

Abstract
Traumatic brain injury (TBI) is a leading cause of mortality and long-term morbidity worldwide. Despite decades of pre-clinical investigation, therapeutic strategies focused on acute neuroprotection failed to improve TBI outcomes. This lack of translational success has necessitated a reassessment of the optimal targets for intervention, including a heightened focus on secondary injury mechanisms. Chronic immune activation correlates with progressive neurodegeneration for decades after TBI; however, significant challenges remain in functionally and mechanistically defining immune activation after TBI. In this review, we explore the burgeoning evidence implicating the acute release of damage associated molecular patterns (DAMPs), such as adenosine 5′-triphosphate (ATP), high mobility group box protein 1 (HMGB1), S100 proteins, and hyaluronic acid in the initiation of progressive neurological injury, including white matter loss after TBI. The role that pattern recognition receptors, including toll-like receptor and purinergic receptors, play in progressive neurological injury after TBI is detailed. Finally, we provide support for the notion that resident and infiltrating macrophages are critical cellular targets linking acute DAMP release with adaptive immune responses and chronic injury after TBI. The therapeutic potential of targeting DAMPs and barriers to clinical translational, in the context of TBI patient management, are discussed.
Keywords: microglia, macrophage, white matter injury, oligodendrocyte, HMGB1, S100, Toll like receptor, T-cell, lymphocyte, leukocyte
1. INTRODUCTION
Traumatic brain injury (TBI) is a significant public health issue, killing or debilitating more individuals than breast cancer, AIDS, multiple sclerosis, and spinal cord injury combined [1]. TBI, defined as a blow or jolt to the head that produces permanent or temporary impairments in neurological function, may affect individuals regardless of gender, ethnicity, age, and socioeconomic status. In spite of improvements in awareness and preventative safety measures, TBI contributes to nearly one-third of injury-related deaths [2] and millions of TBI survivors live with the neurological consequences of a prior TBI [3]. In contrast to other common neurological diseases, such as stroke and Alzheimer’s disease, TBI is more prevalent in younger populations, resulting in substantial loss of productive years and the need for life-long assisted care. This burdens both families and health care systems that provide cognitive, emotional, physical, and psychological support for TBI survivors. Altogether, TBI places an annual $76.5 billion burden on society [4, 5].
Despite the high prevalence and poor outcomes associated with TBI, there is a lack of effective treatment options to improve outcomes. The vast majority of pre-clinical studies to date focused on achieving acute neuroprotection, largely characterized by histological measures of cell death and lesion volume. Unfortunately, many promising pre-clinical and early stage clinical findings ultimately failed to improve long-term outcomes in later phase clinical trials [6]. Given the mechanistically complex and heterogeneous nature of neurological injury, a number of factors were suggested as possible variables that contributed to the lack of translation, including suboptimal experimental models, inadequate randomization and blinding procedures, and selection of drugs with poor pharmacokinetics, limited brain penetration, and/or narrow therapeutic windows [7]. While additional attention to experimental design and target selection will undoubtedly enhance scientific discovery and future drug development, reassessment of neuroprotection as a primary therapeutic target is warranted. In this mini-review, we discuss the emerging association between innate immune activation and chronic white matter injury after TBI. We also identify molecular and cellular targets for the development of therapeutic strategies to limit progressive neurological injury after TBI.
2. CHRONIC NEURODEGENERATION AFTER TBI: IS WHITE MATTER INJURY A VIABLE THERAPEUTIC TARGET?
2.1 Primary Injury and Acute Neuroprotection
Impact and/or coup-contrecoup injuries produce immediate mechanical injury, including axonal shearing and widespread neuronal cell death throughout the cerebral cortex, hippocampus, cerebellum, and thalamus after both clinical and experimental TBI [8–16]. Cell death, ranging from immediate necrosis within the contusion core to delayed apoptosis in the surrounding tissue, is associated with poor neurological outcomes after TBI. A preponderance of experimental investigation over the past several decades focused on elucidating acute injury mechanisms, with the ultimate goal of developing neuroprotective drugs. While a thorough analysis of the shortcomings of all acute neuroprotection studies is beyond our scope herein (for review, see [7]), as just one example, an abundance of pre-clinical literature supported a detrimental role for acute activation of NMDA-type glutamate receptors (NMDA-R) after TBI. Specifically, global NMDA-R antagonists (e.g. MK-801) potently reduced neuronal necrosis in vitro and in rodent models of TBI; however, clinical translational of NMDA-R antagonists were hampered by poor therapeutic windows and intolerable side effects [17]. The repeated lack of success at targeting acute neuroprotection necessitates mechanistically distinct approaches to improve outcomes after TBI. In particular, there is increased attention to defining secondary injury mechanisms, which may provide therapeutic targets that are temporally amenable to clinical intervention.
2.2 Diffuse Axonal Injury
TBI is frequently associated with coup-contrecoup injuries, which produce cerebral contusions and traumatic axonal injury. A coup injury occurs at the site of impact whereas a contrecoup injury occurs on the opposite side of impact [18]. In general, coup injuries are caused by acceleration trauma that occurs when a moving object impacts a stationary head. In contrast, contrecoup injuries are characteristic of deceleration trauma whereby the moving head strikes a stationary object [18]. In addition, cerebrospinal fluid (CSF), which is denser than brain tissue, rapidly moves towards the impact site, displacing the brain to the other side of the skull to produce contrecoup injury [19]. These injuries differentially affect grey matter and white matter and profoundly influence the care and treatment of TBI patients.
Grey matter consists of neuronal cell bodies, dendrites, glia, synapses, and capillaries. In addition, grey matter contains oligodendrocytes, a subtype of glial cell that myelinates up to sixty adjacent axonal segments [20]. Conversely, white matter is primarily comprised of interconnected myelinated axon tracts that allow rapid neurotransmission between different brain regions. In addition to producing focal damage, the differential movement of grey and white matter during acceleration-deceleration results in widespread, diffuse axonal tearing at grey-white matter junctions [21]. As diffuse axonal injury and loss of white matter integrity are strongly associated with chronic sensorimotor, cognitive, and psychiatric dysfunction in both pre-clinical and clinical TBI [22–28], therapies that limit white matter damage and enhance remyelination may profoundly improve long-term clinical outcomes.
White matter injury is undoubtedly initiated during acceleration-deceleration, yet the majority of axonal damage is ultimately not due to physical shearing at the time of TBI [29]. Rather, demyelination and delayed atrophy of white matter tracts develops for over one year post-TBI in rodents [30–33] and progresses for decades in TBI patients [34, 35]. This temporal pattern of delayed white matter injury may therefore provide an amendable therapeutic window; however, the mechanisms that occur between the initial traumatic event and the manifestation of white matter loss remain poorly defined.
3. DAMAGE ASSOCIATED MOLECULAR PATTERNS (DAMPs): MEDIATORS OF PROGRESSIVE WHITE MATTER INJURY?
A prospective study found that 47% of TBI patients exhibited large lesions within the corpus callosum, the largest white matter structure in the brain [36]. The corpus callosum is particularly susceptible to injury due to a rigid attachment to the falx cerebri, a fold of dura mater that descends into the longitudinal fissure, and due to connections with the two independently mobile cerebral hemispheres [21]. A separate study determined that 96% of moderate-severe TBI patients showed atrophy of at least one brain region and 76.8% of TBI patients showed significant loss of corpus callosum volume [34]. Although the mechanisms remain poorly defined, white matter degradation, including a 25% reduction in corpus callosum thickness, was correlated with inflammatory activation at one-year after a single TBI [37]. Similarly, the degree of persistent thalamic inflammatory activation was closely related with thalamo-cortical white matter tract damage in moderate-severe TBI patients with diffuse axonal injury [38]. Thus, defining the regulatory mechanisms of immune activation may identify novel opportunities to affect TBI outcomes.
DAMPs are a diverse set of host molecules that are passively released after trauma or tissue injury. DAMPs, which include proteins such as high mobility group box protein 1 (HMGB1) and heat-shock proteins, extracellular matrix proteins such as hyaluronan fragments, and non-proteins such as adenosine 5′-triphosphoate (ATP), uric acid, heparin sulfate, and DNA [39–43], bind pattern recognition receptors (PRR) to initiate innate immune activation. With respect to brain injury, DAMPs induce pro-inflammatory activation in microglia, astrocytes, and neurons [44–46]. Although the predominance of studies linking DAMPs with the development of white matter injury utilized chronic neurological injury models, which may be mechanistically distinct from TBI, these studies may provide valuable insights to inform future investigations after neurotrauma. Indeed, a number of recent studies observed conserved mechanisms of immune-mediated neurovascular injury between chronic neurodegenerative diseases and TBI [47–50]. In the following subsections, we detail the emerging role for several DAMPs in the development of white matter loss, including references specific to TBI when available.
3.1 Purinergic Signaling
ATP is an essential small molecule that transports chemical energy to facilitate cellular metabolism and intracellular signaling. ATP also is released at low concentrations into the extracellular space as a mechanism of inter-cellular signaling under physiological conditions. In contrast, the passive, extracellular release of high concentrations of ATP initiates innate immune activation following traumatic or ischemic injuries [51]. ATP, a microglial chemoattractant that is rapidly released into the extracellular space from damaged neurons, is associated with poor outcomes after TBI [52, 53], yet the mechanistic and functional significance remains largely undefined. The biological actions of ATP are mediated by ubiquitously expressed purinergic receptors, which are grouped into G-protein coupled P2Y receptors, which bind ATP, ADP, UTP, UDP, and UDP-glucose, and ATP-gated cation permeable P2X receptors.
P2Y receptors are an eight-member receptor family in humans [54], although only P2Y2 and P2Y11 reportedly bind ATP [55]. The role of P2Y receptors is largely unstudied after TBI, though stimulation of ADP-binding P2Y1 receptors reduced edema, attenuated reactive astrogliosis, and enhanced neurological function after TBI [56, 57]. Furthermore, P2Y6 antagonism increased the permeability of the glial limitans and elevated parenchymal cell death following TBI [58]. GPR17, a P2Y-like receptor that mediates neurovascular remodeling in the rodent brain, was strongly co-localized in microglia/macrophages and expressed by proliferated oligodendrocyte precursor cells of TBI patients, suggesting some P2Y family members may represent potential targets for white matter repair after neurotrauma [59, 60]; however, the functions of the ATP binding P2Y members, P2Y2 and P2Y11, remain completely undefined after TBI, providing several exciting areas for future exploration.
In contrast to P2Y, all seven P2X family members bind ATP [54, 61, 62]. Of the P2X members, only P2X7 has received attention after TBI [63–65]. We reported that global genetic or pharmacological blockade of P2X7 reduced neuroinflammation and improved neurobehavioral outcomes in a murine controlled cortical impact model of TBI [64]. This observation was subsequently extended to a weight drop TBI model, where P2X7 activation was associated with the development of post-traumatic cognitive deficits [63, 65]. Although ATP-P2X7 signaling has not been definitely implicated in white matter loss after TBI, ATP mediates reactive gliosis after axonal degeneration via a P2X mediated mechanism [66]. Moreover, ATP signaling directly induced oligodendrocyte death and ischemic white matter damage via activation of P2X7 in a rodent model of multiple sclerosis [67, 68]. Consistent with these reports, P2X7 inhibition reduced anatomic damage, reduced neuronal loss, attenuated local inflammatory responses, and improved outcomes after weight drop induced thoracic spinal cord injury in rats [69, 70]. Another report observed a decrease in P2X7 expression in cultured oligodendrocyte precursor cells after two hours of oxygen-glucose deprivation or in the cerebral cortex, subcortical white matter, and hippocampus after neonatal hypoxia-ischemia, indicative of an endogenous protective mechanism to restrict white matter damage [71]. Similarly, P2X7 inhibition prevented ischemic oligodendrocyte injury and protected myelin after ischemic stroke [72]. Thus, targeted inhibition of P2X family members, including P2X7, may limit white matter injury and improve TBI outcomes.
3.2 Toll-like receptor signaling
Toll-like receptors (TLR), a thirteen-member, evolutionarily-conserved family of single, membrane spanning receptors, initiate innate immune responses after activation by pathogen-associated molecular patterns (PAMPs) or by DAMPs. We reported that acute neuronal release of HMGB1 increased neurovascular injury after experimental TBI via a TLR4-dependent mechanism [73]. We and others also determined that acutely increased plasma or cerebrospinal fluid levels of HMGB1 independently predicted one-year mortality and unfavorable outcomes in severe TBI patients [73–76]. While these data implicate a detrimental role for early HMGB1- TLR4 signaling after TBI, release of HMGB1 from reactive astrocytes, beginning at two weeks after experimental stroke increased the accumulation of endothelial progenitor cells and enhanced peri-infarct angiogenesis within injured white matter via a mechanism that involves activation of receptor for advanced glycation endproducts (RAGE) [77, 78]. Thus, the temporal pattern of HMGB1 release and/or the molecular target of action may determine whether beneficial or detrimental outcomes occur.
Causative studies linking TLR with white matter injury are lacking, yet TLR2, TLR4, HSP70, and MyD88 were localized in macrophages/microglia in lesioned regions and in subcortical white matter after experimental TBI [73, 79]. TLR2, TLR4, and HMGB1 also were increased in reactive glia within spinal cords of patients with amyotrophic lateral sclerosis (ALS) spinal cords [80], a neurodegenerative disorder characterized by motor neuron loss and extensive white matter injury. Although detailed studies remain to be performed after TBI, intravenous injection of the exogenous TLR4 ligand, endotoxin, induced focal periventricular white matter injury, inflammation, and release of S100β (see section 3.3.1), which served as an early biomarker of white matter injury in preterm fetal sheep [81, 82]. In addition, improvements in axonal integrity and reduced oligodendrocyte injury following ischemic preconditioning were eliminated in TLR4−/− mice [83]. Moreover, mice with a global genetic knockout of MyD88, an adapter molecule required for TLR4 signaling, reduced white matter loss in a neonatal hypoxic-ischemic model of brain injury [84]. In agreement with these reports, brain expression of myelination-associated proteins was attenuated in global TLR4−/− mice, as compared to wild-type mice, following chronic ethanol intake [85].
The aforementioned studies documented a detrimental role for TLR in white matter injury, yet a number of contrasting reports implicate TLR in white matter sparing and/or repair. TLR1, TLR2, TLR4, and TLR5 were increased after spinal cord injury, an injury characterized by acute oligodendrocyte loss with the subsequent proliferation of oligodendrocyte progenitor cell to repair damaged tissue. Interestingly, TLR4 mutant mice exhibited sustained locomotor deficits, increased demyelination, astrogliosis, and macrophage activation after SCI [86]. TLR4 activation similarly supported oligodendrocyte lineage cell sparing, long-term oligodendrocyte and oligodendrocyte progenitor replacement, and chronic functional recovery after contusive spinal cord injury [87]. Thus, TLR4 may exhibit protective effects on white matter under certain conditions. In addition to potentially protective effects of TLR4, activation of TLR2 also was associated with reduced degeneration of central myelinated fibers, whereas global TLR2−/− mice exhibited greater axonal injury after experimental spinal cord injury [88]. Taken together, modulation of TLRs may mediate both context-dependent demyelination and endogenous white matter repair. As such, considerable effort must be directed toward identifying endogenous TLR ligands, the temporal pattern of TLR expression/activity, and the cell type(s) mediating the divergent effects of TLR after injury.
3.3 S100 proteins
S100 proteins are a 21-member family of low molecular weight (9–13 kDa), helix E-loop-helix F (EF)-hand calcium-binding proteins [89]. Despite significant sequence and structural homology, S100 family members are implicated in a variety of diverse physiological functions, including the regulation of cellular contraction, cell motility, cellular growth and differentiation, transcription regulation, membrane organization, angiogenesis, and anti-oxidant function [90]. A number of S100 proteins, including S100β, S100A4, S100A8, S100A9, S100A12, and S100A13, are secreted in a tissue- and cell-type specific manner and function as cytokines. Although the extracellular receptors responsible for mediating the biological activity of S100 proteins remain incompletely resolved, S100β [91–93], S100A1 [94], S100A4 [95, 96], S100A5 [95], S100A6 [97], S100A7 [98], S100A8/A9 [99, 100], S100A11 [101, 102], S100A12 [92, 103], S100A13 [104], and S100P [105] reportedly interact with RAGE. In addition, TLR4 serves as a receptor for S100A8/A9 [106], consistent with a role for S100 family members as DAMPs. The following sub-sections detail the established roles for several prominent S100 proteins after TBI and highlight fertile areas for future exploration.
3.3.1 S100β
S100β is highly expressed in the human brain, including prominent expression in astrocytes and in many neural cell types [107]. The physiological release of S100β exerts neurotrophic effects in the brain [94] and intense S100β expression also is observed within white matter tracts, including the corpus callosum [108]. CSF levels of S100β correlated with white matter injury after pneumococcal meningitis [109] and endotoxin-induced periventricular white matter injury in preterm fetal sheep was associated with higher S100β levels [81]. Reduced tissue expression of S100β was observed in the corpus callosum of schizophrenia patients [110] whereas increased serum levels of S100β directly correlated with white matter injury in schizophrenic patients [111] and in asphyxiated preterm babies [112]. In line with these findings, acutely increased serum levels of S100β were associated with patient mortality and poor acute and chronic outcomes after severe TBI [113–119] and were predictive of cognitive function at 4 months after pediatric mild TBI [120]. Increased S100β and S100β auto-antibody serum levels were also observed in football players who had experienced repeated sub-concussive events and correlated with white matter changes and cognitive decline [121]. As extracranial sources of S100β do not affect serum levels in humans [122], reduced tissue expression likely reflects increased extracellular release due to pathological conditions [111]. Functionally, S100β inhibition reduced neurodegeneration [123] and S100β−/− mice or antibody-mediated neutralization of S100β attenuated microglial reactivity and improved memory function after experimental TBI [124]. Thus, S100β may represent a predictive biomarker of outcomes and target for therapeutic development after TBI [125].
3.3.2 S100A4
In addition to S100β, prolonged upregulation of S100A4 was observed in white matter astrocytes in response to degeneration of myelinated axons after peripheral nerve or dorsal root injury [126]. Similarly, expression of S100A4 in astrocytes within the spinal cord or brain white matter was associated with white matter degeneration [127]. Moreover, extracellular application of S100A4 increased the motility of cultured astrocytes, suggesting a role in the formation of a glial scar [128, 129]. Although the functional importance is unexplored after acute neurovascular injury, S100A4 promotes tumor angiogenesis [130] and delayed S100A4 expression at three months post-TBI was linked with improved outcomes [131]. Thus, S100A4 may exert temporal- and/or cell-dependent effects to differentially regulate tissue recovery and injury.
3.3.3 S100A6
S100A6, which is highly expressed in the CNS, interacts with S100β in human astrocytoma and melanoma cells [132, 133]. Whereas the biological actions of S100A6 in the brain remain largely undefined, expression of S100A6 is increased within astrocytes in response to glutamate excitotoxicity within the axotomized hypoglossal nucleus in mice [134]. Notably, reduced S100A6 expression in the hippocampus was associated with subsequent cognitive decline after lateral head acceleration-induced TBI whereas a delayed elevation of S100A6 was associated with cognitive recovery [135]. Thus, expression of either S100A4 or S100A6 may facilitate long-term neurological recovery after TBI.
3.4 Hyaluronic acid
Hyaluronic acid (HA), a non-sulfated glycosaminoglycan, is widely expressed within the nervous system, skin, cartilage, and eye. In addition to serving as a key component of the extracellular matrix, HA also influences tissue hemodynamics and cellular motility via activation of several cell surface receptors, including CD44 and Receptor for Hyaluronan Mediated Motility (RHAMM). Following injury, the extracellular release of HA provides a substrate for hyaluronidases, a seven-member family of enzymes that degrade HA into oligosaccharides and low molecular weight hyaluronan. HA degradation products exhibit pro-angiogenic properties [136, 137], although HA fragments also generate pro-inflammatory responses by activating TLR2 and/or TLR4 on macrophages and dendritic cells [138, 139]. Additionally, HA promotes the recruitment of inflammatory cells into damaged tissue [140].
Hypoxia induced HA production and increased the activity of hyaluronidases, a class of enzymes required for HA degradation [141]. In line with these findings, gene expression for hyaluronan synthases (HAS1) and HAS2, which catalyze the production and extracellular release of hyaluronan, was increased in the ipsilateral brain at 1–3 days post-TBI [142]. Hyaluronidase 1 (HYAL1) also was increased in the contralateral hemisphere at 1–3 days post-TBI [142] whereas expression of the HA receptor, CD44, was dramatically increased around the contusion for up to one week post-TBI [142]. Moreover, a recent genome wide sequencing study from perilesional cortex, thalamus, and hippocampus at three months post-TBI separately revealed a similar upregulation of CD44 after TBI [131], suggesting a dynamic regulation and role for HA metabolism after TBI; however, the precise functions and therapeutic potential of HA remains poorly defined after CNS injury.
Diffusion tensor imaging revealed that defined prefrontal cortical white matter lesions are associated with HA-rich regions in older individuals [143]. Similarly, white matter loss correlated with elevated HA levels following a single episode of hypoxia-ischemia injury in a preterm fetal sheep model of in utero hypoxemia [144]. Consistent with these studies, HA decreased the density of myelin basic protein expressing oligodendrocytes in organotypic forebrain slices from postnatal rat pups [145] and blocked oligodendrocyte lineage progression via a TLR2-dependent mechanism in chronic multiple sclerosis lesions [146, 147]. Notably, hyaluronidase treatment reduced both CD44 and TLR4 expression, attenuated leukocyte infiltration and pro-inflammatory cytokine levels, while simultaneously enhancing oligodendrocyte progenitor cell maturation, increasing myelination, and promoting neurological recovery in a rabbit model of intraventricular hemorrhage [148]. Thus, recombinant human hyaluronidase may provide a novel, efficacious therapy to limit white matter damage after TBI.
4. IS MICROGLIAL ACTIVATION THE CELLULAR LINK BETWEEN DAMP RELEASE AND WHITE MATTER INJURY?
Defining a deleterious role for DAMPs is imperative to validate clinically relevant, prognostic biomarkers and to provide a mechanistic understanding of progressive neurodegeneration after TBI; however, DAMPs are passively released from injured neurons immediately after necrotic injury. As such, strategies that directly block DAMP release likely possess low translational value due to a narrow therapeutic window, as observed with other acute neuroprotectants. In contrast, defining the molecular and cellular mechanisms, downstream from DAMP release, may provide more feasible targets for medical intervention.
Microglia, the resident macrophages of the CNS, exist in a resting state within the healthy brain. Microglia are rapidly activated following CNS injury, chronically persist within the human brain for nearly two decades after a single TBI, and as such, are regarded as a biomarker of injury after TBI [37, 149, 150], but it is unclear whether these changes reflect a causative factor in neurodegeneration or an adaptive response to progressive injury. Interestingly, microglia exhibit morphological and phenotypic changes to generate diverse functions in response to microenvironmental cues [151]. For example, activated microglia phagocytose cellular debris, produce neurotrophic factors, and release anti-inflammatory cytokines, including TGF-β and IL-10, to aid in tissue recovery [152], yet microglia also release pro-inflammatory mediators to disrupt the blood-brain barrier, to recruit peripheral leukocytes into the injured CNS, and to evoke rapid cell death in adjacent oligodendrocytes [153–155]. Moreover, dysregulated microglial activation produces chronic neuroinflammation, including the persistent release of pro-inflammatory cytokines and reactive oxygen species [156], which contribute toward evolving neurodegeneration [157]. Therefore, defining the mechanisms of DAMP-induced microglial activation may identify new opportunities to enhance neurological recovery and/or to attenuate neurodegeneration.
4.1 Microglial TLR Signaling
HMGB1 is associated with deleterious outcomes in a number of injury models, including TBI (see section 3.2). HMGB1 stimulated leukocyte chemotaxis and activation ex vivo [158–160] and induced microglial activation, increased neuroinflammation, and exacerbated neurocognitive deficits, at least in part, via a TLR-dependent mechanism in several non-traumatic injury models [161–163]. The passive release of HMGB1 from NMDA-injured neurons similarly increased pro-inflammatory activation in primary human and murine microglia via a TLR4-dependent mechanism [73, 164]. In addition, neuronal release of HMGB1 in and around the contusion was associated with poor outcomes after experimental TBI, at least in part, via activation of TLR4 on microglia/macrophages [73]. Functionally, anti-HMGB1 antibody therapy or glycyrrhizin, a HMGB1 release inhibitor, reduced inflammatory activation and lessened blood-brain barrier opening after lateral fluid percussion in rats [165, 166]. Similarly, BoxA mediated neutralization of HMGB1 inhibited aging-induced potentiation of microglial pro-inflammatory priming [167]. Thus, microglia are a critical cellular target of HMGB1 within the CNS after TBI.
The specific role of myeloid TLR4 activation remains unexplored after TBI; however, microglial inflammation is associated with axonal injury after experimental autoimmune encephalitis (EAE), a model of multiple sclerosis [168]. Moreover, HMGB1 and myeloid expression of TLR2/TLR4 were elevated within active inflammatory lesions in human and experimental multiple sclerosis [169]. While EAE represents a chronic neurological injury model, activation of microglial TLR4 mediated oligodendrocyte cell death in vitro and in the developing pericallosal white matter of immature rodents [170]. Consistent with these findings, suppression of myeloid TLR4 using CD200-Fc reduced phagocytosis of oligodendrocyte precursor cells by macrophages and enhanced endogenous recovery in a murine model of white matter ischemia [171]. Similarly, fructus mume, a traditional medicine in Asian countries, reduced white matter loss, attenuated both astrocytic and microglial activation and inhibited TLR4/MyD88 dependent signaling after permanent bilateral common carotid artery occlusion [172]. These findings suggest a conserved role microglial TLR4 activation in the initiation of white matter loss in both chronic and acute neurological injuries, a possibility that requires further exploration in experimental models of TBI.
The downstream molecular mediators of microglial activation by HMGB1 are poorly defined. Interestingly, HMGB1 binds to microglial macrophage antigen complex 1 (Mac1) to activate NADPH oxidase, a key enzyme involved in the generation of oxidative stress [173]. We and others observed a biphasic induction of NADPH oxidase expression and activity after TBI, with initial neuronal activity followed by a secondary peak in microglia up to four days later [15, 174, 175]. Inhibition of NADPH oxidase decreased neuronal cell death, attenuated pro-inflammatory microglial activation, reduced blood-brain barrier disruption, limited the accumulation of beta-amyloid, and improved neurocognitive outcomes in multiple experimental models of TBI [15, 174, 176–182]. Oligodendrocytes are extremely susceptible to oxidative damage due to a high metabolic rate [183, 184]. Although a functional association between microglial NADPH oxidase activity and white matter loss remains undetermined after TBI, NADPH oxidase is expressed in microglia and infiltrating macrophages in active multiple sclerosis lesions and NADPH oxidase activity is associated with demyelination, white matter damage, and tissue loss after EAE in mice [185, 186]. Importantly, activation of microglial TLR4 induced pre-oligodendrocyte injury via a NADPH oxidase mediated mechanism in a neonatal rat model of periventricular leukomalacia, which is characterized by white matter loss around the cerebral ventricles [187]. Thus, targeted inhibition of microglial NADPH oxidase may limit white matter loss and improve long-term outcomes downstream from HMGB1-TLR4 activation. This possibility requires further pre-clinical development and exploration.
4.2 Microglial purinergic signaling
ATP, a microglial chemoattractant released from damaged neurons, is associated with poor outcomes after TBI [52, 53]. Amongst the purinergic receptors, P2X1, P2X4, and P2X7 are expressed in amoeboid microglia during development and beginning at postnatal day 30, P2X7 positive microglia are observed throughout the forebrain [188, 189]. Although the functional importance of P2X receptors remain poorly defined after TBI, P2X4 was acutely localized in microglia/macrophages after experimental TBI in rats [190] whereas inhibition of P2X4 reduced hypoxia-induced pro-inflammatory signaling in isolated microglial cells [189]. Although a mechanistic link between P2X7 and white matter injury has not been established to date, genetic or pharmacological P2X7 inhibition reduced the expression and release of the pro-inflammatory cytokine, interleukin-1β (IL-1β) and improved outcomes after TBI [63–65]. The production of IL-1β by amoeboid microglia increased oligodendrocyte injury in the periventricular white matter of rats following neonatal hypoxia [191] and perinatal IL-1β exposure induced ventricular enlargement, increased the loss of mature oligodendrocytes, and produced white matter injury in rats [192]. Neutralization of ATP, an important ligand for P2X7, during the acute injury phase reduced neurological injury after EAE, which is characterized by widespread white matter loss [193]. Consistent with these reports, administration of the selective microglial inhibitor, minocycline, attenuated microglial activation, decreased IL-1β production, protected developing oligodendrocytes, and attenuated white matter injury in neonatal rat brains [194, 195]. Similarly, inhibition of CX3CR1, a receptor involved in microglial chemotaxis, decreased microglial activation, reduced IL-1β production, attenuated white matter injury, and improved neurocognitive function in a pre-clinical model of vascular cognitive impairment [196]. While the aforementioned findings suggest a detrimental role for microglia after TBI, minocycline failed to attenuate traumatic axonal injury, tissue atrophy, neurodegeneration, or spatial learning deficits in a pediatric head trauma model [197]. Thus, additional experimentation is required to ascertain whether the effects of microglial activation are model dependent, time dependent, and/or age specific after TBI.
4.3 Macrophage infiltration: a detrimental consequence of microglia activation after TBI?
The CNS maintains an immunoprivileged status, yet a robust and sequential set of immune responses develops in the hours and days after TBI [198, 199]. In particular, activation of innate immunity, an evolutionarily conserved system of immediate, non-specific host protection, temporally correlates with widespread cellular necrosis after TBI [200]. Although the precise mechanisms remain to be determined, the rapid, local activation of microglia induces the release of powerful chemoattractants that recruit professional phagocytes, such as monocyte-derived macrophages, to sites of injury [201]. In particular, HMGB1-TLR4 signaling is implicated in leukocyte trafficking during trauma [202]. These coordinated changes facilitate the clearance of damaged cells and debris, but chronic immune activation exacerbates secondary injury and worsens outcomes [156, 203, 204]. Thus, early microglial activation after the release of DAMPs may initiate the recruitment of peripheral innate immune mediators after TBI; however, the beneficial and/or detrimental implications of macrophage trafficking into the CNS remain incompletely characterized after TBI.
Myelin is extensively degraded within the cerebrum after pre-clinical TBI [205, 206] and elevated myelin basic protein is observed in CSF of pediatric and adult TBI patients [207–210]. The presence of myelin debris after traumatic axonal injury inhibits axon regeneration and blocks the differentiation of oligodendrocyte progenitor cells, restricting intrinsic remyelination and impeding spontaneous recovery [211]. Early macrophage activation enhanced the phagocytosis of myelin debris after Wallerian degeneration [212, 213] and increased remyelination after spinal cord injury [214], supporting a beneficial effect of acute macrophage infiltration. Conversely, chronic accumulation of pro-inflammatory macrophages within both white and grey matter temporally preceded oligodendrocyte apoptosis, widespread myelin loss, and white matter injury for weeks after experimental TBI [215, 216]. Consistent with the latter deleterious functions, macrophages accumulated within the corpus callosum of >25% patients at 1 year post-TBI and correlated with white matter loss and neurological dysfunction for over 17 years post-TBI [37, 149]. In line with these findings, TLR4 activation accelerated myelin phagocytosis after sciatic nerve lesion [217–219] and activation of myeloid TLR4 mediated oligodendrocyte injury and exacerbated white matter injury in immature rodents [170]. Thus, DAMPs may play a dual role both in myelin clearance and in mediating white matter loss.
In addition to the temporal changes, the activation status of macrophages may contribute toward white matter injury after TBI. Upon activation, macrophages polarize into distinct phenotypes based on the local microenvironment to generate context-dependent functions. Toward this end, “classically activated” (M1) macrophages exhibit a pro-inflammatory phenotype whereas “alternatively activated” (M2) macrophages release anti-inflammatory mediators to dampen immune responses [220, 221]. Consistent with a role for TLR4 in mediating M1 macrophage polarization [222] and in reprogramming M2 macrophages toward a M1 phenotype [223, 224], we reported that both TLR4−/− mice and C3H/HeJ mice, which contain a spontaneous inactivating point mutation in the TLR4 signaling domain, exhibited a reduction in M1 polarization over the first 72h after experimental TBI [225]. Of note, a progressive increase in the ratio of M1:M2 macrophages temporally correlated with evolving neurological injury, including white matter loss and reduced brain volume, after TBI [37, 187, 225, 226]. In line with the aforementioned studies, M1 macrophages accumulated within peri-contusional white matter, within grey matter, and within the contralateral cortex by d3 after TBI, preceding oligodendrocyte apoptosis and widespread myelin loss (hallmarks of white matter injury) over the next several weeks [215]. In contrast, M2 macrophages increased the accumulation of oligodendrocyte progenitor cells and promoted white matter recovery after TBI [215]. Consistent with these findings, conditioned media from M1 macrophages increased oligodendrocyte cell death after oxygen-glucose deprivation whereas conditioned media from alternatively activated (M2) macrophages was protective [187].
5. TRANSLATIONAL IMPLICATIONS
5.1 Model Selection
The selection of an appropriate pre-clinical animal model is essential for target validation and for the development and translation of any therapy. Given the convenience, low cost, and widespread availability of advanced transgenic animals, the majority of TBI research is performed in rodent models of controlled cortical impact, lateral fluid percussion, or weight drop-induced injury. Although each model exhibits distinct strengths and weaknesses [227], no single model fully replicates the complex and heterogeneous pathophysiological of human TBI. In addition, the rodent brain is lissencephalic, is dramatically smaller than the human brain, and is composed of ~10% white matter. Thus, despite the mechanistic benefits of using rodents to identify and screen potential therapeutic targets, such as DAMP associated signaling pathways, lower species remain sub-optimal for the study of progressive white matter loss after TBI. Despite the higher cost and experimental complexities, large animals, such as swine, are gyrencephalic, exhibit a similar ratio of white to grey matter (60:40) to humans, and as such, may provide a more suitable model system to mimic human TBI [228, 229]. In line with possibility, a porcine model of controlled cortical impact was reproducible and clinically relevant with respect to multi-parametric neuromonitoring and histological outcomes [230, 231]. With the advent of improved genetic tools, such as CRISPR/Cas9 technology [232–235], future work will combine the genetic and mechanistic benefits of rodents with the anatomical and physiological advantages of a porcine TBI model.
5.2 Therapeutic development
Future studies will undoubtedly utilize Cre-loxP and targeted gene-silencing techniques establish whether cell-type specific deletion of DAMP receptors improves TBI outcomes. These approaches will identify the precise cellular and molecular mechanisms whereby DAMPs, which are immediately and locally released at the site of impact, contribute to delayed white matter injury spatially distant from the initial TBI; however, these studies possess little translational value beyond target validation. DAMPs are acutely and passively released from damaged cells, including necrotic neurons, immediately after brain injury [73, 236–238]. Thus, despite preclinical reports by our laboratory and others showing either direct HMGB1 neutralization or inhibition of PRR could improve acute TBI outcomes in rodents if administered within the first hours after injury [73, 165, 166, 239–243], a narrow therapeutic window makes successful clinical translation into TBI patient populations seem unlikely. Thus, elucidation of the downstream mechanisms linking DAMP release with poor outcomes is imperative for the development of efficacious approaches with a realistic window for intervention.
5.2.1 Do DAMP-activated macrophages generate adaptive immune responses after TBI?
DAMPs induce pro-inflammatory M1 polarization as a part of the innate immune response after injury. While broad operational definitions for M1/M2 polarization indicate the general inflammatory status after injury, in reality, macrophages polarize along a dynamic continuum rather than rigid, binary polarization phenotypes [244]. Thus, defining the functional implications of DAMP-induced myeloid cell activation may identify novel approaches to improve long-term outcomes after TBI.
Activated macrophages, a component of the mononuclear phagocyte system, exhibit antigen-presenting capability in draining lymph nodes during a primary immune response via a MHC class II-dependent process. The CNS was long believed to lack a classical lymphatic system, yet specialized meningeal lymphatic vessels were recently identified as a conduit for the bi-directional movement of macromolecules and immune cells between the brain and the deep cervical lymph nodes [245, 246]. Indeed, activation of T-lymphocytes occurs within deep cervical lymph nodes, rather than at the site of CNS injury [247].
DAMP-mediated activation of TLR4 increased MHC class II expression [248] and enhanced MHC Class II dependent antigen presentation [249]. Increased expression of myeloid MHC Class II antigens was observed after experimental or clinical TBI [250–252]. Interestingly, HLA-DR, a MHC Class II antigen expressed on macrophages, bound myelin basic protein to initiate adaptive immune responses [253, 254]. As auto-antibodies against myelin basic protein induce demyelination after multiple sclerosis [255], myelin-loaded macrophages may similarly initiate progressive white matter injury after TBI. Coupled with the finding that pharmacological inhibition of MHC Class II-dependent antigen processing reduced neurodegeneration after TBI [256], activated macrophages may link acute TBI with deleterious, long-term adaptive immune responses.
5.2.2 T cell polarization is a consequence of macrophage activation after TBI
DAMP-mediated activation of myeloid cells may, in turn, generate adaptive immune responses to mediate chronic white matter injury. Macrophage activation temporally preceded leukocytosis, brain T-lymphocyte infiltration, parenchymal inflammation, and clinical deterioration after TBI [250, 257, 258]. T-lymphocytes, mediators of adaptive immunity, are comprised of functionally diverse subsets that exert opposing effects. In particular, helper T-cells (TH) cells augment immune responses after antigen recognition by stimulating antibody production and by releasing cytokines to potentiate activation of cytotoxic T-cells and macrophages. Although T-lymphocytes do not routinely cross the blood-brain barrier [259, 260], CNS accumulation of infiltrating macrophages and TH cells was associated with neurodegeneration after experimental TBI and in resected brain tissue from TBI patients [256, 257, 261]. Therefore, elucidating the mechanisms of T-lymphocyte activation may provide novel opportunities for therapeutic intervention, downstream of macrophage activation.
Polarized macrophages release phenotype-specific cytokines to differentiate naïve TH cells into TH subtypes, such as TH1, TH2, and TH17 [262], to provide long-lasting, antigen-specific immunity [249, 263–267]. We showed that monocytes isolated from the CNS after TBI stimulated T-cell proliferation and increased TH1 and TH17 polarization [225]. Moreover, activation of myeloid TLR4 simultaneously increased TH1/TH17 polarization and reduced TREG production in both blood and brain for several weeks after TBI in mice [225]. While the efficacy of TH1/TH17 inhibition remains unexplored after TBI, TH17 cells drove chronic microglial polarization after EAE and co-culture studies showed myelin-specific T-lymphocytes that secreted IL-17 potently activated inflammatory cytokine expression in microglia [268, 269]. Furthermore, attenuation of brain TH17 influx reduced white matter injury, limited brain atrophy, and prevented chronic functional deficits after neonatal hypoxia-ischemic injury [270]. These findings are in line with reports showing myelin-reactive TH17 cells induced demyelination and impaired endogenous remyelination in pre-clinical white matter injury models and in multiple sclerosis patients [271–276]. In addition, curcumin, a curry spice that we found to improve TBI outcomes [243], attenuated expression of RORγT, a transcription factor involved in TH17 differentiation, decreased myelin basic protein-autoreactive T cells, and blocked the production of IL-17, the signature cytokine released by TH17 cells, after EAE [277]. Curcumin also attenuated TH17 production in ovalbumin-sensitized mice and in acute graft vs. host disease [278, 279]. As such, treatment strategies targeting TH17 production and/or activity may provide a potential option to improve long-term outcomes.
Functionally, the M1 macrophage derived, cytokines, IL-6 and IL-23, are essential for TH17 polarization [280–285]. We reported HMGB1 increased IL-6 production in primary human macrophages and after experimental TBI via a TLR4-dependent mechanism [73]. Similarly, the TLR4 agonist, LPS, induced IL-23 expression in peritoneal macrophages via TLR4 activation [286]. Finally, HMGB1-TLR4 signaling also increased myeloid IL-23 expression and subsequent TH17 production/mobilization after myocardial ischemia-reperfusion [287], periodontitis [288], or neonatal hypoxia-ischemia [270], although this issues remains to be fully studied after TBI. Given the temporal delay between TBI and adaptive immune responses, blocking TH polarization, secondary to DAMP-induced macrophage activation, may provide a feasible approach to limit long-term progressive injury. In addition, the presence of TH1/TH17 cells in blood may provide a prospective biomarker for patients that are susceptible for progressive white matter loss. We are actively exploring these interesting and clinically significant questions.
6. CONCLUSIONS
White matter injury is an important component of long-term neurological injury after TBI; however, the limited understanding of progressive white matter damage contributes to a lack of viable therapeutic options. The immune system is increasingly regarded as a critical, yet poorly understood, effector of neurological outcomes after TBI. Thus, defining the protective and detrimental roles of the immune system may improve the diagnosis, management, and treatment of TBI patients. Toward this end, the acute release of DAMPs may promote poor long-term neurological outcomes after TBI, yet potential delays in diagnosis and treatment coupled with narrow therapeutic windows may limit the ultimate clinical utility of DAMP/PRR inhibition. These translational barriers emphasize the need for innovative strategies to attenuate the detrimental long-term actions of acute DAMP release. For example, limiting the myeloid-lymphoid immune transition may provide an unexplored therapeutic avenue to reduce progressive white matter loss and to improve long-term clinical outcomes. Moreover, detection of myelin-directed TH1/TH17 cells may provide a blood biomarker to prospectively diagnose patients at risk of developing chronic neurodegeneration. Despite the inevitable challenges, improved definition of the relationship between immune activation and white matter injury will advance the clinical management and recovery of TBI patients.
FIGURE 1. Proposed mechanism of white matter injury after TBI.
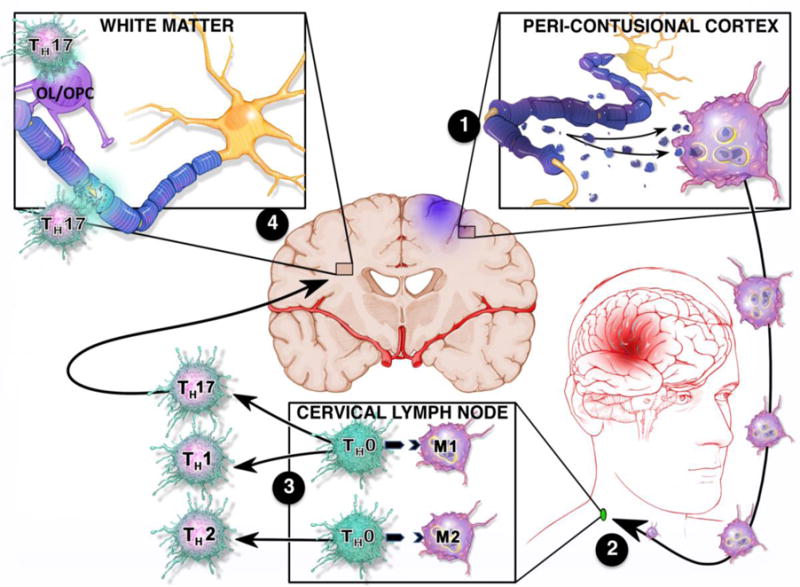
(1) Following TBI, the release of DAMPs from necrotic neurons activate and polarize microglia and infiltrating macrophages toward a pro-inflammatory M1 phenotype. Activated macrophages phagocytose dead/dying cells and myelin debris to initiate the process of neurological recovery. (2) Antigen loaded, M1 macrophages migrate to deep cervical lymph nodes to present antigen (e.g. myelin) to naïve T cells (TH0) cells. (3) M1 macrophages, which predominate after TBI, drive pro-inflammatory TH1/TH17 polarization to provide long-lasting, antigen-specific immunity by direct MHC Class II dependent antigen presentation and/or via the release of phenotype specific cytokines. (4) The subsequent migration of TH1/TH17 cells into the CNS leads to maladaptive immune responses against white matter (e.g. neurons, oligodendrocytes (OL), oligodendrocyte progenitor cells (OPC)). White matter loss results in further tissue destruction, secondary DAMP release, and chronic perpetuation of a pro-inflammatory feedback loop that culminates in neurological dysfunction.
HIGHLIGHTS.
White matter injury induces long-term neurological disability after TBI
Acute release of DAMPs activates myeloid cells after TBI
DAMP-induced myeloid cell activation initiates adaptive immunity after TBI
We provide a mechanistic framework for immune targeting to improve TBI outcomes
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.C. Centers for Disease. Prevention, Prevalence and most common causes of disability among adults–United States, 2005. MMWR Morb Mortal Wkly Rep. 2009;58:421–426. [PubMed] [Google Scholar]
- 2.Faul M, Xu L, Wald MM, Coronado VG. National Center for Injury Prevention and Control. Atlanta, GA: 2010. Traumatic brain injury in the United States: emergency department visits, hospitalizations, and deaths, Centers for Disease Control and Prevention. [Google Scholar]
- 3.Selassie AW, Zaloshnja E, Langlois JA, Miller T, Jones P, Steiner C. Incidence of long-term disability following traumatic brain injury hospitalization, United States, 2003. The Journal of head trauma rehabilitation. 2008;23:123–131. doi: 10.1097/01.HTR.0000314531.30401.39. [DOI] [PubMed] [Google Scholar]
- 4.F. E, C. P, M. T, a. associates. The incidence and economic burden of injuries in the United States. Oxford University Press, Place Published; 2006. [Google Scholar]
- 5.Coronado, McGuire, Faul, Sugerman, Pearson The epidemiology and prevention of TBI. 2012 in press. [Google Scholar]
- 6.Stein DG. Embracing failure: What the Phase III progesterone studies can teach about TBI clinical trials. Brain Injury. 2015;29:1259–1272. doi: 10.3109/02699052.2015.1065344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maas AI, Steyerberg EW, Marmarou A, McHugh GS, Lingsma HF, Butcher I, Lu J, Weir J, Roozenbeek B, Murray GD. IMPACT recommendations for improving the design and analysis of clinical trials in moderate to severe traumatic brain injury, Neurotherapeutics : the journal of the American Society for Experimental. NeuroTherapeutics. 2010;7:127–134. doi: 10.1016/j.nurt.2009.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Adams JH, Doyle D, Graham DI, Lawrence AE, McLellan DR, Gennarelli TA, Pastuszko M, Sakamoto T. The contusion index: a reappraisal in human and experimental non-missile head injury. Neuropathology and applied neurobiology. 1985;11:299–308. doi: 10.1111/j.1365-2990.1985.tb00027.x. [DOI] [PubMed] [Google Scholar]
- 9.Kotapka MJ, Graham DI, Adams JH, Gennarelli TA. Hippocampal pathology in fatal non-missile human head injury. Acta neuropathologica. 1992;83:530–534. doi: 10.1007/BF00310031. [DOI] [PubMed] [Google Scholar]
- 10.Ross DT, Graham DI, Adams JH. Selective loss of neurons from the thalamic reticular nucleus following severe human head injury. Journal of neurotrauma. 1993;10:151–165. doi: 10.1089/neu.1993.10.151. [DOI] [PubMed] [Google Scholar]
- 11.Gennarelli TA. Animate models of human head injury. Journal of neurotrauma. 1994;11:357–368. doi: 10.1089/neu.1994.11.357. [DOI] [PubMed] [Google Scholar]
- 12.Raghupathi R, Conti AC, Graham DI, Krajewski S, Reed JC, Grady MS, Trojanowski JQ, McIntosh TK. Mild traumatic brain injury induces apoptotic cell death in the cortex that is preceded by decreases in cellular Bcl-2 immunoreactivity. Neuroscience. 2002;110:605–616. doi: 10.1016/s0306-4522(01)00461-4. [DOI] [PubMed] [Google Scholar]
- 13.Povlishock JT, Hayes RL, Michel ME, McIntosh TK. Workshop on animal models of traumatic brain injury. Journal of neurotrauma. 1994;11:723–732. doi: 10.1089/neu.1994.11.723. [DOI] [PubMed] [Google Scholar]
- 14.Farook JM, Shields J, Tawfik A, Markand S, Sen T, Smith SB, Brann D, Dhandapani KM, Sen N. GADD34 induces cell death through inactivation of Akt following traumatic brain injury. Cell death & disease. 2013;4:e754. doi: 10.1038/cddis.2013.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang QG, Laird MD, Han D, Nguyen K, Scott E, Dong Y, Dhandapani KM, Brann DW. Critical role of NADPH oxidase in neuronal oxidative damage and microglia activation following traumatic brain injury. PloS one. 2012;7:e34504. doi: 10.1371/journal.pone.0034504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wakade C, Sukumari-Ramesh S, Laird MD, Dhandapani KM, Vender JR. Delayed reduction in hippocampal postsynaptic density protein-95 expression temporally correlates with cognitive dysfunction following controlled cortical impact in mice. Journal of neurosurgery. 2010;113:1195–1201. doi: 10.3171/2010.3.JNS091212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ikonomidou C, Turski L. Why did NMDA receptor antagonists fail clinical trials for stroke and traumatic brain injury? Lancet Neurol. 2002;1:383–386. doi: 10.1016/s1474-4422(02)00164-3. [DOI] [PubMed] [Google Scholar]
- 18.Shaw NA. The neurophysiology of concussion. Progress in Neurobiology. 2002;67:281–344. doi: 10.1016/s0301-0082(02)00018-7. [DOI] [PubMed] [Google Scholar]
- 19.Drew LB, Drew WE. The contrecoup-coup phenomenon. Neurocritical Care. 2004;1:385–390. doi: 10.1385/NCC:1:3:385. [DOI] [PubMed] [Google Scholar]
- 20.Baltan S, Carmichael ST, Matute C, Zhang JH. White Matter Injury in Stroke and CNS Disease. Springer New York: Place Published; 2013. [Google Scholar]
- 21.Evans RW. Diffuse Axonal Injury, Neurology and Trauma. Oxford University Press, USA Place Published; 2006. [Google Scholar]
- 22.Alhilali LM, Delic JA, Gumus S, Fakhran S. Evaluation of White Matter Injury Patterns Underlying Neuropsychiatric Symptoms after Mild Traumatic Brain Injury. Radiology. 2015;277:793–800. doi: 10.1148/radiol.2015142974. [DOI] [PubMed] [Google Scholar]
- 23.Bramlett HM, Dietrich WD. Long-Term Consequences of Traumatic Brain Injury: Current Status of Potential Mechanisms of Injury and Neurological Outcomes. Journal of neurotrauma. 2014 doi: 10.1089/neu.2014.3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cullum CM, Bigler ED. Ventricle size, cortical atrophy and the relationship with neuropsychological status in closed head injury: a quantitative analysis. Journal of clinical and experimental neuropsychology. 1986;8:437–452. doi: 10.1080/01688638608401333. [DOI] [PubMed] [Google Scholar]
- 25.Galanaud D, Perlbarg V, Gupta R, Stevens RD, Sanchez P, Tollard E, de Champfleur NM, Dinkel J, Faivre S, Soto-Ares G, Veber B, Cottenceau V, Masson F, Tourdias T, Andre E, Audibert G, Schmitt E, Ibarrola D, Dailler F, Vanhaudenhuyse A, Tshibanda L, Payen JF, Le Bas JF, Krainik A, Bruder N, Girard N, Laureys S, Benali H, Puybasset L. Assessment of white matter injury and outcome in severe brain trauma: a prospective multicenter cohort. Anesthesiology. 2012;117:1300–1310. doi: 10.1097/ALN.0b013e3182755558. [DOI] [PubMed] [Google Scholar]
- 26.Kinnunen KM, Greenwood R, Powell JH, Leech R, Hawkins PC, Bonnelle V, Patel MC, Counsell SJ, Sharp DJ. White matter damage and cognitive impairment after traumatic brain injury. Brain : a journal of neurology. 2011;134:449–463. doi: 10.1093/brain/awq347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kraus MF, Susmaras T, Caughlin BP, Walker CJ, Sweeney JA, Little DM. White matter integrity and cognition in chronic traumatic brain injury: a diffusion tensor imaging study. Brain : a journal of neurology. 2007;130:2508–2519. doi: 10.1093/brain/awm216. [DOI] [PubMed] [Google Scholar]
- 28.Shi H, Hu X, Leak RK, Shi Y, An C, Suenaga J, Chen J, Gao Y. Demyelination as a rational therapeutic target for ischemic or traumatic brain injury. Experimental neurology. 2015;272:17–25. doi: 10.1016/j.expneurol.2015.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Buki A, Povlishock JT. All roads lead to disconnection?–Traumatic axonal injury revisited. Acta neurochirurgica. 2006;148:181–193. doi: 10.1007/s00701-005-0674-4. discussion 193–184. [DOI] [PubMed] [Google Scholar]
- 30.Su E, Bell M. Diffuse Axonal Injury. In: Laskowitz D, Grant G, editors. Translational Research in Traumatic Brain Injury. Place Published; 2016. [Google Scholar]
- 31.Bramlett HM, Dietrich WD. Quantitative structural changes in white and gray matter 1 year following traumatic brain injury in rats. Acta neuropathologica. 2002;103:607–614. doi: 10.1007/s00401-001-0510-8. [DOI] [PubMed] [Google Scholar]
- 32.Bramlett HM, Kraydieh S, Green EJ, Dietrich WD. Temporal and regional patterns of axonal damage following traumatic brain injury: a beta-amyloid precursor protein immunocytochemical study in rats. Journal of neuropathology and experimental neurology. 1997;56:1132–1141. doi: 10.1097/00005072-199710000-00007. [DOI] [PubMed] [Google Scholar]
- 33.Bramlett HM, Dietrich WD. Progressive damage after brain and spinal cord injury: pathomechanisms and treatment strategies. Progress in brain research. 2007;161:125–141. doi: 10.1016/S0079-6123(06)61009-1. [DOI] [PubMed] [Google Scholar]
- 34.Green RE, Colella B, Maller JJ, Bayley M, Glazer J, Mikulis DJ. Scale and pattern of atrophy in the chronic stages of moderate-severe TBI. Frontiers in human neuroscience. 2014;8:67. doi: 10.3389/fnhum.2014.00067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johnson VE, Stewart W, Smith DH. Axonal pathology in traumatic brain injury. Experimental neurology. 2013;246:35–43. doi: 10.1016/j.expneurol.2012.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gentry LR, Thompson B, Godersky JC. Trauma to the corpus callosum: MR features. American Journal of Neuroradiology. 1988;9:1129–1138. [PMC free article] [PubMed] [Google Scholar]
- 37.Johnson VE, Stewart JE, Begbie FD, Trojanowski JQ, Smith DH, Stewart W. Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain : a journal of neurology. 2013;136:28–42. doi: 10.1093/brain/aws322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scott G, Hellyer PJ, Ramlackhansingh AF, Brooks DJ, Matthews PM, Sharp DJ. Thalamic inflammation after brain trauma is associated with thalamo-cortical white matter damage. Journal of neuroinflammation. 2015;12:224. doi: 10.1186/s12974-015-0445-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Farkas AM, Kilgore TM, Lotze MT. Detecting DNA: getting and begetting cancer. Curr Opin Investig Drugs. 2007;8:981–986. [PubMed] [Google Scholar]
- 40.Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–195. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- 41.Scheibner KA, Lutz MA, Boodoo S, Fenton MJ, Powell JD, Horton MR. Hyaluronan fragments act as an endogenous danger signal by engaging TLR2. J Immunol. 2006;177:1272–1281. doi: 10.4049/jimmunol.177.2.1272. [DOI] [PubMed] [Google Scholar]
- 42.Boeynaems JM, Communi D. Modulation of inflammation by extracellular nucleotides. The Journal of investigative dermatology. 2006;126:943–944. doi: 10.1038/sj.jid.5700233. [DOI] [PubMed] [Google Scholar]
- 43.Gardella S, Andrei C, Ferrera D, Lotti LV, Torrisi MR, Bianchi ME, Rubartelli A. The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO reports. 2002;3:995–1001. doi: 10.1093/embo-reports/kvf198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Farina C, Aloisi F, Meinl E. Astrocytes are active players in cerebral innate immunity. Trends in immunology. 2007;28:138–145. doi: 10.1016/j.it.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 45.Lehnardt S. Innate immunity and neuroinflammation in the CNS: the role of microglia in Toll-like receptor-mediated neuronal injury. Glia. 2010;58:253–263. doi: 10.1002/glia.20928. [DOI] [PubMed] [Google Scholar]
- 46.Rivest S. Regulation of innate immune responses in the brain. Nature reviews Immunology. 2009;9:429–439. doi: 10.1038/nri2565. [DOI] [PubMed] [Google Scholar]
- 47.Franzblau M, Gonzales-Portillo C, Gonzales-Portillo GS, Diamandis T, Borlongan MC, Tajiri N, Borlongan CV. Vascular damage: a persisting pathology common to Alzheimer’s disease and traumatic brain injury. Med Hypotheses. 2013;81:842–845. doi: 10.1016/j.mehy.2013.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Acosta SA, Tajiri N, Sanberg PR, Kaneko Y, Borlongan CV. Increased Amyloid Precursor Protein and Tau Expression Manifests as Key Secondary Cell Death in Chronic Traumatic Brain Injury. J Cell Physiol. 2017;232:665–677. doi: 10.1002/jcp.25629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Acosta SA, Tajiri N, de la Pena I, Bastawrous M, Sanberg PR, Kaneko Y, Borlongan CV. Alpha-synuclein as a pathological link between chronic traumatic brain injury and Parkinson’s disease. J Cell Physiol. 2015;230:1024–1032. doi: 10.1002/jcp.24830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Giunta B, Obregon D, Velisetty R, Sanberg PR, Borlongan CV, Tan J. The immunology of traumatic brain injury: a prime target for Alzheimer’s disease prevention. Journal of neuroinflammation. 2012;9:185. doi: 10.1186/1742-2094-9-185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Russo MV, McGavern DB. Immune Surveillance of the CNS following Infection and Injury. Trends in immunology. 2015;36:637–650. doi: 10.1016/j.it.2015.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fang KM, Yang CS, Sun SH, Tzeng SF. Microglial phagocytosis attenuated by short-term exposure to exogenous ATP through P2X receptor action. Journal of neurochemistry. 2009;111:1225–1237. doi: 10.1111/j.1471-4159.2009.06409.x. [DOI] [PubMed] [Google Scholar]
- 53.Cristofori L, Tavazzi B, Gambin R, Vagnozzi R, Signoretti S, Amorini AM, Fazzina G, Lazzarino G. Biochemical analysis of the cerebrospinal fluid: evidence for catastrophic energy failure and oxidative damage preceding brain death in severe head injury: a case report. Clinical biochemistry. 2005;38:97–100. doi: 10.1016/j.clinbiochem.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 54.Abbracchio MP, Burnstock G, Boeynaems JM, Barnard EA, Boyer JL, Kennedy C, Knight GE, Fumagalli M, Gachet C, Jacobson KA, Weisman GA. International Union of Pharmacology LVIII: update on the P2Y G protein-coupled nucleotide receptors: from molecular mechanisms and pathophysiology to therapy. Pharmacological reviews. 2006;58:281–341. doi: 10.1124/pr.58.3.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Klepeis VE, Weinger I, Kaczmarek E, Trinkaus-Randall V. P2Y receptors play a critical role in epithelial cell communication and migration. J Cell Biochem. 2004;93:1115–1133. doi: 10.1002/jcb.20258. [DOI] [PubMed] [Google Scholar]
- 56.Talley Watts L, Sprague S, Zheng W, Garling RJ, Jimenez D, Digicaylioglu M, Lechleiter J. Purinergic 2Y1 receptor stimulation decreases cerebral edema and reactive gliosis in a traumatic brain injury model. Journal of neurotrauma. 2013;30:55–66. doi: 10.1089/neu.2012.2488. [DOI] [PubMed] [Google Scholar]
- 57.Choo AM, Miller WJ, Chen YC, Nibley P, Patel TP, Goletiani C, Morrison B, 3rd, Kutzing MK, Firestein BL, Sul JY, Haydon PG, Meaney DF. Antagonism of purinergic signalling improves recovery from traumatic brain injury. Brain : a journal of neurology. 2013;136:65–80. doi: 10.1093/brain/aws286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Roth TL, Nayak D, Atanasijevic T, Koretsky AP, Latour LL, McGavern DB. Transcranial amelioration of inflammation and cell death after brain injury. Nature. 2014;505:223–228. doi: 10.1038/nature12808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Franke H, Parravicini C, Lecca D, Zanier ER, Heine C, Bremicker K, Fumagalli M, Rosa P, Longhi L, Stocchetti N, De Simoni MG, Weber M, Abbracchio MP. Changes of the GPR17 receptor, a new target for neurorepair, in neurons and glial cells in patients with traumatic brain injury. Purinergic signalling. 2013;9:451–462. doi: 10.1007/s11302-013-9366-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lecca D, Trincavelli ML, Gelosa P, Sironi L, Ciana P, Fumagalli M, Villa G, Verderio C, Grumelli C, Guerrini U, Tremoli E, Rosa P, Cuboni S, Martini C, Buffo A, Cimino M, Abbracchio MP. The recently identified P2Y-like receptor GPR17 is a sensor of brain damage and a new target for brain repair. PloS one. 2008;3:e3579. doi: 10.1371/journal.pone.0003579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gever JR, Cockayne DA, Dillon MP, Burnstock G, Ford AP. Pharmacology of P2X channels. Pflugers Arch. 2006;452:513–537. doi: 10.1007/s00424-006-0070-9. [DOI] [PubMed] [Google Scholar]
- 62.North RA. Molecular physiology of P2X receptors. Physiol Rev. 2002;82:1013–1067. doi: 10.1152/physrev.00015.2002. [DOI] [PubMed] [Google Scholar]
- 63.Wang YC, Cui Y, Cui JZ, Sun LQ, Cui CM, Zhang HA, Zhu HX, Li R, Tian YX, Gao JL. Neuroprotective effects of brilliant blue G on the brain following traumatic brain injury in rats. Molecular medicine reports. 2015;12:2149–2154. doi: 10.3892/mmr.2015.3607. [DOI] [PubMed] [Google Scholar]
- 64.Kimbler DE, Shields J, Yanasak N, Vender JR, Dhandapani KM. Activation of P2X7 promotes cerebral edema and neurological injury after traumatic brain injury in mice. PloS one. 2012;7:e41229. doi: 10.1371/journal.pone.0041229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sun L, Gao J, Zhao M, Cui J, Li Y, Yang X, Jing X, Wu Z. A novel cognitive impairment mechanism that astrocytic p-connexin 43 promotes neuronic autophagy via activation of P2X7R and down-regulation of GLT-1 expression in the hippocampus following traumatic brain injury in rats. Behavioural brain research. 2015;291:315–324. doi: 10.1016/j.bbr.2015.05.049. [DOI] [PubMed] [Google Scholar]
- 66.James G, Butt AM. Changes in P2Y and P2X purinoceptors in reactive glia following axonal degeneration in the rat optic nerve. Neuroscience letters. 2001;312:33–36. doi: 10.1016/s0304-3940(01)02189-9. [DOI] [PubMed] [Google Scholar]
- 67.Matute C. Glutamate and ATP signalling in white matter pathology. Journal of anatomy. 2011;219:53–64. doi: 10.1111/j.1469-7580.2010.01339.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Matute C. P2X7 receptors in oligodendrocytes: a novel target for neuroprotection. Molecular neurobiology. 2008;38:123–128. doi: 10.1007/s12035-008-8028-x. [DOI] [PubMed] [Google Scholar]
- 69.Peng W, Cotrina ML, Han X, Yu H, Bekar L, Blum L, Takano T, Tian GF, Goldman SA, Nedergaard M. Systemic administration of an antagonist of the ATP-sensitive receptor P2X7 improves recovery after spinal cord injury. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:12489–12493. doi: 10.1073/pnas.0902531106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang X, Arcuino G, Takano T, Lin J, Peng WG, Wan P, Li P, Xu Q, Liu QS, Goldman SA, Nedergaard M. P2X7 receptor inhibition improves recovery after spinal cord injury. Nature medicine. 2004;10:821–827. doi: 10.1038/nm1082. [DOI] [PubMed] [Google Scholar]
- 71.Wang LY, Cai WQ, Chen PH, Deng QY, Zhao CM. Downregulation of P2X7 receptor expression in rat oligodendrocyte precursor cells after hypoxia ischemia. Glia. 2009;57:307–319. doi: 10.1002/glia.20758. [DOI] [PubMed] [Google Scholar]
- 72.Domercq M, Perez-Samartin A, Aparicio D, Alberdi E, Pampliega O, Matute C. P2X7 receptors mediate ischemic damage to oligodendrocytes. Glia. 2010;58:730–740. doi: 10.1002/glia.20958. [DOI] [PubMed] [Google Scholar]
- 73.Laird MD, Shields JS, Sukumari-Ramesh S, Kimbler DE, Fessler RD, Shakir B, Youssef P, Yanasak N, Vender JR, Dhandapani KM. High mobility group box protein-1 promotes cerebral edema after traumatic brain injury via activation of toll-like receptor 4. Glia. 2014;62:26–38. doi: 10.1002/glia.22581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang KY, Yu GF, Zhang ZY, Huang Q, Dong XQ. Plasma high-mobility group box 1 levels and prediction of outcome in patients with traumatic brain injury. Clinica chimica acta; international journal of clinical chemistry. 2012;413:1737–1741. doi: 10.1016/j.cca.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 75.King MD, Laird MD, Ramesh SS, Youssef P, Shakir B, Vender JR, Alleyne CH, Dhandapani KM. Elucidating novel mechanisms of brain injury following subarachnoid hemorrhage: an emerging role for neuroproteomics. Neurosurgical focus. 2010;28:E10. doi: 10.3171/2009.10.FOCUS09223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Au AK, Aneja RK, Bell MJ, Bayir H, Feldman K, Adelson PD, Fink EL, Kochanek PM, Clark RS. Cerebrospinal fluid levels of high-mobility group box 1 and cytochrome C predict outcome after pediatric traumatic brain injury. Journal of neurotrauma. 2012;29:2013–2021. doi: 10.1089/neu.2011.2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hayakawa K, Pham LD, Katusic ZS, Arai K, Lo EH. Astrocytic high-mobility group box 1 promotes endothelial progenitor cell-mediated neurovascular remodeling during stroke recovery. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:7505–7510. doi: 10.1073/pnas.1121146109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hayakawa K, Miyamoto N, Seo JH, Pham LD, Kim KW, Lo EH, Arai K. High-mobility group box 1 from reactive astrocytes enhances the accumulation of endothelial progenitor cells in damaged white matter. Journal of neurochemistry. 2013;125:273–280. doi: 10.1111/jnc.12120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhang Z, Zhang ZY, Wu Y, Schluesener HJ. Immunolocalization of Toll-like receptors 2 and 4 as well as their endogenous ligand, heat shock protein 70, in rat traumatic brain injury. Neuroimmunomodulation. 2012;19:10–19. doi: 10.1159/000326771. [DOI] [PubMed] [Google Scholar]
- 80.Casula M, Iyer AM, Spliet WG, Anink JJ, Steentjes K, Sta M, Troost D, Aronica E. Toll-like receptor signaling in amyotrophic lateral sclerosis spinal cord tissue. Neuroscience. 2011;179:233–243. doi: 10.1016/j.neuroscience.2011.02.001. [DOI] [PubMed] [Google Scholar]
- 81.Garnier Y, Berger R, Alm S, von Duering MU, Coumans AB, Michetti F, Bruschettini M, Lituania M, Hasaart TH, Gazzolo D. Systemic endotoxin administration results in increased S100B protein blood levels and periventricular brain white matter injury in the preterm fetal sheep. European journal of obstetrics, gynecology, and reproductive biology. 2006;124:15–22. doi: 10.1016/j.ejogrb.2005.05.014. [DOI] [PubMed] [Google Scholar]
- 82.Garnier Y, Frigiola A, Li Volti G, Florio P, Frulio R, Berger R, Alm S, von Duering MU, Coumans AB, Reis FM, Petraglia F, Hasaart TH, Abella R, Mufeed H, Gazzolo D. Increased maternal/fetal blood S100B levels following systemic endotoxin administration and periventricular white matter injury in preterm fetal sheep. Reprod Sci. 2009;16:758–766. doi: 10.1177/1933719109335801. [DOI] [PubMed] [Google Scholar]
- 83.Hamner MA, Ye Z, Lee RV, Colman JR, Le T, Gong DC, Ransom BR, Weinstein JR. Ischemic Preconditioning in White Matter: Magnitude and Mechanism. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2015;35:15599–15611. doi: 10.1523/JNEUROSCI.2544-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wang X, Stridh L, Li W, Dean J, Elmgren A, Gan L, Eriksson K, Hagberg H, Mallard C. Lipopolysaccharide sensitizes neonatal hypoxic-ischemic brain injury in a MyD88-dependent manner. J Immunol. 2009;183:7471–7477. doi: 10.4049/jimmunol.0900762. [DOI] [PubMed] [Google Scholar]
- 85.Alfonso-Loeches S, Pascual M, Gomez-Pinedo U, Pascual-Lucas M, Renau-Piqueras J, Guerri C. Toll-like receptor 4 participates in the myelin disruptions associated with chronic alcohol abuse. Glia. 2012;60:948–964. doi: 10.1002/glia.22327. [DOI] [PubMed] [Google Scholar]
- 86.Kigerl KA, Lai W, Rivest S, Hart RP, Satoskar AR, Popovich PG. Toll-like receptor (TLR)-2 and TLR-4 regulate inflammation, gliosis, and myelin sparing after spinal cord injury. Journal of neurochemistry. 2007;102:37–50. doi: 10.1111/j.1471-4159.2007.04524.x. [DOI] [PubMed] [Google Scholar]
- 87.Church JS, Kigerl KA, Lerch JK, Popovich PG, McTigue DM. TLR4 Deficiency Impairs Oligodendrocyte Formation in the Injured Spinal Cord. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2016;36:6352–6364. doi: 10.1523/JNEUROSCI.0353-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Stirling DP, Cummins K, Mishra M, Teo W, Yong VW, Stys P. Toll-like receptor 2-mediated alternative activation of microglia is protective after spinal cord injury. Brain : a journal of neurology. 2014;137:707–723. doi: 10.1093/brain/awt341. [DOI] [PubMed] [Google Scholar]
- 89.Marenholz I, Heizmann CW, Fritz G. S100 proteins in mouse and man: from evolution to function and pathology (including an update of the nomenclature) Biochemical and biophysical research communications. 2004;322:1111–1122. doi: 10.1016/j.bbrc.2004.07.096. [DOI] [PubMed] [Google Scholar]
- 90.Santamaria-Kisiel L, Rintala-Dempsey AC, Shaw GS. Calcium-dependent and -independent interactions of the S100 protein family. The Biochemical journal. 2006;396:201–214. doi: 10.1042/BJ20060195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ostendorp T, Leclerc E, Galichet A, Koch M, Demling N, Weigle B, Heizmann CW, Kroneck PM, Fritz G. Structural and functional insights into RAGE activation by multimeric S100B. EMBO J. 2007;26:3868–3878. doi: 10.1038/sj.emboj.7601805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hofmann MA, Drury S, Fu C, Qu W, Taguchi A, Lu Y, Avila C, Kambham N, Bierhaus A, Nawroth P, Neurath MF, Slattery T, Beach D, McClary J, Nagashima M, Morser J, Stern D, Schmidt AM. RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell. 1999;97:889–901. doi: 10.1016/s0092-8674(00)80801-6. [DOI] [PubMed] [Google Scholar]
- 93.Dattilo BM, Fritz G, Leclerc E, Kooi CW, Heizmann CW, Chazin WJ. The extracellular region of the receptor for advanced glycation end products is composed of two independent structural units. Biochemistry. 2007;46:6957–6970. doi: 10.1021/bi7003735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Huttunen HJ, Kuja-Panula J, Sorci G, Agneletti AL, Donato R, Rauvala H. Coregulation of neurite outgrowth and cell survival by amphoterin and S100 proteins through receptor for advanced glycation end products (RAGE) activation. J Biol Chem. 2000;275:40096–40105. doi: 10.1074/jbc.M006993200. [DOI] [PubMed] [Google Scholar]
- 95.Leclerc E, Fritz G, Vetter SW, Heizmann CW. Binding of S100 proteins to RAGE: an update. Biochim Biophys Acta. 2009;1793:993–1007. doi: 10.1016/j.bbamcr.2008.11.016. [DOI] [PubMed] [Google Scholar]
- 96.Kiryushko D, Novitskaya V, Soroka V, Klingelhofer J, Lukanidin E, Berezin V, Bock E. Molecular mechanisms of Ca(2+) signaling in neurons induced by the S100A4 protein. Mol Cell Biol. 2006;26:3625–3638. doi: 10.1128/MCB.26.9.3625-3638.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Leclerc E, Fritz G, Weibel M, Heizmann CW, Galichet A. S100B and S100A6 differentially modulate cell survival by interacting with distinct RAGE (receptor for advanced glycation end products) immunoglobulin domains. The Journal of biological chemistry. 2007;282:31317–31331. doi: 10.1074/jbc.M703951200. [DOI] [PubMed] [Google Scholar]
- 98.Wolf R, Howard OM, Dong HF, Voscopoulos C, Boeshans K, Winston J, Divi R, Gunsior M, Goldsmith P, Ahvazi B, Chavakis T, Oppenheim JJ, Yuspa SH. Chemotactic activity of S100A7 (Psoriasin) is mediated by the receptor for advanced glycation end products and potentiates inflammation with highly homologous but functionally distinct S100A15. J Immunol. 2008;181:1499–1506. doi: 10.4049/jimmunol.181.2.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Boyd JH, Kan B, Roberts H, Wang Y, Walley KR. S100A8 and S100A9 mediate endotoxin-induced cardiomyocyte dysfunction via the receptor for advanced glycation end products. Circ Res. 2008;102:1239–1246. doi: 10.1161/CIRCRESAHA.107.167544. [DOI] [PubMed] [Google Scholar]
- 100.Ghavami S, Rashedi I, Dattilo BM, Eshraghi M, Chazin WJ, Hashemi M, Wesselborg S, Kerkhoff C, Los M. S100A8/A9 at low concentration promotes tumor cell growth via RAGE ligation and MAP kinase-dependent pathway. Journal of leukocyte biology. 2008;83:1484–1492. doi: 10.1189/jlb.0607397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Cecil DL, Johnson K, Rediske J, Lotz M, Schmidt AM, Terkeltaub R. Inflammation-induced chondrocyte hypertrophy is driven by receptor for advanced glycation end products. J Immunol. 2005;175:8296–8302. doi: 10.4049/jimmunol.175.12.8296. [DOI] [PubMed] [Google Scholar]
- 102.Sakaguchi M, Sonegawa H, Murata H, Kitazoe M, Futami J, Kataoka K, Yamada H, Huh NH. S100A11, an dual mediator for growth regulation of human keratinocytes. Mol Biol Cell. 2008;19:78–85. doi: 10.1091/mbc.E07-07-0682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Xie J, Burz DS, He W, Bronstein IB, Lednev I, Shekhtman A. Hexameric calgranulin C (S100A12) binds to the receptor for advanced glycated end products (RAGE) using symmetric hydrophobic target-binding patches. The Journal of biological chemistry. 2007;282:4218–4231. doi: 10.1074/jbc.M608888200. [DOI] [PubMed] [Google Scholar]
- 104.Hsieh HL, Schafer BW, Weigle B, Heizmann CW. S100 protein translocation in response to extracellular S100 is mediated by receptor for advanced glycation endproducts in human endothelial cells. Biochemical and biophysical research communications. 2004;316:949–959. doi: 10.1016/j.bbrc.2004.02.135. [DOI] [PubMed] [Google Scholar]
- 105.Arumugam T, Simeone DM, Schmidt AM, Logsdon CD. S100P stimulates cell proliferation and survival via receptor for activated glycation end products (RAGE) The Journal of biological chemistry. 2004;279:5059–5065. doi: 10.1074/jbc.M310124200. [DOI] [PubMed] [Google Scholar]
- 106.Vogl T, Tenbrock K, Ludwig S, Leukert N, Ehrhardt C, van Zoelen MA, Nacken W, Foell D, van der Poll T, Sorg C, Roth J. Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nature medicine. 2007;13:1042–1049. doi: 10.1038/nm1638. [DOI] [PubMed] [Google Scholar]
- 107.Steiner J, Bernstein HG, Bielau H, Berndt A, Brisch R, Mawrin C, Keilhoff G, Bogerts B. Evidence for a wide extra-astrocytic distribution of S100B in human brain. BMC neuroscience. 2007;8:2. doi: 10.1186/1471-2202-8-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Streitburger DP, Arelin K, Kratzsch J, Thiery J, Steiner J, Villringer A, Mueller K, Schroeter ML. Validating serum S100B and neuron-specific enolase as biomarkers for the human brain - a combined serum, gene expression and MRI study. PloS one. 2012;7:e43284. doi: 10.1371/journal.pone.0043284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Schmidt H, Gerber J, Stuertz K, Djukic M, Bunkowski S, Fischer FR, Otto M, Nau R. S100B in the cerebrospinal fluid–a marker for glial damage in the rabbit model of pneumococcal meningitis. Neuroscience letters. 2010;475:104–107. doi: 10.1016/j.neulet.2010.03.059. [DOI] [PubMed] [Google Scholar]
- 110.Steiner J, Schmitt A, Schroeter ML, Bogerts B, Falkai P, Turck CW, Martins-de-Souza D. S100B is downregulated in the nuclear proteome of schizophrenia corpus callosum. European archives of psychiatry and clinical neuroscience. 2014;264:311–316. doi: 10.1007/s00406-014-0490-z. [DOI] [PubMed] [Google Scholar]
- 111.Milleit B, Smesny S, Rothermundt M, Preul C, Schroeter ML, von Eiff C, Ponath G, Milleit C, Sauer H, Gaser C. Serum S100B Protein is Specifically Related to White Matter Changes in Schizophrenia. Frontiers in cellular neuroscience. 2016;10:33. doi: 10.3389/fncel.2016.00033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Distefano G, Curreri R, Betta P, Isaja MT, Romeo MG, Amato M. Serial protein S-100 serum levels in preterm babies with perinatal asphyxia and periventricular white matter lesions. American journal of perinatology. 2002;19:317–322. doi: 10.1055/s-2002-34463. [DOI] [PubMed] [Google Scholar]
- 113.Berger RP, Pierce MC, Wisniewski SR, Adelson PD, Kochanek PM. Serum S100B concentrations are increased after closed head injury in children: a preliminary study. Journal of neurotrauma. 2002;19:1405–1409. doi: 10.1089/089771502320914633. [DOI] [PubMed] [Google Scholar]
- 114.Murillo-Cabezas F, Munoz-Sanchez MA, Rincon-Ferrari MD, Martin-Rodriguez JF, Amaya-Villar R, Garcia-Gomez S, Leon-Carrion J. The prognostic value of the temporal course of S100beta protein in post-acute severe brain injury: A prospective and observational study. Brain Inj. 2010;24:609–619. doi: 10.3109/02699051003652823. [DOI] [PubMed] [Google Scholar]
- 115.Egea-Guerrero JJ, Revuelto-Rey J, Murillo-Cabezas F, Munoz-Sanchez MA, Vilches-Arenas A, Sanchez-Linares P, Dominguez-Roldan JM, Leon-Carrion J. Accuracy of the S100beta protein as a marker of brain damage in traumatic brain injury. Brain Inj. 2012;26:76–82. doi: 10.3109/02699052.2011.635360. [DOI] [PubMed] [Google Scholar]
- 116.Mercier E, Boutin A, Lauzier F, Fergusson DA, Simard JF, Zarychanski R, Moore L, McIntyre LA, Archambault P, Lamontagne F, Legare F, Randell E, Nadeau L, Rousseau F, Turgeon AF. Predictive value of S-100beta protein for prognosis in patients with moderate and severe traumatic brain injury: systematic review and meta-analysis. BMJ. 2013;346:f1757. doi: 10.1136/bmj.f1757. [DOI] [PubMed] [Google Scholar]
- 117.Spinella PC, Dominguez T, Drott HR, Huh J, McCormick L, Rajendra A, Argon J, McIntosh T, Helfaer M. S-100beta protein-serum levels in healthy children and its association with outcome in pediatric traumatic brain injury. Critical care medicine. 2003;31:939–945. doi: 10.1097/01.CCM.0000053644.16336.52. [DOI] [PubMed] [Google Scholar]
- 118.Bohmer AE, Oses JP, Schmidt AP, Peron CS, Krebs CL, Oppitz PP, D’Avila TT, Souza DO, Portela LV, Stefani MA. Neuron-specific enolase, S100B, and glial fibrillary acidic protein levels as outcome predictors in patients with severe traumatic brain injury. Neurosurgery. 2011;68:1624–1630. doi: 10.1227/NEU.0b013e318214a81f. discussion 1630–1621. [DOI] [PubMed] [Google Scholar]
- 119.Joseph B, Pandit V, Zangbar B, Kulvatunyou N, Khalil M, Tang A, O’Keeffe T, Gries L, Vercruysse G, Friese RS, Rhee P. Secondary brain injury in trauma patients: the effects of remote ischemic conditioning. J Trauma Acute Care Surg. 2015;78:698–703. doi: 10.1097/TA.0000000000000584. discussion 703–695. [DOI] [PubMed] [Google Scholar]
- 120.Studer M, Simonetti B Goeggel, Heinks T, Steinlin M, Leichtle A, Berger S, Joeris A. Acute S100B in serum is associated with cognitive symptoms and memory performance 4 months after paediatric mild traumatic brain injury. Brain Inj. 2015;29:1667–1673. doi: 10.3109/02699052.2015.1075250. [DOI] [PubMed] [Google Scholar]
- 121.Marchi N, Bazarian JJ, Puvenna V, Janigro M, Ghosh C, Zhong J, Zhu T, Blackman E, Stewart D, Ellis J, Butler R, Janigro D. Consequences of repeated blood-brain barrier disruption in football players. PloS one. 2013;8:e56805. doi: 10.1371/journal.pone.0056805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Pham N, Fazio V, Cucullo L, Teng Q, Biberthaler P, Bazarian JJ, Janigro D. Extracranial sources of S100B do not affect serum levels. PloS one. 2010;5 doi: 10.1371/journal.pone.0012691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hearst SM, Walker LR, Shao Q, Lopez M, Raucher D, Vig PJ. The design and delivery of a thermally responsive peptide to inhibit S100B-mediated neurodegeneration. Neuroscience. 2011;197:369–380. doi: 10.1016/j.neuroscience.2011.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kabadi SV, Stoica BA, Zimmer DB, Afanador L, Duffy KB, Loane DJ, Faden AI. S100B inhibition reduces behavioral and pathologic changes in experimental traumatic brain injury. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2015;35:2010–2020. doi: 10.1038/jcbfm.2015.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Di Battista AP, Buonora JE, Rhind SG, Hutchison MG, Baker AJ, Rizoli SB, Diaz-Arrastia R, Mueller GP. Blood Biomarkers in Moderate-To-Severe Traumatic Brain Injury: Potential Utility of a Multi-Marker Approach in Characterizing Outcome. Frontiers in neurology. 2015;6:110. doi: 10.3389/fneur.2015.00110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kozlova EN, Lukanidin E. Metastasis-associated mts1 (S100A4) protein is selectively expressed in white matter astrocytes and is up-regulated after peripheral nerve or dorsal root injury. Glia. 1999;27:249–258. [PubMed] [Google Scholar]
- 127.Kozlova EN, Lukanidin E. Mts1 protein expression in the central nervous system after injury. Glia. 2002;37:337–348. [PubMed] [Google Scholar]
- 128.Takenaga K, Kozlova EN. Role of intracellular S100A4 for migration of rat astrocytes. Glia. 2006;53:313–321. doi: 10.1002/glia.20284. [DOI] [PubMed] [Google Scholar]
- 129.Fang Z, Duthoit N, Wicher G, Kallskog O, Ambartsumian N, Lukanidin E, Takenaga K, Kozlova EN. Intracellular calcium-binding protein S100A4 influences injury-induced migration of white matter astrocytes. Acta neuropathologica. 2006;111:213–219. doi: 10.1007/s00401-005-0019-7. [DOI] [PubMed] [Google Scholar]
- 130.Ambartsumian N, Klingelhofer J, Grigorian M, Christensen C, Kriajevska M, Tulchinsky E, Georgiev G, Berezin V, Bock E, Rygaard J, Cao R, Cao Y, Lukanidin E. The metastasis-associated Mts1(S100A4) protein could act as an angiogenic factor. Oncogene. 2001;20:4685–4695. doi: 10.1038/sj.onc.1204636. [DOI] [PubMed] [Google Scholar]
- 131.Lipponen A, Paananen J, Puhakka N, Pitkanen A. Analysis of Post-Traumatic Brain Injury Gene Expression Signature Reveals Tubulins, Nfe2l2, Nfkb, Cd44, and S100a4 as Treatment Targets. Scientific reports. 2016;6:31570. doi: 10.1038/srep31570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Deloulme JC, Assard N, Mbele GO, Mangin C, Kuwano R, Baudier J. S100A6 and S100A11 are specific targets of the calcium- and zinc-binding S100B protein in vivo. The Journal of biological chemistry. 2000;275:35302–35310. doi: 10.1074/jbc.M003943200. [DOI] [PubMed] [Google Scholar]
- 133.Yang Q, O’Hanlon D, Heizmann CW, Marks A. Demonstration of heterodimer formation between S100B and S100A6 in the yeast two-hybrid system and human melanoma. Experimental cell research. 1999;246:501–509. doi: 10.1006/excr.1998.4314. [DOI] [PubMed] [Google Scholar]
- 134.Yamada J, Jinno S. Upregulation of calcium binding protein, S100A6, in activated astrocytes is linked to glutamate toxicity. Neuroscience. 2012;226:119–129. doi: 10.1016/j.neuroscience.2012.08.068. [DOI] [PubMed] [Google Scholar]
- 135.Fang B, Liang M, Yang G, Ye Y, Xu H, He X, Huang JH. Expression of S100A6 in rat hippocampus after traumatic brain injury due to lateral head acceleration. International journal of molecular sciences. 2014;15:6378–6390. doi: 10.3390/ijms15046378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Gao F, Yang CX, Mo W, Liu YW, He YQ. Hyaluronan oligosaccharides are potential stimulators to angiogenesis via RHAMM mediated signal pathway in wound healing, Clinical and investigative medicine. Medecine clinique et experimentale. 2008;31:E106–116. doi: 10.25011/cim.v31i3.3467. [DOI] [PubMed] [Google Scholar]
- 137.Matou-Nasri S, Gaffney J, Kumar S, Slevin M. Oligosaccharides of hyaluronan induce angiogenesis through distinct CD44 and RHAMM-mediated signalling pathways involving Cdc2 and gamma-adducin. International journal of oncology. 2009;35:761–773. doi: 10.3892/ijo_00000389. [DOI] [PubMed] [Google Scholar]
- 138.Yung S, Chan TM. Pathophysiology of the peritoneal membrane during peritoneal dialysis: the role of hyaluronan. Journal of biomedicine & biotechnology. 2011;2011:180594. doi: 10.1155/2011/180594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Tesar BM, Jiang D, Liang J, Palmer SM, Noble PW, Goldstein DR. The role of hyaluronan degradation products as innate alloimmune agonists. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2006;6:2622–2635. doi: 10.1111/j.1600-6143.2006.01537.x. [DOI] [PubMed] [Google Scholar]
- 140.Chen WY, Abatangelo G. Functions of hyaluronan in wound repair. Wound repair and regeneration : official publication of the Wound Healing Society [and] the European Tissue Repair Society. 1999;7:79–89. doi: 10.1046/j.1524-475x.1999.00079.x. [DOI] [PubMed] [Google Scholar]
- 141.Linthorst AC. Interactions between corticotropin-releasing hormone and serotonin: implications for the aetiology and treatment of anxiety disorders. Handbook of experimental pharmacology. 2005:181–204. doi: 10.1007/3-540-28082-0_7. [DOI] [PubMed] [Google Scholar]
- 142.Xing G, Ren M, Verma A. Divergent Temporal Expression of Hyaluronan Metabolizing Enzymes and Receptors with Craniotomy vs. Controlled-Cortical Impact Injury in Rat Brain: A Pilot Study. Frontiers in neurology. 2014;5:173. doi: 10.3389/fneur.2014.00173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Back SA, Kroenke CD, Sherman LS, Lawrence G, Gong X, Taber EN, Sonnen JA, Larson EB, Montine TJ. White matter lesions defined by diffusion tensor imaging in older adults. Annals of neurology. 2011;70:465–476. doi: 10.1002/ana.22484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Hagen MW, Riddle A, McClendon E, Gong X, Shaver D, Srivastava T, Dean JM, Bai JZ, Fowke TM, Gunn AJ, Jones DF, Sherman LS, Grafe MR, Hohimer AR, Back SA. Role of recurrent hypoxia-ischemia in preterm white matter injury severity. PloS one. 2014;9:e112800. doi: 10.1371/journal.pone.0112800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Dean JM, Riddle A, Maire J, Hansen KD, Preston M, Barnes AP, Sherman LS, Back SA. An organotypic slice culture model of chronic white matter injury with maturation arrest of oligodendrocyte progenitors. Molecular neurodegeneration. 2011;6:46. doi: 10.1186/1750-1326-6-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Sloane JA, Batt C, Ma Y, Harris ZM, Trapp B, Vartanian T. Hyaluronan blocks oligodendrocyte progenitor maturation and remyelination through TLR2. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:11555–11560. doi: 10.1073/pnas.1006496107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Back SA, Tuohy TM, Chen H, Wallingford N, Craig A, Struve J, Luo NL, Banine F, Liu Y, Chang A, Trapp BD, Bebo BF, Jr, Rao MS, Sherman LS. Hyaluronan accumulates in demyelinated lesions and inhibits oligodendrocyte progenitor maturation. Nature medicine. 2005;11:966–972. doi: 10.1038/nm1279. [DOI] [PubMed] [Google Scholar]
- 148.Vinukonda G, Dohare P, Arshad A, Zia MT, Panda S, Korumilli R, Kayton R, Hascall VC, Lauer ME, Ballabh P. Hyaluronidase and Hyaluronan Oligosaccharides Promote Neurological Recovery after Intraventricular Hemorrhage. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2016;36:872–889. doi: 10.1523/JNEUROSCI.3297-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Ramlackhansingh AF, Brooks DJ, Greenwood RJ, Bose SK, Turkheimer FE, Kinnunen KM, Gentleman S, Heckemann RA, Gunanayagam K, Gelosa G, Sharp DJ. Inflammation after trauma: microglial activation and traumatic brain injury. Annals of neurology. 2011;70:374–383. doi: 10.1002/ana.22455. [DOI] [PubMed] [Google Scholar]
- 150.Hernandez-Ontiveros DG, Tajiri N, Acosta S, Giunta B, Tan J, Borlongan CV. Microglia activation as a biomarker for traumatic brain injury. Frontiers in neurology. 2013;4:30. doi: 10.3389/fneur.2013.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Gehrmann J, Matsumoto Y, Kreutzberg GW. Microglia: Intrinsic immuneffector cell of the brain. Brain Research Reviews. 1995;20:269–287. doi: 10.1016/0165-0173(94)00015-h. [DOI] [PubMed] [Google Scholar]
- 152.Loane DJ, Byrnes KR. Role of microglia in neurotrauma. Neurotherapeutics : the journal of the American Society for Experimental NeuroTherapeutics. 2010;7:366–377. doi: 10.1016/j.nurt.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Town T, Nikolic V, Tan J. The microglial “activation” continuum: from innate to adaptive responses. Journal of neuroinflammation. 2005;2:1–10. doi: 10.1186/1742-2094-2-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Morganti-Kossmann MC, Rancan M, Otto VI, Stahel PF, Kossmann T. Role of cerebral inflammation after traumatic brain injury: a revisited concept. Shock (Augusta, Ga) 2001;16:165–177. doi: 10.1097/00024382-200116030-00001. [DOI] [PubMed] [Google Scholar]
- 155.Sherwin C, Fern R. Acute lipopolysaccharide-mediated injury in neonatal white matter glia: role of TNF-alpha, IL-1beta, and calcium. J Immunol. 2005;175:155–161. doi: 10.4049/jimmunol.175.1.155. [DOI] [PubMed] [Google Scholar]
- 156.Loane DJ, Kumar A. Microglia in the TBI brain: The good, the bad, and the dysregulated. Experimental neurology. 2016;275(Pt 3):316–327. doi: 10.1016/j.expneurol.2015.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Loane DJ, Kumar A, Stoica BA, Cabatbat R, Faden AI. Progressive neurodegeneration after experimental brain trauma: association with chronic microglial activation. Journal of neuropathology and experimental neurology. 2014;73:14–29. doi: 10.1097/NEN.0000000000000021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Venereau E, Casalgrandi M, Schiraldi M, Antoine DJ, Cattaneo A, De Marchis F, Liu J, Antonelli A, Preti A, Raeli L, Shams SS, Yang H, Varani L, Andersson U, Tracey KJ, Bachi A, Uguccioni M, Bianchi ME. Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. The Journal of experimental medicine. 2012;209:1519–1528. doi: 10.1084/jem.20120189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Tang D, Billiar TR, Lotze MT. A Janus tale of two active high mobility group box 1 (HMGB1) redox states. Mol Med. 2012;18:1360–1362. doi: 10.2119/molmed.2012.00314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Schiraldi M, Raucci A, Munoz LM, Livoti E, Celona B, Venereau E, Apuzzo T, De Marchis F, Pedotti M, Bachi A, Thelen M, Varani L, Mellado M, Proudfoot A, Bianchi ME, Uguccioni M. HMGB1 promotes recruitment of inflammatory cells to damaged tissues by forming a complex with CXCL12 and signaling via CXCR4. The Journal of experimental medicine. 2012;209:551–563. doi: 10.1084/jem.20111739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Terrando N, Yang T, Wang X, Fang J, Cao M, Andersson U, Erlandsson HH, Ouyang W, Tong J. Systemic HMGB1 Neutralization Prevents Postoperative Neurocognitive Dysfunction in Aged Rats. Frontiers in immunology. 2016;7:441. doi: 10.3389/fimmu.2016.00441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Lee S, Nam Y, Koo JY, Lim D, Park J, Ock J, Kim J, Suk K, Park SB. A small molecule binding HMGB1 and HMGB2 inhibits microglia-mediated neuroinflammation. Nature chemical biology. 2014;10:1055–1060. doi: 10.1038/nchembio.1669. [DOI] [PubMed] [Google Scholar]
- 163.Takizawa T, Shibata M, Kayama Y, Shimizu T, Toriumi H, Ebine T, Unekawa M, Koh A, Yoshimura A, Suzuki N. High-mobility group box 1 is an important mediator of microglial activation induced by cortical spreading depression. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2016 doi: 10.1177/0271678X16647398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Kim JB, Choi J Sig, Yu YM, Nam K, Piao CS, Kim SW, Lee MH, Han PL, Park JS, Lee JK. HMGB1, a novel cytokine-like mediator linking acute neuronal death and delayed neuroinflammation in the postischemic brain. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2006;26:6413–6421. doi: 10.1523/JNEUROSCI.3815-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Okuma Y, Liu K, Wake H, Zhang J, Maruo T, Date I, Yoshino T, Ohtsuka A, Otani N, Tomura S, Shima K, Yamamoto Y, Yamamoto H, Takahashi HK, Mori S, Nishibori M. Anti-high mobility group box-1 antibody therapy for traumatic brain injury. Annals of neurology. 2012;72:373–384. doi: 10.1002/ana.23602. [DOI] [PubMed] [Google Scholar]
- 166.Okuma Y, Liu K, Wake H, Liu R, Nishimura Y, Hui Z, Teshigawara K, Haruma J, Yamamoto Y, Yamamoto H, Date I, Takahashi HK, Mori S, Nishibori M. Glycyrrhizin inhibits traumatic brain injury by reducing HMGB1-RAGE interaction. Neuropharmacology. 2014;85:18–26. doi: 10.1016/j.neuropharm.2014.05.007. [DOI] [PubMed] [Google Scholar]
- 167.Fonken LK, Frank MG, Kitt MM, D’Angelo HM, Norden DM, Weber MD, Barrientos RM, Godbout JP, Watkins LR, Maier SF. The Alarmin HMGB1 Mediates Age-Induced Neuroinflammatory Priming. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2016;36:7946–7956. doi: 10.1523/JNEUROSCI.1161-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Howell OW, Rundle JL, Garg A, Komada M, Brophy PJ, Reynolds R. Activated microglia mediate axoglial disruption that contributes to axonal injury in multiple sclerosis. Journal of neuropathology and experimental neurology. 2010;69:1017–1033. doi: 10.1097/NEN.0b013e3181f3a5b1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Andersson A, Covacu R, Sunnemark D, Danilov AI, Bianco A Dal, Khademi M, Wallstrom E, Lobell A, Brundin L, Lassmann H, Harris RA. Pivotal advance: HMGB1 expression in active lesions of human and experimental multiple sclerosis. Journal of leukocyte biology. 2008;84:1248–1255. doi: 10.1189/jlb.1207844. [DOI] [PubMed] [Google Scholar]
- 170.Lehnardt S, Lachance C, Patrizi S, Lefebvre S, Follett PL, Jensen FE, Rosenberg PA, Volpe JJ, Vartanian T. The toll-like receptor TLR4 is necessary for lipopolysaccharide-induced oligodendrocyte injury in the CNS. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2002;22:2478–2486. doi: 10.1523/JNEUROSCI.22-07-02478.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Hayakawa K, Pham LD, Seo JH, Miyamoto N, Maki T, Terasaki Y, Sakadzic S, Boas D, van Leyen K, Waeber C, Kim KW, Arai K, Lo EH. CD200 restrains macrophage attack on oligodendrocyte precursors via toll-like receptor 4 downregulation. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2016;36:781–793. doi: 10.1177/0271678X15606148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Lee KM, Bang J, Kim BY, Lee IS, Han JS, Hwang BY, Jeon WK. Fructus mume alleviates chronic cerebral hypoperfusion-induced white matter and hippocampal damage via inhibition of inflammation and downregulation of TLR4 and p38 MAPK signaling. BMC complementary and alternative medicine. 2015;15:125. doi: 10.1186/s12906-015-0652-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Gao HM, Zhou H, Zhang F, Wilson BC, Kam W, Hong JS. HMGB1 acts on microglia Mac1 to mediate chronic neuroinflammation that drives progressive neurodegeneration. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2011;31:1081–1092. doi: 10.1523/JNEUROSCI.3732-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Dohi K, Ohtaki H, Nakamachi T, Yofu S, Satoh K, Miyamoto K, Song D, Tsunawaki S, Shioda S, Aruga T. Gp91phox (NOX2) in classically activated microglia exacerbates traumatic brain injury. Journal of neuroinflammation. 2010;7:41. doi: 10.1186/1742-2094-7-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Ansari MA, Roberts KN, Scheff SW. A time course of NADPH-oxidase up-regulation and endothelial nitric oxide synthase activation in the hippocampus following neurotrauma. Free radical biology & medicine. 2014;77:21–29. doi: 10.1016/j.freeradbiomed.2014.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Choi BY, Jang BG, Kim JH, Lee BE, Sohn M, Song HK, Suh SW. Prevention of traumatic brain injury-induced neuronal death by inhibition of NADPH oxidase activation. Brain research. 2012;1481:49–58. doi: 10.1016/j.brainres.2012.08.032. [DOI] [PubMed] [Google Scholar]
- 177.Loane DJ, Stoica BA, Byrnes KR, Jeong W, Faden AI. Activation of mGluR5 and inhibition of NADPH oxidase improves functional recovery after traumatic brain injury. Journal of neurotrauma. 2013;30:403–412. doi: 10.1089/neu.2012.2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Luo T, Wu J, Kabadi SV, Sabirzhanov B, Guanciale K, Hanscom M, Faden J, Cardiff K, Bengson CJ, Faden AI. Propofol limits microglial activation after experimental brain trauma through inhibition of nicotinamide adenine dinucleotide phosphate oxidase. Anesthesiology. 2013;119:1370–1388. doi: 10.1097/ALN.0000000000000020. [DOI] [PubMed] [Google Scholar]
- 179.Ferreira AP, Rodrigues FS, Della-Pace ID, Mota BC, Oliveira SM, Cde C Velho Gewehr, Bobinski F, de Oliveira CV, Brum JS, Oliveira MS, Furian AF, de Barros CS, Ferreira J, Santos AR, Fighera MR, Royes LF. The effect of NADPH-oxidase inhibitor apocynin on cognitive impairment induced by moderate lateral fluid percussion injury: role of inflammatory and oxidative brain damage. Neurochemistry international. 2013;63:583–593. doi: 10.1016/j.neuint.2013.09.012. [DOI] [PubMed] [Google Scholar]
- 180.Lucke-Wold BP, Naser ZJ, Logsdon AF, Turner RC, Smith KE, Robson MJ, Bailes JE, Lee JM, Rosen CL, Huber JD. Amelioration of nicotinamide adenine dinucleotide phosphate-oxidase mediated stress reduces cell death after blast-induced traumatic brain injury. Translational research : the journal of laboratory and clinical medicine. 2015;166:509–528 e501. doi: 10.1016/j.trsl.2015.08.005. [DOI] [PubMed] [Google Scholar]
- 181.Kumar A, Barrett JP, Alvarez-Croda DM, Stoica BA, Faden AI, Loane DJ. NOX2 drives M1-like microglial/macrophage activation and neurodegeneration following experimental traumatic brain injury. Brain, behavior, and immunity. 2016;58:291–309. doi: 10.1016/j.bbi.2016.07.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Qin L, Liu Y, Wang T, Wei SJ, Block ML, Wilson B, Liu B, Hong JS. NADPH oxidase mediates lipopolysaccharide-induced neurotoxicity and proinflammatory gene expression in activated microglia. The Journal of biological chemistry. 2004;279:1415–1421. doi: 10.1074/jbc.M307657200. [DOI] [PubMed] [Google Scholar]
- 183.Connor JR, Menzies SL. Relationship of iron to oligodendrocytes and myelination. Glia. 1996;17:83–93. doi: 10.1002/(SICI)1098-1136(199606)17:2<83::AID-GLIA1>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 184.Thorburne SK, Juurlink BH. Low glutathione and high iron govern the susceptibility of oligodendroglial precursors to oxidative stress. Journal of neurochemistry. 1996;67:1014–1022. doi: 10.1046/j.1471-4159.1996.67031014.x. [DOI] [PubMed] [Google Scholar]
- 185.Fischer MT, Sharma R, Lim JL, Haider L, Frischer JM, Drexhage J, Mahad D, Bradl M, van Horssen J, Lassmann H. NADPH oxidase expression in active multiple sclerosis lesions in relation to oxidative tissue damage and mitochondrial injury. Brain : a journal of neurology. 2012;135:886–899. doi: 10.1093/brain/aws012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Choi BY, Kim JH, Kho AR, Kim IY, Lee SH, Lee BE, Choi E, Sohn M, Stevenson M, Chung TN, Kauppinen TM, Suh SW. Inhibition of NADPH oxidase activation reduces EAE-induced white matter damage in mice. Journal of neuroinflammation. 2015;12:104. doi: 10.1186/s12974-015-0325-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Wang G, Zhang J, Hu X, Zhang L, Mao L, Jiang X, Liou AK, Leak RK, Gao Y, Chen J. Microglia/macrophage polarization dynamics in white matter after traumatic brain injury. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2013;33:1864–1874. doi: 10.1038/jcbfm.2013.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Xiang Z, Burnstock G. Expression of P2X receptors on rat microglial cells during early development. Glia. 2005;52:119–126. doi: 10.1002/glia.20227. [DOI] [PubMed] [Google Scholar]
- 189.Li F, Wang L, Li JW, Gong M, He L, Feng R, Dai Z, Li SQ. Hypoxia induced amoeboid microglial cell activation in postnatal rat brain is mediated by ATP receptor P2X4. BMC neuroscience. 2011;12:111. doi: 10.1186/1471-2202-12-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Zhang Z, Zhang Z, Artelt M, Burnet M, Schluesener HJ. Dexamethasone attenuates early expression of three molecules associated with microglia/macrophages activation following rat traumatic brain injury. Acta neuropathologica. 2007;113:675–682. doi: 10.1007/s00401-007-0195-8. [DOI] [PubMed] [Google Scholar]
- 191.Deng Y, Lu J, Sivakumar V, Ling EA, Kaur C. Amoeboid microglia in the periventricular white matter induce oligodendrocyte damage through expression of proinflammatory cytokines via MAP kinase signaling pathway in hypoxic neonatal rats. Brain Pathol. 2008;18:387–400. doi: 10.1111/j.1750-3639.2008.00138.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Fan LW, Tien LT, Zheng B, Pang Y, Rhodes PG, Cai Z. Interleukin-1beta-induced brain injury and neurobehavioral dysfunctions in juvenile rats can be attenuated by alpha-phenyl-n-tert-butyl-nitrone. Neuroscience. 2010;168:240–252. doi: 10.1016/j.neuroscience.2010.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Huizinga R, Kreft KL, Onderwater S, Boonstra JG, Brands R, Hintzen RQ, Laman JD. Endotoxin- and ATP-neutralizing activity of alkaline phosphatase as a strategy to limit neuroinflammation. Journal of neuroinflammation. 2012;9:266. doi: 10.1186/1742-2094-9-266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Fan LW, Pang Y, Lin S, Rhodes PG, Cai Z. Minocycline attenuates lipopolysaccharide-induced white matter injury in the neonatal rat brain. Neuroscience. 2005;133:159–168. doi: 10.1016/j.neuroscience.2005.02.016. [DOI] [PubMed] [Google Scholar]
- 195.Cai Z, Lin S, Fan LW, Pang Y, Rhodes PG. Minocycline alleviates hypoxic-ischemic injury to developing oligodendrocytes in the neonatal rat brain. Neuroscience. 2006;137:425–435. doi: 10.1016/j.neuroscience.2005.09.023. [DOI] [PubMed] [Google Scholar]
- 196.Liu Y, Wu XM, Luo QQ, Huang S, Yang QW, Wang FX, Ke Y, Qian ZM. CX3CL1/CX3CR1-mediated microglia activation plays a detrimental role in ischemic mice brain via p38MAPK/PKC pathway. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2015;35:1623–1631. doi: 10.1038/jcbfm.2015.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Hanlon LA, Huh JW, Raghupathi R. Minocycline Transiently Reduces Microglia/Macrophage Activation but Exacerbates Cognitive Deficits Following Repetitive Traumatic Brain Injury in the Neonatal Rat. Journal of neuropathology and experimental neurology. 2016;75:214–226. doi: 10.1093/jnen/nlv021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Ransohoff RM, Engelhardt B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nature reviews Immunology. 2012;12:623–635. doi: 10.1038/nri3265. [DOI] [PubMed] [Google Scholar]
- 199.Morganti-Kossmann MC, Satgunaseelan L, Bye N, Kossmann T. Modulation of immune response by head injury. Injury. 2007;38:1392–1400. doi: 10.1016/j.injury.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 200.Czigner A, Mihaly A, Farkas O, Buki A, Krisztin-Peva B, Dobo E, Barzo P. Kinetics of the cellular immune response following closed head injury. Acta neurochirurgica. 2007;149:281–289. doi: 10.1007/s00701-006-1095-8. [DOI] [PubMed] [Google Scholar]
- 201.Israelsson C, Bengtsson H, Kylberg A, Kullander K, Lewen A, Hillered L, Ebendal T. Distinct cellular patterns of upregulated chemokine expression supporting a prominent inflammatory role in traumatic brain injury. Journal of neurotrauma. 2008;25:959–974. doi: 10.1089/neu.2008.0562. [DOI] [PubMed] [Google Scholar]
- 202.Venereau E, Schiraldi M, Uguccioni M, Bianchi ME. HMGB1 and leukocyte migration during trauma and sterile inflammation. Molecular immunology. 2013;55:76–82. doi: 10.1016/j.molimm.2012.10.037. [DOI] [PubMed] [Google Scholar]
- 203.Hinson HE, Rowell S, Schreiber M. Clinical evidence of inflammation driving secondary brain injury: A systematic review. The journal of trauma and acute care surgery. 2015;78:184–191. doi: 10.1097/TA.0000000000000468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Chodobski A, Zink BJ, Szmydynger-Chodobska J. Blood-brain barrier pathophysiology in traumatic brain injury. Translational stroke research. 2011;2:492–516. doi: 10.1007/s12975-011-0125-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Ottens AK, Golden EC, Bustamante L, Hayes RL, Denslow ND, Wang KK. Proteolysis of multiple myelin basic protein isoforms after neurotrauma: characterization by mass spectrometry. Journal of neurochemistry. 2008;104:1404–1414. doi: 10.1111/j.1471-4159.2007.05086.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Liu MC, Akle V, Zheng W, Kitlen J, O’Steen B, Larner SF, Dave JR, Tortella FC, Hayes RL, Wang KK. Extensive degradation of myelin basic protein isoforms by calpain following traumatic brain injury. Journal of neurochemistry. 2006;98:700–712. doi: 10.1111/j.1471-4159.2006.03882.x. [DOI] [PubMed] [Google Scholar]
- 207.Berger RP, Dulani T, Adelson PD, Leventhal JM, Richichi R, Kochanek PM. Identification of inflicted traumatic brain injury in well-appearing infants using serum and cerebrospinal markers: a possible screening tool. Pediatrics. 2006;117:325–332. doi: 10.1542/peds.2005-0711. [DOI] [PubMed] [Google Scholar]
- 208.Papa L, Robertson CS, Wang KK, Brophy GM, Hannay HJ, Heaton S, Schmalfuss I, Gabrielli A, Hayes RL, Robicsek SA. Biomarkers improve clinical outcome predictors of mortality following non-penetrating severe traumatic brain injury. Neurocrit Care. 2015;22:52–64. doi: 10.1007/s12028-014-0028-2. [DOI] [PubMed] [Google Scholar]
- 209.Berger RP, Adelson PD, Pierce MC, Dulani T, Cassidy LD, Kochanek PM. Serum neuron-specific enolase, S100B, and myelin basic protein concentrations after inflicted and noninflicted traumatic brain injury in children. Journal of neurosurgery. 2005;103:61–68. doi: 10.3171/ped.2005.103.1.0061. [DOI] [PubMed] [Google Scholar]
- 210.Su E, Bell MJ, Kochanek PM, Wisniewski SR, Bayir H, Clark RS, Adelson PD, Tyler-Kabara EC, Janesko-Feldman KL, Berger RP. Increased CSF concentrations of myelin basic protein after TBI in infants and children: absence of significant effect of therapeutic hypothermia. Neurocrit Care. 2012;17:401–407. doi: 10.1007/s12028-012-9767-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Kotter MR, Li WW, Zhao C, Franklin RJ. Myelin impairs CNS remyelination by inhibiting oligodendrocyte precursor cell differentiation. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2006;26:328–332. doi: 10.1523/JNEUROSCI.2615-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Siebert H, Sachse A, Kuziel WA, Maeda N, Bruck W. The chemokine receptor CCR2 is involved in macrophage recruitment to the injured peripheral nervous system. Journal of neuroimmunology. 2000;110:177–185. doi: 10.1016/s0165-5728(00)00343-x. [DOI] [PubMed] [Google Scholar]
- 213.Perrin FE, Lacroix S, Aviles-Trigueros M, David S. Involvement of monocyte chemoattractant protein-1, macrophage inflammatory protein-1alpha and interleukin-1beta in Wallerian degeneration. Brain : a journal of neurology. 2005;128:854–866. doi: 10.1093/brain/awh407. [DOI] [PubMed] [Google Scholar]
- 214.Kotter MR, Zhao C, van Rooijen N, Franklin RJ. Macrophage-depletion induced impairment of experimental CNS remyelination is associated with a reduced oligodendrocyte progenitor cell response and altered growth factor expression. Neurobiology of disease. 2005;18:166–175. doi: 10.1016/j.nbd.2004.09.019. [DOI] [PubMed] [Google Scholar]
- 215.Flygt J, Djupsjo A, Lenne F, Marklund N. Myelin loss and oligodendrocyte pathology in white matter tracts following traumatic brain injury in the rat. The European journal of neuroscience. 2013;38:2153–2165. doi: 10.1111/ejn.12179. [DOI] [PubMed] [Google Scholar]
- 216.Glushakova OY, Johnson D, Hayes RL. Delayed increases in microvascular pathology after experimental traumatic brain injury are associated with prolonged inflammation, blood-brain barrier disruption, and progressive white matter damage. Journal of neurotrauma. 2014;31:1180–1193. doi: 10.1089/neu.2013.3080. [DOI] [PubMed] [Google Scholar]
- 217.Karanth S, Yang G, Yeh J, Richardson PM. Nature of signals that initiate the immune response during Wallerian degeneration of peripheral nerves. Experimental neurology. 2006;202:161–166. doi: 10.1016/j.expneurol.2006.05.024. [DOI] [PubMed] [Google Scholar]
- 218.Boivin A, Pineau I, Barrette B, Filali M, Vallieres N, Rivest S, Lacroix S. Toll-like receptor signaling is critical for Wallerian degeneration and functional recovery after peripheral nerve injury. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2007;27:12565–12576. doi: 10.1523/JNEUROSCI.3027-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Vallieres N, Berard JL, David S, Lacroix S. Systemic injections of lipopolysaccharide accelerates myelin phagocytosis during Wallerian degeneration in the injured mouse spinal cord. Glia. 2006;53:103–113. doi: 10.1002/glia.20266. [DOI] [PubMed] [Google Scholar]
- 220.Mantovani A, Sica A, Locati M. Macrophage polarization comes of age. Immunity. 2005;23:344–346. doi: 10.1016/j.immuni.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 221.Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. 2010;32:593–604. doi: 10.1016/j.immuni.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 222.Qin H, Holdbrooks AT, Liu Y, Reynolds SL, Yanagisawa LL, Benveniste EN. SOCS3 deficiency promotes M1 macrophage polarization and inflammation. J Immunol. 2012;189:3439–3448. doi: 10.4049/jimmunol.1201168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Krausgruber T, Blazek K, Smallie T, Alzabin S, Lockstone H, Sahgal N, Hussell T, Feldmann M, Udalova IA. IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nature immunology. 2011;12:231–238. doi: 10.1038/ni.1990. [DOI] [PubMed] [Google Scholar]
- 224.Duan M, Li WC, Vlahos R, Maxwell MJ, Anderson GP, Hibbs ML. Distinct macrophage subpopulations characterize acute infection and chronic inflammatory lung disease. J Immunol. 2012;189:946–955. doi: 10.4049/jimmunol.1200660. [DOI] [PubMed] [Google Scholar]
- 225.Braun M, Vaibhav K, Saad N, Fatima S, Brann DW, Vender JR, Wang LP, Hoda MN, Baban B, Dhandapani KM. Activation of Myeloid TLR4 Mediates T Lymphocyte Polarization after Traumatic Brain Injury. J Immunol. 2017;198:3615–3626. doi: 10.4049/jimmunol.1601948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Kumar A, Stoica BA, Sabirzhanov B, Burns MP, Faden AI, Loane DJ. Traumatic brain injury in aged animals increases lesion size and chronically alters microglial/macrophage classical and alternative activation states. Neurobiology of aging. 2013;34:1397–1411. doi: 10.1016/j.neurobiolaging.2012.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Xiong Y, Mahmood A, Chopp M. Animal models of traumatic brain injury. Nat Rev Neurosci. 2013;14:128–142. doi: 10.1038/nrn3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Morales DM, Marklund N, Lebold D, Thompson HJ, Pitkanen A, Maxwell WL, Longhi L, Laurer H, Maegele M, Neugebauer E, Graham DI, Stocchetti N, McIntosh TK. Experimental models of traumatic brain injury: do we really need to build a better mousetrap? Neuroscience. 2005;136:971–989. doi: 10.1016/j.neuroscience.2005.08.030. [DOI] [PubMed] [Google Scholar]
- 229.Laurer HL, McIntosh TK. Experimental models of brain trauma. Curr Opin Neurol. 1999;12:715–721. doi: 10.1097/00019052-199912000-00010. [DOI] [PubMed] [Google Scholar]
- 230.Manley GT, Rosenthal G, Lam M, Morabito D, Yan D, Derugin N, Bollen A, Knudson MM, Panter SS. Controlled cortical impact in swine: pathophysiology and biomechanics. Journal of neurotrauma. 2006;23:128–139. doi: 10.1089/neu.2006.23.128. [DOI] [PubMed] [Google Scholar]
- 231.Alessandri B, Heimann A, Filippi R, Kopacz L, Kempski O. Moderate controlled cortical contusion in pigs: effects on multi-parametric neuromonitoring and clinical relevance. Journal of neurotrauma. 2003;20:1293–1305. doi: 10.1089/089771503322686094. [DOI] [PubMed] [Google Scholar]
- 232.Holm IE, Alstrup AK, Luo Y. Genetically modified pig models for neurodegenerative disorders. J Pathol. 2016;238:267–287. doi: 10.1002/path.4654. [DOI] [PubMed] [Google Scholar]
- 233.Yan S, Tu Z, Li S, Li XJ. Use of CRISPR/Cas9 to model brain diseases. Prog Neuropsychopharmacol Biol Psychiatry. 2017 doi: 10.1016/j.pnpbp.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Yang W, Tu Z, Sun Q, Li XJ. CRISPR/Cas9: Implications for Modeling and Therapy of Neurodegenerative Diseases. Front Mol Neurosci. 2016;9:30. doi: 10.3389/fnmol.2016.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Tu Z, Yang W, Yan S, Guo X, Li XJ. CRISPR/Cas9: a powerful genetic engineering tool for establishing large animal models of neurodegenerative diseases. Molecular neurodegeneration. 2015;10:35. doi: 10.1186/s13024-015-0031-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Faraco G, Fossati S, Bianchi ME, Patrone M, Pedrazzi M, Sparatore B, Moroni F, Chiarugi A. High mobility group box 1 protein is released by neural cells upon different stresses and worsens ischemic neurodegeneration in vitro and in vivo. Journal of neurochemistry. 2007;103:590–603. doi: 10.1111/j.1471-4159.2007.04788.x. [DOI] [PubMed] [Google Scholar]
- 237.Qiu J, Nishimura M, Wang Y, Sims JR, Qiu S, Savitz SI, Salomone S, Moskowitz MA. Early release of HMGB-1 from neurons after the onset of brain ischemia. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2008;28:927–938. doi: 10.1038/sj.jcbfm.9600582. [DOI] [PubMed] [Google Scholar]
- 238.Kawabata H, Setoguchi T, Yone K, Souda M, Yoshida H, Kawahara K, Maruyama I, Komiya S. High mobility group box 1 is upregulated after spinal cord injury and is associated with neuronal cell apoptosis. Spine (Phila Pa 1976) 2010;35:1109–1115. doi: 10.1097/BRS.0b013e3181bd14b6. [DOI] [PubMed] [Google Scholar]
- 239.Gu XJ, Xu J, Ma BY, Chen G, Gu PY, Wei D, Hu WX. Effect of glycyrrhizin on traumatic brain injury in rats and its mechanism. Chin J Traumatol. 2014;17:1–7. [PubMed] [Google Scholar]
- 240.Su X, Wang H, Zhao J, Pan H, Mao L. Beneficial effects of ethyl pyruvate through inhibiting high-mobility group box 1 expression and TLR4/NF-kappaB pathway after traumatic brain injury in the rat. Mediators Inflamm. 2011;2011:807142. doi: 10.1155/2011/807142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Ahmad A, Crupi R, Campolo M, Genovese T, Esposito E, Cuzzocrea S. Absence of TLR4 reduces neurovascular unit and secondary inflammatory process after traumatic brain injury in mice. PloS one. 2013;8:e57208. doi: 10.1371/journal.pone.0057208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Zhu HT, Bian C, Yuan JC, Chu WH, Xiang X, Chen F, Wang CS, Feng H, Lin JK. Curcumin attenuates acute inflammatory injury by inhibiting the TLR4/MyD88/NF-kappaB signaling pathway in experimental traumatic brain injury. Journal of neuroinflammation. 2014;11:59. doi: 10.1186/1742-2094-11-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Laird MD, Sukumari-Ramesh S, Swift AE, Meiler SE, Vender JR, Dhandapani KM. Curcumin attenuates cerebral edema following traumatic brain injury in mice: a possible role for aquaporin-4? Journal of neurochemistry. 2010;113:637–648. doi: 10.1111/j.1471-4159.2010.06630.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122:787–795. doi: 10.1172/JCI59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, Derecki NC, Castle D, Mandell JW, Lee KS, Harris TH, Kipnis J. Structural and functional features of central nervous system lymphatic vessels. Nature. 2015;523:337–341. doi: 10.1038/nature14432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Aspelund A, Antila S, Proulx ST, Karlsen TV, Karaman S, Detmar M, Wiig H, Alitalo K. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J Exp Med. 2015;212:991–999. doi: 10.1084/jem.20142290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Walsh JT, Zheng J, Smirnov I, Lorenz U, Tung K, Kipnis J. Regulatory T cells in central nervous system injury: a double-edged sword. J Immunol. 2014;193:5013–5022. doi: 10.4049/jimmunol.1302401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Kokkola R, Andersson A, Mullins G, Ostberg T, Treutiger CJ, Arnold B, Nawroth P, Andersson U, Harris RA, Harris HE. RAGE is the major receptor for the proinflammatory activity of HMGB1 in rodent macrophages. Scandinavian journal of immunology. 2005;61:1–9. doi: 10.1111/j.0300-9475.2005.01534.x. [DOI] [PubMed] [Google Scholar]
- 249.Kabelitz D, Medzhitov R. Innate immunity–cross-talk with adaptive immunity through pattern recognition receptors and cytokines. Current opinion in immunology. 2007;19:1–3. doi: 10.1016/j.coi.2006.11.018. [DOI] [PubMed] [Google Scholar]
- 250.Holmin S, Mathiesen T, Shetye J, Biberfeld P. Intracerebral inflammatory response to experimental brain contusion. Acta neurochirurgica. 1995;132:110–119. doi: 10.1007/BF01404857. [DOI] [PubMed] [Google Scholar]
- 251.Holmin S, Schalling M, Hojeberg B, Nordqvist AC, Skeftruna AK, Mathiesen T. Delayed cytokine expression in rat brain following experimental contusion. Journal of neurosurgery. 1997;86:493–504. doi: 10.3171/jns.1997.86.3.0493. [DOI] [PubMed] [Google Scholar]
- 252.Oehmichen M, Jakob S, Mann S, Saternus KS, Pedal I, Meissner C. Macrophage subsets in mechanical brain injury (MBI)–a contribution to timing of MBI based on immunohistochemical methods: a pilot study. Leg Med (Tokyo) 2009;11:118–124. doi: 10.1016/j.legalmed.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 253.Pizzolla A, Gelderman KA, Hultqvist M, Vestberg M, Gustafsson K, Mattsson R, Holmdahl R. CD68-expressing cells can prime T cells and initiate autoimmune arthritis in the absence of reactive oxygen species. European journal of immunology. 2011;41:403–412. doi: 10.1002/eji.201040598. [DOI] [PubMed] [Google Scholar]
- 254.Vergelli M, Pinet V, Vogt AB, Kalbus M, Malnati M, Riccio P, Long EO, Martin R. HLA-DR-restricted presentation of purified myelin basic protein is independent of intracellular processing. European journal of immunology. 1997;27:941–951. doi: 10.1002/eji.1830270421. [DOI] [PubMed] [Google Scholar]
- 255.Berger T, Rubner P, Schautzer F, Egg R, Ulmer H, Mayringer I, Dilitz E, Deisenhammer F, Reindl M. Antimyelin antibodies as a predictor of clinically definite multiple sclerosis after a first demyelinating event. The New England journal of medicine. 2003;349:139–145. doi: 10.1056/NEJMoa022328. [DOI] [PubMed] [Google Scholar]
- 256.Tobin RP, Mukherjee S, Kain JM, Rogers SK, Henderson SK, Motal HL, Rogers MK, Shapiro LA. Traumatic brain injury causes selective, CD74-dependent peripheral lymphocyte activation that exacerbates neurodegeneration. Acta neuropathologica communications. 2014;2:143. doi: 10.1186/s40478-014-0143-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Holmin S, Soderlund J, Biberfeld P, Mathiesen T. Intracerebral inflammation after human brain contusion. Neurosurgery. 1998;42:291–298. doi: 10.1097/00006123-199802000-00047. discussion 298–299. [DOI] [PubMed] [Google Scholar]
- 258.Unterberg A, Kiening K, Schmiedek P, Lanksch W. Long-term observations of intracranial pressure after severe head injury. The phenomenon of secondary rise of intracranial pressure. Neurosurgery. 1993;32:17–23. doi: 10.1227/00006123-199301000-00003. discussion 23–14. [DOI] [PubMed] [Google Scholar]
- 259.Mosley RL, Hutter-Saunders JA, Stone DK, Gendelman HE. Inflammation and adaptive immunity in Parkinson’s disease. Cold Spring Harbor perspectives in medicine. 2012;2:a009381. doi: 10.1101/cshperspect.a009381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Hickey WF, Hsu BL, Kimura H. T-lymphocyte entry into the central nervous system. Journal of neuroscience research. 1991;28:254–260. doi: 10.1002/jnr.490280213. [DOI] [PubMed] [Google Scholar]
- 261.Hua R, Mao SS, Zhang YM, Chen FX, Zhou ZH, Liu JQ. Effects of pituitary adenylate cyclase activating polypeptide on CD4(+)/CD8(+)T cell levels after traumatic brain injury in a rat model. World journal of emergency medicine. 2012;3:294–298. doi: 10.5847/wjem.j.issn.1920-8642.2012.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Gutcher I, Becher B. APC-derived cytokines and T cell polarization in autoimmune inflammation. J Clin Invest. 2007;117:1119–1127. doi: 10.1172/JCI31720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Gowans JL. The recirculation of lymphocytes from blood to lymph in the rat. The Journal of physiology. 1959;146:54–69. doi: 10.1113/jphysiol.1959.sp006177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Mora JR, von Andrian UH. T-cell homing specificity and plasticity: new concepts and future challenges. Trends in immunology. 2006;27:235–243. doi: 10.1016/j.it.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 265.Fu H, Wang A, Mauro C, Marelli-Berg F. T lymphocyte trafficking: molecules and mechanisms. Front Biosci (Landmark Ed) 2013;18:422–440. doi: 10.2741/4111. [DOI] [PubMed] [Google Scholar]
- 266.Marelli-Berg FM, Cannella L, Dazzi F, Mirenda V. The highway code of T cell trafficking. J Pathol. 2008;214:179–189. doi: 10.1002/path.2269. [DOI] [PubMed] [Google Scholar]
- 267.Fearon DT, Locksley RM. The instructive role of innate immunity in the acquired immune response. Science. 1996;272:50–53. doi: 10.1126/science.272.5258.50. [DOI] [PubMed] [Google Scholar]
- 268.Murphy AC, Lalor SJ, Lynch MA, Mills KH. Infiltration of Th1 and Th17 cells and activation of microglia in the CNS during the course of experimental autoimmune encephalomyelitis. Brain, behavior, and immunity. 2010;24:641–651. doi: 10.1016/j.bbi.2010.01.014. [DOI] [PubMed] [Google Scholar]
- 269.Rostami A, Ciric B. Role of Th17 cells in the pathogenesis of CNS inflammatory demyelination. Journal of the neurological sciences. 2013;333:76–87. doi: 10.1016/j.jns.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Yang D, Sun YY, Bhaumik SK, Li Y, Baumann JM, Lin X, Zhang Y, Lin SH, Dunn RS, Liu CY, Shie FS, Lee YH, Wills-Karp M, Chougnet CA, Kallapur SG, Lewkowich IP, Lindquist DM, Murali-Krishna K, Kuan CY. Blocking lymphocyte trafficking with FTY720 prevents inflammation-sensitized hypoxic-ischemic brain injury in newborns. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2014;34:16467–16481. doi: 10.1523/JNEUROSCI.2582-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Baxi EG, DeBruin J, Tosi DM, Grishkan IV, Smith MD, Kirby LA, Strasburger HJ, Fairchild AN, Calabresi PA, Gocke AR. Transfer of myelin-reactive th17 cells impairs endogenous remyelination in the central nervous system of cuprizone-fed mice. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2015;35:8626–8639. doi: 10.1523/JNEUROSCI.3817-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Montes M, Zhang X, Berthelot L, Laplaud DA, Brouard S, Jin J, Rogan S, Armao D, Jewells V, Soulillou JP, Markovic-Plese S. Oligoclonal myelin-reactive T-cell infiltrates derived from multiple sclerosis lesions are enriched in Th17 cells. Clin Immunol. 2009;130:133–144. doi: 10.1016/j.clim.2008.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Cao Y, Goods BA, Raddassi K, Nepom GT, Kwok WW, Love JC, Hafler DA. Functional inflammatory profiles distinguish myelin-reactive T cells from patients with multiple sclerosis. Science translational medicine. 2015;7:287ra274. doi: 10.1126/scitranslmed.aaa8038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Grifka-Walk HM, Lalor SJ, Segal BM. Highly polarized Th17 cells induce EAE via a T-bet independent mechanism. European journal of immunology. 2013;43:2824–2831. doi: 10.1002/eji.201343723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Becher B, Segal BM. T(H)17 cytokines in autoimmune neuro-inflammation. Current opinion in immunology. 2011;23:707–712. doi: 10.1016/j.coi.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Kroenke MA, Segal BM. IL-23 modulated myelin-specific T cells induce EAE via an IFNgamma driven, IL-17 independent pathway. Brain, behavior, and immunity. 2011;25:932–937. doi: 10.1016/j.bbi.2010.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Xie L, Li XK, Funeshima-Fuji N, Kimura H, Matsumoto Y, Isaka Y, Takahara S. Amelioration of experimental autoimmune encephalomyelitis by curcumin treatment through inhibition of IL-17 production. International immunopharmacology. 2009;9:575–581. doi: 10.1016/j.intimp.2009.01.025. [DOI] [PubMed] [Google Scholar]
- 278.Ma C, Ma Z, Fu Q, Ma S. Curcumin attenuates allergic airway inflammation by regulation of CD4+CD25+ regulatory T cells (Tregs)/Th17 balance in ovalbumin-sensitized mice. Fitoterapia. 2013;87:57–64. doi: 10.1016/j.fitote.2013.02.014. [DOI] [PubMed] [Google Scholar]
- 279.Park MJ, Moon SJ, Lee SH, Yang EJ, Min JK, Cho SG, Yang CW, Park SH, Kim HY, Cho ML. Curcumin attenuates acute graft-versus-host disease severity via in vivo regulations on Th1, Th17 and regulatory T cells. PloS one. 2013;8:e67171. doi: 10.1371/journal.pone.0067171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 281.Iwakura Y, Ishigame H. The IL-23/IL-17 axis in inflammation. J Clin Invest. 2006;116:1218–1222. doi: 10.1172/JCI28508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Bettelli E, Korn T, Kuchroo VK. Th17: the third member of the effector T cell trilogy. Current opinion in immunology. 2007;19:652–657. doi: 10.1016/j.coi.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Annunziato F, Cosmi L, Liotta F, Maggi E, Romagnani S. The phenotype of human Th17 cells and their precursors, the cytokines that mediate their differentiation and the role of Th17 cells in inflammation. International immunology. 2008;20:1361–1368. doi: 10.1093/intimm/dxn106. [DOI] [PubMed] [Google Scholar]
- 284.Oppmann B, Lesley R, Blom B, Timans JC, Xu Y, Hunte B, Vega F, Yu N, Wang J, Singh K, Zonin F, Vaisberg E, Churakova T, Liu M, Gorman D, Wagner J, Zurawski S, Liu Y, Abrams JS, Moore KW, Rennick D, de Waal-Malefyt R, Hannum C, Bazan JF, Kastelein RA. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. 2000;13:715–725. doi: 10.1016/s1074-7613(00)00070-4. [DOI] [PubMed] [Google Scholar]
- 285.Hoeve MA, Savage ND, de Boer T, Langenberg DM, de Waal Malefyt R, Ottenhoff TH, Verreck FA. Divergent effects of IL-12 and IL-23 on the production of IL-17 by human T cells. European journal of immunology. 2006;36:661–670. doi: 10.1002/eji.200535239. [DOI] [PubMed] [Google Scholar]
- 286.Liu W, Ouyang X, Yang J, Liu J, Li Q, Gu Y, Fukata M, Lin T, He JC, Abreu M, Unkeless JC, Mayer L, Xiong H. AP-1 activated by toll-like receptors regulates expression of IL-23 p19. J Biol Chem. 2009;284:24006–24016. doi: 10.1074/jbc.M109.025528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Zhu H, Li J, Wang S, Liu K, Wang L, Huang L. Hmgb1-TLR4-IL-23-IL-17A axis promote ischemia-reperfusion injury in a cardiac transplantation model. Transplantation. 2013;95:1448–1454. doi: 10.1097/TP.0b013e318293b7e1. [DOI] [PubMed] [Google Scholar]
- 288.Allam JP, Duan Y, Heinemann F, Winter J, Gotz W, Deschner J, Wenghoefer M, Bieber T, Jepsen S, Novak N. IL-23-producing CD68(+) macrophage-like cells predominate within an IL-17- polarized infiltrate in chronic periodontitis lesions. Journal of clinical periodontology. 2011;38:879–886. doi: 10.1111/j.1600-051X.2011.01752.x. [DOI] [PubMed] [Google Scholar]