

Abstract
Both insulin signaling disruption and Ca2+ dysregulation are closely related to memory loss during aging and increase the vulnerability to Alzheimer's disease (AD). In hippocampal neurons, aging-related changes in calcium regulatory pathways have been shown to lead to higher intracellular calcium levels and an increase in the Ca2+-dependent afterhyperpolarization (AHP), which is associated with cognitive decline. Recent studies suggest that insulin reduces the Ca2+-dependent AHP. Given the sensitivity of neurons to insulin and evidence that brain insulin signaling is reduced with age, insulin-mediated alterations in calcium homeostasis may underlie the beneficial actions of insulin in the brain. Indeed, increasing insulin signaling in the brain via intranasal delivery has yielded promising results such as improving memory in both clinical and animal studies. However, while several mechanisms have been proposed, few have focused on regulation on intracellular Ca2+. In the present study, we further examined the effects of acute insulin on calcium pathways in primary hippocampal neurons in culture. Using the whole-cell patch-clamp technique, we found that acute insulin delivery reduced voltage-gated calcium currents. Fura-2 imaging was used to also address acute insulin effects on spontaneous and depolarization-mediated Ca2+ transients. Results indicate that insulin reduced Ca2+ transients, which appears to have involved a reduction in ryanodine receptor function. Together, these results suggest insulin regulates pathways that control intracellular Ca2+ which may reduce the AHP and improve memory. This may be one mechanism contributing to improved memory recall in response to intranasal insulin therapy in the clinic.
Keywords: diabetes, intranasal, excitability, imaging, electrophysiology, aging
Introduction
Aging is a major risk factor for Alzheimer's disease (AD), and both brain aging and AD are characterized by a progressive decline in cognitive and memory function. A promising approach for the treatment of cognitive impairment in AD seeks to maintain or enhance insulin signaling in the brain. This approach is based on nearly two decades of research that provides support for strong associations between diabetes, cognitive decline, and AD (Ott et al., 1996; Leibson et al., 1997; Luchsinger, 2010; Bosco et al., 2011; De Felice, 2013). Other support comes from studies showing that insulin can act as a cognitive and neural modulator (Baura et al., 1993; Cholerton et al., 2011; Heni et al., 2014a; Sartorius et al., 2015). Insulin acting in the brain is synthesized by pancreatic β-cells and gains access to the brain via the blood brain barrier. In the periphery and in the brain, insulin has been shown to bind to insulin receptors and to regulate glucose uptake by inducing translocation of glucose transporters to the plasma membrane (McNay et al., 2013).
In aging or AD, declining insulin levels, insulin receptor numbers, and/or glucose transporters can lead to dysregulation in glucose uptake which may underlie cognitive decline (Schioth et al., 2012a). Indeed, there is evidence that insulin receptor numbers and their functions are decreased in aging and AD animal models (Zaia & Piantanelli, 1996; Frolich et al., 1998; Zhao et al., 2004; Zhao et al., 2008). At the cellular level, brain insulin deficiency and reduction in insulin signaling, perhaps mediated by insulin resistance, could represent one of the altered pathways linked to altered memory function or synaptic communication during aging and AD (Craft et al., 1996; Rasgon & Jarvik, 2004; de la Monte et al., 2006; Talbot et al., 2012; De Felice, 2013; Sasaoka et al., 2014; Maimaiti et al., 2016). One approach to combat this reduction in insulin signaling that has received much interest clinically is the use of intranasal insulin delivery to selectively increase insulin concentration in the brain (Born et al., 2002).
The intranasal route of insulin administration raises insulin acutely in the central nervous system without much risk of peripheral hypoglycemia (Kern et al., 1999; Ott et al., 2014; Lochhead et al., 2015; Anderson et al., 2016; Maimaiti et al., 2016). Intranasal insulin therapy has been shown to improve memory function in AD patients and in healthy individuals (Benedict et al., 2004; Reger et al., 2006; Hanson & Frey, 2008; Reger et al., 2008; Craft et al., 2012; Schioth et al., 2012b; Freiherr et al., 2013; Craft et al., 2017). Further, animal models of aging and AD also show the positive impact of intranasal insulin in combating cognitive decline (Francis et al., 2008; Marks et al., 2009; Apostolatos et al., 2012; de la Monte, 2013; Adzovic et al., 2015; Salameh et al., 2015; Anderson et al., 2016; Maimaiti et al., 2016). Despite these promising outcomes, the mechanisms of action in the brain, and specifically on neurons of the hippocampus where insulin plays a recognizable role in learning and memory (Zhao et al., 1999; Zhao et al., 2004), remain largely unknown.
Because there is compelling evidence that aspects of cognitive aging may be due to calcium dysregulation that results in enhanced calcium levels (Porter et al., 1997; Thibault et al., 2001; Clodfelter et al., 2002; Hemond & Jaffe, 2005; Gant et al., 2006; Oh et al., 2013) and voltage-gated calcium channels (VGCC) activity (Moyer et al., 1992; Thibault & Landfield, 1996; Thibault et al., 2001; Nunez-Santana et al., 2014), we focused on studying two well-characterized calcium sources in hippocampal neurons. The rationale for this approach was based on recent evidence from our lab that insulin acutely reduces the calcium-dependent afterhyperpolarization (AHP) in neurons recorded from hippocampal slices (Pancani et al., 2013; Maimaiti et al., 2016). The AHP is a hyperpolarization potential that is enhanced in aging, limits neuronal firing, and is associated with cognitive decline (Disterhoft et al., 1996; Tombaugh et al., 2005; Kadish et al., 2009; Pancani et al., 2013). The larger AHP seen in aging, is mediated in part, by an increase in the density of L-VGCCs (Thibault & Landfield, 1996) and by calcium-induced calcium-release (CICR) through activation of ryanodine receptors (RyRs) (Kumar & Foster, 2005; Gant et al., 2006; Gant et al., 2013). Because we have also previously characterized these two calcium sources using neuronal depolarization protocols that evoke increases in intracellular calcium in hippocampal neurons (Attucci et al., 2002; Clodfelter et al., 2002; Gant et al., 2011), we sought to quantify their sensitivity to treatment with acute insulin. Here we used KCl-mediated long-lasting neuronal depolarization to activate large amplitude calcium transients under imaging conditions and also directly measure calcium influx in patch clamping experiments.
We tested the hypothesis that acute insulin (glulisine, zinc-free and fast acting) would reduce calcium levels, or promote calcium homeostasis by altering VGCC and/ or RyR function in cultured neurons. To address these questions, we used patch clamp recording of VGCCs with rapid drug delivery, as well as Fura-2 imaging during KCl-mediated depolarization in the presence or absence of a RyR blocker. Our results indicate that insulin is able to reduce intracellular calcium levels during periods of neuronal depolarization and that this is mediated, at least in part, by reductions in VGCC and RyR function. These two aging-sensitive neuronal Ca2+ targets, therefore, are sensitive to the actions of insulin and could represent novel therapeutic targets for cognitive decline in aging and/ or AD. Re-establishing Ca2+ homeostasis represents a mechanism by which insulin and by extension, its targeted delivery to the brain (i.e., intranasal insulin), may offset learning and memory dysregulation in vivo.
Experimental Procedures
Cell culture
Hippocampal mixed (neuron/glia) cultures were prepared as described previously (Porter et al., 1997; Pancani et al., 2009; Pancani et al., 2011) and established from (E18) Sprague-Dawley rats. E18 pups and hippocampi were dissected under a microscope in ice-cold Hank's balanced salt solution (Thermo Fisher Scientific Inc., MA, USA) supplemented with 4.2 mM NaHCO3, 10 mg/L gentamicin and 12 mM HEPES (pH 7.3). Hippocampi were transferred to a 37°C 0.25 % Trypsin EDTA solution (Thermo Fisher) and left at room temperature for 11 minutes. Trypsin was removed and the hippocampi were washed three times with Minimum Essential Medium (MEM). Hippocampi were then titrated, and diluted with MEM to the desired final concentration (5-7 × 105 neurons/mL) before being plated onto 35 mm poly-L-lysine coated dishes. Cultured neurons were incubated (36°C, 5% CO2, 95% O2) for 24 h before the first medium exchange. At this time, half of the medium was replaced with 90% SMEM supplemented with 10% Horse serum. After three days in vitro (DIV), half of the medium was replaced with SMEM, horse serum, 5-Fluoro-2-Dioxyuridine and uridine to stop glial cell growth. At DIV 10, a sodium bicarbonate solution (200 μL) was added to help maintain pH and limit evaporation.
For whole-cell recording experiments, plastic culture dishes were used (Corning Inc., Corning, NY, USA) and for calcium imaging experiments, glass bottom culture dishes (Mattek Crop., Ashland MA, USA) were used. All data presented were collected between DIV 13 and 17, when neuronal connections, the density of VGCCs, and measures of cell survival are relatively stable (Porter et al., 1997; Blalock et al., 1999). All experiments were conducted following 24 h exposure to a lower glucose-containing MEM (5.5 mM; MEM with no added glucose). This was done to maintain relatively normal glucose oxidation rates and insulin sensitivity (Pancani et al., 2011). All data presented were obtained at room temperature.
VGCC recording solution
External recording solution of VGCC currents was as follows (in mM): 111 NaCl, 5 BaCl.H2O, 5 CsCl, 2 MgCl2, 10 glucose, 10 HEPES, 20 TEA.Cl.H2O, pH 7.35 with NaOH, and 500 nM tetrodotoxin (TTX) was added before recording to inhibit Na+ channels. The internal pipette solution (in mM): 145 CH4O3S-methanesulfonic acid, 10 HEPES, 3 MgCl2, 11 EGTA, 1 CaCl2, 13 TEA.Cl.H2O, 14 phosphocreatine Tris-salt, 4 Tris-ATP, 0.3 Tris-GTP, pH 7.3 with CsOH. All solutions were sterile filtered.
Drugs and solution application
External solutions were delivered using a rapid solution exchange system (SF77A - Warner Instruments Corp. Hamden, CT, USA) positioned approximately 400 μm above the cell being recorded. Flow rate was set to 0.3 mL/min and a control solution supplemented with TTX was used to establish baseline recording (∼10 min) during which passive cell membrane properties were obtained (e.g., membrane resistance, capacitance, access resistance), and currents were allowed to run up. Rapidly switching the position of the tubing delivering the external solutions above the recorded or imaged cell allowed us to control the environment and deliver either insulin, KCl (50 mM), ryanodine (20 μM), a high-affinity small peptide interacting with insulin (affibody, ab31906, Abcam, Cambridge, MA, USA; 100 g/mL, 500 ng/mL, or 1 μg/mL), or an insulin receptor antibody (S961, Phoenix Pharmaceuticals Inc., Burlingame, CA., USA; 500 ng/mL). Insulin glulisine (10 nM, a fast acting, zinc-free insulin) was prepared weekly in external recording solution from a 6 μM stock (Apidra®, Sanofi-Avantis US. LLC, diluted in sterile saline). This concentration was chosen based on previous results from our lab (Pancani et al., 2013; Maimaiti et al., 2016).
Whole-cell recording and analysis
To minimize capacitance artifacts, whole-cell patch-clamp electrodes were coated with polystyrene Q-dope before recording (see Table 1 for membrane and electrode properties). The culture dish was rinsed with external solution twice and then supplemented with TTX. Current/voltage (I-V) relationships (−60 to + 30 mV) were used to identify the voltage step eliciting the maximal current amplitude. This voltage was used for each cell in the study in order to compare data at the peak of the I-V relationship (150 ms depolarization). Cells were either held at −70 mV or −40 mV and currents were elicited (every 30 s) at the maximal peak response. All currents were leak subtracted using 5-8 scaled hyperpolarizing sub-pulses. Currents were recorded once stable and for 10 min of either control or insulin solution (last 2 min of each condition was averaged). Because insulin may alter cell size, we report on measures of current densities (pA/pF), derived from dividing peak current amplitude by membrane capacitance (measured in pClamp) for each cell. All recordings were conducted on the stage of an epifluorescence microscope (E600FN - Nikon Inc., Melville, NY, USA) placed on an anti-vibration table. An amplifier (Axopatch 1D - Molecular Devices, Sunnyvale, CA, USA) in combination with an A/D board (Digidata 1200 - Molecular Devices) and acquisition software (pClamp 7 - Molecular Devices) were used for electrophysiology acquisition. Data were digitized at 5-10 KHz, low-pass filtered at 2-5 KHz, and were quantified in Clampfit 7 (Molecular Devices).
Table 1. Passive membrane properties and patch clamping parameters.
All data presented are means ± SEM on measurements from patched neurons in Figure 1. Capacitance, membrane resistance, access resistance, and holding current measures were not significantly different across the groups.
| Group | Capacitance (pF) | Membrane resistance (MΩ) | Access resistance (MΩ) | Holding current (pA) |
|---|---|---|---|---|
| Time Control (n=17) | 50.8 ± 3.9 | 432.9 ± 32.0 | 10.6 ± 0.9 | −102 ± 8.6 |
| Insulin (n=19) | 56.2 ± 3.5 | 415 ± 34.8 | 10.7 ± 0.8 | −100 ± 8.7 |
Ca2+ imaging and analysis
Cultures were incubated in Ca2+ imaging solution (in mM): 145 NaCl, 2.5 KCl, 10 HEPES, 10 D-glucose, 2 CaCl2, 1MgCl2, 0.01 glycine, pH 7.3 with NaOH for 30 min in the dark. This solution contained 2 μM Fura-2 AM (F1221 – Invitrogen) that was made monthly with fresh DMSO (0.085%) and Pluronic® F-127 (0.015%, weight/ volume). Each culture dish was then rinsed three times with Ca2+ imaging solution containing no indicator and placed in the dark for 20 min (de-esterification period). The dish was then placed on the stage of the microscope. Intracellular Ca2+ transients were visualized by exciting Fura-2 at 340 +/−20 nm and 380 +/− 20 nm using a high-speed filter changer (Lambda DG4, Sutter instruments, Novato, CA, USA) to obtain a ratiometric value independent of indicator concentration. Emitted light was passed through a dichroic filter (400 nm high pass) and an emitter filter (520 +/− 30 nm; Chroma Technology) and was digitized onto the sensor of an EMCCD camera (iXon-Andor Technology, Belfast, Ireland). Acquisition speed for spontaneous Ca2+ transients (Figure 2) and resting Ca2+ measures (Figure 3) was set to one ratio every 30 s. In order to better define the peak of the Ca2+ transient during KCl-mediated depolarization, acquisition speed was increased to one ratio every 5 s. For measures on spontaneous calcium transients (AUC, delta peak ratio, and number of events) all data were normalized to baseline (periods of non-activity). We integrated the AUC during 10 minutes of control solution perfusion, and insulin perfusion, and also quantified the amplitude of the largest calcium transient (delta peak) and the number of events in those time periods. Events were counted if their amplitude was greater than 25% of the peak calcium transient during a 10 min period. All calcium measures were analyzed using Imaging Workbench 5.0 (INDEC BioSystems, Santa Clara, CA) as previously published (Pancani et al., 2009; Pancani et al., 2011). Measures were taken during two 10 min periods of imaging solution perfusion (Time Control) or during a 10 min period of control solution, followed by 10 min of insulin perfusion (Insulin). The greater ratio (340/380 nm) values reported reflect higher Ca2+ levels. The gray value for both excitation wavelengths of each cell measured (region-of-interest - ROI drawn around soma) was background subtracted from an area devoid of cellular components. When imaging calcium during periods of depolarization (50 mM KCl), the Ca2+ imaging solution was modified with a reduction in NaCl to 97 mM in order to control for osmolarity changes.
Figure 2.

Spontaneous Ca2+ transients are not altered in response to acute insulin exposure. A. Example of neurons and ROI selection (green circles and numbered arrows- left). Calibration bar = 10 μm. Right shows spontaneous activity in the three ROIs (used to quantify insulin-mediated changes). Measures were taken during two 10 min periods of imaging solution perfusion (Time Control) or during a 10 min period of control solution followed by 10 min of insulin perfusion (Insulin). B.-D. Insulin perfusion did not change spontaneous calcium activity based on measures of AUC, peak transient amplitude, or the number of events detected. Data are presented as mean +/− SEM except for data in D, which represent median with interquartile ranges.
Figure 3.
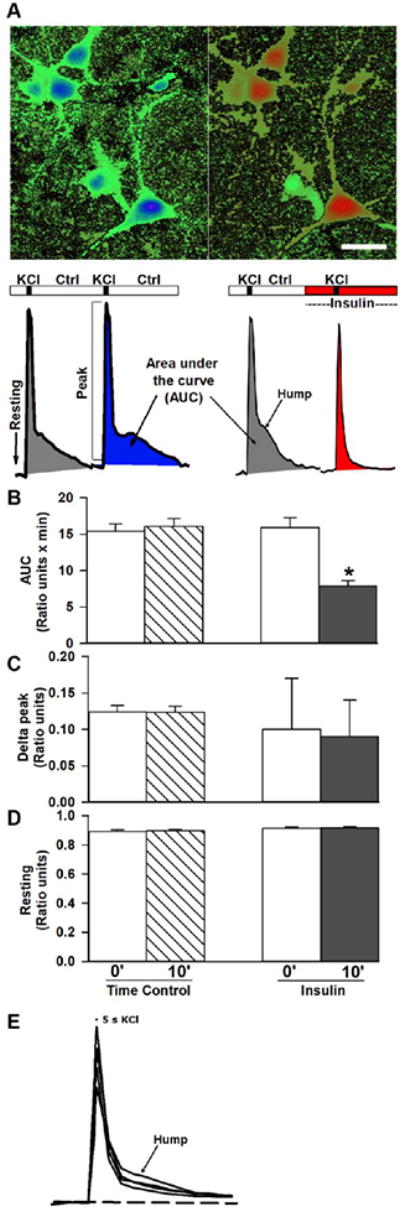
Effects of insulin on KCl-induced depolarization. A. Pseudocolored images of cells in culture during Fura-2 imaged at rest (left) and during peak depolarization (right). Calibration bar = 10 μm. Red colors indicate greater levels of calcium (higher ratios). Two sequential KCl depolarizations were separated by 10 minutes of either control solution or insulin. Representative calcium transients are shown and highlight measures of resting calcium, peak calcium and AUC. Note the selective effect of insulin on reducing the late phase of the calcium transient, essentially eliminating the “hump”. B. Ratiometric quantification during time control experiments or after insulin shows the AUC resulting from KCl-induced depolarization was significantly decreased by insulin. C.-D. Peak ratio or resting ratio measures were not altered by insulin. Data are presented as mean +/− SEM except for data in C, right, which represent median with interquartile ranges. Asterisks indicate significance at the p<0.05 level.
Statistics
Data were analyzed using Prism 5.0 (GraphPad Software, La Jolla, CA, USA). Because all variables analyzed were obtained before and after control or insulin solutions, either paired t-tests or ANOVAs (1-way or repeated) were used to test for group differences. When using ANOVAs to test for significance, Bonferroni's multiple comparison test were used as post-hoc tests. We tested for normal distributions and equal variances of all data presented, and equal variance was found in all groups. In cases where the normal distribution criterion was not met (Figures 1C left, 1F right; 2D, and 3C right), a Wilcoxon signed rank test was used and the median with interquartile range are presented. Significance was set at p< 0.05. For electrophysiology experiments, data from each recorded cell (solution perfusion precluded recording more than one cell per dish) was considered a single data point (i.e., n=1). For calcium imaging data, 5-10 cells in the field of view were averaged and considered a single data point (i.e., n=1). The data presented here are derived from 32 dams and over 550 imaged or recorded neurons in culture.
Figure 1.
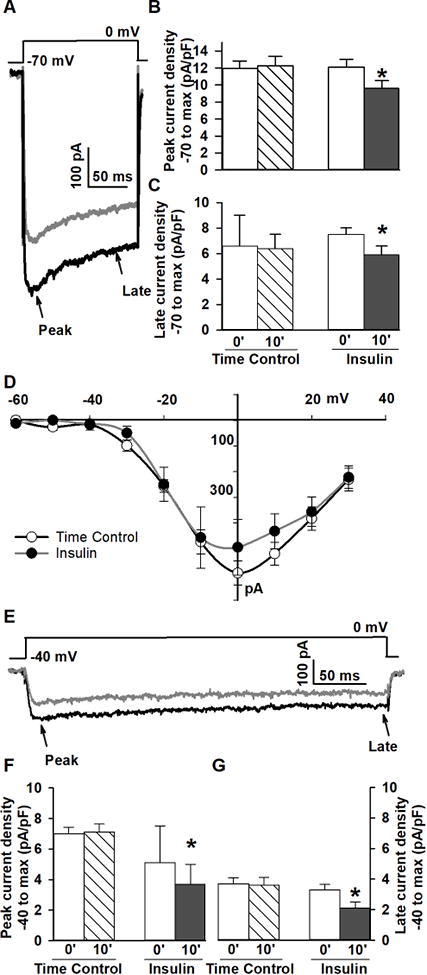
Calcium channel currents are reduced in response to acute insulin application in mixed hippocampal cultured neurons. A. Example of VGCC currents recorded during recording solution perfusion (black) and after 10 min insulin (grey) during maximal step depolarization from −70 mV. B.-C. Quantification across groups of cells following 10 min of insulin perfusion (Insulin) versus recording solution perfusion (Time Control) shows peak and late Ca2+ currents were significantly reduced (p<0.05). D. In a subgroup of cells, current-voltage relationships were not significantly altered. E. Example of the effects of insulin on L-VGCCs recorded from a holding potential of −40 mV during recording solution perfusion (black) and after 10 min insulin perfusion (gray). F.-G. Compared to time control, recordings from −40 mV, max peak (F) and late Ca2+ current (G) were significantly reduced by insulin (p<0.05). The effect of insulin on VGCC does not appear to be L-VGCC selective. Data are presented as mean +/− SEM except for data in F, right, which represent median with interquartile ranges. Data are normalized to cell size (density, pA/pF) and asterisks indicate significance at the p<0.05 level.
Results
Effect of insulin on whole-cell Ca2+currents
Ca2+ currents were recorded before and after a 10 min perfusion of either external solution (time control; n=19), or external solution supplemented with 10 nM insulin (n=17) using the SF77A fast perfusion system. Cells were held at −70 mV and were stepped to the peak voltage determined from the I-V relationship. Peak and late currents (obtained immediately prior to termination of the voltage step) were quantified and are presented normalized to cell size (pA/pF). A significant decrease in peak (−20%; p<0.0001; paired t-test) and late (−31%, p<0.002; paired t-test) Ca2+ currents in response to acute insulin application was seen (Figure 1A-C). Based on a subset of the cells recorded (n=7 per group), there was no change in the I-V relationship (Figure 1D). Recovery experiments (10-15 min washout following interruption of insulin) did not succeed, likely due to current rundown during this extended recording period.
To determine if the actions of insulin were selective for different Ca2+ channels, cells were held at −40 mV and stepped (350 ms) to peak voltage. Analysis indicated that similarly to reductions seen from a holding potential of −70 mV, L-type-enriched Ca2+ currents were significantly reduced (Figure 1E and F) both at the peak (−22%; p<0.0004; Wilcoxon rank test) and during the late current phase (−33%; p<0.005; paired t-test). The data also suggest that irrespective of holding potential, insulin did not alter the inactivation rate of currents (Figures 1A and 1E), and did not show a greater inhibitory effect on currents elicited from −40 mV. Together, these results indicate insulin does not selectively reduce L-VGCCs but instead, may reduce all VGCCs.
Effect of insulin on spontaneous Ca2+ transients
We then used ratiometric calcium imaging to test whether insulin could alter spontaneous activity in cultured neurons. We used a similar experimental time frame as presented in Figure 1 and quantified calcium oscillations during 10 min of Ca2+ imaging solution perfusion (time control; n=11) and during 10 minutes when the solution was supplemented with 10 nM insulin (n=14). Insulin did not change spontaneous network activity (no TTX) derived from measures of spontaneous Ca2+ events (Figure 2A – right). Those include measures of cumulative area-under-the curve measures during 10 minute periods either under control or insulin presentation (AUC; Figure 2B; p>0.05; paired t-test), peak transient amplitude (Figure 2C; p>0.05; paired t-test), and the number of spontaneous events detected (Figure 2D; p>0.05; Wilcoxon rank test). These spontaneous Ca2+ transients were completeley blocked by TTX (data not shown).
Effect of insulin on KCl-mediated Ca2+ transients
To induce larger and more predictable calcium transients, we used a potassium-mediated depolarization (30 s) in Ca2+ imaging experimental conditions and investigated the impact of insulin. This approach causes membrane depolarization with subsequent activation of VGCCs and calcium influx through the plasma membrane, which in turn, activates calcium-induced calcium-release (CICR) from the endoplasmic reticulum through activation of RyRs (Sukhareva et al., 2002; Berrout & Isokawa, 2009; Pancani et al., 2011). Potassium-induced Ca2+ transients were reduced by 10 min of insulin perfusion (n=23) compared to 10 min of Ca2+ imaging perfusion (time control; n=14; Figure 3A and B). Consistent with the results on spontaneous calcium fluctuations, insulin did not reduce resting Ca2+ levels measured immediately prior to the depolarization (Figure 3D), or the peak of the calcium responses (Figure 3C). However, measures of the AUC of the calcium transient unmasked a significant insulin-mediate reduction (Figure 3B; p<0.0001; paired t-test). Attempts to reverse this effect by washing out insulin for 10-20 minutes did not succeed, indicating the impact of insulin may be long-lasting under these conditions. Of interest, the reduction in the potassium-mediated Ca2+ response seemed to selectively reduce the delayed calcium component or “hump”, present after the peak response (Figure 3A, bottom right). Because in a subset of cells, shorter potassium depolarizations (5 s) also induced a hump (see examples in Figure 3E), and small humps are also occasionally seen under spontaneous oscillations conditions (i.e., Figure 2), we believe this delayed response is not an artifact of long duration depolarizations. Instead, we believe this highlights a potential link to the activation of CICR mechanisms, given that this response by nature is often delayed compared to an initiating and rapid calcium spike most likely triggered by VGCCs.
Insulin appears to reduce CICR function
To test whether the reduction in the “hump” during potassium-induced depolarization reflected inhibition of the CICR component, we used high concentrations of ryanodine (20 uM) which significantly reduce CICR (Clodfelter et al., 2002; Gant et al., 2006). These experiments required three repeated potassium depolarizations each separated by 10 min (time control; Figure 4). To test for the impact of ryanodine and/ or insulin, the first depolarization was triggered after 2 min of imaging solution perfusion, the second depolarization occurred after 10 min of ryanodine perfusion, and the third depolarization was after an additional 10 min of insulin perfusion (Figure 4A). As shown in Figure 4A and C, blocking CICR with ryanodine during potassium mediated depolarization (n=10) significantly reduced the “hump” after the initial high amplitude transient (Figure 4C, F(2,29) = 53.8; p <0.0001; RM ANOVA). As consistently shown throughout, time control experiments show no significant difference in the variables measured (n=11).
Fig 4.
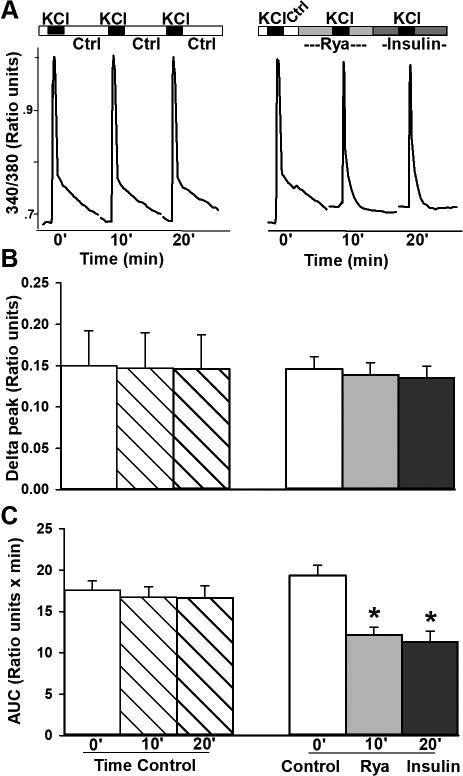
Ryanodine receptors are likely targets of insulin actions. A. Three sequential KCl-mediated depolarizations were used in these experiments and were triggered under imaging solution perfusion (left) and during perfusion with ryanodine, or insulin (right). Representative ratiometric calcium transients highlight the impact of inhibiting CICR with ryanodine on the “hump”. Addition of insulin did not further reduce the “hump”. B. Peak ratios were not different across groups. C. Quantification across the groups of cells imaged shows ryanodine (Rya) significantly reduced the “hump”, indicating ryanodine and insulin may both be working on inhibition of RyRs. All data represent mean +/− SEM. Asterisks indicate significance at the p<0.05 level.
No further reduction in the hump was seen when insulin was added to ryanodine treated cells, indicating that high ryanodine concentration may have occluded the impact of insulin. The lack of an additive effect between insulin and ryanodine actions suggests insulin may reduce CICR by inhibiting ryanodine receptors. Analysis of the peak calcium transient heights (delta peak Figure 4B) shows no significant differences across conditions. While this is surprising due to the results of Figure 1 showing insulin reduces VGCC, it is important to note that VGCC and RyR are in series and signals tend to merge in calcium imaging experiments. Further, the relatively rapid inactivating nature of VGCCs (calcium and/ or voltage) combined with a relatively slow calcium acquisition speed in these conditions (one ratio/ 5 s) preclude us from reliably identify the VGCC component during imaging experiments.
Insulin actions occur via occupation of the insulin receptor
To investigate whether 1) components other than insulin in the glulisine formulation, or whether 2) the actions of insulin were mediated via occupation of the insulin receptor, we used an insulin binding peptide (i.e., affibody) and an anti-insulin receptor antibody, respectively. For these experiments, we used the same experimental protocol as presented in Figure 3 with two potassium-mediated depolarization 10 min apart. Either the affibody peptide (n=6-16 per condition) or the antibody (n=9) was used. The results are again compared to time controls with perfusion of imaging solution (n=16). While lower doses of the affibody (100 ng and 500 ng/mL) did not prevent the insulin-mediated reductions in the calcium AUC (Figure 5A, p<0.01 and p<0.05, respectively, ANOVA), a higher dose (1 μg/ml) was able to completely inhibit this effect such that the reduction in AUC was no longer present (p>0.05; ANOVA). Doses used are modelled after in vivo studies (Paranjape et al., 2010; Luckett et al., 2013). Given that we did not perform similar blocking experiments on recorded cells (e.g., Figure 1), we cannot address whether the impact of insulin on calcium currents requires insulin binding to its own receptor. Nevertheless, the results presented here using imaging conditions suggest that the effects of insulin on hippocampal Ca2+ levels were mediated through activation of insulin receptors (Figure 5B - right).
Fig 5.

Anti-insulin affibody peptide and anti-insulin receptor antibody inhibited insulin actions on KCl-mediated Ca2+ transients. Experiments were conducted as in Figure 3, with 2 sequential KCl depolarizations. A. High-dose (1 μg/mL) anti-insulin affibody neutralized the effect of insulin on KCl-induced Ca2+ transients while lower concentrations (100 ng/mL and 500 ng/mL) did not. B. 500 ng/mL insulin receptor antibody also was able to inhibit insulin actions on the KCl-mediated Ca2+ transients. These results indicate the actions reported here on calcium homeostasis are mediated by insulin working on insulin receptors. All data represent mean +/− SEM. Asterisks indicate significance at the p<0.05 level.
Discussion
The experiments described here characterized the impact of acute insulin exposure on neuronal calcium currents and calcium levels. The techniques used provide the most direct approach for analyses on the impact of insulin on hippocampal neurons without the confounding interactions of blood vessels and other cell types. Cultured neurons provide a simplified environment where acute drug exposure is rapid and direct, free from the diffusion hindrance of the brain parenchyma or vascular interactions. Similar approaches in our lab and others have revealed significant connections between the actions of hormones and neuronal function (Clodfelter et al., 2002; Pancani et al., 2011). Our results provide evidence that Apidra, a zinc-free insulin formulation, is able to reduce VGCC currents and calcium levels attained during neuronal depolarization. The mechanism is dependent on insulin receptor activation, and also appears to engage ryanodine receptors as CICR contributions were reduced in response to acute insulin treatment.
Calcium currents are a target of acute insulin in neurons
The insulin-mediated reduction in whole-cell calcium currents does not appear to be specific to one subtype of VGCC. This is supported by evidence of currents recorded from −70 mV and −40 mV before and approximately 10 minutes after the addition of 10 nM insulin, showing the same degree of inhibition for the peak and late currents (i.e., similar inactivation rates before and after insulin in Figures 1A and 1E). This similar current decay argues for the reduction of a noninactivating current. Furthermore, the insulin-induced reduction in currents elicited from a holding potential of −40 mV, a potential favoring recording of L-type calcium currents, was not significantly greater in amplitude compared to the inhibition seen from −70 mV, arguing against a selective effect of insulin on L-type calcium channels. If N-type currents were selectively reduced by insulin, one would expect reductions from −70 mV but not from a holding potential of −40 mV. Conversely, if L-VGCC were selectively reduced by insulin, one would expect a great reduction in current from −40 mV (enriched in L-type since N-type are voltage inactivated), compared to holding at −70 mV. Previous work on regulation of VGCC in different cell types also notes that the effect of insulin may not be selective for a particular type of VGCC (e.g., N- or L-type VGCCs; (Strauss et al., 1997; Wijetunge et al., 2002).
This insulin mediated reduction in VGCC was not accompanied by a shift in the I-V relationship (Figure 1D), indicating the mechanism likely impacts flux of the charge carrier through the channels rather than alterations in the voltage sensitivity or inactivation properties of the channels. Insulin may mediate VGCC reduction through a mechanism involving tyrosine phosphorylation. Indeed, prior work in pinealocytes and photoreceptors, showing that tyrosine phosphorylation can reduce L-VGCC function (Chik et al., 1997; Stella et al., 2001). Evidence also suggests that the pore-forming alpha subunit of some VGCCs can be regulated directly by tyrosine residue phosphorylation (Strauss et al., 1997; Potier & Rovira, 1999; Wijetunge et al., 2002). Prior work has shown that acute insulin can alter other ion channels including large conductance Ca2+-activated potassium channels (BK-type). Under these conditions, insulin reduced somatic calcium levels during spontaneous oscillations in cultured hippocampal neurons (O'Malley & Harvey, 2007). Further, given the evidence that reductions in Src binding to the alpha 1 subunit of the L-VGCC significantly reduces channel current amplitude in cardiac myocytes (Dubuis et al., 2006) and that insulin can reduce Src activity (Arbet-Engels et al., 1999), this signaling pathway likely highlights another potential mechanism by which insulin reduces VGCC current flux.
Still, it should be noted that some cells respond to tyrosine kinase phosphorylation with enhanced VGCC function and potentiated calcium levels (Bence-Hanulec et al., 2000; Paez et al., 2010), and that given the complexity of second messenger systems working through many kinases and phosphatases (Dolphin, 2016), the exact impact of insulin on VGCC likely depends on the presence and state of activation of other interacting pathways (i.e., PKA, PKC, AKAP, phosphatases, etc.). Irrespective of the mechanism of inhibition of VGCC by insulin, it is clear that VGCC are a target of insulin in neurons. The effect described here, therefore, is likely to mediate the beneficial impact of the hormone on cognitive function following intranasal delivery in animal models (Apostolatos et al., 2012; Adzovic et al., 2015; Anderson et al., 2016; Maimaiti et al., 2016) and in early AD subjects (Reger et al., 2006; Reger et al., 2008; Craft et al., 2012; Freiherr et al., 2013; Rosenbloom et al., 2014; Claxton et al., 2015). Indeed, according to the calcium hypothesis of aging and dementia (Landfield, 1987; Khachaturian, 1989; Alzheimer's Association Calcium Hypothesis, 2017), reducing calcium levels in postsynaptic neurons is a favorable approach to offset cognitive decline in aging and AD (see below).
Other potential targets of insulin actions
Previous work shows insulin can increase cell surface expression of NMDA receptors (Skeberdis et al., 2001) and can reduce surface expression of AMPA receptors (Lin et al., 2000; Man et al., 2000). These studies employed similar insulin exposure time as used in the current study, but concentrations were 50-1000 fold higher. Thus, it is not clear whether transient changes in NMDA and AMPA surface expression would manifest using 10 nM insulin. As reported in Figure 2, the nearly physiological insulin concentration used here does not necessarily alter network excitability.
This is evidenced by a lack of insulin action on peak transient amplitude of the spontaneous calcium responses, the AUC of the spontaneous events, as well as the number of spontaneous events. Our work, therefore, may suggest that higher concentrations of insulin are needed to alter network oscillations, overall excitability and spontaneous activity. Alternatively, the ability to detect calcium changes using ratiometric Fura-2 imaging is somewhat restricted when compared to the speed of electrophysiological acquisition systems, and this may limit the ability to detect the more subtle and transient impact of insulin on network spontaneous activity. Although not tested directly here, our results also do not provide evidence that insulin can increase surface expression of GABAA receptors (Wan et al., 1997; Vetiska et al., 2007) which should have reduced excitability. Within the period tested (∼10 minutes) and at the low concentration used, we did not see evidence of depression in spontaneous calcium events (Figure 2). Thus, further studies focusing on insulin concentration-responses and time of exposure are needed to clarify these discrepancies. Of course, it will also be important to address the different types of available insulin formulations and to include the adequate zinc controls, given that almost all insulin formulations are zinc-based.
While insulin did not alter spontaneous calcium activity and calcium events amplitudes within the network of cells in culture (Figure 2), we did notice a significant alteration in the shape of the potassium-mediated calcium responses following insulin treatment (Figure 3). The disappearance of the “hump” following the peak calcium rise revealed the possibility that RyRs might be sensitive to insulin. We tested this hypothesis by pretreating the cultures with high concentrations of ryanodine to inhibit calcium-induced calcium release (CICR). As shown here, ryanodine pretreatment precluded further insulin-mediated inhibition of the calcium response to depolarization (Figure 4). This suggests insulin is capable of reducing CICR. This result is in line with prior evidence that neuronal store-operated Ca2+ channels (SOCCs) responsible for a small but significant secondary calcium entry pathway for replenishing the endoplasmic reticulum, are also a target of protein tyrosine phosphorylation (Koss et al., 2009), although it appears tyrosine phosphorylation may enhance this function. Of interest, RyR are rapidly tyrosine phosphorylated upon activation in activated T-lymphocytes (Guse et al., 2001) and growth hormone can tyrosine phosphorylate RyR in a β-cell-derived cell line (Zhang et al., 2004). Prior work has also shown that insulin can elicit an increase in calcium release through activation of RyR in muscle cells (Contreras-Ferrat et al., 2014). Future studies are necessary to identify if RyR subtypes can respond differently to insulin, whether these effects are tissue specific, dependent on particular signaling pathways, or whether sarcoplasmic-endoplasmic Ca2+-ATPases (SERCAs) function also is sensitive to insulin.
We suggest that the novel finding of insulin actions on CICR could be highly beneficial to neurons and to calcium dysregulation seen in aging and/ or AD, and that this new target potentially offers a secondary level of protection from sustained calcium-related actions in neurons. While it is not clear that the reduction in the “hump” (Figure 4) may provide a neuroprotective effect to neurons, the insulin-mediated reduction in depolarization observed during this calcium response could well keep neurons engaged during periods of activation.
Learning and memory selective actions of insulin in the brain
Past work studying insulin function in the brain identified critical links to learning and memory. Whether exogenous insulin on hippocampal neurons worked through changes in GABA, NMDA, AMPA, or even glucose transporters, it was understood that insulin could have a specific and selective impact on learning and memory and conversely, that learning also could have an impact on insulin receptors (Zhao et al., 1999; Dou et al., 2005). More recent analyses of insulin actions provide evidence that large and diverse areas of the CNS are indeed sensitive to insulin. Previous work shows insulin is a dynamic hormone with a rich influence on brain function.
Insulin stimulates the translocation of glucose transporter-4 to plasma membranes of hippocampal neurons within 15-30 min (Grillo et al., 2009). This rapid process is likely to regulate neuronal metabolic demands and the energy needed for learning and memory processing. In addition, insulin enhances synaptic plasticity by increasing the expression of dendritic postsynaptic density scaffolding protein (PSD-95), a key element of postsynaptic junctions in hippocampal neurons (Lee et al., 2005). Together these functions and their sensitivity to insulin undoubtedly contribute to improved encoding of synaptic information for the storage, and perhaps, the retrieval of memories.
Equally significant examples of the links between insulin and memory come from evidence that insulin can acutely reduce the hippocampal AHP, a calcium-dependent hyperpolarizing potential, which maintains hyperpolarization and precludes action potential threshold from being reached (Pancani et al., 2013; Maimaiti et al., 2016). The AHP is larger in aged compared to young animals (Landfield & Pitler, 1984; Potier et al., 1992; Disterhoft et al., 1996; Hemond & Jaffe, 2005; Tombaugh et al., 2005; Kumar & Foster, 2007; Gant et al., 2011), thus reducing the amplitude or duration of this hyperpolarizing potential may help combat age-related memory dysregulation by allowing cells to participate within a functional network (Gant & Thibault, 2009). Indeed, it has been shown that elevating potassium channel activity impairs learning (McKay et al., 2012), while lowering L-VGCC contribution to the AHP reduces the AHP (Moyer et al., 1992; Gamelli et al., 2011) and enhances learning (Deyo et al., 1989; Moyer et al., 1992). Moreover, increasing cholinergic neurotransmission which enhances cellular excitability also can enhance learning (Saar et al., 1998; Oh et al., 1999; Saar et al., 2001; Disterhoft & Oh, 2006).
The evidence of robust insulin actions linked to memory processes or processes capable of facilitating memory encoding (e.g., metabolism, blood flow), does not detract from the traditional role of insulin in glucose homeostasis (Vianna & Coppari, 2011; Heni et al., 2014b). Indeed, it is becoming very clear that once in the brain, the hormone is not limited in function to a single or even a few target areas (Kullmann et al., 2013; Kullmann et al., 2015), strengthening its position as a potential therapeutic target which requires further research emphasis.
Conclusions
We used imaging and electrophysiology techniques to show that zinc-free insulin has rapid and selective effects on calcium sensitive functions in neurons, and that the effects are dependent on insulin activating the insulin receptor (Figure 5). This clearly engages a series of intracellular signals that through uncharacterized pathways or mechanisms, lead to rapid calcium reductions during neuronal depolarization. More research will need to be conducted to fully characterize the underlying mechanism and to address the selectivity of the effect using different insulin formulations. Importantly, our research and those of others show that reducing calcium dysregulation in brain aging, AD and in neuronal cultures has generally been associated with positive and neuroprotective outcomes, from improving cognitive functions to reducing cellular toxicity. We present evidence here that insulin could well be an endogenous modulator of these functions.
Reductions in insulin levels or in insulin signaling in the brain during aging and in AD may highlight one of the mechanisms that could reduce metabolism, thereby reducing cognitive function. However, our results also suggest that replacing insulin or enhancing insulin signaling in the brain of aging, or early AD subjects may be a valuable approach. We emphasize that the novel impact of insulin on neurons presented here could well underlie the beneficial impact of intranasal insulin on cognitive enhancement in the clinic and in animal studies. An important caveat is that it is not clear from our data whether insulin is able to alter calcium homeostasis in vivo, nor is it clear that KCl depolarizations used here reflect activation of processes that are also initiated in the hippocampus during synaptic activation, albeit in more restricted time periods (i.e., single action potentials or bursts of activity). If insulin is able to reduce calcium levels in vivo, the noted reductions in insulin-sensitive calcium responses may offset brain aging processes and ultimately, offset the impact of dementia on brain function.
Highlights.
Acute insulin reduces VGCC currents and calcium levels in hippocampal neurons
Ryanodine receptors appear to be sensitive to insulin
Insulin-mediated interactions with calcium signaling pathways may underlie its cognitive enhancement effects
Acknowledgments
This work was supported by the National Institutes of Health research grant to OT (R01AG033649) and a training grant to HF (T32DK007778).
Abbreviations
- AHP
afterhyperpolarization
- VGCC
voltage-gated Ca2+ channel
- AD
Alzheimer's disease
- KCl
potassium chloride
- RyR
ryanodine receptor
- Rya
ryanodine
- SEM
standard error of the mean
- TTX
tetrodotoxin, CICR, calcium-induced calcium-release
- AUC
area-under-the-curve
- ROI
region-of-interest
- SERCA
sarcoplasmic-endoplasmic Ca2+-ATPase
- L-VGCC
L-type voltage-gated Ca2+ current
- SOCCs
store-operated calcium channels
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adzovic L, Lynn AE, D'Angelo HM, Crockett AM, Kaercher RM, Royer SE, Hopp SC, Wenk GL. Insulin improves memory and reduces chronic neuroinflammation in the hippocampus of young but not aged brains. J Neuroinflammation. 2015;12:63. doi: 10.1186/s12974-015-0282-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alzheimer's Association Calcium Hypothesis, W. Calcium Hypothesis of Alzheimer's disease and brain aging: A framework for integrating new evidence into a comprehensive theory of pathogenesis. Alzheimers Dement. 2017 doi: 10.1016/j.jalz.2016.12.006. [DOI] [PubMed] [Google Scholar]
- Anderson KL, Frazier HN, Maimaiti S, Bakshi VV, Majeed ZR, Brewer LD, Porter NM, Lin AL, Thibault O. Impact of Single or Repeated Dose Intranasal Zinc-free Insulin in Young and Aged F344 Rats on Cognition, Signaling, and Brain Metabolism. J Gerontol A Biol Sci Med Sci. 2016 doi: 10.1093/gerona/glw065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apostolatos A, Song S, Acosta S, Peart M, Watson JE, Bickford P, Cooper DR, Patel NA. Insulin promotes neuronal survival via the alternatively spliced protein kinase CdeltaII isoform. J Biol Chem. 2012;287:9299–9310. doi: 10.1074/jbc.M111.313080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbet-Engels C, Tartare-Deckert S, Eckhart W. C-terminal Src kinase associates with ligand-stimulated insulin-like growth factor-I receptor. J Biol Chem. 1999;274:5422–5428. doi: 10.1074/jbc.274.9.5422. [DOI] [PubMed] [Google Scholar]
- Attucci S, Clodfelter GV, Thibault O, Staton J, Moroni F, Landfield PW, Porter NM. Group I metabotropic glutamate receptor inhibition selectively blocks a prolonged Ca(2+) elevation associated with age-dependent excitotoxicity. Neuroscience. 2002;112:183–194. doi: 10.1016/s0306-4522(02)00002-7. [DOI] [PubMed] [Google Scholar]
- Baura GD, Foster DM, Porte D, Jr, Kahn SE, Bergman RN, Cobelli C, Schwartz MW. Saturable transport of insulin from plasma into the central nervous system of dogs in vivo. A mechanism for regulated insulin delivery to the brain. J Clin Invest. 1993;92:1824–1830. doi: 10.1172/JCI116773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bence-Hanulec KK, Marshall J, Blair LA. Potentiation of neuronal L calcium channels by IGF-1 requires phosphorylation of the alpha1 subunit on a specific tyrosine residue. Neuron. 2000;27:121–131. doi: 10.1016/s0896-6273(00)00014-3. [DOI] [PubMed] [Google Scholar]
- Benedict C, Hallschmid M, Hatke A, Schultes B, Fehm HL, Born J, Kern W. Intranasal insulin improves memory in humans. Psychoneuroendocrinology. 2004;29:1326–1334. doi: 10.1016/j.psyneuen.2004.04.003. [DOI] [PubMed] [Google Scholar]
- Berrout J, Isokawa M. Homeostatic and stimulus-induced coupling of the L-type Ca2+ channel to the ryanodine receptor in the hippocampal neuron in slices. Cell Calcium. 2009;46:30–38. doi: 10.1016/j.ceca.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blalock EM, Porter NM, Landfield PW. Decreased G-protein-mediated regulation and shift in calcium channel types with age in hippocampal cultures. J Neurosci. 1999;19:8674–8684. doi: 10.1523/JNEUROSCI.19-19-08674.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Born J, Lange T, Kern W, McGregor GP, Bickel U, Fehm HL. Sniffing neuropeptides: a transnasal approach to the human brain. Nat Neurosci. 2002;5:514–516. doi: 10.1038/nn849. [DOI] [PubMed] [Google Scholar]
- Bosco D, Fava A, Plastino M, Montalcini T, Pujia A. Possible implications of insulin resistance and glucose metabolism in Alzheimer's disease pathogenesis. Journal of cellular and molecular medicine. 2011;15:1807–1821. doi: 10.1111/j.1582-4934.2011.01318.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chik CL, Li B, Karpinski E, Ho AK. Insulin and insulin-like growth factor-I inhibit the L-type calcium channel current in rat pinealocytes. Endocrinology. 1997;138:2033–2042. doi: 10.1210/endo.138.5.5129. [DOI] [PubMed] [Google Scholar]
- Cholerton B, Baker LD, Craft S. Insulin resistance and pathological brain ageing. Diabet Med. 2011;28:1463–1475. doi: 10.1111/j.1464-5491.2011.03464.x. [DOI] [PubMed] [Google Scholar]
- Claxton A, Baker LD, Hanson A, Trittschuh EH, Cholerton B, Morgan A, Callaghan M, Arbuckle M, Behl C, Craft S. Long-acting intranasal insulin detemir improves cognition for adults with mild cognitive impairment or early-stage Alzheimer's disease dementia. J Alzheimers Dis. 2015;44:897–906. doi: 10.3233/JAD-141791. [DOI] [PubMed] [Google Scholar]
- Clodfelter GV, Porter NM, Landfield PW, Thibault O. Sustained Ca2+-induced Ca2+-release underlies the post-glutamate lethal Ca2+ plateau in older cultured hippocampal neurons. Eur J Pharmacol. 2002;447:189–200. doi: 10.1016/s0014-2999(02)01843-5. [DOI] [PubMed] [Google Scholar]
- Contreras-Ferrat A, Lavandero S, Jaimovich E, Klip A. Calcium signaling in insulin action on striated muscle. Cell Calcium. 2014;56:390–396. doi: 10.1016/j.ceca.2014.08.012. [DOI] [PubMed] [Google Scholar]
- Craft S, Baker LD, Montine TJ, Minoshima S, Watson GS, Claxton A, Arbuckle M, Callaghan M, Tsai E, Plymate SR, Green PS, Leverenz J, Cross D, Gerton B. Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: a pilot clinical trial. Arch Neurol. 2012;69:29–38. doi: 10.1001/archneurol.2011.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craft S, Claxton A, Baker LD, Hanson AJ, Cholerton B, Trittschuh EH, Dahl D, Caulder E, Neth B, Montine TJ, Jung Y, Maldjian J, Whitlow C, Friedman S. Effects of Regular and Long-Acting Insulin on Cognition and Alzheimer's Disease Biomarkers: A Pilot Clinical Trial. J Alzheimers Dis. 2017;57:1325–1334. doi: 10.3233/JAD-161256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craft S, Newcomer J, Kanne S, Dagogo-Jack S, Cryer P, Sheline Y, Luby J, Dagogo-Jack A, Alderson A. Memory improvement following induced hyperinsulinemia in Alzheimer's disease. Neurobiol Aging. 1996;17:123–130. doi: 10.1016/0197-4580(95)02002-0. [DOI] [PubMed] [Google Scholar]
- De Felice FG. Alzheimer's disease and insulin resistance: translating basic science into clinical applications. J Clin Invest. 2013;123:531–539. doi: 10.1172/JCI64595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Monte SM. Intranasal insulin therapy for cognitive impairment and neurodegeneration: current state of the art. Expert Opin Drug Deliv. 2013;10:1699–1709. doi: 10.1517/17425247.2013.856877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Monte SM, Tong M, Lester-Coll N, Plater M, Jr, Wands JR. Therapeutic rescue of neurodegeneration in experimental type 3 diabetes: Relevance to Alzheimer's disease. J Alzheimers Dis. 2006;10:89–109. doi: 10.3233/jad-2006-10113. [DOI] [PubMed] [Google Scholar]
- Deyo RA, Straube KT, Disterhoft JF. Nimodipine facilitates associative learning in aging rabbits. Science. 1989;243:809–811. doi: 10.1126/science.2916127. [DOI] [PubMed] [Google Scholar]
- Disterhoft JF, Oh MM. Pharmacological and molecular enhancement of learning in aging and Alzheimer's disease. J Physiol Paris. 2006;99:180–192. doi: 10.1016/j.jphysparis.2005.12.079. [DOI] [PubMed] [Google Scholar]
- Disterhoft JF, Thompson LT, Moyer JR, Jr, Mogul DJ. Calcium-dependent afterhyperpolarization and learning in young and aging hippocampus. Life Sci. 1996;59:413–420. doi: 10.1016/0024-3205(96)00320-7. [DOI] [PubMed] [Google Scholar]
- Dolphin AC. Voltage-gated calcium channels and their auxiliary subunits: physiology and pathophysiology and pharmacology. J Physiol. 2016;594:5369–5390. doi: 10.1113/JP272262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou JT, Chen M, Dufour F, Alkon DL, Zhao WQ. Insulin receptor signaling in long-term memory consolidation following spatial learning. Learn Mem. 2005;12:646–655. doi: 10.1101/lm.88005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubuis E, Rockliffe N, Hussain M, Boyett M, Wray D, Gawler D. Evidence for multiple Src binding sites on the alpha1c L-type Ca2+ channel and their roles in activity regulation. Cardiovasc Res. 2006;69:391–401. doi: 10.1016/j.cardiores.2005.11.006. [DOI] [PubMed] [Google Scholar]
- Francis GJ, Martinez JA, Liu WQ, Xu K, Ayer A, Fine J, Tuor UI, Glazner G, Hanson LR, Frey WH, 2nd, Toth C. Intranasal insulin prevents cognitive decline, cerebral atrophy and white matter changes in murine type I diabetic encephalopathy. Brain. 2008;131:3311–3334. doi: 10.1093/brain/awn288. [DOI] [PubMed] [Google Scholar]
- Freiherr J, Hallschmid M, Frey WH, 2nd, Brunner YF, Chapman CD, Holscher C, Craft S, De Felice FG, Benedict C. Intranasal insulin as a treatment for Alzheimer's disease: a review of basic research and clinical evidence. CNS Drugs. 2013;27:505–514. doi: 10.1007/s40263-013-0076-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frolich L, Blum-Degen D, Bernstein HG, Engelsberger S, Humrich J, Laufer S, Muschner D, Thalheimer A, Turk A, Hoyer S, Zochling R, Boissl KW, Jellinger K, Riederer P. Brain insulin and insulin receptors in aging and sporadic Alzheimer's disease. J Neural Transm. 1998;105:423–438. doi: 10.1007/s007020050068. [DOI] [PubMed] [Google Scholar]
- Gamelli AE, McKinney BC, White JA, Murphy GG. Deletion of the L-type calcium channel Ca(V) 1.3 but not Ca(V) 1.2 results in a diminished sAHP in mouse CA1 pyramidal neurons. Hippocampus. 2011;21:133–141. doi: 10.1002/hipo.20728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gant JC, Blalock EM, Chen KC, Kadish I, Porter NM, Norris CM, Thibault O, Landfield PW. FK506-binding protein 1b/12.6: A key to aging-related hippocampal Ca dysregulation? Eur J Pharmacol. 2013 doi: 10.1016/j.ejphar.2013.10.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gant JC, Chen KC, Norris CM, Kadish I, Thibault O, Blalock EM, Porter NM, Landfield PW. Disrupting function of FK506-binding protein 1b/12.6 induces the Ca(2)+-dysregulation aging phenotype in hippocampal neurons. J Neurosci. 2011;31:1693–1703. doi: 10.1523/JNEUROSCI.4805-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gant JC, Sama MM, Landfield PW, Thibault O. Early and simultaneous emergence of multiple hippocampal biomarkers of aging is mediated by Ca2+-induced Ca2+ release. J Neurosci. 2006;26:3482–3490. doi: 10.1523/JNEUROSCI.4171-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gant JC, Thibault O. Action potential throughput in aged rat hippocampal neurons: regulation by selective forms of hyperpolarization. Neurobiol Aging. 2009;30:2053–2064. doi: 10.1016/j.neurobiolaging.2008.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grillo CA, Piroli GG, Hendry RM, Reagan LP. Insulin-stimulated translocation of GLUT4 to the plasma membrane in rat hippocampus is PI3-kinase dependent. Brain Res. 2009;1296:35–45. doi: 10.1016/j.brainres.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guse AH, Tsygankov AY, Weber K, Mayr GW. Transient tyrosine phosphorylation of human ryanodine receptor upon T cell stimulation. J Biol Chem. 2001;276:34722–34727. doi: 10.1074/jbc.M100715200. [DOI] [PubMed] [Google Scholar]
- Hanson LR, Frey WH., 2nd Intranasal delivery bypasses the blood-brain barrier to target therapeutic agents to the central nervous system and treat neurodegenerative disease. BMC Neurosci. 2008;9 Suppl 3:S5. doi: 10.1186/1471-2202-9-S3-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemond P, Jaffe DB. Caloric restriction prevents aging-associated changes in spike-mediated Ca2+ accumulation and the slow afterhyperpolarization in hippocampal CA1 pyramidal neurons. Neuroscience. 2005;135:413–420. doi: 10.1016/j.neuroscience.2005.05.044. [DOI] [PubMed] [Google Scholar]
- Heni M, Schopfer P, Peter A, Sartorius T, Fritsche A, Synofzik M, Haring HU, Maetzler W, Hennige AM. Evidence for altered transport of insulin across the blood-brain barrier in insulin-resistant humans. Acta Diabetol. 2014a;51:679–681. doi: 10.1007/s00592-013-0546-y. [DOI] [PubMed] [Google Scholar]
- Heni M, Wagner R, Kullmann S, Veit R, Mat Husin H, Linder K, Benkendorff C, Peter A, Stefan N, Haring HU, Preissl H, Fritsche A. Central insulin administration improves whole-body insulin sensitivity via hypothalamus and parasympathetic outputs in men. Diabetes. 2014b;63:4083–4088. doi: 10.2337/db14-0477. [DOI] [PubMed] [Google Scholar]
- Kadish I, Thibault O, Blalock EM, Chen KC, Gant JC, Porter NM, Landfield PW. Hippocampal and cognitive aging across the lifespan: a bioenergetic shift precedes and increased cholesterol trafficking parallels memory impairment. J Neurosci. 2009;29:1805–1816. doi: 10.1523/JNEUROSCI.4599-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern W, Born J, Schreiber H, Fehm HL. Central nervous system effects of intranasally administered insulin during euglycemia in men. Diabetes. 1999;48:557–563. doi: 10.2337/diabetes.48.3.557. [DOI] [PubMed] [Google Scholar]
- Khachaturian ZS. Calcium, membranes, aging, and Alzheimer's disease. Introduction and overview. Ann N Y Acad Sci. 1989;568:1–4. doi: 10.1111/j.1749-6632.1989.tb12485.x. [DOI] [PubMed] [Google Scholar]
- Koss DJ, Riedel G, Platt B. Intracellular Ca2+ stores modulate SOCCs and NMDA receptors via tyrosine kinases in rat hippocampal neurons. Cell Calcium. 2009;46:39–48. doi: 10.1016/j.ceca.2009.04.001. [DOI] [PubMed] [Google Scholar]
- Kullmann S, Frank S, Heni M, Ketterer C, Veit R, Haring HU, Fritsche A, Preissl H. Intranasal insulin modulates intrinsic reward and prefrontal circuitry of the human brain in lean women. Neuroendocrinology. 2013;97:176–182. doi: 10.1159/000341406. [DOI] [PubMed] [Google Scholar]
- Kullmann S, Heni M, Veit R, Scheffler K, Machann J, Haring HU, Fritsche A, Preissl H. Selective insulin resistance in homeostatic and cognitive control brain areas in overweight and obese adults. Diabetes Care. 2015;38:1044–1050. doi: 10.2337/dc14-2319. [DOI] [PubMed] [Google Scholar]
- Kumar A, Foster T. Environmental enrichment decreases the afterhyperpolarization in senescent rats. Brain Res. 2007;1130:103–107. doi: 10.1016/j.brainres.2006.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Foster TC. Intracellular calcium stores contribute to increased susceptibility to LTD induction during aging. Brain Res. 2005;1031:125–128. doi: 10.1016/j.brainres.2004.10.023. [DOI] [PubMed] [Google Scholar]
- Landfield PW. ‘Increased calcium-current’ hypothesis of brain aging. Neurobiol Aging. 1987;8:346–347. doi: 10.1016/0197-4580(87)90074-1. [DOI] [PubMed] [Google Scholar]
- Landfield PW, Pitler TA. Prolonged Ca2+-dependent afterhyperpolarizations in hippocampal neurons of aged rats. Science. 1984;226:1089–1092. doi: 10.1126/science.6494926. [DOI] [PubMed] [Google Scholar]
- Lee CC, Huang CC, Wu MY, Hsu KS. Insulin stimulates postsynaptic density-95 protein translation via the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway. J Biol Chem. 2005;280:18543–18550. doi: 10.1074/jbc.M414112200. [DOI] [PubMed] [Google Scholar]
- Leibson CL, Rocca WA, Hanson VA, Cha R, Kokmen E, O'Brien PC, Palumbo PJ. Risk of dementia among persons with diabetes mellitus: a population-based cohort study. Am J Epidemiol. 1997;145:301–308. doi: 10.1093/oxfordjournals.aje.a009106. [DOI] [PubMed] [Google Scholar]
- Lin JW, Ju W, Foster K, Lee SH, Ahmadian G, Wyszynski M, Wang YT, Sheng M. Distinct molecular mechanisms and divergent endocytotic pathways of AMPA receptor internalization. Nat Neurosci. 2000;3:1282–1290. doi: 10.1038/81814. [DOI] [PubMed] [Google Scholar]
- Lochhead JJ, Wolak DJ, Pizzo ME, Thorne RG. Rapid transport within cerebral perivascular spaces underlies widespread tracer distribution in the brain after intranasal administration. J Cereb Blood Flow Metab. 2015;35:371–381. doi: 10.1038/jcbfm.2014.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchsinger JA. Type 2 diabetes, related conditions, in relation and dementia: an opportunity for prevention? Journal of Alzheimer's disease : JAD. 2010;20:723–736. doi: 10.3233/JAD-2010-091687. [DOI] [PubMed] [Google Scholar]
- Luckett BS, Frielle JL, Wolfgang L, Stocker SD. Arcuate nucleus injection of an anti-insulin affibody prevents the sympathetic response to insulin. Am J Physiol Heart Circ Physiol. 2013;304:H1538–1546. doi: 10.1152/ajpheart.00081.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maimaiti S, Anderson KL, DeMoll C, Brewer LD, Rauh BA, Gant JC, Blalock EM, Porter NM, Thibault O. Intranasal Insulin Improves Age-Related Cognitive Deficits and Reverses Electrophysiological Correlates of Brain Aging. J Gerontol A Biol Sci Med Sci. 2016;71:30–39. doi: 10.1093/gerona/glu314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man HY, Lin JW, Ju WH, Ahmadian G, Liu L, Becker LE, Sheng M, Wang YT. Regulation of AMPA receptor-mediated synaptic transmission by clathrin-dependent receptor internalization. Neuron. 2000;25:649–662. doi: 10.1016/s0896-6273(00)81067-3. [DOI] [PubMed] [Google Scholar]
- Marks DR, Tucker K, Cavallin MA, Mast TG, Fadool DA. Awake intranasal insulin delivery modifies protein complexes and alters memory, anxiety, and olfactory behaviors. J Neurosci. 2009;29:6734–6751. doi: 10.1523/JNEUROSCI.1350-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay BM, Oh MM, Galvez R, Burgdorf J, Kroes RA, Weiss C, Adelman JP, Moskal JR, Disterhoft JF. Increasing SK2 channel activity impairs associative learning. J Neurophysiol. 2012;108:863–870. doi: 10.1152/jn.00025.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNay EC, Sandusky LA, Pearson-Leary J. Hippocampal insulin microinjection and in vivo microdialysis during spatial memory testing. J Vis Exp. 2013:e4451. doi: 10.3791/4451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyer JR, Jr, Thompson LT, Black JP, Disterhoft JF. Nimodipine increases excitability of rabbit CA1 pyramidal neurons in an age- and concentration-dependent manner. J Neurophysiol. 1992;68:2100–2109. doi: 10.1152/jn.1992.68.6.2100. [DOI] [PubMed] [Google Scholar]
- Nunez-Santana FL, Oh MM, Antion MD, Lee A, Hell JW, Disterhoft JF. Surface L-type Ca2+ channel expression levels are increased in aged hippocampus. Aging Cell. 2014;13:111–120. doi: 10.1111/acel.12157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Malley D, Harvey J. MAPK-dependent actin cytoskeletal reorganization underlies BK channel activation by insulin. Eur J Neurosci. 2007;25:673–682. doi: 10.1111/j.1460-9568.2007.05347.x. [DOI] [PubMed] [Google Scholar]
- Oh MM, Oliveira FA, Waters J, Disterhoft JF. Altered calcium metabolism in aging CA1 hippocampal pyramidal neurons. J Neurosci. 2013;33:7905–7911. doi: 10.1523/JNEUROSCI.5457-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh MM, Power JM, Thompson LT, Moriearty PL, Disterhoft JF. Metrifonate increases neuronal excitability in CA1 pyramidal neurons from both young and aging rabbit hippocampus. J Neurosci. 1999;19:1814–1823. doi: 10.1523/JNEUROSCI.19-05-01814.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott A, Stolk RP, Hofman A, van Harskamp F, Grobbee DE, Breteler MM. Association of diabetes mellitus and dementia: the Rotterdam Study. Diabetologia. 1996;39:1392–1397. doi: 10.1007/s001250050588. [DOI] [PubMed] [Google Scholar]
- Ott V, Lehnert H, Staub J, Wonne K, Born J, Hallschmid M. Central nervous insulin administration does not potentiate the acute glucoregulatory impact of concurrent mild hyperinsulinemia. Diabetes. 2014 doi: 10.2337/db14-0931. [DOI] [PubMed] [Google Scholar]
- Paez PM, Fulton DJ, Spreur V, Handley V, Campagnoni AT. Multiple kinase pathways regulate voltage-dependent Ca2+ influx and migration in oligodendrocyte precursor cells. J Neurosci. 2010;30:6422–6433. doi: 10.1523/JNEUROSCI.5086-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pancani T, Anderson KL, Brewer LD, Kadish I, DeMoll C, Landfield PW, Blalock EM, Porter NM, Thibault O. Effect of high-fat diet on metabolic indices, cognition, and neuronal physiology in aging F344 rats. Neurobiol Aging. 2013;34:1977–1987. doi: 10.1016/j.neurobiolaging.2013.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pancani T, Anderson KL, Porter NM, Thibault O. Imaging of a glucose analog, calcium and NADH in neurons and astrocytes: Dynamic responses to depolarization and sensitivity to pioglitazone. Cell Calcium. 2011;50:548–558. doi: 10.1016/j.ceca.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pancani T, Phelps JT, Searcy JL, Kilgore MW, Chen KC, Porter NM, Thibault O. Distinct modulation of voltage-gated and ligand-gated Ca2+ currents by PPAR-gamma agonists in cultured hippocampal neurons. J Neurochem. 2009;109:1800–1811. doi: 10.1111/j.1471-4159.2009.06107.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paranjape SA, Chan O, Zhu W, Horblitt AM, McNay EC, Cresswell JA, Bogan JS, McCrimmon RJ, Sherwin RS. Influence of insulin in the ventromedial hypothalamus on pancreatic glucagon secretion in vivo. Diabetes. 2010;59:1521–1527. doi: 10.2337/db10-0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter NM, Thibault O, Thibault V, Chen KC, Landfield PW. Calcium channel density and hippocampal cell death with age in long-term culture. J Neurosci. 1997;17:5629–5639. doi: 10.1523/JNEUROSCI.17-14-05629.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potier B, Rascol O, Jazat F, Lamour Y, Dutar P. Alterations in the properties of hippocampal pyramidal neurons in the aged rat. Neuroscience. 1992;48:793–806. doi: 10.1016/0306-4522(92)90267-6. [DOI] [PubMed] [Google Scholar]
- Potier B, Rovira C. Protein tyrosine kinase inhibitors reduce high-voltage activating calcium currents in CA1 pyramidal neurones from rat hippocampal slices. Brain Res. 1999;816:587–597. doi: 10.1016/s0006-8993(98)01241-4. [DOI] [PubMed] [Google Scholar]
- Rasgon N, Jarvik L. Insulin resistance, affective disorders, and Alzheimer's disease: review and hypothesis. J Gerontol A Biol Sci Med Sci. 2004;59:178–183. doi: 10.1093/gerona/59.2.m178. discussion 184-192. [DOI] [PubMed] [Google Scholar]
- Reger MA, Watson GS, Frey WH, 2nd, Baker LD, Cholerton B, Keeling ML, Belongia DA, Fishel MA, Plymate SR, Schellenberg GD, Cherrier MM, Craft S. Effects of intranasal insulin on cognition in memory-impaired older adults: modulation by APOE genotype. Neurobiol Aging. 2006;27:451–458. doi: 10.1016/j.neurobiolaging.2005.03.016. [DOI] [PubMed] [Google Scholar]
- Reger MA, Watson GS, Green PS, Baker LD, Cholerton B, Fishel MA, Plymate SR, Cherrier MM, Schellenberg GD, Frey Ii WH, Craft S. Intranasal Insulin Administration Dose-Dependently Modulates Verbal Memory and Plasma Amyloid-beta in Memory-Impaired Older Adults. J Alzheimers Dis. 2008;13:323–331. doi: 10.3233/jad-2008-13309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbloom MH, Barclay TR, Pyle M, Owens BL, Cagan AB, Anderson CP, Frey WH, 2nd, Hanson LR. A Single-Dose Pilot Trial of Intranasal Rapid-Acting Insulin in Apolipoprotein E4 Carriers with Mild-Moderate Alzheimer's Disease. CNS Drugs. 2014 doi: 10.1007/s40263-014-0214-y. [DOI] [PubMed] [Google Scholar]
- Saar D, Grossman Y, Barkai E. Reduced after-hyperpolarization in rat piriform cortex pyramidal neurons is associated with increased learning capability during operant conditioning. Eur J Neurosci. 1998;10:1518–1523. doi: 10.1046/j.1460-9568.1998.00149.x. [DOI] [PubMed] [Google Scholar]
- Saar D, Grossman Y, Barkai E. Long-lasting cholinergic modulation underlies rule learning in rats. J Neurosci. 2001;21:1385–1392. doi: 10.1523/JNEUROSCI.21-04-01385.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salameh TS, Bullock KM, Hujoel IA, Niehoff ML, Wolden-Hanson T, Kim J, Morley JE, Farr SA, Banks WA. Central Nervous System Delivery of Intranasal Insulin: Mechanisms of Uptake and Effects on Cognition. J Alzheimers Dis. 2015;47:715–728. doi: 10.3233/JAD-150307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartorius T, Peter A, Heni M, Maetzler W, Fritsche A, Haring HU, Hennige AM. The brain response to peripheral insulin declines with age: a contribution of the blood-brain barrier? PLoS One. 2015;10:e0126804. doi: 10.1371/journal.pone.0126804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaoka T, Wada T, Tsuneki H. Insulin resistance and cognitive function. Nihon rinsho Japanese journal of clinical medicine. 2014;72:633–640. [PubMed] [Google Scholar]
- Schioth HB, Craft S, Brooks SJ, Frey WH, 2nd, Benedict C. Brain insulin signaling and Alzheimer's disease: current evidence and future directions. Mol Neurobiol. 2012a;46:4–10. doi: 10.1007/s12035-011-8229-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schioth HB, Frey WH, Brooks SJ, Benedict C. Insulin to treat Alzheimer's disease: just follow your nose? Expert Rev Clin Pharmacol. 2012b;5:17–20. doi: 10.1586/ecp.11.70. [DOI] [PubMed] [Google Scholar]
- Skeberdis VA, Lan J, Zheng X, Zukin RS, Bennett MV. Insulin promotes rapid delivery of N-methyl-D- aspartate receptors to the cell surface by exocytosis. Proc Natl Acad Sci U S A. 2001;98:3561–3566. doi: 10.1073/pnas.051634698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stella SL, Jr, Bryson EJ, Thoreson WB. Insulin inhibits voltage-dependent calcium influx into rod photoreceptors. Neuroreport. 2001;12:947–951. doi: 10.1097/00001756-200104170-00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss O, Mergler S, Wiederholt M. Regulation of L-type calcium channels by protein tyrosine kinase and protein kinase C in cultured rat and human retinal pigment epithelial cells. FASEB J. 1997;11:859–867. doi: 10.1096/fasebj.11.11.9285484. [DOI] [PubMed] [Google Scholar]
- Sukhareva M, Smith SV, Maric D, Barker JL. Functional properties of ryanodine receptors in hippocampal neurons change during early differentiation in culture. J Neurophysiol. 2002;88:1077–1087. doi: 10.1152/jn.2002.88.3.1077. [DOI] [PubMed] [Google Scholar]
- Talbot K, Wang HY, Kazi H, Han LY, Bakshi KP, Stucky A, Fuino RL, Kawaguchi KR, Samoyedny AJ, Wilson RS, Arvanitakis Z, Schneider JA, Wolf BA, Bennett DA, Trojanowski JQ, Arnold SE. Demonstrated brain insulin resistance in Alzheimer's disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Invest. 2012;122:1316–1338. doi: 10.1172/JCI59903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibault O, Hadley R, Landfield PW. Elevated postsynaptic [Ca2+]i and L-type calcium channel activity in aged hippocampal neurons: relationship to impaired synaptic plasticity. J Neurosci. 2001;21:9744–9756. doi: 10.1523/JNEUROSCI.21-24-09744.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibault O, Landfield PW. Increase in single L-type calcium channels in hippocampal neurons during aging. Science. 1996;272:1017–1020. doi: 10.1126/science.272.5264.1017. [DOI] [PubMed] [Google Scholar]
- Tombaugh GC, Rowe WB, Rose GM. The slow afterhyperpolarization in hippocampal CA1 neurons covaries with spatial learning ability in aged Fisher 344 rats. J Neurosci. 2005;25:2609–2616. doi: 10.1523/JNEUROSCI.5023-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetiska SM, Ahmadian G, Ju W, Liu L, Wymann MP, Wang YT. GABAA receptor-associated phosphoinositide 3-kinase is required for insulin-induced recruitment of postsynaptic GABAA receptors. Neuropharmacology. 2007;52:146–155. doi: 10.1016/j.neuropharm.2006.06.023. [DOI] [PubMed] [Google Scholar]
- Vianna CR, Coppari R. A treasure trove of hypothalamic neurocircuitries governing body weight homeostasis. Endocrinology. 2011;152:11–18. doi: 10.1210/en.2010-0778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Q, Xiong ZG, Man HY, Ackerley CA, Braunton J, Lu WY, Becker LE, MacDonald JF, Wang YT. Recruitment of functional GABA(A) receptors to postsynaptic domains by insulin. Nature. 1997;388:686–690. doi: 10.1038/41792. [DOI] [PubMed] [Google Scholar]
- Wijetunge S, Dolphin AC, Hughes AD. Tyrosine kinases act directly on the alpha1 subunit to modulate Ca(v)2.2 calcium channels. Biochem Biophys Res Commun. 2002;290:1246–1249. doi: 10.1006/bbrc.2001.6248. [DOI] [PubMed] [Google Scholar]
- Zaia A, Piantanelli L. Alterations of brain insulin receptor characteristics in aging mice. Archives of gerontology and geriatrics. 1996;23:27–37. doi: 10.1016/0167-4943(96)00705-4. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Kohler M, Yang SN, Zhang F, Larsson O, Berggren PO. Growth hormone promotes Ca(2+)-induced Ca2+ release in insulin-secreting cells by ryanodine receptor tyrosine phosphorylation. Mol Endocrinol. 2004;18:1658–1669. doi: 10.1210/me.2004-0044. [DOI] [PubMed] [Google Scholar]
- Zhao W, Chen H, Xu H, Moore E, Meiri N, Quon MJ, Alkon DL. Brain insulin receptors and spatial memory. Correlated changes in gene expression, tyrosine phosphorylation, and signaling molecules in the hippocampus of water maze trained rats. J Biol Chem. 1999;274:34893–34902. doi: 10.1074/jbc.274.49.34893. [DOI] [PubMed] [Google Scholar]
- Zhao WQ, Chen H, Quon MJ, Alkon DL. Insulin and the insulin receptor in experimental models of learning and memory. Eur J Pharmacol. 2004;490:71–81. doi: 10.1016/j.ejphar.2004.02.045. [DOI] [PubMed] [Google Scholar]
- Zhao WQ, De Felice FG, Fernandez S, Chen H, Lambert MP, Quon MJ, Krafft GA, Klein WL. Amyloid beta oligomers induce impairment of neuronal insulin receptors. FASEB J. 2008;22:246–260. doi: 10.1096/fj.06-7703com. [DOI] [PubMed] [Google Scholar]