute illness such as sepsis. Much has been described about the chronic medical problems seen with obesity [21], but there has been limited study of how obesity affects the response to acute illness. Data from both humans and mice show mixed results in regards to whether or not obesity is detrimental in acute states such as trauma or sepsis. Overweight and obese patients may have protection from critical illness in certain diseases, the obesity paradox, although equipoise remains regarding this concept. The results from this current study, together with published data from our laboratory, suggest that obesity is detrimental during murine sepsis [6, 7].
In our current study, obese mice develop decreased left ventricular fractional shortening compared to non-obese mice and elevated cTnI levels, a marker of cardiac tissue injury. Sepsis causes an increase in cardiac tissue injury in non-obese mice at levels similar to obese mice. Surprisingly, sepsis did not increase cTnI levels in obese mice as these levels remained elevated, suggesting a maximum level of injury occurred. In addition to cardiac myocyte injury, sepsis increased cardiac neutrophil infiltration in both obese and non-obese mice. This increase in inflammatory response may prove necessary and beneficial to the reparative response in sepsis. Han et al. demonstrated that an acute inflammatory response is necessary for cardiac regeneration after cardiac injury [22]. In fact, mice that received immunosuppression with dexamethasone failed to regenerate lost myocardium after apical resection.
Cardiac repair occurs through activation of STAT3, an acute phase reactant that regulates many genes involved in inflammation, apoptosis and the stress response [23, 24]. Similar to the findings of dexamethasone treatment, ablation of STAT3 in cardiomyocytes impaired the proliferative response after apical resection in neonatal mice [22]. Our study also demonstrates changes in STAT3 phosphorylation at serine 727 were increased at baseline in obese mice, consistent with chronic inflammation in obesity. After sepsis phosphorylation of STAT3 at serine 727 increased in both non-obese and obese mice and there was a significant dietary effect. In contrast, phosphorylation of STAT3 at tyrosine 705 was increased in cardiac tissue in both septic obese and non-obese mice but there was no dietary effect. The importance of the different phosphorylation sites on STAT3 may impact cellular localization and function. Phosphorylation of STAT3 at tyrosine 705 promotes translocation to the nucleus however phosphorylation of STAT3 at serine 727 is associated with mitochondrial localization [25]. Shulga et al. demonstrated that TNFα stimulation of mouse fibrosarcoma cells, in combination with a caspase inhibitor, increased phosphorylation of STAT3 at serine 727 and led to its accumulation in the mitochondria resulting in increased reactive oxygen species and cell death. Although not investigated in the current study the increased phosphorylation of STAT3 at serine 727 may contribute to the increased cTnI at baseline in obese mice as a result of mitochondrial damage and generation of reactive oxygen species but more work is needed in this area to investigate this process. Taken together, acute inflammation and mechanical stress through the STAT3 pathway are important for cardiac regeneration during sepsis and altered by obesity.
We found that IL-17a expression was the most differentially regulated gene in obese septic mice compared to non-obese septic mice. IL-17a promotes the production of pro-inflammatory cytokines and chemokines, induces neutrophil chemotaxis, and is increased in human inflammatory diseases [26–28]. IL-17a also drives cardiac damage and fibrosis and may provide a link between adipose tissue and inflammation [29–31]. It was not surprising that Csf2, also known as granulocyte-macrophage colony stimulating factor, was significantly increased in obese septic mice compared to non-obese septic mice as IL-17a induces Csf2 in cardiac fibroblasts to induce inflammation [30]. These findings are in line with other studies in murine sepsis. Increased IL-17 production correlated with STAT3 binding to the IL-17 promoter in CD4 T cells during sepsis [32]. STAT3 is also necessary for the production of IL-17a and mice that lack STAT3 in T cells are unable to generate Th17 cells [33, 34]. These findings suggest cardiac expression of IL-17a may affect STAT3 expression and provide further evidence that IL-17 may serve as a link between adipose tissue and inflammation but more work is needed to understand the importance of cardiac IL-17a in obese mice during sepsis.
SOCS3 acts as a negative inhibitor for STAT3 and inhibits phosphorylation and subsequent activation of STAT3. SOCS 3 is also important for left ventricular remodeling [35]. In our study, it is possible that the increase in SOCS3 expression in obese mice contributed to cardiac remodeling, although more studies are necessary to demonstrate a direct link. In non-obese mice we found a significant increase in SOCS3 expression after sepsis, which is an expected response to the increase in cytokine-induced STAT3 signaling. In a normal state this is an adaptive response as STAT3 has protective properties on the heart after an acute insult. In contrast, obese mice do not have the additional increase in SOCS3 expression after sepsis, which may be a result of elevated baseline levels of cardiac SOCS3. Since SOCS3 is a negative regulator of STAT3, it is conceivable that the chronic inhibition of the STAT3 pathway impairs the STAT3 protection of the myocardium during sepsis.
We found obese septic mice had decreased expression of gp130 compared to non-obese septic mice. Gp130 is associated with regulation of cardiac growth and development [36]. Binding of IL-6 and other cytokines to the receptor can lead to dimerization with gp130 and subsequent downstream activation of target proteins including STAT3. STAT3 is phosphorylated by JAK proteins bound to the cytoplasmic portion of gp130 [37]. Targeted deletion of the gp130 gene in mice is embryonically lethal and at necropsy mice have hypoplastic ventricular myocardium [38]. Mice with cardiac myocyte-restricted deletion of gp130 fail to develop compensatory hypertrophy and progress to cardiac dilation and failure [39]. It is possible that decreased gp130 expression in obese septic mice affected remodeling and contributed to the lower left ventricular mass found by echocardiography.
A limitation of our study is the focus on early time points after sepsis. Published data from our laboratory demonstrates a higher mortality in obese mice after CLP compared to non-obese [6]. Mice start dying around 6 hours after CLP and by 24 hours only ~20% of obese mice (compared to ~40% in non-obese mice) remain alive [6]. Therefore, for the current study, we focused on the mechanistic changes that occur in the heart early after CLP (at 6h). We investigated cardiac function by echocardiography at a later time point (18h after CLP) to determine the impact of the changes found early in sepsis. Examination of mechanistic changes that occur at later time points may show differences that are not demonstrated early in sepsis. More studies are necessary to examine the effects of obesity on the cardiac STAT3 pathway at later time points after sepsis.
In conclusion, high fat diet-induced obesity causes significant effects on the heart during sepsis. Cardiac serine phosphorylation of STAT3 may lead to necroptosis and cellular injury. In the non-obese septic state, in addition to the STAT3 serine effects, tyrosine STAT3 phosphorylation leads to increased SOCS3 expression which acts as a negative and compensatory regulator of this pathway controlling the inflammatory response and cardiac injury. These studies support the findings that obese mice have alteration in the STAT3 pathway and significant cardiac inflammation during sepsis and may partially explain the higher mortality in obese mice during sepsis. However, even with the new data presented here, there are still complexities that are not understood, leading to contradictory evidence on the role of obesity on heart function in sepsis.
Highlights.
High fat diet-induced obesity leads to elevated cardiac troponin I.
Obesity has deleterious effects on the heart following sepsis.
These effects may occur as a result of activation of the IL17a and STAT3 signaling pathways.
Acknowledgments
The current institution for Theodore DeMartini is Division of Critical Care, Department of Pediatrics, Pennsylvania State Milton S. Hershey Medical Center, Hershey, Pennsylvania.
Funding
This work was supported, in part, by the National Institutes of Health grants K08 GM093135 (JK), and P30DK078392.
Abbreviations
- CLP
cecal ligation and puncture
- cTnI
cardiac troponin
- STAT
signal transducer and activator of transcription
- SOCS3
suppressor of the cytokine signaling family
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
JK reports financial support to the institution for work on DSMB of a clinical trial (Eli Lilly) not related in any way to the work in the manuscript.
References
- 1.Martin GS, Mannino DM, Eaton S, Moss M. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med. 2003;348:1546–1554. doi: 10.1056/NEJMoa022139. [DOI] [PubMed] [Google Scholar]
- 2.Clermont G, Angus DC, Kalassian KG, Linde-Zwirble WT, Ramakrishnan N, Linden PK, Pinsky MR. Reassessing the value of short-term mortality in sepsis: comparing conventional approaches to modeling. Crit Care Med. 2003;31:2627–2633. doi: 10.1097/01.CCM.0000094233.35059.81. [DOI] [PubMed] [Google Scholar]
- 3.Vachharajani V. Influence of obesity on sepsis. Pathophysiology. 2008;15:123–134. doi: 10.1016/j.pathophys.2008.04.008. [DOI] [PubMed] [Google Scholar]
- 4.Prescott HC, Chang VW, O’Brien JM, Jr, Langa KM, Iwashyna T. Obesity and 1-Year Outcomes in Older Americans With Severe Sepsis. Crit Care Med. 2014 doi: 10.1097/CCM.0000000000000336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Winfield RD, Delano MJ, Cuenca AG, Cendan JC, Lottenberg L, Efron PA, Maier RV, Remick DG, Moldawer LL, Cuschieri J. Obese patients show a depressed cytokine profile following severe blunt injury. Shock. 2012;37:253–256. doi: 10.1097/SHK.0b013e3182449c0e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaplan JM, Nowell M, Lahni P, Shen H, Shanmukhappa SK, Zingarelli B. Obesity enhances sepsis-induced liver inflammation and injury in mice. Obesity (Silver Spring) 2016;24:1480–1488. doi: 10.1002/oby.21504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kaplan JM, Nowell M, Lahni P, O’Connor MP, Hake PW, Zingarelli B. Short-term high fat feeding increases organ injury and mortality after polymicrobial sepsis. Obesity (Silver Spring) 2012;20:1995–2002. doi: 10.1038/oby.2012.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peterson LR, Waggoner AD, Schechtman KB, Meyer T, Gropler RJ, Barzilai B, Davila-Roman VG. Alterations in left ventricular structure and function in young healthy obese women: assessment by echocardiography and tissue Doppler imaging. J Am Coll Cardiol. 2004;43:1399–1404. doi: 10.1016/j.jacc.2003.10.062. [DOI] [PubMed] [Google Scholar]
- 9.Mitra MS, Donthamsetty S, White B, Mehendale HM. High fat diet-fed obese rats are highly sensitive to doxorubicin-induced cardiotoxicity. Toxicology and applied pharmacology. 2008;231:413–422. doi: 10.1016/j.taap.2008.05.006. [DOI] [PubMed] [Google Scholar]
- 10.Yu L, Zhao Y, Xu S, Jin C, Wang M, Fu G. Leptin confers protection against TNF-alpha-induced apoptosis in rat cardiomyocytes. Biochem Biophys Res Commun. 2014;455:126–132. doi: 10.1016/j.bbrc.2014.10.134. [DOI] [PubMed] [Google Scholar]
- 11.Oreopoulos A, Padwal R, Kalantar-Zadeh K, Fonarow GC, Norris CM, McAlister FA. Body mass index and mortality in heart failure: a meta-analysis. Am Heart J. 2008;156:13–22. doi: 10.1016/j.ahj.2008.02.014. [DOI] [PubMed] [Google Scholar]
- 12.Horwich TB, Fonarow GC, Hamilton MA, MacLellan WR, Woo MA, Tillisch JH. The relationship between obesity and mortality in patients with heart failure. J Am Coll Cardiol. 2001;38:789–795. doi: 10.1016/s0735-1097(01)01448-6. [DOI] [PubMed] [Google Scholar]
- 13.Zhang W, Qu X, Chen B, Snyder M, Wang M, Li B, Tang Y, Chen H, Zhu W, Zhan L, Yin N, Li D, Xie L, Liu Y, Zhang JJ, Fu XY, Rubart M, Song LS, Huang XY, Shou W. Critical Roles of STAT3 in beta-Adrenergic Functions in the Heart. Circulation. 2016;133:48–61. doi: 10.1161/CIRCULATIONAHA.115.017472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xia J, Zhang Y, Xin L, Kong S, Chen Y, Yang S, Li K. Global Transcriptomic Profiling of Cardiac Hypertrophy and Fatty Heart Induced by Long-Term High-Energy Diet in Bama Miniature Pigs. PLoS One. 2015;10:e0132420. doi: 10.1371/journal.pone.0132420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Larsen L, Ropke C. Suppressors of cytokine signalling: SOCS, APMIS. 2002;110:833–844. doi: 10.1034/j.1600-0463.2002.1101201.x. [DOI] [PubMed] [Google Scholar]
- 16.C. National Research Council Committee for the Update of the Guide for the, A. Use of Laboratory, (2011).
- 17.Zingarelli B, Piraino G, Hake PW, O’Connor M, Denenberg A, Fan H, Cook JA. Peroxisome proliferator-activated receptor {delta} regulates inflammation via NF-{kappa}B signaling in polymicrobial sepsis. Am J Pathol. 2010;177:1834–1847. doi: 10.2353/ajpath.2010.091010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zingarelli B, Sheehan M, Hake PW, O’Connor M, Denenberg A, Cook JA. Peroxisome proliferator activator receptor-gamma ligands, 15-deoxy-Delta(12,14)-prostaglandin J2 and ciglitazone, reduce systemic inflammation in polymicrobial sepsis by modulation of signal transduction pathways. J Immunol. 2003;171:6827–6837. doi: 10.4049/jimmunol.171.12.6827. [DOI] [PubMed] [Google Scholar]
- 19.Acehan D, Vaz F, Houtkooper RH, James J, Moore V, Tokunaga C, Kulik W, Wansapura J, Toth MJ, Strauss A, Khuchua Z. Cardiac and skeletal muscle defects in a mouse model of human Barth syndrome. J Biol Chem. 2011;286:899–908. doi: 10.1074/jbc.M110.171439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaplan JM, Cook JA, Hake PW, O’Connor M, Burroughs TJ, Zingarelli B. 15-Deoxy-delta(12,14)-prostaglandin J(2) (15D-PGJ(2)), a peroxisome proliferator activated receptor gamma ligand, reduces tissue leukosequestration and mortality in endotoxic shock. Shock. 2005;24:59–65. doi: 10.1097/01.shk.0000167108.88376.f2. [DOI] [PubMed] [Google Scholar]
- 21.Guh DP, Zhang W, Bansback N, Amarsi Z, Birmingham CL, Anis AH. The incidence of co-morbidities related to obesity and overweight: a systematic review and meta-analysis, BMC public health. 2009;9:88. doi: 10.1186/1471-2458-9-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Han C, Nie Y, Lian H, Liu R, He F, Huang H, Hu S. Acute inflammation stimulates a regenerative response in the neonatal mouse heart. Cell Res. 2015;25:1137–1151. doi: 10.1038/cr.2015.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Darnell JE., Jr STATs and gene regulation. Science. 1997;277:1630–1635. doi: 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- 24.Sakamori R, Takehara T, Ohnishi C, Tatsumi T, Ohkawa K, Takeda K, Akira S, Hayashi N. Signal transducer and activator of transcription 3 signaling within hepatocytes attenuates systemic inflammatory response and lethality in septic mice. Hepatology. 2007;46:1564–1573. doi: 10.1002/hep.21837. [DOI] [PubMed] [Google Scholar]
- 25.Shulga N, Pastorino JG. GRIM-19-mediated translocation of STAT3 to mitochondria is necessary for TNF-induced necroptosis. J Cell Sci. 2012;125:2995–3003. doi: 10.1242/jcs.103093. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 26.Matusevicius D, Kivisakk P, He B, Kostulas N, Ozenci V, Fredrikson S, Link H. Interleukin-17 mRNA expression in blood and CSF mononuclear cells is augmented in multiple sclerosis. Mult Scler. 1999;5:101–104. doi: 10.1177/135245859900500206. [DOI] [PubMed] [Google Scholar]
- 27.Amadi-Obi A, Yu CR, Liu X, Mahdi RM, Clarke GL, Nussenblatt RB, Gery I, Lee YS, Egwuagu CE. TH17 cells contribute to uveitis and scleritis and are expanded by IL-2 and inhibited by IL-27/STAT1. Nat Med. 2007;13:711–718. doi: 10.1038/nm1585. [DOI] [PubMed] [Google Scholar]
- 28.Onishi RM, Gaffen SL. Interleukin-17 and its target genes: mechanisms of interleukin-17 function in disease. Immunology. 2010;129:311–321. doi: 10.1111/j.1365-2567.2009.03240.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barry SP, Ounzain S, McCormick J, Scarabelli TM, Chen-Scarabelli C, Saravolatz LI, Faggian G, Mazzucco A, Suzuki H, Thiemermann C, Knight RA, Latchman DS, Stephanou A. Enhanced IL-17 signalling following myocardial ischaemia/reperfusion injury. Int J Cardiol. 2013;163:326–334. [Google Scholar]
- 30.Wu L, Ong S, Talor MV, Barin JG, Baldeviano GC, Kass DA, Bedja D, Zhang H, Sheikh A, Margolick JB, Iwakura Y, Rose NR, Cihakova D. Cardiac fibroblasts mediate IL-17A-driven inflammatory dilated cardiomyopathy. J Exp Med. 2014;211:1449–1464. doi: 10.1084/jem.20132126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ahmed M, Gaffen SL. IL-17 in obesity and adipogenesis. Cytokine Growth Factor Rev. 2010;21:449–453. doi: 10.1016/j.cytogfr.2010.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mukherjee S, Allen RM, Lukacs NW, Kunkel SL, Carson WFt. STAT3-mediated IL-17 production by postseptic T cells exacerbates viral immunopathology of the lung. Shock. 2012;38:515–523. doi: 10.1097/SHK.0b013e31826f862c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang XO, Panopoulos AD, Nurieva R, Chang SH, Wang D, Watowich SS, Dong C. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J Biol Chem. 2007;282:9358–9363. doi: 10.1074/jbc.C600321200. [DOI] [PubMed] [Google Scholar]
- 34.Yang XP, Ghoreschi K, Steward-Tharp SM, Rodriguez-Canales J, Zhu J, Grainger JR, Hirahara K, Sun HW, Wei L, Vahedi G, Kanno Y, O’Shea JJ, Laurence A. Opposing regulation of the locus encoding IL-17 through direct, reciprocal actions of STAT3 and STAT5. Nat Immunol. 2011;12:247–254. doi: 10.1038/ni.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oba T, Yasukawa H, Hoshijima M, Sasaki K, Futamata N, Fukui D, Mawatari K, Nagata T, Kyogoku S, Ohshima H, Minami T, Nakamura K, Kang D, Yajima T, Knowlton KU, Imaizumi T. Cardiac-specific deletion of SOCS-3 prevents development of left ventricular remodeling after acute myocardial infarction. J Am Coll Cardiol. 2012;59:838–852. doi: 10.1016/j.jacc.2011.10.887. [DOI] [PubMed] [Google Scholar]
- 36.Ji JD, Kim HJ, Rho YH, Choi SJ, Lee YH, Cheon HJ, Sohn J, Song GG. Inhibition of IL-10-induced STAT3 activation by 15-deoxy-Delta12,14-prostaglandin J2. Rheumatology (Oxford) 2005;44:983–988. doi: 10.1093/rheumatology/keh657. [DOI] [PubMed] [Google Scholar]
- 37.Jones SA, Scheller J, Rose-John S. Therapeutic strategies for the clinical blockade of IL-6/gp130 signaling. J Clin Invest. 2011;121:3375–3383. doi: 10.1172/JCI57158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yoshida K, Taga T, Saito M, Suematsu S, Kumanogoh A, Tanaka T, Fujiwara H, Hirata M, Yamagami T, Nakahata T, Hirabayashi T, Yoneda Y, Tanaka K, Wang WZ, Mori C, Shiota K, Yoshida N, Kishimoto T. Targeted disruption of gp130, a common signal transducer for the interleukin 6 family of cytokines, leads to myocardial and hematological disorders. Proc Natl Acad Sci U S A. 1996;93:407–411. doi: 10.1073/pnas.93.1.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hirota H, Chen J, Betz UA, Rajewsky K, Gu Y, Ross J, Jr, Muller W, Chien KR. Loss of a gp130 cardiac muscle cell survival pathway is a critical event in the onset of heart failure during biomechanical stress. Cell. 1999;97:189–198. doi: 10.1016/s0092-8674(00)80729-1. [DOI] [PubMed] [Google Scholar]