

Abstract
Schizophrenia (SCZ) is characterized not only by psychosis, but also by working memory and executive functioning deficiencies, processes that rely on the prefrontal cortex (PFC). Because these cognitive impairments emerge prior to psychosis onset, we investigated synaptic function during development in the neurodevelopmental methylazoxymethanol acetate (MAM) model for SCZ. Specifically, we hypothesize that NMDAR hypofunction is attributable to reductions in the NR2B subunit through aberrant epigenetic regulation of gene expression, resulting in deficient synaptic physiology and PFC-dependent cognitive dysfunction, a hallmark of SCZ. Using Western blot and whole-cell patch-clamp electrophysiology, we found that the levels of synaptic NR2B protein are significantly decreased in juvenile MAM animals, and the function of NMDARs is substantially compromised. Both NMDA-mEPSCs and synaptic NMDA-eEPSCs are significantly reduced in prelimbic PFC (plPFC). This protein loss during the juvenile period is correlated with an aberrant increase in enrichment of the epigenetic transcriptional repressor REST and the repressive histone marker H3K27me3 at the Grin2b promoter, as assayed by ChIP-qPCR. Glutamate hypofunction has been a prominent hypothesis in the understanding of SCZ pathology; however, little attention has been given to the NMDAR system in the developing PFC in models for SCZ. Our work is the first to confirm that NMDAR hypofunction is a feature of early postnatal development, with epigenetic hyper-repression of the Grin2b promoter being a contributing factor. The selective loss of NR2B protein and subsequent synaptic dysfunction weakens plPFC function during development and may underlie early cognitive impairments in SCZ models and patients.
Keywords: NMDA receptors, repressor protein REST/NRSF, histone modification H3K27me3, ChIP, animal model, schizophrenia, juvenile
Graphical abstract
In This Issue
Glutamate hypofunction has been a prominent hypothesis in schizophrenia pathology; however, when and how NMDAR dysfunction occurs is unknown. We report that NMDAR hypofunction is a feature of early postnatal development, with epigenetic hyper-repression of the Grin2b promoter, and subsequent loss of NR2B protein and synaptic NMDAR hypofunction, thus weakening plPFC function. This may underlie early cognitive impairments in schizophrenia.
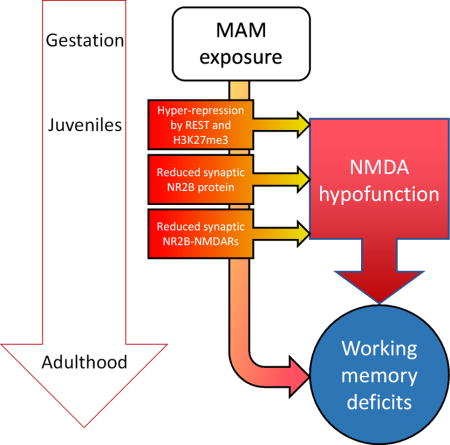
Introduction
Glutamatergic hypofunction in the prefrontal cortex (PFC) is speculated to underlie cognitive symptoms (Lipska and Weinberger, 2000; Gainetdinov et al., 2001; Kantrowitz and Javitt, 2012) and contribute to the pathophysiology of schizophrenia (SCZ) (Coyle et al., 2002; Lau and Zukin, 2007; Snyder and Gao, 2013). Particularly, the N-methyl-D-aspartate receptor (NMDAR), a major glutamate receptor subtype, demonstrates alterations in post-mortem SCZ brain that support this long-standing hypothesis (Meador-Woodruff and Healy, 2000; Martucci et al., 2006; Beneyto and Meador-Woodruff, 2008; Kristiansen et al., 2010; Weickert et al., 2013). NMDARs are heterotetrameric complexes composed of the requisite NR1 subunit, as well as two subunits of either NR2 (A-D) or NR3 (A-B) (Paoletti et al., 2013). The subunit composition of NMDARs dictates their physiological properties, pharmacological sensitivity, and functional complexity (Paoletti et al., 2013). NR2A and NR2B subunits are predominantly expressed in the adult brain and are essential for synaptic plasticity (Monyer et al., 1994). In the PFC, NR2B-containing NMDARs (NR2B-NMDARs) on pyramidal neurons are necessary for the sustained firing activity required to maintain working memory function in response to incoming stimuli (Goldman-Rakic, 1995; Wang et al., 2008). Moreover, NR2B subunits are persistently expressed throughout PFC maturation (Wang et al., 2008), whereas other brain regions undergo an NR2B-to-NR2A subunit switch. This switch is an essential element of experience-dependent maturation of many brain areas (Dumas, 2005); however, PFC maturation is characterized by different developmental milestones (Monaco et al., 2015).
Deficits in NMDAR function, particularly NR2B-NMDARs, impairs working memory (Wang et al., 2013), which is a constituent process of many higher-order cognitive capacities including cognitive flexibility and executive functioning (Goldman-Rakic, 1995). These cognitive deficits, among others, are commonly reported in the SCZ population (Reichenberg, 2010) and are the best predictor of functional outcome (Bowie and Harvey, 2006). Notably, behavioral manifestations of impaired cognition are evident prior to psychosis in patients (Jones et al., 1994; Cannon et al., 2000; Rosso et al., 2000). Thus, a greater understanding of the developmental vulnerabilities that produce this early cognitive dysfunction is critical.
We use the methylazoxymethanol acetate (MAM) neurodevelopmental model for SCZ to investigate PFC postnatal development, with a focus on NMDAR expression (Gilmour et al., 2012). Lending both face and predictive validities to this model for SCZ, MAM animals demonstrate a greater degree of MK801-induced hyper-locomotion that is sensitive to treatment with antipsychotics (Moore et al., 2006; Lodge and Grace, 2009). Additionally, deficiences in PFC-dependent cognition, including working memory and cognitive flexibility, are evident in this model (Gourevitch et al., 2004; Flagstad et al., 2005; Lavin et al., 2005; Moore et al., 2006). Furthermore, MAM animals have altered hippocampal NMDAR expression, and impaired learning and memory function in juvenile and adolescent stages of development (Hradetzky et al., 2012; Snyder et al., 2013), demonstrating the relevance of this model in assessing NMDAR hypofunction in early postnatal development.
Epigenetic mechanisms regulate glutamatergic gene expression in normal brain development (Stadler et al., 2005), with alterations in histone and DNA modifications implicated in neuropsychiatric disorders, including SCZ (Akbarian, 2010a; b; 2014; Nestler et al., 2016). A possible mechanism underlying NMDAR hypofunction is repression of glutamatergic genes within the PFC, as reported in post-mortem SCZ brain tissue (Numata et al., 2014). In fact, in the MAM model, aberrant histone modification patterns are evident in the developing medial PFC (mPFC) (Mackowiak et al., 2014). However, two major questions remain: (1) is NMDAR hypofunction evident in early postnatal development of the PFC in this SCZ model, and (2) is there an epigenetic mechanism that may contribute to this endophenotype? In the following experiments, we seek to address these questions by characterizing the expression profile, synaptic physiology, and regulation by epigenetic mechanisms of NMDARs in the PFC of juvenile MAM and saline animals.
Materials and Methods
Animals
All animal procedures were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Drexel University College of Medicine Institutional Animal Care and Use Committee. Adult pregnant Sprague-Dawley rats were purchased from Charles River Laboratories International, Inc. (RRID:RGD_734476; Wilmington, MA) and, upon arrival, animals were kept on a normal light/dark cycle (lights on 7:00, lights off 19:00) in 21°C±1°C with humidity at 30–70%, and ad libitum access to food (PicoLab® Rodent Diet 20, Labdiet) and water. Dams were singly housed upon arrival and after giving birth. Pregnant Sprague-Dawley females were arbitrarily selected for intraperitoneal (I.P.) injection with either 25 mg/kg methylazoxymethanol acetate (MAM) or vehicle (saline) on embryonic day 17 (E17), as previously reported (Hradetzky et al., 2012; Snyder et al., 2013; Xing et al., 2016; Li et al., 2017). Pups were usually born on E21/P0, and were weaned and rehoused on postnatal day 21 (P21). Animals were separated according to gender such that at least two but no more than four animals were in each cage. Male animals aged P17-23 (classified as juveniles (Spear, 2000; Wang and Gao, 2009; 2010)) in each litter were arbitrarily used and multiple litters (3–5) were included in all experiments.
Tissue collection and western blot for synaptosomal proteins
Animals were anesthetized with Euthasol (0.2 ml kg−1, I.P.) until unresponsive to toe pinch, and then the brain was extracted. Brain tissue containing the medial PFC (mPFC) was dissected, homogenized in sucrose buffer (in mM: 320 sucrose, 4 HEPES-NaOH buffer, pH 7.4, 2 EGTA, 1 sodium orthovanadate, 0.1 phenylmethylsulfonyl fluoride, 50 sodium fluoride, 10 sodium pyrophosphate, 20 glycerophosphate, with 1 μg/ml leupeptin and 1 μg/ml aprotinin), and then centrifuged at 1000 g for 10 minutes to remove large cell fragments and nuclear materials. The resulting supernatant was centrifuged at 17,000 g for 15 minutes to yield cytoplasmic proteins in the supernatant. The pellet from this spin was re-suspended in homogenization buffer and centrifuged at 17,000 g for an additional 15 minutes to yield washed synaptosomes. The synaptosomal fraction then was hypo-osmotically lysed and centrifuged at 25,000 g for 20 minutes to yield synaptosomal plasma membranes in the pellet.
A bicinchoninic acid (BCA) protein assay was performed to determine protein concentration. The protein sample was mixed with laemmli sample buffer, boiled for 5 minutes, and separated on a 7.5% SDS-PAGE gel. After electrophoresis, proteins were transferred to polyvinylidene difluoride membranes (EMD-Millipore, Billerica, MA). The membrane was blocked in 5% nonfat milk and probed with primary anti-serum. Each blot was probed for anti-mouse NR1 (RRID:AB_2533060; Invitrogen, 32-0500, 1:5000, Carlsbad, CA), anti-rabbit NR2A (RRID:AB_1163481; EMD-Millipore, 04-901, 1:4000), anti-mouse NR2B (RRID:AB_417391; EMD-Millipore, 05-920, 1:2000), anti-rabbit NR3A (RRID:AB_2112620; EMD-Millipore, 07-356, 1:4000), anti-rabbit NR3B (RRID:AB_10240972; Tocris, 2060, 1:4000), and anti-mouse actin (RRID:AB_476743; Sigma, A5316, 1:100,000, St Louis, MO), which was used as a loading control. The blots were incubated with horseradish peroxidase-coupled anti-rabbit or anti-mouse IgG secondary antibody (Vector Laboratories, Burlingame, CA), and proteins were visualized using enhanced chemiluminescence (ECL Plus, Amersham Biosciences, Piscataway, NJ). Protein expression of each subunit was evaluated by densitometry using Image-J software. Additionally, samples from each animal were run at least 4 times to minimize interblot variance. Raw values for NMDA subunit proteins were normalized to actin and this ratio was further normalized to the first usable band on each membrane. The normalized values for each protein were averaged per animal to yield the mean and standard error per group.
Whole-cell patch-clamp electrophysiology
Male juvenile (aged P17-23) saline and MAM animals were anesthetized with Euthasol (0.2 ml kg−1, I.P.), the brains were removed, and coronal slices containing the prelimbic PFC (plPFC) were cut (300 μm) into an ice-cold bath of oxygenated artificial cerebrospinal fluid (aCSF, in mM: 124 NaCl, 2.5 KCl, 1.25 NaH2PO4, 2 CaCl2, 1 MgSO4, 26 NaHCO3, and 10 dextrose, pH 7.4) using a VT-1200S vibratome tissue slicer (Leica Microsystems, Wetzlar, Germany). Slices were transferred to a holding chamber, submerged in oxygenated aCSF at 35°C for one hour and then remained at room temperature until used for recording. Slices were placed into a recording chamber mounted on an Olympus upright microscope (BX51, Olympus America, Center Valley, PA), where they were continuously bathed in oxygenated aCSF. Neurons were visualized with infrared differential interference video microscopy. All experiments were conducted with an Axon MultiClamp 700B amplifier (Molecular Devices), and data were acquired using pCLAMP 9.2 software and analyzed using Clampfit 10.2 software (Molecular Devices, Novato, CA).
Spontaneous and miniature excitatory postsynaptic currents
To record AMPA- and NMDA-mediated spontaneous or miniature excitatory postsynaptic currents (EPSCs), somatic whole-cell voltage-clamp recordings were obtained from layer V pyramidal cells in plPFC using patch electrodes with an open tip resistance of 5–7 MΩ (internal solution, in mM: 110 D-gluconic acid, 110 CsOH, 10 CsCl2, 1 EGTA, 1 CaCl2, 5 QX-314, 1 ATP-Mg, 10 HEPES, at pH 7.3, adjusted with CsOH). In voltage-clamp mode, the membrane potentials were held at -70 mV in the presence of picrotoxin (50 μM) to record AMPA-mediated spontaneous EPSCs (AMPA-sEPSCs), or with both picrotoxin and tetrodotoxin (TTX, 0.5 μM) for miniature EPSCs (AMPA-mEPSCs). mEPSCs were recorded for at least 5 minutes and then DNQX (20 μM) was washed on to block AMPARs. When all currents ceased, the membrane potential was slowly ramped up to 60 mV. NMDA-mediated mEPSCs were then recorded for at least 5 minutes. A typical s/mEPSC event was selected to create a sample template for event detections within a 5-min period for each data file. The frequency (number of events) and amplitude of the individual events were examined with the threshold set at the medium level (i.e., 5 within a range of 1 to 9) in Clampfit. The detected events were then visually inspected to ensure specificity.
Evoked excitatory postsynaptic currents
To record evoked AMPA-mediated EPSCs (AMPA-eEPSCs) and NMDA-mediated EPSCs (NMDA-eEPSCs), a bipolar stimulating electrode was placed in layer II/III of the plPFC approximately 200–300 μm medial to visually-identified layer V pyramidal cells. Picrotoxin (50 μM) was used to block GABAAR-mediated neurotransmission. Cells were held at 60 mV to record NMDA-eEPSCs for 5 min before and 5 min after bath application of a selective NR2B antagonist Ro25-6981 (Ro25, Sigma-Aldrich, R7150, 1 μM). EPSCs were evoked through the stimulating electrode using a physiologically relevant 10-pulse, 20 Hz train with a 0.1 Hz stimulating interval. Stimulus intensity was adjusted until 50–100 pA NMDA-eEPSC amplitude responses were achieved. Cells with an unstable baseline or unusual response patterns throughout recording were removed from the analysis. The EPSC recordings were monitored throughout each experiment by implementing a -100 pA current pulse with a 200 ms duration. Traces were analyzed for amplitude relative to the first post-stimulus baseline and cumulative charge (charge transfer, nA*ms), as well as paired-pulse ratio (PPR).
Western blot for histone proteins
mPFC tissue was collected as described above. The Epiquik Total Histone Extraction Kit (OP-0006, EpiGentek, Farmingdale, NY) was used to isolate the histone fraction. A BCA assay was used to determine protein concentration. After boiling in laemmli buffer for 5 minutes, 15 μg of protein were loaded on a 15% SDS-PAGE gel. After electrophoresis, blots were transferred to pore size 0.20 μm polyvinylidene difluoride membranes (Millipore, Billerica, MA). Membranes were blocked with 5% nonfat milk and separately probed with the following primary antibodies: H3K9ac (RRID:AB_297491; Abcam, ab10812, 1:5000, Cambridge, UK), H3K18ac (RRID:AB_298692; Abcam, ab1191, 1:5000), H3K27ac (RRID:AB_2118291; Abcam, ab4729, 1:5000), H3K27me3 (RRID:AB_305237; Abcam, ab6002, 1:2000), and total histone H3 (RRID:AB_10001790; Novus, NB500-171, 1:100,000) was used as a loading control. Blots were incubated with horseradish peroxidase-coupled anti-rabbit or anti-mouse IgG secondary antibody (Vector Laboratories, Burlingame, CA), and proteins were visualized using enhanced chemiluminescence (ECL Plus, Amersham Biosciences, Piscataway, NJ). Protein expression for each histone modification was evaluated by densitometry using Image-J software. Samples from each animal were run at least 4 times to minimize interblot variance.
Chromatin immunoprecipitation-qPCR assay
mPFC tissue was collected from saline and MAM animals at P21 and pooled for the chromatin immunoprecipitation (ChIP) assays performed using the truChIP™ Chromatin Shearing Kit (Covaris, PN 520083). Three separate reactions with 50 mg of sheared chromatin were carried out using: (i) 2 μg of REST antibody (RRID:AB_310728; Millipore; 07-579), (ii) 1 μg H3K27me3 antibody (Abcam; ab6002), and (iii) a mock reaction containing all reagents with IgG as a control. The chromatin was immunoprecipitated using the EZ-Magna ChIP™ A-Chromatin Kit (Millipore; 17-408). The eluted material was purified using a QIAquick PCR kit (Qiagen; 28104) and was directly used for qPCR. Positive and negative control primer sets were purchased from Active Motif (71027, 71024). PCRs were performed in a 20-μL reaction containing purified chromatin DNA, 1 mΜ Power SYBR Green PCR Master Mix (Applied Biosystems; 4367659), and 10 mM of both forward and reverse primers, reported by Rodenas-Ruano et al. (2012) and shown in Table 1. PCR was performed in triplicate using an ABI 7500 system (Applied Biosystems), yielding a final sample size of 6 data points. Briefly, a standard curve was generated from serial 10-fold dilutions of input DNA. The Ct values were used to estimate DNA quantity of the ChIP and mock (no antibody) control samples. Fold enrichment levels were calculated as a ratio of the DNA quantity in the ChIP samples to control samples.
Table 1.
Forward and reverse primers sequences for Grin2b (proximal promoter region), Grin2a, and Grin1 (from Rodenas-Ruano et al. (2012)).
| Forward primers | Reverse primers | |
|---|---|---|
| Grin2b | GGTCAAGCTGCCTCTCCAT | GCAGAGCAGAAGGAAATGTATTCG |
| Grin2a | TCCGGAGTGGAACAGAAAGC | CTCATCCAGCCCCATGCT |
| Grin1 | TCCCTGCTTCCTCTCTTGGA | AATGACTGCTGGGAGCAAGAC |
Data analysis
Power analysis (G*Power 3.1.9.2) was used to determine sample sizes for experiments from preliminary data (α=0.05). Ten cells per group were found to be sufficient for electrophysiological experiments. For biochemical analyses, 4–6 animals were sufficient. In some experiments, the experimenter was blind to group (saline or MAM) until after data analysis was complete. Statistical analyses were carried out using SPSS Statistics version 24.0 (IBM, Armonk, NY). Normality and homogeneity of variance testing determined the use of parametric or non-parametric tests for each dataset. Outliers were identified by z-score testing, and data points that may have been compromised by experimental error were excluded. Protein expression analyses, AMPA- and NMDA-s/mEPSC amplitude and frequency, and ChIP-qPCR enrichment levels were compared using unpaired Student’s t-test or Mann-Whitney U tests. Enrichment of REST and H3K27me3 at Grin1, Grin2a, and Grin2b between groups and across promoter regions were analyzed using a One-Way ANOVA with a Tukey’s post-hoc. Evoked data were analyzed using repeated measures ANOVA to compare amplitude or charge transfer between groups and across pulses. Simple main effects were determined if a significant interaction effect was found. Otherwise, Student’s t-test or Mann-Whitney U tests were used for post-hoc analysis. All data are presented as mean±standard error (S.E.). Single, double, and triple asterisks represent p<0.05, p<0.01, and p<0.001, respectively.
Results
NMDAR protein levels are reduced in the mPFC of juvenile MAM rats
To determine if NMDARs are altered during prefrontal development in this SCZ model, we first examined how prenatal MAM exposure affects NMDAR protein levels in juvenile (P21) animals (Figure 1). We collected synaptic plasma membrane fractions from saline and MAM animals and probed for NMDAR subunit levels via Western blot. Actin was used as a loading control. We found no significant differences between saline and MAM animals in NR1, NR2A, NR2B, NR3A, or NR3B subunit levels at P14 (data not shown; Saline n=3–5 rats, MAM n=3–5 rats; p=0.651, p=0.360, p=0.892, p=0.172, and p=0.230, respectively). However, in the mPFC of late juvenile (P21) MAM animals, there was a significant decrease in NR2B protein compared to saline animals (Figure 1; saline 0.76±0.07, n=4; MAM 0.53±0.06, n=5; p=0.045). No difference was found for NR1, NR2A, NR3A, or NR3B levels at P21 (Saline n=4, MAM n=5; p=0.842, 0.170, 0.994, and 0.681, respectively). Together, these data indicate that prenatal MAM exposure alters NMDAR protein levels during the critical juvenile period of the developing mPFC. Moving forward, experiments in juvenile animals encompassed the late juvenile (P21) time point.
Figure 1.
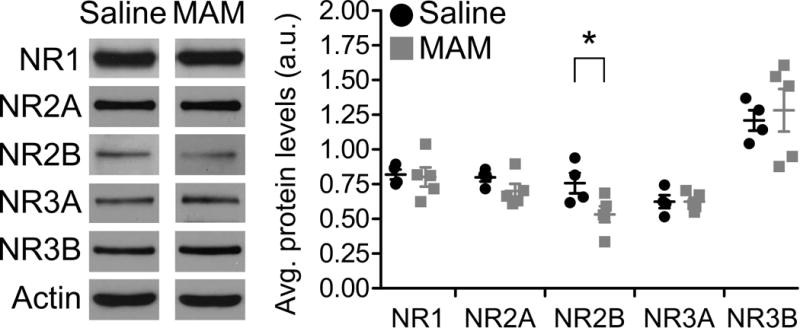
NR2B protein is reduced in synaptosomes from mPFC of juvenile MAM animals. Representative Western blots (left) and summary scatterplot (right) demonstrating significant loss of synaptic NR2B protein in mPFC from MAM-exposed animals compared to saline-exposed controls. * p<0.05
NMDA-mEPSCs, but not AMPA-s/mEPSCs, are diminished in juvenile MAM mPFC
We next sought to determine if the reduced NMDAR subunit protein levels in juvenile MAM animals resulted in altered NMDAR functionality. We utilized whole-cell patch-clamp electrophysiology to record from layer V pyramidal neurons in the plPFC and examined both NMDAR- and AMPAR-mediated EPSCs. This approach allowed us to understand the functional state of NMDARs and AMPARs in response to inputs from multiple brain regions. We first recorded sEPSCs that presumably resulted from action potentials of presynaptic neurons, as well as the spontaneous release of neurotransmitter at synapses, and then washed on tetrodotoxin to block action potentials in the slice and recorded mEPSCs. We held the cell at −70 mV with GABAAR function blocked to assess if MAM exposure had an effect on functional AMPARs and if there were changes in presynaptic function. We found no differences in either the frequency or amplitude of the AMPA-sEPSCs or –mEPSCs in juvenile (P17-23) MAM and saline rats (Figure 2A–B; sEPSCs: frequency, Hz, saline 2.12±0.42, n=14 cells; MAM 1.99±0.31, n=23 cells; p=0.731. Amplitude, pA, saline 17.80±1.29; MAM 16.26±0.54, p=0.506. mEPSCS: frequency, Hz, saline 1.34±0.32, n=13; MAM 2.28±0.66, n=15; p=0.387. Amplitude, pA, saline 14.27±0.59; MAM 14.33±1.02, p=0.961).
Figure 2.
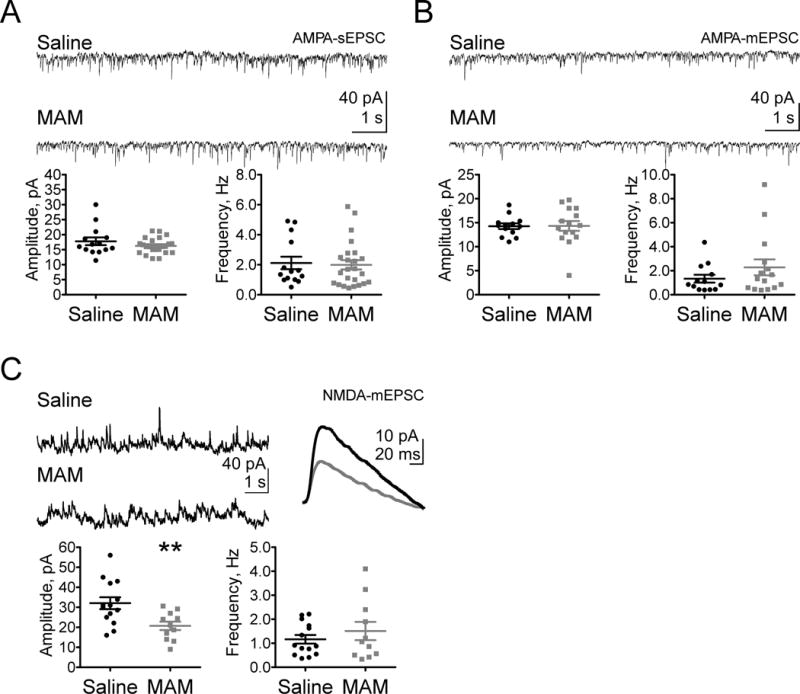
NMDAR-mediated neurotransmission in plPFC is selectively impaired in juveniles following prenatal MAM exposure. Representative traces (top) and summary scatterplots (bottom) illustrate (A) AMPA-sEPSCs and (B) –mEPSCs are not significantly affected by prenatal MAM exposure. (C) Juvenile NMDA-mEPSCs have significantly reduced amplitude (top representative traces), but no change in frequency of currents. ** p<0.01
We next held the cell at 60 mV while blocking GABAARs and AMPARs to examine changes in NMDAR function. In juveniles, we saw a significant decrease of NMDAR-mEPSC amplitude, but not frequency, in MAM plPFC neurons compared with those in saline animals (Figure 2C; mEPSCs: frequency, Hz, saline 1.16±0.18, n=14; MAM 1.51±0.38, n=11; p=0.424. Amplitude, pA, saline 32.06±2.98, MAM 20.73±2.12; p=0.007). Thus, prenatal MAM exposure impairs functional NMDARs, but not AMPARs, in the developing plPFC; however, it is unclear whether pre- or post-synaptic dysfunction contributes to deficient NMDAR neurotransmission.
Juvenile MAM animals have a significant reduction in post-synaptic NR2B-NMDARs in plPFC layer V pyramidal neurons
To determine whether synaptic function of plPFC pyramidal cells is affected by prenatal MAM exposure, we recorded NMDAR-mediated EPSCs evoked by stimulating layer II/III afferents with a 10-pulse, 20 Hz train. To evaluate the functional state of NMDARs during development, we compared the amplitude and charge transfer of the evoked EPSCs between saline and MAM animals. GABAAR-mediated neurotransmission was blocked with picrotoxin, and stimulation was adjusted until EPSC amplitudes of 50–100 pA were achieved. We found that amplitudes of NMDAR-eEPSCs are reduced in juvenile MAM animals (Figure 3A; amplitude, pA, Saline n=10, MAM n=10, Repeated Measures ANOVA: group: F(1,18)=9.64, p=0.006, pulse: F(1.69, 30.43)=5.39, p=0.013; interaction: F(1.69, 30.43)=1.17, p=0.316). Simple main effects analysis reveals amplitude is significantly diminished in MAM animals at all 10 pulses (p<0.05). Similarly, charge transfer of NMDA-eEPSCs is significantly reduced in MAM animals (Figure 3A, charge transfer, nA*ms, Repeated Measures ANOVA: group: F(1,18)=7.36, p=0.014, pulse: F(1.06, 19.08)=55.51, p<0.001; interaction: F(1.06, 19.08)=6.25, p=0.020). Simple main effects analysis shows charge transfer is significantly decreased in juvenile MAM plPFC at all pulses (p<0.05). We did not find any changes in paired-pulse ratio (PPR), a measure of presynaptic release probability, which indicates these reductions in synaptic transmission are restricted to alterations in post-synaptic receptor function (Figure 3A; t(18)=1.08, p>0.05). We also did not find any significant differences in stimulation intensity between the saline and MAM groups (Figure 3A; U=42; p>0.05). Together, these data demonstrate a loss of post-synaptic NMDARs in layer V pyramidal neurons in juvenile MAM plPFC.
Figure 3.
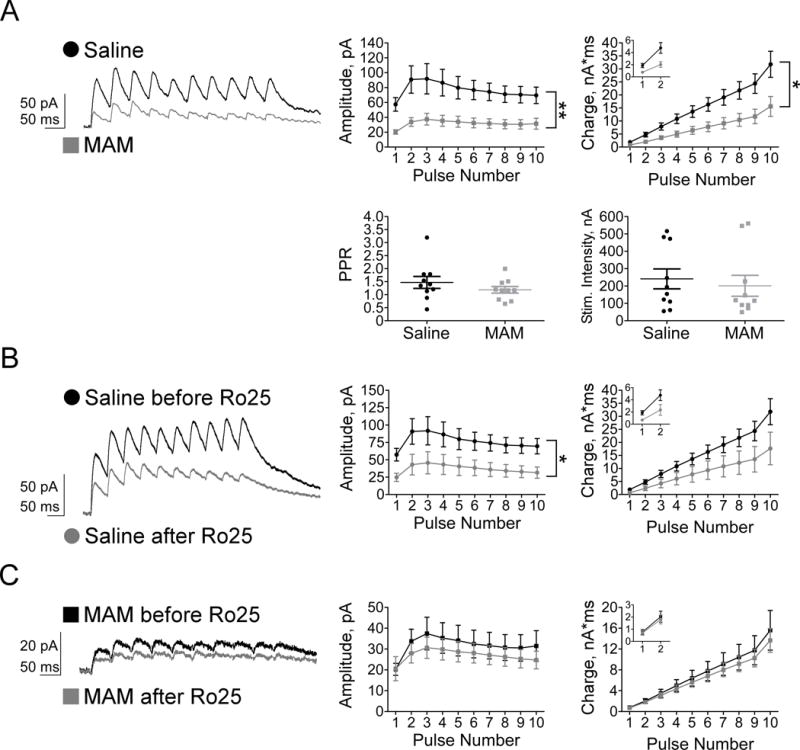
Synaptic NMDARs are diminished in juvenile MAM plPFC, specifically due to a loss of NR2B subunits. Representative traces (right) and summary graphs illustrate (A) a significant group effect of prenatal MAM exposure on both amplitude and charge transfer of NMDAR-eEPSCs, but no significant differences between groups in PPR or stimulation intensity. (B) Ro25, a selective NR2B antagonist, moderately decreases the amplitude of saline NMDAR-eEPSCs, with strongly trending effects on charge transfer. (C) The loss of synaptic NR2B-NMDARs in MAM plPFC occludes the effect of Ro25 on amplitude and charge transfer of NMDAR-eEPSCs. * p<0.05, ** p<0.01
The NR2B protein loss seen in Western blot experiments and the reduction of synaptic NMDAR-eEPSC responses may be reflective of a reduction in postsynaptic NR2B-NMDARs specifically. To test this hypothesis, we bath-applied Ro25-6981 (Ro25, 1 μM), a selective NR2B antagonist, while holding the cell at 60 mV. Reductions in evoked NMDA amplitude and charge transfer following Ro25 treatment indicate a blockade of solely NR2B-NMDARs. Following bath application of Ro25 in saline cells, we found a moderate but significant reduction of NMDAR-eEPSC amplitude (Figure 3B; amplitude, pA; Saline before Ro n=10, Saline after Ro n=8, group: F(1, 16)=5.04; p=0.039; pulse: F(1.36, 21.69)=5.32, p=0.022; interaction: F(1.36, 21.69)=0.38, p=0.608; post-hoc: pulses 3–5, p<0.05, pulses 1–2, 6–10, p>0.05). Surprisingly, charge transfer is only marginally diminished following Ro25 wash-on in saline cells (Figure 3B; charge transfer, nA*ms; group: F(1, 16)=3.06, p=0.10; pulse: F(1.05, 16.73)=38.02, p<0.001; interaction: F(1.05, 16.73)=2.88, p=0.107; post-hoc: pulses 1&4, p<0.05, pulses 2–3, 5–10, p>0.05). However, bath application of Ro25 does not reduce amplitude or charge transfer in juvenile MAM animals, likely due to the significant loss of NMDARs (Figure 3C; amplitude, pA; MAM before Ro n=10, MAM after Ro n=9, group: F(1, 17)=0.43; p=0.519; pulse: F(1.83, 31.18)=3.42, p=0.049, interaction: F(1.83, 31.18)=0.25, p=0.763; charge transfer, nA*ms; group: F(1, 17)=0.19, p=0.671; pulse: F(1.05, 17.79)=38.85, p<0.001; interaction: F(1.05, 17.79)=13.90, p=0.691). Post-hoc analyses reveal no significant differences in amplitude or charge transfer between saline or MAM animals after Ro25 treatment. We can conclude synaptic NR2B-NMDARs are reduced in juvenile MAM animals as confirmed by the occlusion of Ro25’s effect on layer V pyramidal cells in juvenile MAM plPFC.
To further confirm AMPAR function is unchanged following prenatal MAM exposure, we evaluated the effect of layer II/III stimulation on AMPAR-eEPSCs in saline and MAM plPFC. To record AMPAR-eEPSCs, the cells were held at −70 mV in the presence of picrotoxin (50 μM) to block GABAAR-mediated neurotransmission. EPSCs were evoked as described above; stimulation intensity was increased until 50–100 pA responses were elicited. We found no significant differences in amplitude or charge between juvenile saline and MAM animals (data not shown; amplitude, pA, Saline n=11, MAM n=9, Repeated Measures ANOVA: group: F(1,18)=3.09, p=0.096, pulse: F(2.49, 44.84)=17.16, p<0.001; interaction: F(2.49, 44.84)=0.88, p=0.441. Charge transfer, nA*ms, Repeated Measures ANOVA: group: F(1,18)=0.77, p=0.391, pulse: F(1.03, 18.57)=64.09, p<0.001; interaction: F(1.03, 18.57)=0.77, p=0.395). Further, there were no significant differences in PPR or stimulation intensity between the two groups (data not shown; PPR: saline 1.03±0.05, MAM 0.89±0.11, p=0.236. Stimulation intensity: saline 247.36±45.40, MAM 256.44±79.65, p=0.370). Together, these data confirm that pre-synaptic release mechanisms and AMPARs are unaffected by prenatal MAM exposure; however, hypofunction of NR2B-NMDARs is evident in juvenile MAM plPFC.
H3K27me3 is selectively increased in the mPFC of juvenile MAM rats
To evaluate whether epigenetic mechanisms may be implicated in regulating NMDAR expression during postnatal development, we assayed a number of histone modifications relevant to learning and memory processes. This exploratory approach would lend insight into whether a misregulation of the epigenome underlies juvenile NMDAR hypofunction in the MAM model for SCZ. We collected mPFC tissue from saline and MAM animals at P21 and processed the tissue to isolate the histone fraction. Using Western blotting to determine levels of acetylation and methylation on histone H3, we found juvenile MAM animals had a significant increase in H3K27me3 (Figure 4; Saline 0.93±0.04, n=5; MAM 1.05±0.01, n=6; p=0.045), but no change in the acetylation markers assayed (H3K9ac, p=0.263; H3K18ac, p=0.524; H3K27ac, p=0.294). These data suggest epigenetic modifications are facilitating the NR2B protein loss observed in the juvenile MAM mPFC. We further tested this possibility using ChIP-qPCR as described below.
Figure 4.
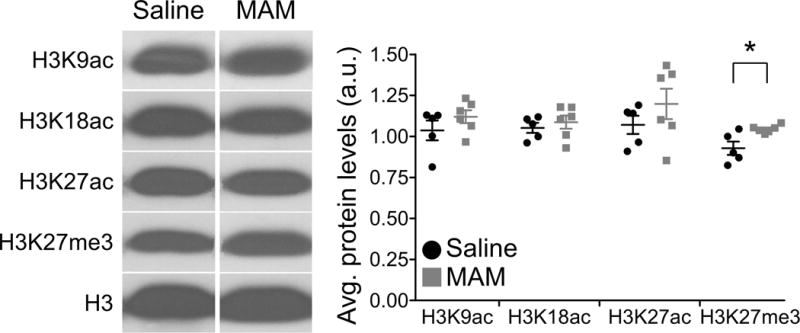
H3K27me3 protein levels are selectively increased in developing MAM mPFC. Representative Western blots (left) and summary scatterplot (right) demonstrates the selective upregulation of this histone modification in MAM compared to saline animals. * p<0.05
The proximal promoter region of Grin2b is highly enriched in the transcriptional repressor REST and the histone marker H3K27me3 in juvenile MAM mPFC
Our findings thus far reveal a marked increase of the repressive epigenetic marker H3K27me3 with concomitant reduction of NR2B protein levels and functional NR2B-NMDARs in juvenile MAM mPFC and plPFC, respectively. Using the evidence of increased H3K27me3 in juvenile MAM mPFC, we carried out ChIP-qPCR assays to measure the enrichment levels of the RE1-silencing transcription factor (REST) and repressive histone marker H3K27me3 at the proximal promoter region of the Grin2b gene (NR2B-encoding). Grin2b has response element (RE1) motifs within its promoter region to which REST can be recruited (Rodenas-Ruano et al., 2012). To test the specificity of REST-mediated repression, we also assessed the enrichment levels of REST at the RE1 sites of Grin1 (NR1-encoding) and Grin2a (NR2A-encoding). For saline controls, we found REST was significantly enriched at Grin2b, but not at Grin1 or Grin2a RE1 sites (Figure 5A; saline REST, One-Way ANOVA, group: Grin2b 20.86±5.23, n=4 rats yielding 6 data points from qPCR; Grin2a 3.85±1.04; Grin1 3.05±1.13; F(2, 17)=9.17, p=0.003; Tukey’s post-hoc: saline Grin2b vs Grin2a, p=0.005; saline Grin2b vs Grin1, p=0.007; saline Grin2a vs Grin1, p=0.985). Similarly, repressive marker H3K27me3 was significantly enriched at Grin2b, but not at the Grin1 or Grin2a RE1 sites in saline PFC (Figure 5B; saline H3K27me3, One-Way ANOVA, group: Grin2b 230.03±19.24, n=4; Grin2a 102.82±28.49; Grin1 57.88±12.41; F(2, 17)=17.91, p<0.001; Tukey’s post-hoc: saline Grin2b vs Grin2a, p=0.002; saline Grin2b vs Grin1, p<0.001; saline Grin2a vs Grin1, p=0.316). Together, these data demonstrate that REST is normally more highly enriched within the proximal promoter region of Grin2b than at either Grin1 or Grin2a RE1 sites. This confirms the selective role of REST in regulating Grin2b expression, as has been shown in the hippocampus (Rodenas-Ruano et al., 2012). Further, repressive marker H3K27me3 enrichment at the Grin2b promoter region, but not at Grin1 or Grin2a, lends evidence to the notion that Grin2b expression is tightly regulated by these mechanisms compared to other major NMDAR subunit proteins.
Figure 5.
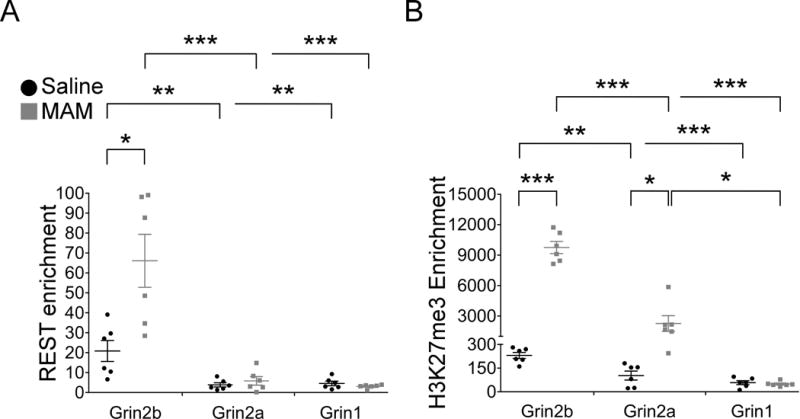
PCR was performed in triplicate yielding a final sample size of 6 data points. A standard curve was generated from serial 10-fold dilutions of input DNA. The Ct values were used to estimate DNA quantity of the ChIP and mock (no antibody) control samples. Fold enrichment levels were calculated as a ratio of the DNA quantity in the ChIP samples to control samples. (A) REST-mediated repression of the proximal promoter of Grin2b contributes to reduced synaptic NR2B protein in juvenile MAM animals. Representative scatterplots depicting elevated REST enrichment at Grin2b in saline and MAM animals compared to other promoter regions, Grin2a and Grin1, confirming regulation of gene expression by REST is specific to Grin2b in the juvenile mPFC. Moreover, REST enrichment is significantly greater at Grin2b in MAM compared to saline mPFC. (B) H3K27me3 enrichment is significantly elevated at Grin2b and Grin2a in MAM animals, but not at Grin1, compared to saline animals. * p<0.05, ** p<0.01, *** p<0.001
In contrast, Grin2b in MAM animals has a significant increase of REST enrichment in the mPFC compared to saline controls (Figure 5A; REST Grin2b: saline 20.86 ±5.23, n=4; MAM 66.10 ± 13.28, n=4; p=0.017). We found that enhanced enrichment of REST binding at Grin2b is accompanied by a significant increase in H3K27me3 levels (Figure 5B; H3K27me3 Grin2b: saline 230.03 ± 19.24; MAM 9752.23 ± 598.3; p<0.001). Of note, we did observe an increased level of H3K27me3 in the MAM mPFC at the Grin2a RE1 site (Figure 5B; H3K27me3 Grin2a: saline 102.82 ± 28.49, MAM 2258.43 ± 774.18; p=0.019). Such enrichment is not associated with increased REST enrichment at this Grin2a RE1 site (Figure 5A; REST Grin2a: saline 3.85±1.04, MAM 5.85±2.14; p=0.420). Moreover, we did not observe a significant difference in REST or H3K27me3 enrichment at the Grin1 promoter region in saline versus MAM animals, suggesting MAM exposure does not induce transcriptional regulation at this Grin1 RE1 site (Figure 5A–B; REST Grin1: saline 4.59±1.13, MAM 3.05±0.40; p=0.230; H3K27me3 Grin1: saline 57.88±12.41, MAM 49.18±6.27; p=0.546). Thus, in comparing levels of REST and H3K27me3 enrichment at RE1 sites of NMDAR-expressing genes Grin1, Grin2a, and Grin2b between saline and MAM animals, it is evident that the extent of REST and H3K27me3 enrichment is far greater than that observed in control PFC, indicating that the Grin2b proximal promoter region is in a hyper-repressed state in juvenile MAM mPFC. We conclude that this hyper-repression in MAM mPFC in part contributes to the decrease in synaptic NR2B protein and NR2B-NMDAR hypofunction evident in plPFC in prior experiments (Figure 1B and 3C).
Discussion
In these experiments, we explored the glutamate hypofunction hypothesis by measuring NMDAR subunit expression and function in the developing mPFC using the neurodevelopmental MAM rat model for SCZ. Our findings provide the first direct evidence that NMDAR hypofunction is occurring at an early (juvenile) stage of development in the MAM model for SCZ. Specifically, synaptic NR2B protein expression is significantly decreased in the mPFC. Concomitantly, NMDAR-mediated neurotransmission is deficient as evidenced by reduced amplitude of NMDAR-mEPSCs and diminished synaptic NMDAR-mediated current in juvenile MAM plPFC pyramidal neurons. Mechanistically, epigenetic mechanisms demonstrate a key role in this NR2B-related dysfunction. REST repressor protein is preferentially localized to the RE1 site in the Grin2b proximal promoter, but not to RE1 sites in the promoters of other major NMDAR subunit genes, Grin1 or Grin2a. Additionally, a functional marker of transcription, H3K27me3, is significantly increased at the Grin2b promoter in juvenile MAM mPFC. Together, these data suggest that epigenetic mis-regulation of NR2B expression underlies NMDAR hypofunction in the plPFC of juvenile animals in the MAM model for SCZ.
Pharmacological, neurodevelopmental, and transgenic mouse models are used extensively to understand the role of NMDARs in SCZ (Kantrowitz and Javitt, 2010; Gilmour et al., 2012; Kantrowitz and Javitt, 2012; Snyder and Gao, 2013). However, conclusions drawn from previous work remain incomplete given that this research often focuses on investigation of the adult brain, albeit cognitive dysfunction emerging early in development and preceding psychosis (Jones et al., 1994; Cannon et al., 2000; Rosso et al., 2000). Indeed, transient antagonism of NMDARs during the juvenile period can elicit commonly reported SCZ-like endophenotypes in adulthood (Jeevakumar et al., 2015). These results corroborate our findings in the MAM model; NMDAR hypofunction in juveniles, in the form of reduced NR2B-NMDARs, precedes dopaminergic hypersensitivity, diminished social interaction (Flagstad et al., 2004), sensorimotor gating deficits, and perseverative behavior in cognitive flexibility (Moore et al., 2006), akin to positive, negative, and cognitive symptoms reported in SCZ patients (Elliott et al., 1995; Laruelle, 2000; Geyer et al., 2001). This is an important indicator that NMDAR hypofunction may emerge in early postnatal development, such as the juvenile stage of mPFC development, in SCZ.
NMDAR hypofunction in juvenile MAM mPFC is specifically due to reduced NR2B protein expression and function at the post-synaptic site, as evidenced by the smaller amplitude of NMDAR-mEPSCs and evoked NMDAR currents. The NR2B subunit is integral for promoting persistent activity, which is the proposed molecular correlate of working memory function (Goldman-Rakic, 1995; Wang et al., 2013). Because NR2B levels remain persistently high in the PFC throughout development and adulthood (Wang et al., 2008), the selective loss of NR2B-NMDARs in MAM plPFC results in a vulnerability to working memory impairments (Snyder et al., 2013; Xing et al., 2016; Li et al., 2017). NR2B protein levels are substantially reduced, as shown by Western blot analysis, but this approach cannot distinguish the regional or laminar specificity of this deficiency. To this end, we used a selective blocker of NR2B, Ro25, to confirm the loss of synaptic NR2B-NMDARs in pyramidal neurons of the plPFC. In the saline group, Ro25 treatment reduces the amplitude of NMDAR-mediated currents by 57% at the first pulse, and approximately 50% at each proceeding pulse (range 49–55%). However, in the MAM group, NMDAR-mediated amplitude is reduced by only 22% (range 14–26%). The significant loss of post-synaptic NMDARs in MAM cells occludes the predicted effect of Ro25 compared to saline cells, confirming a significant reduction in specifically NR2B-NMDARs in MAM plPFC (Figure 6).
Figure 6.
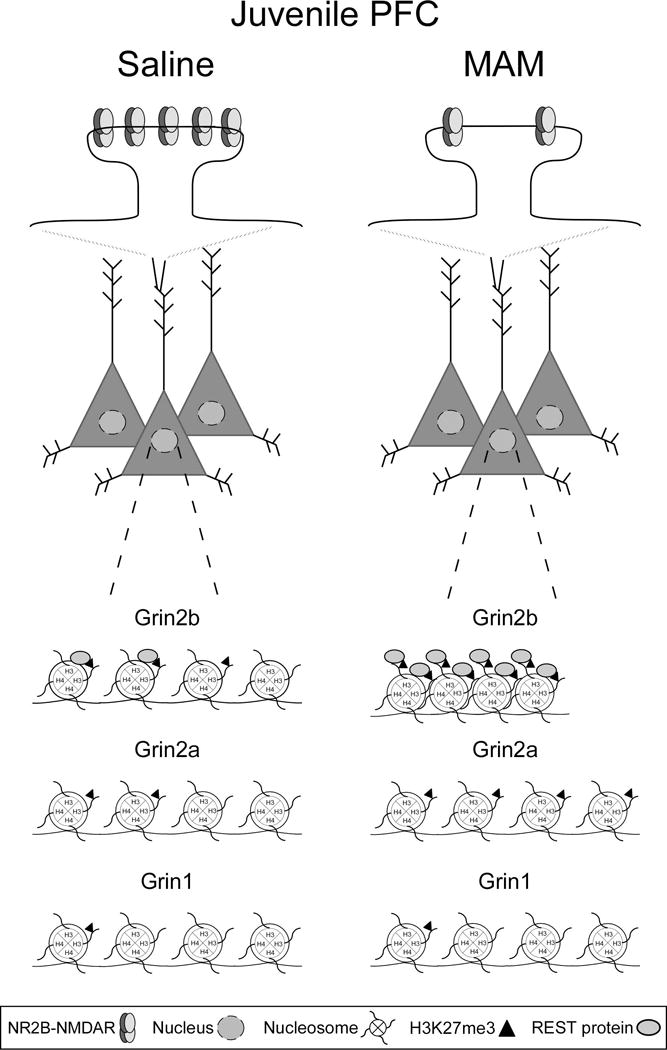
A schematic representation of NR2B-NMDAR loss in the MAM juvenile mPFC, and REST (gray ovals) and H3K27me3 (black triangles) enrichment at response element (RE1) sites in the promoter regions of Grin2b, Grin2a, and Grin1 in juvenile saline and MAM mPFC. Top panel, NR2B-NMDAR levels are persistently high in the developing mPFC and play a critical role in synaptic plasticity and PFC-dependent cognitive functions. However, MAM animals are vulnerable to cognitive dysfunction due to the significant loss of synaptic NR2B-NMDARs in the critical juvenile period. Bottom panel, REST and H3K27me3 enrichment levels in saline mPFC are higher at the Grin2b RE1 site compared to the Grin1 and Grin2a RE1 sites, demonstrating selective REST regulation of Grin2b is an endogenous repressor mechanism. In the MAM mPFC, REST and H3K27me3 enrichment levels are greatly elevated compared to the saline group, demonstrating the hyper-repression of the proximal promoter region of Grin2b. Levels of REST and H3K27me3 enrichment are comparable between saline and MAM animals at the Grin1 and Grin2a RE1 sites, with the exception of a surprising elevation of H3K27me3 enrichment at the Grin2a promoter in MAM mPFC.
Since glutamate hypofunction emerged as a potential neurobiological underpinning of SCZ, efforts have focused on identifying the mechanism underlying this molecular disruption. Single-nucleotide polymorphisms (SNPs) and other genetic disturbances implicate Grin2b in psychiatric disorders, including SCZ (Qin et al., 2005; Endele et al., 2010; Jiang et al., 2010; Jia et al., 2012; Weickert et al., 2013; Akbarian, 2014; Fromer et al., 2014). Indeed, complex higher-order chromatin conformations are involved in regulating gene expression and eventually, cognitive function. For example, rare SNPs in distal gene locations can, in fact, disrupt Grin2b expression and affect working memory function in both humans and mice (Bharadwaj et al., 2014). Therefore, a highly complex network of genetic and epigenetic processes is implicated in how glutamatergic genes may become dysregulated in SCZ.
We sought to identify how the NR2B subunit was diminished in MAM mPFC without substantial alteration of other NMDAR subunit protein levels. To address this question, we looked to a well-characterized molecular process, the NR2B-to-NR2A subunit switch, in which NR2B protein levels are significantly decreased while NR2A protein expression is augmented to promote functional maturation of critical brain regions (Dumas, 2005; Rodenas-Ruano et al., 2012; Monaco et al., 2015). This process is tightly controlled by an epigenetic mechanism that is dependent upon the repressor REST and its corepressor protein partners (Tamminga and Zukin, 2015). Though it is unclear what dictates the specificity of REST for Grin2b compared to other NMDAR promoters, this pattern has been shown in these experiments and others (Rodenas-Ruano et al., 2012). Thus, we hypothesized that this endogenous repressive mechanism of Grin2b expression is responsible, at least in part, for the selective reduction in NR2B protein observed in juvenile MAM animals via aberrant hyper-repression of the Grin2b promoter. Our work is the first to reveal REST is a selective regulator of Grin2b expression in the juvenile mPFC, wherein the typical NR2B-to-NR2A subunit switch is absent, with no significant enrichment at the Grin1 or Grin2a promoters (Figure 6). Further, aberrant REST enrichment selectively at Grin2b contributes to NR2B protein loss in juvenile MAM mPFC.
Histone modifications can serve as functional readouts of chromatin state (Akbarian, 2010b). Altered histone modification patterns have been reported in the developing PFC of MAM animals (Mackowiak et al., 2014), indicating aberrant epigenetic processes contribute to abnormal PFC development in this model. Specifically, the repressive histone modification H3K27me3 is implicated in cellular vulnerability in SCZ (Kano et al., 2013), reinforcing the contribution of this modification to neural dysfunction in the human condition. However, information regarding the specific location of these histone modification changes in the genetic landscape is lacking. Here, we confirm that the increase in H3K27me3 in juvenile MAM mPFC, as shown in Western blots, is also evident at the Grin2b proximal promoter site. This is likely due to the REST-mediated recruitment of Polycomb complex PRC2, which contains a catalytic enzyme domain, EZH2, responsible for the H3K27me3 modification (Kim and Kim, 2012). Importantly, these processes are transient regulators of gene expression. Thus, our focus was on the intermediate stage (p21) of juvenile development in order to measure H3K27me3 enrichment levels concomitant with NR2B protein loss in the mPFC.
Surprisingly, there was also a significant increase of H3K27me3 enrichment at Grin2a in MAM mPFC, although the levels of REST are not significantly enriched at this promoter, and protein levels of NR2A are not altered in MAM animals. As evident in our ChIP experiments, a pattern of H3K27me3 enrichment exists in saline animals across promoters; that is, levels of this histone modification are highest at Grin2b, lower at Grin2a, and lowest at Grin1 (Grin2b > Grin2a > Grin1). Interestingly, MAM animals show a similar pattern of H3K27me3 enrichment in the mPFC, although to a much higher degree. Compared to saline mPFC, the level of H3K27me3 enrichment is 41-fold higher at Grin2b, while the enrichment at Grin2a is only 21-fold higher in MAM mPFC. We hypothesize that levels of H3K27me3 are elevated by a REST-independent mechanism at the Grin2a promoter. In fact, shRNA-mediated knockdown of REST does not significantly affect NR2A mRNA levels (Rodenas-Ruano et al., 2012), supporting the notion that significant enrichment of H3K27me3 levels at Grin2a in MAM mPFC are likely independent of REST. Alternatively, histone modifications such as H3K4me3 and H3K36me3 balance the chromatin state by mitigating the effects of repressive histone modifications, prohibiting alterations to gene expression (Aranda et al., 2015). We propose an initial increase of H3K27me3 levels at the Grin2b promoter disrupts the balance of histone modifications, eventually recruiting REST for further EZH2-mediated enrichment of H3K27me3, resulting in reduced NR2B protein levels. However, at the Grin2a promoter, this threshold was not surpassed by the 21-fold increase in H3K27me3. Further, the levels of other histone modifications, including the mutually exclusive marker H3K27ac, are unknown at these promoter regions. Therefore, more complex mechanisms are likely regulating expression of Grin2a, cumulatively resulting in no change to NR2A protein levels.
We have previously demonstrated the persistent expression of NR2B in PFC compared to other cortical regions (Wang et al., 2008). Therefore, alterations in NR2B protein in the mPFC throughout development and adulthood can have a great impact on circuitry and behavior (Tang et al., 1999; Cui et al., 2011; Monaco et al., 2015). The aberrant decrease in synaptic NR2B-NMDARs during juvenile development of MAM animals can affect signaling cascade activation, facilitation in response to incoming stimuli, and working memory and higher-order cognitive abilities (Monaco et al., 2015). These disruptions to the NMDAR system in the mPFC and hippocampus (Snyder et al., 2013), particularly during brain development, impose a vulnerability upon the critical fine-tuning of the network necessary for normal cognition in adulthood. Given the importance of the hippocampus-mPFC pathway in cognitive functioning (Marquis et al., 2006; Marquis et al., 2008), future experiments will explore the cognitive profile of juvenile MAM rats with a focus on mPFC-dependent processes, such as working memory and cognitive flexibility. Due to the continued lack of successful treatments for cognitive impairments in SCZ (Lett et al., 2014), even in the face of improved positive and negative symptoms, these deficits will persist as will the psychological and monetary burden of this disorder. These data are a contribution to the ever-growing force to understand the neurobiological underpinnings of the highly complex and devastating etiology of SCZ.
Acknowledgments
The authors acknowledge Yan-Chun Li, Bo Xing, and Brielle R. Ferguson for assistance with electrophysiology experiments, and Sarah A. Monaco for helpful comments on the manuscript.
Funding and Disclosure
Funding for the current study was provided by the NIH R01MH085666 and NARSAD Independent Investigator Award 2015 to W.J. Gao. The authors report no biomedical financial interests or potential conflicts of interest. We reaffirm that the content of this manuscript is not under consideration for publication elsewhere nor has the information been previously published. All of the authors have fulfilled all conditions required for authorship and have approved the submission.
List of Abbreviations
- aCSF
artificial cerebrospinal fluid
- AMPAR
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor
- BCA
bicinchoninic acid
- ChIP
chromatin immunoprecipitation
- DNQX
6,7-dinitroquinoxaline-2,3-dione
- E
embryonic day
- eEPSC
evoked excitatory post-synaptic current
- EPSC
excitatory post-synaptic current
- GABAR
gamma-aminobutyric acid receptor
- IP
intraperitoneal
- MAM
methylazoxymethanol
- mEPSC
miniature excitatory post-synaptic current
- mPFC
medial prefrontal cortex
- NMDAR
N-methyl-D-aspartate receptor
- NR2B-NMDAR
NR2B-containing NMDA receptor
- P
postnatal day
- PFC
prefrontal cortex
- plPFC
prelimbic prefrontal cortex
- PPR
paired-pulse ratio
- qPCR
quantitative polymerase chain reaction
- RE1
response element 1
- REST
RE1-silencing transcription factor
- SCZ
schizophrenia
- sEPSC
spontaneous excitatory post-synaptic current
- TTX
tetrodotoxin
Footnotes
DR. WEN-JUN GAO (Orcid ID : 0000-0002-8585-3082)
Author Contributions
Y.G. designed and carried out the experiments and data analysis of Western blots, electrophysiological recordings, and ChIP. She also wrote the paper. S.J.X. conducted the ChIP experiments and analyzed the data. M.A.S. also designed and carried out experiments and analysis of Western blot and electrophysiology data. She wrote part of the manuscript. E.F. supervised the ChIP experiments and consulted on the project. W.J.G. conceived the study, supervised the project, and finalized the manuscript.
Involves human subjects: No
If yes: Informed consent & ethics approval achieved:
=> if yes, please ensure that the info “Informed consent was achieved for all subjects, and the experiments were approved by the local ethics committee.” is included in the Methods.
ARRIVE guidelines have been followed:
Yes
=> if No or if it is a Review or Editorial, skip complete sentence => if Yes, insert “All experiments were conducted in compliance with the ARRIVE guidelines.” unless it is a Review or Editorial
Conflicts of interest: None
=> if ‘none’, insert “The authors have no conflict of interest to declare.”
=> otherwise insert info unless it is already included
References
- Akbarian S. Epigenetics of schizophrenia. Curr Top Behav Neurosci. 2010a;4:611–628. doi: 10.1007/7854_2010_38. [DOI] [PubMed] [Google Scholar]
- Akbarian S. The molecular pathology of schizophrenia–focus on histone and DNA modifications. Brain Res Bull. 2010b;83(3–4):103–107. doi: 10.1016/j.brainresbull.2009.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akbarian S. Epigenetic mechanisms in schizophrenia. Dialogues Clin Neurosci. 2014;16(3):405–417. doi: 10.31887/DCNS.2014.16.3/sakbarian. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aranda S, Mas G, Di Croce L. Regulation of gene transcription by Polycomb proteins. Sci Adv. 2015;1(11):e1500737. doi: 10.1126/sciadv.1500737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beneyto M, Meador-Woodruff JH. Lamina-specific abnormalities of NMDA receptor-associated postsynaptic protein transcripts in the prefrontal cortex in schizophrenia and bipolar disorder. Neuropsychopharmacology. 2008;33(9):2175–2186. doi: 10.1038/sj.npp.1301604. [DOI] [PubMed] [Google Scholar]
- Bharadwaj R, Peter CJ, Jiang Y, Roussos P, Vogel-Ciernia A, Shen EY, et al. Conserved higher-order chromatin regulates NMDA receptor gene expression and cognition. Neuron. 2014;84(5):997–1008. doi: 10.1016/j.neuron.2014.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowie CR, Harvey PD. Cognitive deficits and functional outcome in schizophrenia. Neuropsychiatr Dis Treat. 2006;2(4):531–536. doi: 10.2147/nedt.2006.2.4.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon TD, Bearden CE, Hollister JM, Rosso IM, Sanchez LE, Hadley T. Childhood cognitive functioning in schizophrenia patients and their unaffected siblings: a prospective cohort study. Schizophr Bull. 2000;26(2):379–393. doi: 10.1093/oxfordjournals.schbul.a033460. [DOI] [PubMed] [Google Scholar]
- Coyle JT, Tsai G, Goff DC. Ionotropic glutamate receptors as therapeutic targets in schizophrenia. Curr Drug Targets CNS Neurol Disord. 2002;1(2):183–189. doi: 10.2174/1568007024606212. [DOI] [PubMed] [Google Scholar]
- Cui Y, Jin J, Zhang X, Xu H, Yang L, Du D, et al. Forebrain NR2B overexpression facilitating the prefrontal cortex long-term potentiation and enhancing working memory function in mice. PloS one. 2011;6(5) doi: 10.1371/journal.pone.0020312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumas TC. Developmental regulation of cognitive abilities: modified composition of a molecular switch turns on associative learning. Prog Neurobiol. 2005;76(3):189–211. doi: 10.1016/j.pneurobio.2005.08.002. [DOI] [PubMed] [Google Scholar]
- Elliott R, McKenna PJ, Robbins TW, Sahakian BJ. Neuropsychological evidence for frontostriatal dysfunction in schizophrenia. Psychol Med. 1995;25(3):619–630. doi: 10.1017/s0033291700033523. [DOI] [PubMed] [Google Scholar]
- Endele S, Rosenberger G, Geider K, Popp B, Tamer C, Stefanova I, et al. Mutations in GRIN2A and GRIN2B encoding regulatory subunits of NMDA receptors cause variable neurodevelopmental phenotypes. Nat Genet. 2010;42(11):1021–1026. doi: 10.1038/ng.677. [DOI] [PubMed] [Google Scholar]
- Flagstad P, Glenthoj BY, Didriksen M. Cognitive deficits caused by late gestational disruption of neurogenesis in rats: a preclinical model of schizophrenia. Neuropsychopharmacology. 2005;30(2):250–260. doi: 10.1038/sj.npp.1300625. [DOI] [PubMed] [Google Scholar]
- Flagstad P, Mork A, Glenthoj BY, van Beek J, Michael-Titus AT, Didriksen M. Disruption of neurogenesis on gestational day 17 in the rat causes behavioral changes relevant to positive and negative schizophrenia symptoms and alters amphetamine-induced dopamine release in nucleus accumbens. Neuropsychopharmacology. 2004;29(11):2052–2064. doi: 10.1038/sj.npp.1300516. [DOI] [PubMed] [Google Scholar]
- Fromer M, Pocklington AJ, Kavanagh DH, Williams HJ, Dwyer S, Gormley P, et al. De novo mutations in schizophrenia implicate synaptic networks. Nature. 2014;506(7487):179–184. doi: 10.1038/nature12929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gainetdinov RR, Mohn AR, Caron MG. Genetic animal models: focus on schizophrenia. Trends Neurosci. 2001;24(9):527–533. doi: 10.1016/s0166-2236(00)01886-5. [DOI] [PubMed] [Google Scholar]
- Geyer MA, Krebs-Thomson K, Braff DL, Swerdlow NR. Pharmacological studies of prepulse inhibition models of sensorimotor gating deficits in schizophrenia: a decade in review. Psychopharmacology (Berl) 2001;156(2–3):117–154. doi: 10.1007/s002130100811. [DOI] [PubMed] [Google Scholar]
- Gilmour G, Dix S, Fellini L, Gastambide F, Plath N, Steckler T, et al. NMDA receptors, cognition and schizophrenia–testing the validity of the NMDA receptor hypofunction hypothesis. Neuropharmacology. 2012;62(3):1401–1412. doi: 10.1016/j.neuropharm.2011.03.015. [DOI] [PubMed] [Google Scholar]
- Goldman-Rakic PS. Cellular basis of working memory. Neuron. 1995;14(3):477–485. doi: 10.1016/0896-6273(95)90304-6. [DOI] [PubMed] [Google Scholar]
- Gourevitch R, Rocher C, Le Pen G, Krebs MO, Jay TM. Working memory deficits in adult rats after prenatal disruption of neurogenesis. Behav Pharmacol. 2004;15(4):287–292. doi: 10.1097/01.fbp.0000135703.48799.71. [DOI] [PubMed] [Google Scholar]
- Hradetzky E, Sanderson TM, Tsang TM, Sherwood JL, Fitzjohn SM, Lakics V, et al. The methylazoxymethanol acetate (MAM-E17) rat model: molecular and functional effects in the hippocampus. Neuropsychopharmacology. 2012;37(2):364–377. doi: 10.1038/npp.2011.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeevakumar V, Driskill C, Paine A, Sobhanian M, Vakil H, Morris B, et al. Ketamine administration during the second postnatal week induces enduring schizophrenia-like behavioral symptoms and reduces parvalbumin expression in the medial prefrontal cortex of adult mice. Behav Brain Res. 2015;282:165–175. doi: 10.1016/j.bbr.2015.01.010. [DOI] [PubMed] [Google Scholar]
- Jia P, Wang L, Fanous AH, Pato CN, Edwards TL, International Schizophrenia, C et al. Network-assisted investigation of combined causal signals from genome-wide association studies in schizophrenia. PLoS Comput Biol. 2012;8(7):e1002587. doi: 10.1371/journal.pcbi.1002587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Jakovcevski M, Bharadwaj R, Connor C, Schroeder FA, Lin CL, et al. Setdb1 histone methyltransferase regulates mood-related behaviors and expression of the NMDA receptor subunit NR2B. J Neurosci. 2010;30(21):7152–7167. doi: 10.1523/jneurosci.1314-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P, Rodgers B, Murray R, Marmot M. Child development risk factors for adult schizophrenia in the British 1946 birth cohort. Lancet. 1994;344(8934):1398–1402. doi: 10.1016/s0140-6736(94)90569-x. [DOI] [PubMed] [Google Scholar]
- Kano S, Colantuoni C, Han F, Zhou Z, Yuan Q, Wilson A, et al. Genome-wide profiling of multiple histone methylations in olfactory cells: further implications for cellular susceptibility to oxidative stress in schizophrenia. Mol Psychiatry. 2013;18(7):740–742. doi: 10.1038/mp.2012.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantrowitz J, Javitt DC. Glutamatergic transmission in schizophrenia: from basic research to clinical practice. Curr Opin Psychiatry. 2012;25(2):96–102. doi: 10.1097/YCO.0b013e32835035b2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantrowitz JT, Javitt DC. N-methyl-d-aspartate (NMDA) receptor dysfunction or dysregulation: the final common pathway on the road to schizophrenia? Brain Res Bull. 2010;83(3–4):108–121. doi: 10.1016/j.brainresbull.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kim H. Recruitment and biological consequences of histone modification of H3K27me3 and H3K9me3. ILAR J. 2012;53(3–4):232–239. doi: 10.1093/ilar.53.3-4.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristiansen LV, Bakir B, Haroutunian V, Meador-Woodruff JH. Expression of the NR2B-NMDA receptor trafficking complex in prefrontal cortex from a group of elderly patients with schizophrenia. Schizophr Res. 2010;119(1–3):198–209. doi: 10.1016/j.schres.2010.02.1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laruelle M. The role of endogenous sensitization in the pathophysiology of schizophrenia: implications from recent brain imaging studies. Brain Res Brain Res Rev. 2000;31(2–3):371–384. doi: 10.1016/s0165-0173(99)00054-5. [DOI] [PubMed] [Google Scholar]
- Lau CG, Zukin RS. NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat Rev Neurosci. 2007;8(6):413–426. doi: 10.1038/nrn2153. [DOI] [PubMed] [Google Scholar]
- Lavin A, Moore HM, Grace AA. Prenatal disruption of neocortical development alters prefrontal cortical neuron responses to dopamine in adult rats. Neuropsychopharmacology. 2005;30(8):1426–1435. doi: 10.1038/sj.npp.1300696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lett TA, Voineskos AN, Kennedy JL, Levine B, Daskalakis ZJ. Treating working memory deficits in schizophrenia: a review of the neurobiology. Biol Psychiatry. 2014;75(5):361–370. doi: 10.1016/j.biopsych.2013.07.026. [DOI] [PubMed] [Google Scholar]
- Li ML, Gulchina Y, Monaco SA, Xing B, Ferguson BR, Li YC, et al. Juvenile treatment with a novel mGluR2 agonist/mGluR3 antagonist compound, LY395756, reverses learning deficits and cognitive flexibility impairments in adults in a neurodevelopmental model of schizophrenia. Neurobiol Learn Mem. 2017;140:52–61. doi: 10.1016/j.nlm.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipska BK, Weinberger DR. To model a psychiatric disorder in animals: schizophrenia as a reality test. Neuropsychopharmacology. 2000;23(3):223–239. doi: 10.1016/S0893-133X(00)00137-8. [DOI] [PubMed] [Google Scholar]
- Lodge DJ, Grace AA. Gestational methylazoxymethanol acetate administration: a developmental disruption model of schizophrenia. Behav Brain Res. 2009;204(2):306–312. doi: 10.1016/j.bbr.2009.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackowiak M, Bator E, Latusz J, Mordalska P, Wedzony K. Prenatal MAM administration affects histone H3 methylation in postnatal life in the rat medial prefrontal cortex. Eur Neuropsychopharmacol. 2014;24(2):271–289. doi: 10.1016/j.euroneuro.2013.05.013. [DOI] [PubMed] [Google Scholar]
- Marquis JPP, Goulet S, Doré FYY. Neonatal ventral hippocampus lesions disrupt extra-dimensional shift and alter dendritic spine density in the medial prefrontal cortex of juvenile rats. Neurobiology of learning and memory. 2008;90(2):339–346. doi: 10.1016/j.nlm.2008.04.005. [DOI] [PubMed] [Google Scholar]
- Marquis JP, Goulet S, Dore FY. Neonatal lesions of the ventral hippocampus in rats lead to prefrontal cognitive deficits at two maturational stages. Neuroscience. 2006;140(3):759–767. doi: 10.1016/j.neuroscience.2006.02.048. [DOI] [PubMed] [Google Scholar]
- Martucci L, Wong AH, De Luca V, Likhodi O, Wong GW, King N, et al. N-methyl-D-aspartate receptor NR2B subunit gene GRIN2B in schizophrenia and bipolar disorder: Polymorphisms and mRNA levels. Schizophr Res. 2006;84(2–3):214–221. doi: 10.1016/j.schres.2006.02.001. [DOI] [PubMed] [Google Scholar]
- Meador-Woodruff JH, Healy DJ. Glutamate receptor expression in schizophrenic brain. Brain Res Brain Res Rev. 2000;31(2–3):288–294. doi: 10.1016/s0165-0173(99)00044-2. [DOI] [PubMed] [Google Scholar]
- Monaco SA, Gulchina Y, Gao WJ. NR2B subunit in the prefrontal cortex: A double-edged sword for working memory function and psychiatric disorders. Neurosci Biobehav Rev. 2015;56:127–138. doi: 10.1016/j.neubiorev.2015.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 1994;12(3):529–540. doi: 10.1016/0896-6273(94)90210-0. [DOI] [PubMed] [Google Scholar]
- Moore H, Jentsch JD, Ghajarnia M, Geyer MA, Grace AA. A neurobehavioral systems analysis of adult rats exposed to methylazoxymethanol acetate on E17: implications for the neuropathology of schizophrenia. Biol Psychiatry. 2006;60(3):253–264. doi: 10.1016/j.biopsych.2006.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler EJ, Pena CJ, Kundakovic M, Mitchell A, Akbarian S. Epigenetic Basis of Mental Illness. Neuroscientist. 2016;22(5):447–463. doi: 10.1177/1073858415608147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numata S, Ye T, Herman M, Lipska BK. DNA methylation changes in the postmortem dorsolateral prefrontal cortex of patients with schizophrenia. Front Genet. 2014;5:280. doi: 10.3389/fgene.2014.00280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletti P, Bellone C, Zhou Q. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci. 2013;14(6):383–400. doi: 10.1038/nrn3504. [DOI] [PubMed] [Google Scholar]
- Qin S, Zhao X, Pan Y, Liu J, Feng G, Fu J, et al. An association study of the N-methyl-D-aspartate receptor NR1 subunit gene (GRIN1) and NR2B subunit gene (GRIN2B) in schizophrenia with universal DNA microarray. Eur J Hum Genet. 2005;13(7):807–814. doi: 10.1038/sj.ejhg.5201418. [DOI] [PubMed] [Google Scholar]
- Reichenberg A. The assessment of neuropsychological functioning in schizophrenia. Dialogues Clin Neurosci. 2010;12(3):383–392. doi: 10.31887/DCNS.2010.12.3/areichenberg. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodenas-Ruano A, Chavez AE, Cossio MJ, Castillo PE, Zukin RS. REST-dependent epigenetic remodeling promotes the developmental switch in synaptic NMDA receptors. Nat Neurosci. 2012;15(10):1382–1390. doi: 10.1038/nn.3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosso IM, Bearden CE, Hollister JM, Gasperoni TL, Sanchez LE, Hadley T, et al. Childhood neuromotor dysfunction in schizophrenia patients and their unaffected siblings: a prospective cohort study. Schizophr Bull. 2000;26(2):367–378. doi: 10.1093/oxfordjournals.schbul.a033459. [DOI] [PubMed] [Google Scholar]
- Snyder MA, Adelman AE, Gao WJ. Gestational methylazoxymethanol exposure leads to NMDAR dysfunction in hippocampus during early development and lasting deficits in learning. Neuropsychopharmacology. 2013;38(2):328–340. doi: 10.1038/npp.2012.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder MA, Gao WJ. NMDA hypofunction as a convergence point for progression and symptoms of schizophrenia. Front Cell Neurosci. 2013;7:31. doi: 10.3389/fncel.2013.00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spear LP. The adolescent brain and age-related behavioral manifestations. Neurosci Biobehav Rev. 2000;24(4):417–463. doi: 10.1016/s0149-7634(00)00014-2. [DOI] [PubMed] [Google Scholar]
- Stadler F, Kolb G, Rubusch L, Baker SP, Jones EG, Akbarian S. Histone methylation at gene promoters is associated with developmental regulation and region-specific expression of ionotropic and metabotropic glutamate receptors in human brain. J Neurochem. 2005;94(2):324–336. doi: 10.1111/j.1471-4159.2005.03190.x. [DOI] [PubMed] [Google Scholar]
- Tamminga CA, Zukin RS. Schizophrenia: Evidence implicating hippocampal GluN2B protein and REST epigenetics in psychosis pathophysiology. Neuroscience. 2015;309:233–242. doi: 10.1016/j.neuroscience.2015.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang YP, Shimizu E, Dube GR, Rampon C, Kerchner GA, Zhuo M, et al. Genetic enhancement of learning and memory in mice. Nature. 1999;401(6748):63–69. doi: 10.1038/43432. [DOI] [PubMed] [Google Scholar]
- Wang H, Stradtman GG, 3rd, Wang XJ, Gao WJ. A specialized NMDA receptor function in layer 5 recurrent microcircuitry of the adult rat prefrontal cortex. Proc Natl Acad Sci U S A. 2008;105(43):16791–16796. doi: 10.1073/pnas.0804318105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HX, Gao WJ. Cell type-specific development of NMDA receptors in the interneurons of rat prefrontal cortex. Neuropsychopharmacology. 2009;34(8):2028–2040. doi: 10.1038/npp.2009.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HX, Gao WJ. Development of calcium-permeable AMPA receptors and their correlation with NMDA receptors in fast-spiking interneurons of rat prefrontal cortex. J Physiol. 2010;588(Pt 15):2823–2838. doi: 10.1113/jphysiol.2010.187591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Yang Y, Wang CJ, Gamo NJ, Jin LE, Mazer JA, et al. NMDA receptors subserve persistent neuronal firing during working memory in dorsolateral prefrontal cortex. Neuron. 2013;77(4):736–749. doi: 10.1016/j.neuron.2012.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weickert CS, Fung SJ, Catts VS, Schofield PR, Allen KM, Moore LT, et al. Molecular evidence of N-methyl-D-aspartate receptor hypofunction in schizophrenia. Mol Psychiatry. 2013;18(11):1185–1192. doi: 10.1038/mp.2012.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing B, Li YC, Gao WJ. GSK3beta hyperactivity during an early critical period impairs prefrontal synaptic plasticity and induces lasting deficits in spine morphology and working memory. Neuropsychopharmacology. 2016;41:3003–3015. doi: 10.1038/npp.2016.110. [DOI] [PMC free article] [PubMed] [Google Scholar]