

Abstract
Sepsis-induced myeloid-derived suppressor cells (MDSCs) contribute to immunosuppression associated with sepsis. We reported that the CCAAT enhancer-binding protein C/EBPβ activates microRNA (miR)-21 and miR-181b expressions, which induce transcription factor NFI-A to support the generation and expansion of MDSCs in the bone marrow and spleens of septic mice. Here, using a conditional knockout mouse model lacking C/EBPβ in the myeloid lineage, we find that without C/EBPβ, myeloid progenitor cells could not express miR-21 or miR-181b, and ectopic expression of C/EBPβ in the C/EBPβ-deficient myeloid progenitors activated the expression of the two miRNAs. Moreover, C/EBPβ-reconstituted myeloid cells expressed IL-10 and reduced T cell proliferation and function, similar to control MDSCs that express C/EBPβ. Exogenous expression of miR-21 and miR-181b in the C/EBPβ-deficient myeloid progenitors from septic mice produced similar results. Notably, NFI-A-dependent transactivation of NF-kB MDSC generating pathway was reversed in the C/EBPβ-deficient myeloid progenitors from septic mice. Together, these results support that decreasing C/EBPβ expression prevents MDSC generation and decreases immunosuppression in septic mice, providing a target for sepsis treatment.
Keywords: C/EBPβ, Immunosuppression, MDSC, Sepsis
Graphical Abstract

1. Introduction
The early inflammatory response to sepsis is characterized by a hyperinflammatory reaction that, if not treated early, progresses to a protracted immunosuppressive phase, which alters both innate and adaptive immunity (Hotchkiss et al., 2013b; Liu et al., 2012; Remick, 2007). At the cellular level, sepsis-induced immunosuppression is associated with tolerance of monocytes/macrophages and neutrophils as demonstrated by their decreased responses to bacterial toxins and failure to clear microbial infections (Alves-Filho et al., 2010; Ellaban et al., 2004), depletion of adaptive immune effector cells by apoptosis (Hotchkiss et al., 2013b; Oberholzer et al., 2001; Wesche et al., 2005), and suppression of T cell proliferation and function (Boomer et al., 2011; Guignant et al., 2011).
Our previous studies in a mouse model of polymicrobial sepsis induced by cecal ligation and puncture, which develops into early and late sepsis phases, revealed substantial expansion of myeloid-derived suppressor cells (MDSCs) within the bone marrow and spleen during the late septic phase (Brudecki et al., 2012a). We reported that immature Gr1+CD11b+ myeloid cell from late septic mice (i.e., MDSCs) produce immunosuppressive mediators, inhibit T cell activation and proliferation, and are unable to differentiate into mature immune cells (McClure et al., 2016; McClure et al., 2014). MicroRNA (miR)-21 and miR-181b inhibit MDSCs differentiation during sespsis by inducing the myeloid cell differentiation-related transcription factor NFI-A (McClure et al., 2016). In vivo inhibition of miR-21 and miR-181b in septic mice decreases repressor MDSCs and improves late sepsis survival (McClure et al., 2014).
The CCAAT enhancer-binding transcription factor C/EBPβ supports inflammation-induced proliferation of myeloid cells, where it accelerates myelopoiesis induced by cytokines or infections (Hirai et al., 2006; Marigo et al., 2010). For example, cytokines and growth factors, such as IL-6, G-CSF and GM-CSF, produced during infection or in tumor microenvironment can induces C/EBPβ expression (Manz and Boettcher, 2014; Marigo et al., 2010; Zhang et al., 2010). Previous studies have shown that activation of the transcription factor Stat3 by G-CSF or IL-6 is required for induction of C/EBPβ transcription in mouse myeloid progenitors (Hirai et al., 2006; Niehof et al., 2001). In addition, the transcription factor CREB (cAMP-response element-binding protein) is also required for the IL-6 mediated induction of C/EBPβ transcription by tethering Stat3 at the C/EBPβ promoter (Niehof et al., 2001). In support of the important role of Stat3 in the induction of C/EBPβ, recent studies have shown that the phenotype of bone marrow conditional Stat3 knockout mice resembles C/EBPβ deficiency since it exhibits impaired accelerated myelpoiesis under the pathophysiological conditions (Hirai et al., 2006; Panopoulos et al., 2006).
We previously reported that C/EBPβ in conjunction with Stat3 binds to and activates the promoters of both miR-21 and miR-181b in naive Gr1+CD11b+ cells (McClure et al., 2017), which are phenotypically similar to MDSCs. We hypothesized that C/EBPβ triggers the miR-21 and miR-181b dependent path that generates MDSCs during late sepsis immunosuppression. In this study, we used a mouse model with conditional deletion of Cebpb gene in the myeloid lineage to further investigate the mechanism of C/EBPβ-induced generation of MDSCs in sepsis. We find that C/EBPβ induces miR-21 and miR-181b expression to drive MDSCs during sepsis. Notable, myeloid precursors generated in the C/EBPβ conditional knockout mice during sepsis differentiate into competent innate immune cells, supporting that targeting the C/EBPβ-mediated pathway may prevent late sepsis immunsuppression.
2. Materials and Methods
2.1. Mice
Generation of BALB/c Cebpb conditional, myeloid cell-specific knockout mice have been described previously (McPeak et al., 2017). The Cebpbflox/flox;Lyz2Cre/+ mice, where the expression of the Cre recombinase inactivates the floxed Cebpb allele in the myeloid lineage cells, served as our myeloid-specific knockout. The Cebpbflox/flox;Lyz2+/+ mice, which do not express the Cre recombinase and thus the floxed Cebpb allele is still expressed in the myeloid lineage cells, served as controls.
The mice were bred and housed in a pathogen-free facility in the Division of Laboratory Animal Resources. Male mice, 8–10 weeks old were used. All experiments were conducted in accordance with National Institutes of Health guidelines and were approved by the East Tennessee State University Animal Care and Use Committee
2.2. Induction of sepsis
Polymicrobial sepsis was induced by cecal ligation and puncture (CLP). CLP was performed using a 21-gauge needle and two punctures, and mice were administered antibiotic to generate early/acute and late/chronic septic phases as described previously (Brudecki et al., 2012b). This model creates a prolonged infection with 100% mortality over 4 weeks. To generate late sepsis, mice were subcutaneously administered antibiotic (Imipenem; 25 mg/kg body weight) or an equivalent volume of 0.9% saline. To establish intra-abdominal infection and approximate the clinical situation of early human sepsis where there often is a delay between the onset of sepsis and the delivery of therapy (Mazuski et al., 2002), injections of Imipenem were given at 8 and 16 hr after CLP, which results in high mortality (~60–70%) during the late/chronic phase (Brudecki et al., 2012b). The presence of early sepsis was confirmed by transient systemic bacteremia and elevated cytokine levels in the first 5 days after CLP. Late/chronic sepsis (after day 5) was confirmed by enhanced peritoneal bacterial overgrowth and reduced circulating proinflammatory cytokines.
2.3. Isolation of Gr1+CD11b+ cells
Gr1+CD11b+ cells were isolated from the bone marrow using magnetically assisted cell sorting according to the manufacturer’s protocol (Miltenyi Biotech, Auburn, CA). The bone marrow cells were flushed out of the femurs with RPMI-1640 medium (without serum) under aseptic conditions (Brudecki et al., 2012b). A single cell suspension of the bone marrow was made by pipetting up and down and filtering through a 70-μm nylon strainer, followed by incubation with erythrocyte lysis buffer. After washing, total Gr1+CD11b+ cells were purified by subjecting the single cell suspension to positive selection of the Gr1+CD11b+ cells by incubating with biotin-coupled mouse anti-Gr1 antibody (Clone RB6-8C5; eBioscience, San Diego, CA) for 15 min at 4°C. Cells were then incubated with anti-biotin magnetic beads for 20 min at 4°C and subsequently passed over a MS column. Purified Gr1+CD11b+ cells were then washed and resuspended in sterile saline. The cell purity was determined by flow cytometry. Typically ~90% Gr1+CD11b+ cells were obtained by this procedure.
Gr1+CD11b+ cells were cultured in RPMI-1640 medium (Invitrogen, Carlsbad, CA) supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM L-glutamine (all from Hyclone Laboratories, Logan, UT), and 10% fetal bovine serum (Atlanta Biologicals, Lawrenceville, GA) at 37°C and 5% CO2.
2.4. ELISA
Cytokine production was determined using enzyme-linked immunosorbent assay (ELISA) kits (eBioscience) according to the manufacturer’s instructions. Each sample was run in duplicate.
2.5. C/EBPβ expression plasmid
Full length mouse Cebpb cDNA (transcript variant 1) was cloned in pEZ-M02 expression vector downstream of the CMV promoter, and C/EBPβ protein expression was verified by western blotting. A pReceiver-M02 vector served as a negative control.
2.6. Transfection of C/EBPβ plasmid and miRNA precusors
Plasmid DNA was suspended in HiPerFect reagent (Qiagen, Valencia, CA) (final concentration: 0.5 μg/ml). For miR-21 and miR-181b overexpression, negative control precursor or miR-21 or miR-181b precursor (Ambion) were suspended in a HiPerFect reagent at 50 nM final concentration. Gr1+CD11b+ cells were transfected using the Gene Pulser MXCell system (Bio-Rad, Herclues, CA). After 24 hr, portions of the cells were removed and either used for RNA isolation and miRNA measurements by PCR or stimulated for 12 hr with 1 μg/ml of LPS, and culture supernatants were used for cytokine measurements by ELISA. The remainder of the cells was differentiated for 6 days with M-CSF plus rIL-4 and then analyzed by flow cytometry.
For Stat3 knockdown, Gr1+CD11b+ cells were transfected with pools of Stat3-specific or scrambled (control) siRNAs (Santa Cruz Biotechnology) at a 0.5 μM final concentration as described above and then incubated for 36 hr.
2.7. miRNA measurement
Expression levels of miR-21 and miR-181b were determined by quatitative real-time PCR (RT-qPCR) using miRNA-enriched RNA and miScript SYBR Green PCR kit with miScript Primer Assays specific to miR-21 and miR-181b according to the manufacturer’s instructions (Qiagen). The relative expression was calculated using the 2−ΔΔCt cycle threshold method after normalization to the endogenous U6 RNA as an internal control.
2.8. Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) was performed to assess in vivo DNA-protein interactions at the miR-21 and miR-181b promoters using ChIP-IT Express Enzymatic Shearing kit according to the manufacturer’s instructions (Active Motif, Carlsbad, CA). Briefly, Gr1+CD11b+ cells were harvested from the bone marrow and protein-DNA complexes were cross-linked by fixation in 1% formaldehyde in minimal culture medium for 10 min at room temperature. After washing with cold PBS, cells were lysed in 1x lysis buffer containing protease inhibitor cocktail. Cell lysate was cleared by centrifugation at 5,000 rpm for 10 min at 4°C. The pelleted nuclei were then resuspended in digestion buffer and incubated with the enzymatic shearing cocktail at 37°C for 10 min. The sheared chromatin solution was recovered by centrifugation at 15,000rpm for 10 min at 4°C. Ten microliter of the chromatin solution was reserved as “input” DNA sample. The remaining chromatin solution was immunoprecipitated overnight at 4°C with protein G magnetic beads and 3 μg of antibody specific to p-Stat3, C/EBPα, C/EBPβ, or isotype control antibody (Santa Cruz Biotechnology). The chromatin/antibody complexes captured on the beads were washed three times in ChIP buffer and then eluted by incubation for 15 min in 50 μl elution buffer. Next, the DNA-protein cross-links were reveresed by incubating the eluted chromatin with 50 μl of reverese cross-linking buffer. The supernatant containing the DNA was then incubated, along with the “input” DNA samples at 95°C for 15 min. After treatment with proteinase K for 1 h at 37°C, the reaction was stopped, and the resulting DNA was extracted and stored at −20°C.
Enrichment of the miR-21 and miR-181b promoter sequences in the ChIPed DNA was measured by quantitative real-time PCR using primer and fluorescently labeled internal probe sequences (Integrated DNA Technologies, Coralville, IA) specific to the miR-21 and miR-181b promoters, as described previously (McClure et al., 2017).
2.9. Flow cytometry
Cells were stained by incubation for 30 min on ice in staining buffer (PBS plus 2% FBS) with the following antibodies: anti-Gr1 conjugated to FITC, anti-CD11b conjugated to phycoerythrin (PE), anti-F4/80 conjugated to allophycocyanin (APC), anti-CD11c conjugated to PE, anti-MHC II conjugated to FITC, anti-CD4 conjugated to PE (all from eBioscience, San Diego, CA). An appropriate isotype-matched control was used for each antibody. After washing, the samples were analyzed by a FACSCaliber flow cytometer (BD Biosciences, Sparks, MD). About 25,000 events were acquired and analyzed using the CellQuest Pro software (BD Biosciences).
2.10. Gr1+CD11b+ cell differentiation
Gr1+CD11b+ cells were cultured for 6 days with complete RPMI-1640 medium in the presence of 10 ng/ml of M-CSF (PeproTech Inc., Rocky Hill, NJ) and 10 ng/ml rIL-4 (eBioscience, San Diego, CA). The cell phenotypes were analyzed by flow cytometry.
2.11. T cell proliferation
Gr1+CD11b+ cells were cultured with CD4+ T cells to determine their effects on T cell proliferation and IFN-γ production. Briefly, spleen CD4+ T cells were isolated by positive selection using biotinylated anti-CD4 magnetic beads (Miltenyi). Cells were fluorescently labeled with carboxy-fluorosceindiacetate, succinimidyl ester (CFSE) dye using the Vybrant CFDA SE Cell Tacer Kit (Invitrogen Molecular Probes, Eugene, OR). Cells were incubated for 10 min at room temperature with 10 μM CFSE dye and then co-cultured (at 1:1 ratio) with Gr1+CD11b+ cells. T cell proliferation was induced by the stimulation with an anti-CD3 plus an anti-CD28 (R&D Systems, Minneapolis, MN) antibody (1 μg/ml/each). After 3 days, cells were harvested and CD4+ T cell proliferation was determined by the step-wise dilution of CFSE dye in dividing, CD3-gated CD4+ cells using flow cytometry. Culture supernatants were collected for the IFN-γ measurement.
2.12. Western blot
Equal amounts of protein extracts were mixed with 5x Laemmeli sample buffer, separated by a SDS-10% polyacrylamide gel (Bio-Rad) and subsequently transferred to nitrocellulose membranes (Thermo Scientific, Waltham, MA). After blocking with 5% milk in Tris-buffered saline/Tween-20 for 1 hr at room temperature, membranes were probed overnight at 4°C with the appropriate primary antibodies. After washing, blots were incubated with the appropriate HRP-conjugated secondary antibody (Life Technologies, Grand Island, NY) for 2 hr at room temperature. Proteins were detected with the enhanced chemiluminescence detection system (Thermo Fisher Scientific). The developed bands were visualized using the ChemiDoc XRS Detection System (Bio-Rad) and the images were captured with the Image Lab Software V3.0. Membranes were stripped and re-probed with b-actin antibody (Sigma-Aldrich) as a loading control.
2.13. Statistical analysis
All data are expressed as mean ± s.d. and were analyzed by Microsoft Excel, V3.0. Differences among groups were analyzed by a two-tailed student’s t test for two groups and by ANOVA for multiple groups. Statistical significance was designated at p < 0.05.
3. Results
3.1. Expression of miR-21 and miR-181b is lost in C/EBPβ-deficient Gr1+CD11b+ cells
Our previous studies showed that myeloid cell-specific inactivation of the Cebpb gene expression prevents the Gr1+CD11b+ MDSCs generation during sepsis and increases survival by 80% (McPeak et al., 2017). Morever, mice with C/EBPβ conditional knockout survive late sepsis and generate Gr1+CD11b+ immune competent myeloid progenitors (McPeak et al., 2017). C/EBPβ induces miR-21 and miR-181b to generate immunosuppressive Gr1+CD11b+ in the bone marrow of septic mice (McClure et al., 2017). In this study, we hypothesized that genetic deletion Cebpb will disrupt the path that leads to Gr1+CD11b+ MDSC generation. To test this, we first determined miR-21 and miR-181b expressions in sepsis Gr1+CD11b+ cells isolated from the bone marrow of control and C/EBPβ knockout mice. As shown in Fig. 1A, miR-21 and miR-181b expression was induced in early sepsis and further increased in late sepsis cells. However, C/EBPβ-deficient Gr1+CD11b+ cells from septic mice did not express miR-21 and miR-181b similar to cells from sham mice (lower panel).
Figure 1. Loss of miR-21 and miR-181b expression in C/EBPβ-deficient Gr1+CD11b+ cells during sepsis.

Sepsis was induced by cecal ligation and puncture (CLP) using a 23-gauge needle, and mice were given antibiotics (imipenem) with fluid resuscitation, which produced early (days 1–5) and late (days 6–28) sepsis phases. The Gr1+CD11b+ cells were isolated from the bone marrow of sham and septic mice using magnetic beads. (A) Measurments of miR-21 and miR-181b expression. MiRNA-enriched RNA was isolated, and levels of miR-21 and miR-181b were measured by RT-qPCR using miR-21 and miR-181b specific assay primer sets. Values were normalized to U6 RNA as an internal control. Data are expressed as mean ± s.d. (*p < 0.05) of 5 mice per group and are presented relative to values from, control sham cells (set at 1-fold). Data are from one of two experiments. *, compare with sham. (B) Transcription factor binding to the miRNA promoters. Gr1+CD11b+ cells were isolated and pooled (n = 3 mice per group) from the bone marrow of sham and septic mice. Cells were fixed in 1% formaldehyde to cross-link protein-DNA complexes. Cells were lyzed and the pelleted nuclei were digested with chromatin shearing enzymatic cocktail. The chromatin solution was then immunoprecipitated with antibodies specific to p-Stat3 (Tyr705), C/EBPβ, C/EBPα, or IgG isotype control antibody. Next, chromatin cross-links were reversed to recover the protein-bound DNA. The purified DNA was amplified by real-time PCR to measure the level of enrichment of miR-21 and miR-181b sequences in the immunoprecipitated complexes using promoter-specific primer/probe sets. PCR reactions were performed in triplicate. Samples were normalized to the “input” DNA (i.e., DNA isolated before immunoprecipitation) and are presented as fold enrichment relative to the IgG-immunoprecipitated samples (set at 1-fold). Data are expressed as mean ± s.d. (*p < 0.05) of 5 mice per group and represent one of two experiments. *, compared with IgG. cKO, conditional knockout.
We also reported that C/EBPα, and not C/EBPβ, binds the same consensus motif at miR-21 and miR-181b promoters and is required for generating immune competent Gr1+CD11b+ cells (McClure et al., 2017), thus disrupting expression of miR-21 and miR-181b. In this study and using Chromatin immunoprecipitation (ChIP) assay, we show that C/EBPβ binding at the miR-21 promoter in sepsis but not sham Gr1+CD11b+ cells from the control mice, which further increased in late sepsis cells (Fig. 1B), concurrent with the miRNA induction. In sepsis Gr1+CD11b+ cells from the C/EBPβ knockout mice, C/EBPα binding simulated cells from sham mice (Fig. 1B, lower panel). These results demonstrate that in the absence of C/EBPβ expression, C/EBPα binds to and activates the miR-21 and miR-181b promoters in sepsis Gr1+CD11b+ cells.
3.2. C/EBPβ plays an essential role in limiting Gr1+CD11b MDSC differentiation and maturation during sepsis
To confirm the role of C/EBPβ in the activation of miR-21 and miR-181b expressions in sepsis Gr1+CD11b+ cells, we ectopically expressed C/EBPβ in the C/EBPβ-deficient cells from late septic mice. As shown in Fig. 2A, we detected C/EBPβ protein at the miR-21 promoter with its binding pattern, Stat3, similar to that seen in control Gr1+CD11b+ cells. C/EBPβ binding paralleled miR-21 expression (Fig. 2B) and miR-181b expression (data not shown). Of note, C/EBPβ attenuated the Gr1+CD11b+ cells differentiation into macrophages and dendritic cells (Fig. 2C).
Figure 2. Expression of C/EBPβ in C/EBPβ-deficient Gr1+CD11b+ cells from late septic mice reactivates miRNA expression and attenuates cell differentiation.

Bone marrow Gr1+CD11b+ cells were isolated from control or C/EBPβ conditional knockout mice during late sepsis. Cells were transfected with a C/EBPβ expression plasmid or an empty vector. After 36 hr, portions of the cells were harvested and used for ChIP assay and PCR. (A) Transcription factor binding to the miR-21 promoter. Enrichment of the miR-21 promoter sequences in the ChIP DNA was measurec as described in Fig. 1. Right panel show western blotting of C/EBPβ protein levels after transfection with C/EBPβ. *, compare with IgG. (B) miR-21 expression was determined by real-time PCR as in Fig. 1. (C) The remainder of the cells (2×106) was differentiated for 6 days with M-CSF plus rIL-4 (10 ng/ml/each), and numbers of differentiated cells were determined by flow cytometry. Percentages of differentiated cells (left) and absolute numbers of cells (right) in the culture after 6-day incubation are shown. Data are expressed as mean ± s.d. (*/#p < 0.05) of 7 mice per group and represent one of two experiments. *, compared with cells from C/EBPβ control mice; #, compared with cells from C/EBPβ cKO mice and transfected with vector. cKO, conditional knockout.
We reported that late sepsis Gr1+CD11b+ cells are immunosuppressive producers of IL-10 (MDSCs) and inhibit T cell proliferation and activation (Brudecki et al., 2012; McClure et al., 2016). Here, septic control mice transfected with empty vector, Gr1+CD11b+ cells from the C/EBPβ conditional knockout mice reciprocally produced more TNFα and less IL-10 and did not suppress CD4+ T cell proliferation or IFNγ production (Fig. 3). Moreover, ectopic expression of C/EBPβ reprogrammed these cells into the immunosuppressive phenotype (Fig. 3). Together, these results demonstrate that C/EBPβ plays a pivotal role in supporting miR-21 and miR-181b expression in Gr1+CD11b+ immunosuppressive cells during sepsis.
Figure 3. Expression of C/EBPβ in C/EBPβ-deficient Gr1+CD11b+ cells from late septic mice switches them from the immunocompetent to the immunosuppressive phenotype.

Gr1+CD11b+ cells were isolated from the bone marrows of control and C/EBPβ conditional knockout mice during late sepsis and transfected with a C/EBPβ expression plasmid or an empty vector for 36 hr in the presence of 10 ng/ml of recombinant mouse IL-10 in order to activate Stat3. (A) C/EBPβ expression inhibits TNFα while inducing IL-10 production in C/EBPβ-deficient cells. Cells (1×106) were cultured with 1 μg/ml LPS for 12 h. Supernatants were collected and levels of TNFα and IL-10 were determined by ELISA. (B) C/EBPβ-deficient cells reconstituted with C/EBPβ suppress T cell proliferation. Gr1+CD11b+ cells were co-cultured (at 1:1 ratio) with spleen CD4+ T cells isolated from normal (naive) mice were labeled with the fluorescent dye CFSE for 10 min at room temperature. The cells were then incubated in the presence of anti-CD3 plus anti-CD28 antibodies (1 μg/ml/each). After 3 days, CD4+ T cell proliferation was determined by the step-wise dilution of CFSE dye in dividing CD4+ T cells by flow cytometry. Percentages of cell proliferation were calculated as follow: % cell proliferation = 100 x (count from T cell + Gr1+CD11b+ cell culture/count from T cell culture). Data are expressed as mean ± s.d. (*/#p < 0.05) of 6 mice per group and represent one of two experiments. *, compared with cells isolated from control mice; #, compared with cells isolated from C/EBPβ cKO mice and transfected with vector. cKO, conditional knockout.
3.3. C/EBPβ primarily generates immunosuppressive Gr1+CD11b+ MDSCs in sepsis by inducing miR-21 and miR-181b
We next investigated the essential role of miR-21 and miR-181b in generating immunosuppressive Gr1+CD11b+ cells in sepsis, using C/EBPβ-deficient cells without miR-21 and miR-181b expression (Fig. 1A). Transfection of miR-21 and miR-181b precursors elevated both miRNAs to a level even higher than C/EBPβ-expressing mice and subsequently attenuated their differentiation into macrophages and dendritic cells (Fig. 4). We then examined Gr1+CD11b+ cell cytokine production and immunosuppression of CD4+ T cells. miR-21 and miR-181b transfection into the sepsis C/EBPβ-deficient Gr1+CD11b+ cells diminished the TNFα levels and increased IL-10 production (Fig. 5A) and repressed CD4+ T cell proliferation and activation, as demonstrated by IFNγ production (Fig. 5B). Transfection with precursors of either miRNA alone did not elicit any significant effects (data not shown). These data support that, miR-21 and miR-181b expression is dominant and lies downstream of C/EBPβ in generating Gr1+CD11b+ MDSCs.
Figure 4. Expression of miR-21 and miR-181 in C/EBPβ-deficient Gr1+CD11b+ cells from late septic mice attenuates their differentiation capacity.

Gr1+CD11b+ cells were isolated from the bone marrow of control and C/EBPβ conditional knockout mice during late sepsis. Cells were transfected with negative control, miR-21 or miR-181b precursor at 50 nM final concentration. (A) Measurement of miR-21 and miR-181b levels. After 24 hr in culture, portion of the cells were used for RNA isolation and miRNA measurment by PCR. The results are presented as fold change relative to cells from control, sham mice (set at 1-fold). (B) Cell differentiation. The remainder of the cells was differentiated for 6 days with M-CSF plus rIL-4, and percentages of macrophage (F4/80+CD11b+) and dendritic cell (CD11c+MHC II+) were determined by flow cytometry. Data are expressed as mean ± s.d. (*/#p < 0.05) of 4 mice per group and represent one of three experiments. *, compared with cells isolated from control mice; #, compared with cells isolated from C/EBPβ cKO mice and transfected with control precursor. cKO, conditional knockout.
Figure 5. Expression of miR-21 and miR-181 in C/EBPβ-deficient Gr1+CD11b+ cells from late septic mice switches them into the immunosuppressive phenotype.

Gr1+CD11b+ cells were isolated from the bone marrow of control and C/EBPβ conditional knockout mice during late sepsis. Cells were transfected with negative control, miR-21 or miR-181b precursor (50 nM final concentration) in the presence of 10 ng/ml of recombinant IL-10. (A) Cytokine production. Cells (1×106) were cultured with 1 μg/ml LPS for 12 h. Supernatants were collected and levels of TNFα and IL-10 were determined by ELISA. (B) Effect on T cell proliferation. Gr1+CD11b+ cells were co-cultured (at 1:1 ratio) with spleen CD4+ T cells that have been isolated from normal (naive) mice and labeled with the fluorescent dye CFSE for 10 min at room temperature. The cells then were incubated in the presence of anti-CD3 plus anti-CD28 antibodies (1 μg/ml/each). After 3 days, CD4+ T cell proliferation was determined by the step-wise dilution of CFSE dye in dividing CD4+ cells by flow cytometry. Percentages of cell proliferation were calculated as follow: % cell proliferation = 100 x (count from T cell + Gr1+CD11b+ cell culture/count from T cell culture). Data are expressed as mean ± s.d. (*/#p < 0.05) of 5 mice per group and represent one of two experiments. *, compared with cells isolated from control mice; #, compared with cells isolated from C/EBPβ cKO mice and transfected control precursor. cKO, conditional knockout.
3.4. C/EBPβ conditional knockout reverses the immunosuppressive Gr1+CD11b+ MDSC development during sepsis
We previously reported that NFI-A induction by miR-21 and miR-181b drives Gr1+CD11b+ MDSC development in sepsis (McClure et al., 2016), by a complex pathway involving cyclin-dependent kinase (cdk4) and its inhibitor p21, PTEN, and NF-kB proteins (McClure et al., 2016). Here, we show that NFI-A expression was completely inhibited in the C/EBPβ-deficient cells with increased p21 levels, similar to cells from sham mice (Fig. 6A). Furthermore, reversing NFI-A and p21 expression patterns disrupted cdk4 protein complex and inactivated NF-kB (Fig. 6B), a terminal supporter of Gr1+CD11b+ cell expansion (McClure et al., 2016). Thus, C/EBPβ is a key and proximally drives Gr1+CD11b+ MDSC development in sepsis.
Figure 6. C/EBPβ conditional knockout leads to differential changes in expression of the proteins involved in Gr1+CD11b+ cell expansion in sepsis.
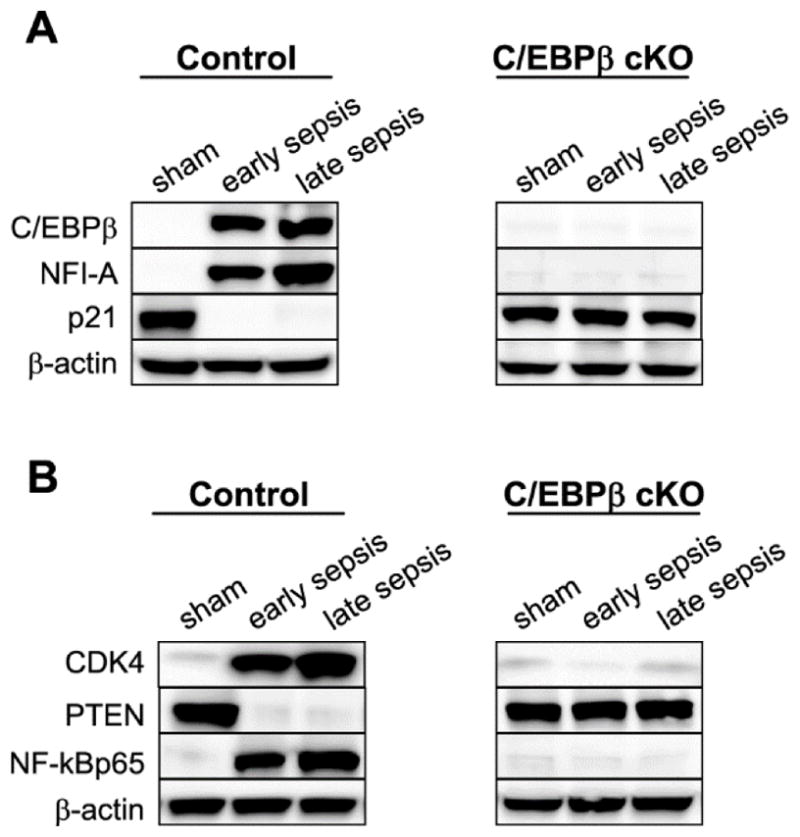
Gr1+CD11b+ cells were isolated from the bone marrows of control and C/EBPβ conditional knockout mice. Levels of the indicated proteins were determined by western blotting. The results represent one of three experiments. cKO, conditional knockout.
We also examined the p-Stat3 and miRNA levels in the presence or absence of exogeneous C/EBPβ protein. Both total and phosphorylated (active) Stat3 were expressed in the C/EBPβ-deficient cells (data not shown), and ectopic expression of C/EBPβ did not affect p-Stat3 levels, and meantime activated the miR-21 and miR-181b expressions (Fig. 7A). To confirm the above results, we ectopically expressed the C/EBPβ protein in sepsis C/EBPβ-deficient cells with or without Stat3 knockdown. As shown in Fig. 7B, lack of p-Stat3 prevented the miR-21 and miR-181b expression, despite the presence of the C/EBPβ protein. These results extend our previous findings of a synergism between p-Stat3 and C/EBPβ for supporting miR-21 and miR-181b expressions.
Figure 7. Induction of miR-21 and miR-181b expression ex vivo in C/EBPβ-deficient cells by C/EBPβ and Stat3.

Gr1+CD11b+ cells were isolated from the bone marrow of C/EBPβ conditional knockout mice during late sepsis. Cells were transfected either with a C/EBPβ expression plasmid or an empty vector alone (A) or plus Stat3-specific siRNA or control siRNA (B). After 36 hr, cells were stimulated with 10 ng/ml of recombinant mouse IL-6 to activate Stat3. Portions of the cells were harvested and levels of C/EBPβ and p-Stat3 were determined by western blotting. Levels of miR-21 and miR-181b were determined by real-time PCR. Data are expressed as mean ± s.d. (*p < 0.05) of 3 mice per group and represent one of three experiments. The results are presented as fold change relative to cells from control, sham mice (set at 1-fold) as in Fig. 1. *, compared with cells transfected with vector.
4. Discussion
We previously reported that C/EBPβ plays an essential and proximal role in generating immunosuppressive Gr1+CD11b+ myeloid cells (McClure et al., 2017; McPeak et al., 2017). Taking a genetic approach, we confirm and extend support of the concept that C/EBPβ epigenetically controls miR-21 and miR-181b expression to drive MDSC reprogramming to the repressor phenotype so important in sepsis outcome. Immunosuppressive Gr1+CD11b+ cells (i.e., MDSCs) secrete IL-10 to repress CD4+ T cells and likely innate immune phagocytes during sepsis. Our genetic based study further supports the clinically important concept that MDSC generation bridges repressor responses of innate and adaptive immune during sepsis—thereby promoting uncontrolled infection with many microbes, including fungi (Hotchkiss et al., 2013a; Hotchkiss et al., 2013b). Since late/chronic sepsis survival correlates with resolution of the immunosuppressive state, this study supports C/EBPβ targeting as a novel therapy for improving human sepsis outcomes. Most human sepsis deaths occur during the late stage of sepsis (Hotchkiss et al., 2013a; Mira et al., 2017).
Steady-state myelopoiesis maintains innate immunity cells (Ueda et al., 2005). However, sepsis accelerates myelopoiesis. The rapid mobilization of innate immunity cells, especially granulocytes, from the bone marrow in response to the microbial products and damage-associated molecular patterns creates niches in the bone marrow (Manz et al., 2014; Ueda et al., 2005), which under the stress of sepsis preferentially differentiate toward the myeloid lineage and more immature (progenitor) myeloid cells accumulate (Furze and Rankin, 2008; Manz et al., 2014). While this “emergency” myelpoiesis may aim to repopulate the innate immune system, in sepsis the cells do not mature normally. Because MDSCs are intermediate between myeloid progenitors arising from the hematopoietic stem cells and mature myeloid cells, a block in differentiation and maturation may account for MDSC generation (Dilek et al., 2012; Gabrilovich and Nagaraj, 2009; Talmadge and Gabrilovich, 2013). We have identified C/EBPβ as a key checkpoint for arresting Gr1+CD11b+ cells differentiation and maturation and promoting immune repressor cells. The numbers of the myeloid progenitors in the C/EBPβ conditional knockout mice are similar to the control, C/EBPβ-expressing mice (McPeak et al., 2017), suggesting that C/EBPβ is not involved in the steady-state myelopoieis during sepsis-driven immune suppression.
MiR-21 and miR-181b expression in Gr1+CD11b+ cells acts distal to C/EBPβ during sepsis to arrest differentiation and maturation (McClure et al., 2014). Mechanistically, C/EBPβ, jointly with p-Stat3, bind to and activate the miR-21 and miR-181b promoters (McClure et al., 2017). In line with this, expression of miR-21 and miR-181b was inhibited in C/EBPβ-deficient cells. In addition, we have previously shown that C/EBPβ and p-Stat3 proteins binds the miRNA promoters in a single complex (McClure et al., 2017). Together these data identify an epigenetic cooperative that includes NF-kB/RelB to silence selective genes by forming heterochromatin (El Gazzar et al., 2009; McCall et al., 2010; Chen et al., 2009). Importantly, this epigenetic reprogram is reversible and can be a target for promoting sepsis resolution.
There are potential extensions and implications of this study. Immunosuppression with disrupted development and differentiation of innate and adaptive immunity is a major part of sepsis, but the latest sepsis III 2016 conference report places organ failure as a primary cause of death and dysregulated inflammation and immunity as a major contributor (Singer et al., 2016). It will be important to determine if the C/EBPβ pathway, a ubiquitous transcription factor, disrupts organ physiology during sepsis.
Figure 8. A model for sepsis-induced myelopoiesis and generation of MDSCs.

During the immune response to sepsis, inflammatory cytokines such as IL-6 and IL-10, and bacterial endotoxins induce expression the of C/EBPβ, which synergizes with Stat3 to activate the miR-21 and miR-181b promoters in the Gr1+CD11b+ myeloid progenitors. The miRNAs then induce NFI-A expression, which attenuates the cell differentiation and maturation via decreasing p21, increasing cdk4, and decreasing PTEN protein levels (not shown). These persistently activate the NF-kB and thus accelerate myelopoiesis and expand numbers of Gr1+CD11b+ cells. During the early phase of sepsis, these cells are immunocompetent like Gr1+CD11b+ cells generated under steady-state conditions. During the late phase, Gr1+CD11b+ cells are reprogrammed into the immunosuppressive phenotype, i.e., MDSCs. The myeloid cell-specific knockout of C/EBPβ blocks this process during sepsis and thus prevents immunosuppression.
Highlights.
A mechanism for MDSC expansion during sepsis is proposed.
The mecanism involves induction of miR-21 and miR-181b by C/EBPβ.
Inhibiting C/EBPβ expression can prevent MDSC expansion and sepsis immunosuppression.
Acknowledgments
This work was supported by a National Institutes of Health Grant R01GM103887 (to M.E.).
Footnotes
Conflicts of interest
None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Alves-Filho JC, Spiller F, Cunha FQ. Neutrophil paralysis in sepsis. Shock. 2010;34(Suppl 1):15–21. doi: 10.1097/SHK.0b013e3181e7e61b. [DOI] [PubMed] [Google Scholar]
- 2.Boomer JS, To K, Chang KC, Takasu O, Osborne DF, Walton AH, Bricker TL, Jarman SD, Kreisel D, Krupnick AS, Srivastava A, Swanson PE, Green JM, Hotchkiss RS. Immunosuppression in patients who die of sepsis and multiple organ failure. JAMA. 2011;306:2594–2605. doi: 10.1001/jama.2011.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brudecki L, Ferguson DA, McCall CE, El Gazzar M. Myeloid-derived suppressor cells evolve during sepsis and can enhance or attenuate the systemic inflammatory response. Infect Immun. 2012a;80:2026–2034. doi: 10.1128/IAI.00239-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brudecki L, Ferguson DA, Yin D, Lesage GD, McCall CE, El Gazzar M. Hematopoietic stem-progenitor cells restore immunoreactivity and improve survival in late sepsis. Infect Immun. 2012b;80:602–611. doi: 10.1128/IAI.05480-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen X, Yoza BK, El Gazzar M, Hu JY, Cousart SL, McCall CE. RelB sustains IkappaBalpha expression during endotoxin tolerance. Clin Vaccine Immunol. 2009;16:104–110. doi: 10.1128/CVI.00320-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dilek N, Vuillefroy de SR, Blancho G, Vanhove B. Myeloid-derived suppressor cells: mechanisms of action and recent advances in their role in transplant tolerance. Front Immunol. 2012;3:208. doi: 10.3389/fimmu.2012.00208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.El Gazzar M, Yoza BK, Chen X, Garcia BA, Young NL, McCall CE. Chromatin-specific remodeling by HMGB1 and linker histone H1 silences proinflammatory genes during endotoxin tolerance. Mol Cell Biol. 2009;29:1959–1971. doi: 10.1128/MCB.01862-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ellaban E, Bolgos G, Remick D. Selective macrophage suppression during sepsis. Cell Immunol. 2004;231:103–111. doi: 10.1016/j.cellimm.2004.12.010. [DOI] [PubMed] [Google Scholar]
- 9.Furze RC, Rankin SM. Neutrophil mobilization and clearance in the bone marrow. Immunology. 2008;125:281–288. doi: 10.1111/j.1365-2567.2008.02950.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guignant C, Lepape A, Huang X, Kherouf H, Denis L, Poitevin F, Malcus C, Cheron A, Allaouchiche B, Gueyffier F, Ayala A, Monneret G, Venet F. Programmed death-1 levels correlate with increased mortality, nosocomial infection and immune dysfunctions in septic shock patients. Crit Care. 2011;15:R99. doi: 10.1186/cc10112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hirai H, Zhang P, Dayaram T, Hetherington CJ, Mizuno S, Imanishi J, Akashi K, Tenen DG. C/EBPbeta is required for ‘emergency’ granulopoiesis. Nat Immunol. 2006;7:732–739. doi: 10.1038/ni1354. [DOI] [PubMed] [Google Scholar]
- 13.Hotchkiss RS, Monneret G, Payen D. Immunosuppression in sepsis: a novel understanding of the disorder and a new therapeutic approach. Lancet Infect Dis. 2013a;13:260–268. doi: 10.1016/S1473-3099(13)70001-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hotchkiss RS, Monneret G, Payen D. Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat Rev Immunol. 2013b;13:862–874. doi: 10.1038/nri3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu TF, Brown CM, El Gazzar M, McPhail L, Millet P, Rao A, Vachharajani VT, Yoza BK, McCall CE. Fueling the flame: bioenergy couples metabolism and inflammation. J Leukoc Biol. 2012;92:499–507. doi: 10.1189/jlb.0212078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Manz MG, Boettcher S. Emergency granulopoiesis. Nat Rev Immunol. 2014;14:302–314. doi: 10.1038/nri3660. [DOI] [PubMed] [Google Scholar]
- 17.Marigo I, Bosio E, Solito S, Mesa C, Fernandez A, Dolcetti L, Ugel S, Sonda N, Bicciato S, Falisi E, Calabrese F, Basso G, Zanovello P, Cozzi E, Mandruzzato S, Bronte V. Tumor-induced tolerance and immune suppression depend on the C/EBPbeta transcription factor. Immunity. 2010;32:790–802. doi: 10.1016/j.immuni.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 18.Mazuski JE, Sawyer RG, Nathens AB, DiPiro JT, Schein M, Kudsk KA, Yowler C. The Surgical Infection Society guidelines on antimicrobial therapy for intra-abdominal infections: an executive summary. Surg Infect (Larchmt) 2002;3:161–173. doi: 10.1089/109629602761624171. [DOI] [PubMed] [Google Scholar]
- 19.McCall CE, Yoza B, Liu T, El Gazzar M. Gene-specific epigenetic regulation in serious infections with systemic inflammation. J Innate Immun. 2010;2:395–405. doi: 10.1159/000314077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McClure C, Ali E, Youssef D, Yao ZQ, McCall CE, El Gazzar M. NFI-A disrupts myeloid cell differentiation and maturation in septic mice. J Leukoc Biol. 2016;99:201–211. doi: 10.1189/jlb.4A0415-171RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McClure C, Brudecki L, Ferguson DA, Yao ZQ, Moorman JP, McCall CE, El Gazzar M. MicroRNA 21 (miR-21) and miR-181b couple with NFI-A to generate myeloid-derived suppressor cells and promote immunosuppression in late sepsis. Infect Immun. 2014;82:3816–3825. doi: 10.1128/IAI.01495-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McClure C, McPeak MB, Youssef D, Yao ZQ, McCall CE, El Gazzar M. Stat3 and C/EBPbeta synergize to induce miR-21 and miR-181b expression during sepsis. Immunol Cell Biol. 2017;95:42–55. doi: 10.1038/icb.2016.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McPeak MB, Youssef D, Williams DA, Pritchett CL, Yao ZQ, McCall CE, El Gazzar M. Frontline Science: Myeloid cell-specific deletion of Cebpb decreases sepsis-induced immunosuppression in mice. J Leukoc Biol. 2017;102:191–200. doi: 10.1189/jlb.4HI1216-537R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mira JC, Gentile LF, Mathias BJ, Efron PA, Brakenridge SC, Mohr AM, Moore FA, Moldawer LL. Sepsis Pathophysiology, Chronic Critical Illness, and Persistent Inflammation-Immunosuppression and Catabolism Syndrome. Crit Care Med. 2017;45:253–262. doi: 10.1097/CCM.0000000000002074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Niehof M, Streetz K, Rakemann T, Bischoff SC, Manns MP, Horn F, Trautwein C. Interleukin-6-induced tethering of STAT3 to the LAP/C/EBPbeta promoter suggests a new mechanism of transcriptional regulation by STAT3. J Biol Chem. 2001;276:9016–9027. doi: 10.1074/jbc.M009284200. [DOI] [PubMed] [Google Scholar]
- 26.Oberholzer A, Oberholzer C, Moldawer LL. Sepsis syndromes: understanding the role of innate and acquired immunity. Shock. 2001;16:83–96. doi: 10.1097/00024382-200116020-00001. [DOI] [PubMed] [Google Scholar]
- 27.Panopoulos AD, Zhang L, Snow JW, Jones DM, Smith AM, El Kasmi KC, Liu F, Goldsmith MA, Link DC, Murray PJ, Watowich SS. STAT3 governs distinct pathways in emergency granulopoiesis and mature neutrophils. Blood. 2006;108:3682–3690. doi: 10.1182/blood-2006-02-003012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Remick DG. Pathophysiology of sepsis. Am J Pathol. 2007;170:1435–1444. doi: 10.2353/ajpath.2007.060872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche JD, Coopersmith CM, Hotchkiss RS, Levy MM, Marshall JC, Martin GS, Opal SM, Rubenfeld GD, van der Poll T, Vincent JL, Angus DC. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3) JAMA. 2016;315:801–810. doi: 10.1001/jama.2016.0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Talmadge JE, Gabrilovich DI. History of myeloid-derived suppressor cells. Nat Rev Cancer. 2013;13:739–752. doi: 10.1038/nrc3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ueda Y, Kondo M, Kelsoe G. Inflammation and the reciprocal production of granulocytes and lymphocytes in bone marrow. J Exp Med. 2005;201:1771–1780. doi: 10.1084/jem.20041419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wesche DE, Lomas-Neira JL, Perl M, Chung CS, Ayala A. Leukocyte apoptosis and its significance in sepsis and shock. J Leukoc Biol. 2005;78:325–337. doi: 10.1189/jlb.0105017. [DOI] [PubMed] [Google Scholar]
- 33.Zhang H, Nguyen-Jackson H, Panopoulos AD, Li HS, Murray PJ, Watowich SS. STAT3 controls myeloid progenitor growth during emergency granulopoiesis. Blood. 2010;116:2462–2471. doi: 10.1182/blood-2009-12-259630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao JL, Rao DS, Boldin MP, Taganov KD, O’Connell RM, Baltimore D. NF-kappaB dysregulation in microRNA-146a-deficient mice drives the development of myeloid malignancies. Proc Natl Acad Sci U S A. 2011;108:9184–9189. doi: 10.1073/pnas.1105398108. [DOI] [PMC free article] [PubMed] [Google Scholar]