

Abstract
Precise regulation of lipid biosynthesis, transport, and storage is key to the homeostasis of cells and organisms. Cells rely on a sophisticated but poorly understood network of vesicular and non-vesicular transport mechanisms to ensure efficient delivery of lipids to target organelles. The lysosome stands at the crossroads of this network due to its ability to process and sort exogenous and endogenous lipids. The lipid sorting function of the lysosome is intimately connected to its recently discovered role as a metabolic command-and-control center, which relays multiple nutrient cues to the master growth regulator, mechanistic Target of Rapamycin Complex 1 (mTORC1) kinase. In turn, mTORC1 potently drives anabolic processes, including de novo lipid synthesis, while inhibiting lipid catabolism. Here we describe the dual role of the lysosome in lipid transport and biogenesis, and we discuss how integration of these two processes may play important roles both in normal physiology and in disease.
Keywords: Lysosome, lipid metabolism, membrane contact sites, cholesterol, mTORC1
Lipid homeostasis is key for the function of cells, tissues, and organisms
Lipid balance is intricately coordinated at multiple levels, including cellular uptake, synthesis, storage, metabolism, and export. All lipid classes, including fatty acids, sphingolipids, sterols and phospholipids undergo dynamic regulation, the extent of which depends on the specific tissue type and nutrient status of the cell (reviewed in (van Meer et al., 2008)). A prominent and relatively well-studied example is cholesterol, a lipid with unique biophysical properties that support its essential role in regulating membrane structure and function (reviewed in (Goldstein and Brown, 2015)). Cholesterol modulates the activity of transmembrane proteins such as G protein-coupled receptors, and facilitates signal transduction and trafficking processes. In addition, cholesterol is the precursor for steroid hormones and bile acids, which function in signal transduction and absorption of dietary lipids, respectively.
At the systemic level, organs perform specialized roles in maintaining cholesterol homeostasis. Dietary cholesterol is absorbed from the gut by intestinal enterocytes, which express a cholesterol-translocating membrane protein known as Niemann-Pick C1-like 1 (NPC1L1) in their apical membranes (reviewed in (Chang et al., 2006, Ikonen, 2008)). Enterocytes package diet-derived lipids into chylomicrons, which are transported to the liver. There, hepatocytes package cholesterol, phospholipids and triglycerides into blood-soluble protein-containing particles known as very low-density lipoprotein (VLDL). Upon remodeling of their lipid and protein contents, VLDL mature into low-density lipoprotein (LDL), which deliver esterified cholesterol to peripheral tissues. The liver is responsible for the bulk of cholesterol biosynthesis, although the ability to synthesize endogenous cholesterol is common to most cell types(Chang et al., 2006, Goldstein and Brown, 2015, Ikonen, 2008). Endogenous sterol synthesis is critical in the brain, which is isolated from the circulating plasma cholesterol due to the inability of lipoproteins to traverse the blood-brain barrier.
Failure to maintain the levels of circulating plasma cholesterol within a tight range promotes the development of atherosclerotic plaques in the arteries, resulting in high blood pressure and increased risk of cardiovascular disease. Plasma cholesterol levels are buffered by the uptake of LDL by extrahepatic tissues. Adipose tissue, in particular, serves as a reservoir for excess cholesterol through its incorporation into intracellular storage compartments known as lipid droplets. Cholesterol accumulation in extrahepatic tissues is counteracted by a process known as reverse cholesterol transport. In this case, cholesterol is transported across the plasma membrane via ABCA1, packaged into high-density lipoprotein (HDL) at the cell surface, and returned through the plasma to the liver for biliary secretion. Oxidation of cholesterol to bile acids and their subsequent excretion is a liver-specific process, allowing for bulk elimination of sterols from the body (reviewed in (Goldstein and Brown, 2015)).
At the cellular level, membrane-bound organelles are distinguished by their characteristic lipid compositions. Cholesterol is distributed heterogeneously on cellular membranes. A majority of cholesterol is found at the plasma membrane, while significant populations also exist at the endocytic recycling compartment (ERC) and the trans-Golgi network(van Meer et al., 2008). The endoplasmic reticulum, mitochondria, and lysosomes contain a relatively minimal amount of cholesterol. Within any given membrane, cholesterol is distributed heterogeneously. Through tight association with sphingolipids, cholesterol becomes enriched in ‘raft-like’ microdomains that are thought to play important roles in signal transduction (Das et al., 2014). Furthermore, organelles also serve diverse functional roles in maintaining cholesterol homeostasis. Endogenous cholesterol synthesis occurs in the endoplasmic reticulum, under the control of the rate-limiting enzyme HMG-CoA reductase (HMG-CoAR) (reviewed in (Espenshade and Hughes, 2007)). Excess cholesterol in the ER is esterified via acetyl-CoA acetyltransferase (ACAT) 1 and stored in lipid droplets (reviewed in (Thiam et al., 2013)). In steroidogenic tissues such as gonads and adrenal glands, mitochondria import cholesterol into the inner mitochondrial membrane, where it is used as a substrate for steroid hormone synthesis. Lysosomes serve a key role in maintaining cholesterol homeostasis by acting as a processing center for LDL-derived cholesterol. Thus, cholesterol homeostasis is a highly organized process that requires cross-talk among many membranous compartments.
The lysosome as a cellular trafficking station
Lysosomes are small (100–500 nm diameter) organelles characterized by an acidic interior, which enables the activity of up to 60 different hydrolytic enzymes including proteases, lipases and nucleases. Its limiting membrane is spanned by numerous transmembrane proteins, which possess highly glycosylated luminal domains that protect the proteins and lipids of the limiting membrane from hydrolytic digestion (reviewed in (Perera and Zoncu, 2016, Saftig and Klumperman, 2009, Settembre et al., 2013b)). Lysosomal transmembrane proteins include an ATP-driven proton pump, known as the vacuolar H+ (v-ATPase), responsible for the acidification of the lysosome interior, as well as numerous transporters that mediate the export of lysosomal digestion products to the cytoplasm (Perera and Zoncu, 2016, Saftig and Klumperman, 2009, Settembre et al., 2013b). The lysosome is the end-point of numerous vesicle trafficking pathways including the endocytic, phagocytic and autophagic pathways. Its ability to fuse with endosomes, phagosomes and autophagosomes enables the lysosome to break down a wide range of both endogenous and exogenous cargo including macromolecules, certain pathogens and old or damaged organelles (reviewed in (He and Klionsky, 2009, Hurley and Schulman, 2014, Mizushima et al., 2008, Singh and Cuervo, 2011)). In addition to its basic role as the cellular ‘recycle bin’, the lysosome plays additional roles in processes such as nutrient sensing, pathogen sensing and membrane repair. A deeper understanding of these new functions of the lysosome has elevated this previously neglected organelle to the center stage in disease-related research, including neurodegeneration, cancer and aging (reviewed in (Perera and Zoncu, 2016, Saxton and Sabatini, 2017, Settembre et al., 2013b)).
The convergence of several trafficking pathways at the lysosome makes this organelle a key player in coordinating the sorting and delivery of lipids to various membrane compartments. For instance, exogenous sterols, triglycerides, and phospholipids carried by LDL enter the cell via receptor-mediated endocytosis and are processed by the lysosome (Goldstein and Brown, 2015). The lysosome also sorts and recycles endogenous lipids. For example, fusion of lysosomes with double-membraned autophagosomes brings the autophagosomal internal membrane, along with engulfed organellar cargo, inside the lysosomal lumen. During endocytosis, a protein machinery known as the ESCRT complex mediates the inward budding of vesicles from the limiting membrane of endosomes (reviewed in(Schoneberg et al., 2017)). As these structures mature into lysosomes (either via direct conversion or fusion), the lysosomal lumen is progressively filled with vesicles. These vesicles eventually coalesce into membrane sheets or ‘whorls’ as their lipid composition is modified in the internal acidic environment of the lysosome. These exogenous or endogenous lipids must be removed from the lumen and inserted into the lysosomal limiting membrane. From there, the lipids can be further moved to other organelles such as the ER, where they may be esterified and stored in lipid droplets, or the mitochondria, where simple lipids such as cholesterol may be converted into steroid hormones.
Mechanisms of lipid transport within the lysosome
A well-characterized lipid transport process involves the uptake and processing of LDL by the lysosome (reviewed in(Goldstein and Brown, 2015, Mesmin et al., 2013a)). LDL particles bind to the LDL receptor (LDLR) on the plasma membrane of target cells, and are taken up via clathrin-mediated endocytosis. Following clathrin coated vesicle uncoating, the LDL-receptor complexes enter the endosomal pathway. As the endosomal lumen progressively acidifies, LDL particles separate from LDLR and remain in the endosomal lumen, whereas the LDLR are sorted back to the plasma membrane. As endosomes mature into lysosomes, LDL particles are attacked by the lysosomal acid lipase type A (LIPA), a hydrolase that de-esterifies the cholesterol and triglyceride molecules as they are liberated from LDL particles (Chang et al., 2006).
The transport of LDL-derived, de-esterified cholesterol out of the lysosomal lumen has been studied in great detail. Free cholesterol is highly insoluble and cannot diffuse within the aqueous environment of the lysosomal lumen. Thus, it requires the concerted action of two proteins known as Niemann-Pick type C (NPC) 1 and 2(Infante et al., 2008, Mesmin et al., 2013a). NPC2 is a luminal protein that plays an obligate role in shuttling cholesterol through the lysosomal lumen. Structural studies have revealed that NPC2 harbors a cholesterol-binding pocket lined with hydrophobic residues. The highly hydrophobic iso-octyl chain is buried within the pocket, whereas the 3b-hydroxyl group on the first tetracyclic ring is exposed to the solvent(Xu et al., 2007). NPC2 is thought to deliver cholesterol to the luminal N-terminal domain of NPC1, although direct deposition of cholesterol into the limiting membrane has also been proposed(Deffieu and Pfeffer, 2011, Infante et al., 2008, McCauliff et al., 2015). NPC1 is a polytopic transmembrane protein that harbors an N-terminal cholesterol-binding domain (NTD) protruding into the lysosomal lumen. The crystal structure of the NPC1-NTD revealed a hydrophobic pocket in which cholesterol is oriented with the 3b-hydroxyl group inside and the iso-octyl chain facing out(Kwon et al., 2009). This orientation is opposite from the one in the NPC2 sterol binding pocket, supporting a model in which cholesterol is transferred in a direct manner from NPC2 to NPC1, via coordination between the respective sterol binding pockets.
Recent structural studies based on cryo-electron microscopy (cryo-EM) and X-ray crystallography have shed light on the overall structural organization of NPC1. In particular, the models suggest cooperation between the NTD and the middle luminal domain to allow docking of NPC2 and subsequent cholesterol transfer to the NTD (Gong et al., 2016, Li et al., 2016b). This work also provided for the first time a structural view of the transmembrane sterol-sensing domain (SSD) of NPC1, which is homologous to that of other sterol-regulated proteins such as SCAP, Patched and HMG-CoAR (Brown et al., 2002, Martin et al., 2001, Millard et al., 2005, Sever et al., 2003, Yabe et al., 2002). Whereas the NPC1 SSD has no assigned function so far, an attractive model is that it could be involved in accepting cholesterol from the NTD and promoting its insertion into the limiting membrane.
Recently, the lysosomal proteins LAMP1 and LAMP2 were shown to bind to cholesterol with high affinity and specificity via their luminal domain 1. This domain was also capable of interacting with NPC1 and NPC2. Based on these data, a model emerges where LAMP1/2 proteins, which are highly abundant at the lysosomal membrane, may accept cholesterol from NPC2 and possibly facilitate the flow of cholesterol through the NPC2-NPC1 export pathway (Li and Pfeffer, 2016).
Mechanisms of lipid export from the lysosome (LE/LY)
Once cholesterol has been deposited on the lysosomal membrane, its transport to extra-lysosomal locations is thought to occur rapidly (Mesmin et al., 2013a, Mobius et al., 2002). Emerging evidence indicates that sterol export relies on cooperation between vesicular and non-vesicular routes. The extensive lysosomal fission and fusion that has been observed in multiple cell types may serve to distribute lipids among lysosomes or deliver them to downstream membranes and organelles. Trafficking routes connect the LE/LY to the plasma membrane, the Golgi apparatus and the endoplasmic reticulum.
In addition to vesicular traffic, a major development in recent years has been identification of non-vesicular routes for intracellular lipid transport. Non-vesicular trafficking is mediated by several classes of lipid-binding proteins that localize at sites of physical proximity between different organelles both in mammalian and yeast cells (reviewed in (Daniele and Schiaffino, 2016, Lahiri et al., 2015, Phillips and Voeltz, 2016). These protein families derive their names from characteristic lipid-binding domains they harbor, which include the Oxysterol Binding Protein (OSBP)-Related Proteins (ORPs, also known as Osh proteins in yeast) (Mesmin et al., 2013a, Phillips and Voeltz, 2016), the ‘Steroidogenic Acute Regulatory Protein Related Lipid Transfer’ (STARTs) (Alpy and Tomasetto, 2014), the related VAD1 Analog of Star Related Lipid Transfer (VASt) and the Synaptotagmin-like Mitochondrial Lipid Binding Protein (SMP) (Reinisch and De Camilli, 2016). With the exception of the START proteins, which are only found in metazoans, these domains are present in all eukaryotes. The defining feature of these domains is a hydrophobic cavity where lipid ligands can be accommodated and shielded from the aqueous environment of the cytoplasm(Im et al., 2005, Schauder et al., 2014, Tong et al., 2013, Tsujishita and Hurley, 2000). ORPs have been shown to bind to cholesterol and its oxysterol derivatives as well as phospholipids(Im et al., 2005, Maeda et al., 2013, Mesmin et al., 2013b, Tong et al., 2013), whereas SMP domain proteins appear to mainly transport phospholipids.
The founding member of the ORPs, OSBP, has been shown to localize to contact regions between the Golgi and ER (Ridgway et al., 1992). At these locations, OSBP mediates the exchange of cholesterol and phosphatidylinositol-4-phosphate (PI4P) between the two organelles. The ORP domain of OSBP and related Osh proteins can bind to either cholesterol or PI4P(Im et al., 2005, Tong et al., 2013). It has been proposed that OSBP exploits a Golgi-to-ER concentration gradient of PI4P, which is established by PI4-Kinase type III-mediated PI4P synthesis on Golgi membranes and its hydrolysis on ER membranes by the Sac1 phosphatase. In turn, this gradient is thought to power the transport of cholesterol in the ER-to-Golgi direction (Mesmin et al., 2013a, Mesmin et al., 2013b). Additionally, several Osh proteins were shown to deliver newly-synthesized phosphatidylserine (PS) from the ER to the plasma membrane in yeast (Maeda et al., 2013). SMP proteins also play key roles in lipid homeostasis. In yeast, an SMP-containing protein complex known as the ER-Mitochondria Encounter Structure (ERMES) operates the transfer of ER-synthesized phospholipids to mitochondria, an obligate step for the synthesis of the mitochondria-specific phospholipid, cardiolipin (Kornmann et al., 2009). Thus, transport by ORP/Osh and SMP proteins may be key to enable different organelles to achieve the lipid composition that supports their function.
A growing number of reports indicate that the lysosome may also rely on membrane contacts for lipid transfer with other organelles, particularly the ER. The STAR Related Lipid Transfer Domain-3 (STARD-3) protein, also known as MLN-64, localizes to the LE/LY membrane and is thought to interact with VAPA/B on the ER membranes (Alpy et al., 2013, Tsujishita and Hurley, 2000). Overexpressing STARD-3/MLN-64 is sufficient to enhance the contacts between LE/LY and ER in a VAP-dependent manner, and to enhance cholesterol transfer in the ER-to-lysosome direction (Alpy et al., 2013, Wilhelm et al., 2017). Interestingly, STARD-3/MLN-64 was also shown to transfer cholesterol from LE/LY to mitochondria, providing the precursor for steroid hormones synthesized in the mitochondria (Charman et al., 2010). OSBP-related protein 5 (ORP5) was proposed to mediate cholesterol export from LE/LY. ORP5 has a single transmembrane helix inserted in the ER membrane, and appears to be able to interact in trans with NPC1. Cholesterol accumulation in lysosomes of cells lacking ORP5, suggests that ORP5 may be an acceptor for cholesterol mobilized by the NPC2-NPC1 system (Du et al., 2011). The OSBP-related protein 1L (ORP1L), which was previously implicated in lysosomal positioning in response to cholesterol levels (Rocha et al., 2009), was recently found to mediate transport of cholesterol from LE/LY to the ER in a VAPA/B dependent manner (Zhao and Ridgway, 2017). Cells lacking ORP1L showed significant accumulation of cholesterol at the lysosomal limiting membrane, with concomitant reduction of cholesterol esterification in the ER, suggesting blockage of an important route for cholesterol delivery to the ER. There is also evidence that, in yeast, contacts between the vacuole (the equivalent structure to the lysosome in yeast) and the ER may be involved in trafficking of sterols and other lipids. A member of the VASt family known as Ltc1 (homologous to the VASt-containing GRAM proteins in humans), is inserted in the membrane of the ER via a single transmembrane helix and localizes to junctions between the ER and the vacuole via physical interaction with the vacuolar protein Vac8 (Murley et al., 2015). Interestingly, elevated Ltc1 activity was shown to be associated with the formation of sterol-rich microdomains on the vacuolar surface. These domains appear to play roles in response to stresses such as acute glucose withdrawal (Murley et al., 2015, Seo et al., 2017, Toulmay and Prinz, 2013).
In addition to the ER, other organelles can accept lysosome-derived cholesterol. Using a cleverly designed high throughput screening strategy, it was found that a fraction of LDL-derived cholesterol is transferred to peroxisomes in an NPC1-dependent manner. Live-cell imaging experiments showed that lysosomes engage in transient physical contacts with peroxisomes and that these contacts play an important role in enabling lysosomal cholesterol egress. Lysosome-localized synaptotagmin 7 served as the tethering factor by binding to PI(4,5)P2 on the peroxisomal membrane(Chu et al., 2015). As further genetic and biochemical studies are carried out, it is likely that additional routes for lipid transfer as well as the proteins mediating this transport will be discovered.
The lysosome as a metabolic signaling center
The traditional view of the lysosome as the cellular ‘recycle bin’ has been turned on its head by a number of recent discoveries, sparked by the identification of a physical and functional association of this organelle with the mTORC1 kinase, a master regulator of growth and metabolism (reviewed in (Perera and Zoncu, 2016, Saxton and Sabatini, 2017, Settembre et al., 2013b)). In response to amino acids, mTORC1 translocates from the cytoplasm to the lysosomal surface, where it becomes competent to phosphorylate its downstream substrates. This amino acid-dependent recruitment is mediated by the Rag guanosine triphosphatases (GTPases), which exist as heterodimers of RagA or RagB in complex with RagC or RagD(Kim et al., 2008, Sancak et al., 2010, Sancak et al., 2008). A crucial property of the Rag GTPases is that their nucleotide state changes as a function of the amino acid status of the cell. When amino acid levels are low, it is thought that RagA/B is loaded with GDP, whereas RagC/D is loaded with GTP. The resulting RagAGDP-RagCGTP complex cannot bind to mTORC1, which therefore remains inactive in the cytoplasm. Amino acids cause the switch to the RagAGTP-RagCGDP state, which is competent for mTORC1 binding and causes rapid translocation of mTORC1 to the lysosomal surface (Kim et al., 2008, Sancak et al., 2010, Sancak et al., 2008). Switching of the Rag GTPases from the inactive to the active state is mediated by the concerted action of Guanine Nucleotide Exchange Factors (GEFs) and GTPase Activating Proteins (GAPs) that act either on RagA/B or on RagC/D. The emerging picture is that the GEFs and GAPs for Rag GTPases are activated by an array of specialized sensor proteins located on the lysosomal limiting membrane and in the cytoplasm. These sensors appear to be strategically placed in order to integrate the availability of two amino acid pools, one in the cytoplasm and the other within the lysosomal lumen(Perera and Zoncu, 2016, Saxton and Sabatini, 2017, Settembre et al., 2013b). The integrated action of these two pools is essential to achieve full recruitment and activation of mTORC1 at the lysosomal surface (Box 1).
BOX 1. Mechanisms of nutrient-mediated mTORC1 activation at the lysosome.
A large body of work has shed light on the mechanisms via which amino acids cause the Rag GTPases to switch from the inactive to the active state. This occurs via the action of dedicated GEFs and GAPs. The pentameric Ragulator complex is thought to be a GEF that mediates the loading of RagA/B with GTP, thus promoting mTORC1 recruitment to the lysosome(Bar-Peled et al., 2012, Sancak et al., 2010). Ragulator, along with its orthologue in S. cerevisiae known as the Ego 1–3 complex, also plays an essential role in anchoring the Rag GTPases (and their yeast orthologs Gtr1 and Gtr2), which lack any lipidation signal, to the lysosomal surface, so that in its absence both the Rags and mTORC1 are constitutively cytoplasmic(Binda et al., 2009, Powis et al., 2015, Sancak et al., 2010). Conversely, the trimeric GATOR1 complex (SEACIT in S. cerevisiae) was shown to function as a RagA/B specific GAP that enables mTORC1 inactivation when amino acid levels are low(Bar-Peled et al., 2013, Panchaud et al., 2013, Wolfson et al., 2017). GATOR1 was recently shown to be anchored to the lysosomal surface by the recently described SZT2 protein, which is part of a GATOR1-regulating protein complex known as KICSTOR (Peng et al., 2017, Wolfson et al., 2017). The Folliculin (FLCN)/FNIP complex (Lst4/7 in yeast) is a GAP that converts RagC/D to the GDP-bound form, this step appears especially critical for the ability of the Rag heterodimer to bind to mTORC1(Peli-Gulli et al., 2015, Petit et al., 2013, Tsun et al., 2013). A GEF for RagC/D has not been yet identified. An intriguing question is why the Rag GTPases are dimeric, whereas most signaling GTPases such as Ras, Rheb and Rab GTPases are monomers. Possibly each Rag monomer plays a specialized role, such as binding to mTORC1 or to Ragulator. Also having two GTPases, each with its own GAP and GEF, may allow fine-tuning of mTORC1 lysosomal recruitment by multiple nutrient inputs.
Two amino acid pools are thought to cooperate in switching the Rag GTPases to the active state. In the cytoplasm, leucine-binding protein Sestrin2 and an arginine-binding Castor1/2 interact with the Gator2 complex and block its ability to inhibit Gator1 when they are free from their respective amino acid ligands(Chantranupong et al., 2016, Saxton et al., 2016a, Saxton et al., 2016b, Wolfson et al., 2016). By binding to Sestrin2 and Castor1/2, respectively, leucine and arginine enable Gator2 to block the GAP activity of Gator1, thus causing RagA/B to remain in the GTP-bound state. Thus, mTORC1 binds to the lysosome in response to high micromolar concentrations of leucine and arginine in the cytoplasm(Chantranupong et al., 2016, Saxton et al., 2016a, Saxton et al., 2016b, Wolfson et al., 2016). The lysosomal lumen also contains amino acids, which are generated from proteolysis of both intracellular and extracellular substrates, and may also be imported from the cytoplasm by permeases on the lysosomal limiting membrane. The putative amino acid permease SLC38A9, which specifically localizes to the lysosome, has been implicated in mTORC1 activation by lysosomal amino acids(Jung et al., 2015, Rebsamen et al., 2015, Wang et al., 2015). Moreover, the v-ATPase is also essential for mTORC1 recruitment to the lysosome in response to amino acids(Zoncu et al., 2011). Both SLC38A9 and the v-ATPase physically interact with the Ragulator complex, and may trigger its GEF activity toward RagA/B in response to high amino acid concentrations within the lysosomal lumen(Bar-Peled et al., 2012, Wang et al., 2015, Zoncu et al., 2011).
Recent evidence indicates that mTORC1 recruitment to the lysosome is regulated at the transcriptional level. The mTORC1 substrate TFEB strongly drives RagD expression, leading to enhanced lysosomal localization and signaling activity of mTORC1. This feedback mechanism may enable rapid resumption of growth as cells transition from starved to nutrient-replete states(Di Malta et al., 2017).
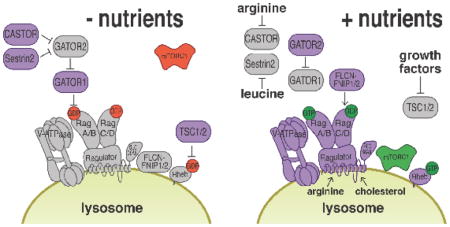
Translocation to the lysosome enables mTORC1 to interact with another small GTPase known as Ras homolog enriched in brain (Rheb). Genetic and biochemical studies have shown that Rheb plays an essential role in priming the kinase activity of mTORC1 toward its substrates, although the precise mechanism of action of Rheb is still mysterious(Long et al., 2005, Tee et al., 2003). Rheb functions downstream of the phosphatidylinositol 3-kinase (PI3K)/Akt pathway, which is activated by growth factors and hormones such as insulin. By binding to the insulin receptor, insulin causes the activation of the serine/threonine kinase Akt. In turn, Akt phosphorylates and inhibits Tuberous Sclerosis Complex (TSC), composed of TSC1, TSC2, and TBC1D7, which functions as a GAP for Rheb(Inoki et al., 2003, Tee et al., 2003). Interestingly, TSC also appears to localize to the lysosomal surface, and phosphorylation by Akt in response to insulin causes its detachment from the lysosome, which may prevent it from inactivating Rheb(Menon et al., 2014). Thus, insulin and growth factors directly control mTORC1 kinase activity by de-inhibiting Rheb. This is an elegant example of coincidence detection, in which the local availability of amino acids places mTORC1 in a physical location, the lysosome, where long-range signals initiated by growth factors trigger its kinase function.
Following its activation by the combined action of nutrients and growth factors, mTORC1 phosphorylates a number of substrates that either promote biosynthetic processes or inhibit catabolic ones. Although the list of mTORC1 substrates keeps growing, here we only list a few important examples. mTORC1 phosphorylates and activates S6-kinase 1 (S6K1), which drives numerous pro-growth processes including ribosome biogenesis, along with lipid and nucleotide biosynthesis(Ben-Sahra et al., 2013, Ben-Sahra et al., 2016, Duvel et al., 2010). mTORC1 phosphorylates and inhibits eukaryotic translation initiation factor 4E-binding proteins (4E-BP) 1 and 2. 4E-BPs block mRNA translation initiation by binding to eIF4E, a component of a multisubunit complex that recruits 40S ribosomal subunits to the 5′ end of mRNAs; thus, their phosphorylation by mTORC1 triggers translation of a class of mRNAs that contain a 5′ terminal oligopyrimidine (TOP) motif(Ma and Blenis, 2009, Thoreen et al., 2012).
The serine/threonine kinase ULK1 promotes initiation of autophagy by phosphorylating several proteins required for the initiation and the expansion of the phagophore(Egan et al., 2015, Lin and Hurley, 2016). ULK1 is inactivated by mTORC1-mediated phosphorylation, resulting in suppression of autophagy under nutrient-replete conditions. A class of recently identified mTORC1 substrates is the MiT/TFE family, basic-helix loop helix (bHLH) transcription factors TFEB, MiTF and TFE3. These proteins were shown to function as transcriptional activators of lysosomal biogenesis and autophagy, via their ability to bind to a DNA consensus sequence, known as the Coordinated Lysosomal Expression And Regulation (CLEAR) motif, which is found upstream of their target genes(Sardiello et al., 2009, Settembre et al., 2011). mTORC1 was shown to directly phosphorylate TFEB. This phosphorylation triggers binding of 14-3-3 proteins to TFEB, leading to its exclusion from the nucleus (Martina et al., 2012, Martina et al., 2014, Roczniak-Ferguson et al., 2012, Settembre et al., 2012). Thus, in high nutrient states, mTORC1-mediated phosphorylation of MiT/TFE factors serves to block the transcriptional activation of pro-catabolic gene programs.
Lysosomal mTORC1 signaling in control of lipid biogenesis and catabolism
The activation of mTORC1 at the lysosomal surface has important consequences for cellular lipid homeostasis. mTORC1 promotes de novo lipid synthesis while at the same time suppressing lipid catabolism. Both actions synergize in promoting membrane biosynthesis during proliferation, as well as building lipid stores for future use when precursor nutrients are plentiful (reviewed in(Caron et al., 2015, Ricoult and Manning, 2013)).
mTORC1 strongly drives the expression, trafficking and proteolytic processing of sterol-responsive element-binding proteins (SREBPs), SREBP1c and SREBP2, which are master regulators for the synthesis of fatty acids and sterols, respectively(Duvel et al., 2010, Owen et al., 2012, Yecies et al., 2011). In cellular and animal models, hyper-activating mTORC1 (e.g. by deletion of TSC2) resulted in enhanced activation of SREBPs and their target genes(Duvel et al., 2010, Wang et al., 2012). Conversely, ablating mTORC1 activity via rapamycin treatment or liver-specific deletion of its essential subunit Raptor led to suppression of SREBP-dependent lipid biogenesis(Peterson et al., 2011, Porstmann et al., 2008). At least some of the pro-lipogenic activities of mTORC1 are mediated by its substrate S6-Kinase 1 (S6K1). S6K1 was shown to promote SREBP1/2 activation and the downstream biosynthetic machinery for fatty acids and sterols(Duvel et al., 2010, Owen et al., 2012).
A further mechanism for SREBP regulation is provided by lipin1, a phosphatidic acid-phosphatase involved in the synthesis of glycerolipids. In mammals lipin1 has been proposed to function as an mTORC1 substrate that suppresses the activity of the SREBPs. Liver-specific inactivation of mTORC1 protected mice from lipid accumulation in the liver (a process known as hepatic steatosis) caused by high fat diet (HFD), but this protective effect was largely lost upon knock down of lipin1 (Peterson et al., 2011). Thus, lipin1 appears to be a key mediator of mTORC1-dependent lipogenesis.
The ability of mTORC1 to promote lipid synthesis is particularly evident during adipogenesis, a developmental process in which precursor cells mature into full adipocytes though the enhanced synthesis and accumulation of triglycerides (Caron et al., 2015, Laplante et al., 2012, Polak et al., 2008, Zhang et al., 2009). Adipogenesis is induced by high circulating levels of insulin and nutrients in the bloodstream. Insulin and nutrients synergistically promote the synthesis and uptake of free fatty acids in adipocytes, and their subsequent esterification to glycerol in the ER of these cells. The resulting triglycerides are stored in lipid droplets, which fill the cytoplasm of mature adipocytes. In in vitro models for adipogenesis, inhibiting mTORC1 by rapamycin treatment completely abolishes insulin-induced adipocyte differentiation by preventing the activation of peroxisome proliferator-activated receptor γ (PPAR-γ), a nuclear receptor that promotes pro-adipogenic and lipogenic transcriptional programs (Caron et al., 2015, Kim and Chen, 2004).
When glucose and amino acids are scarce, cells turn to fat as a primary energy source. In adipose cells, triglycerides within lipid droplets are hydrolyzed to free fatty acids (FFA), which are released into the bloodstream and are taken up by tissues such as skeletal muscle, heart and liver. In these tissues, a process known as beta-oxidation, which takes place inside mitochondria and peroxisomes, converts FFAs to acetyl-CoA while reducing NAD+ and FAD molecules to NADH and FADH2, respectively. In the liver, beta-oxidation is required to trigger gluconeogenesis and maintain glycemia during fasting. This process is also essential to support the production of ketone bodies, which are released into the bloodstream and used by the brain (which is unable to perform beta-oxidation) during prolonged fasting.
True to its role as a suppressor of catabolism, mTORC1 potently suppresses lipid droplet (LD) breakdown and peripheral beta-oxidation. In liver, accumulating evidence suggests that a specialized form of autophagy known as lipophagy participates in lipid store breakdown by targeting LDs for degradation in autolysosomes (Schulze et al., 2013, Singh et al., 2009). There, the lysosomal acidic lipase LIPA de-esterifies triglycerides and releases FFAs, which are then transported to mitochondria for beta-oxidation (Singh and Cuervo, 2011). Consistent with this model, treating hepatocytes with rapamycin was sufficient to induce an autophagy-dependent reduction in LD number and a spike in the rate of beta-oxidation (Singh et al., 2009). Although this paradigm of autophagy-mediated LD consumption seems to hold true in liver cells, an alternative model has been put forward whereby autophagy actually promotes LD formation, possibly by recycling fatty acids and sterols from other membrane compartments. In this model, mTORC1 inhibition would be expected to increase LD number rather than causing LD disappearance (Nguyen et al., 2017, Rambold et al., 2015). How autophagy-derived lipids are funneled into LD formation, and how the subsequent transfer of LD-derived FFAs to mitochondria occurs are the subjects of active investigation (Barbosa and Siniossoglou, 2017).
TFEB has recently emerged as a major regulator of lipid catabolism in whole-organism settings (reviewed in (Settembre and Ballabio, 2014)). Mice carrying a liver-specific deletion of TFEB (TFEB LiKO) failed to degrade LDs upon starvation, and displayed high circulating FFAs and defective peripheral beta-oxidation. Moreover, these animals gained more weight than control littermates when challenged with a HFD. Conversely, mice overexpressing TFEB in the liver displayed increased rates of beta-oxidation relative to wild-type littermates, had lower circulating FFAs and were strongly protected from weight gain induced by HFD(Settembre et al., 2013a). These actions of TFEB were due, at least in part, to its ability to control the expression of a transcriptional regulator of lipid catabolism, peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α), which, together with downstream nuclear receptor peroxisome proliferator-activated receptor α (PPAR-α), promotes the expression of numerous genes for FFA uptake and beta-oxidation in liver. TFEB also contributes to LD breakdown in the liver via its ability to directly induce the expression of autophagic genes. Similar findings in C. elegans show that the TFEB orthologue, HLH-30, promotes lipid catabolism, thereby influencing metabolic homeostasis and lifespan (O’Rourke and Ruvkun, 2013). Thus, TFEB (possibly along with other MiT-TFE family members) exerts a powerful influence on lipid catabolic programs in the liver. Due to the role of mTORC1 as a gatekeeper of TFEB activity, the nutrients-mTORC1-TFEB axis emerges as a key mediator of the fasting response at the whole organism level (Perera and Zoncu, 2016, Settembre and Ballabio, 2014).
Feedback regulation of mTORC1 activity by lipid-derived signals
Cells have evolved sophisticated mechanisms that sense the availability of specific lipid ligands. Upon binding of lipids to dedicated receptors, signaling responses are triggered which fine tune the rate of lipid synthesis, transport and storage in order to maintain membrane homeostasis. Perhaps the best understood example is the cholesterol sensing system in the ER membrane, which was discovered by Brown and Goldstein two decades ago (Goldstein and Brown, 2015). This system is centered on a sterol-sensing transmembrane protein in the ER, known as SREBP Cleavage Activating Protein (SCAP) (Yang et al., 2002). The function of SCAP is to promote the loading of the membrane-anchored SREBP1/2 transcription factors onto COPII vesicles, which then transport SREBP to the Golgi. At the Golgi, dedicated site-1 and site-2 proteases cleave the transmembrane anchor of SREBPs (Sakai et al., 1998) and allow them to move into the nucleus, where the SREBPs bind to sterol responsive elements (SREs) on the promoter of genes involved in cholesterol uptake (e.g. LDL Receptor) and for de novo cholesterol synthesis (e.g, HMG-CoAR) (Sudhof et al., 1987). When cholesterol levels in the ER membrane are high, translocation and activation of SREBPs to the Golgi is inhibited (Radhakrishnan et al., 2004, Yang et al., 2002). Conversely, when cholesterol levels drop, SCAP undergoes a conformational change that causes its dissociation from Insig and allows SREBP to be loaded onto COPII vesicles (Radhakrishnan et al., 2008). In addition to cholesterol, SREBP1 integrates information about other lipid species, such as phosphatidylcholine (PC), to fine-tune cellular lipid balance (Smulan et al., 2016, Walker et al., 2011).
Membrane biogenesis is essential for cellular growth and proliferation. Thus, one would expect that mTORC1 would be able to sense available lipids and facilitate their utilization as well as the production of new lipid species for membrane production. Consistent with this possibility, feeding mice with a HFD boosts mTORC1 signaling in multiple tissues(Dibble and Manning, 2013, Khamzina et al., 2005, Korsheninnikova et al., 2006, Um et al., 2004). However, whether this response is an indirect consequence of hyperinsulinemia triggered by lipid-driven insulin resistance, or if it reflects dedicated, cell autonomous mechanisms for lipid sensing remains unclear.
There is evidence for cell-autonomous regulation of mTORC1 by phospholipids, particularly phosphatidic acid (PA) (Fang et al., 2001, Foster et al., 2014, Yoon et al., 2011). PA was shown to activate mTORC1 signaling in multiple cell types, and knocking down the main enzyme responsible for PA synthesis, phospholipase D (PLD) 1, suppressed mitogen-induced mTORC1 activation (Fang et al., 2001, Sun et al., 2008). Mechanistically, PA was proposed to bind to the FKBP-Rapamycin Binding (FRB) domain of mTORC1, which is immediately N-terminal to the kinase domain and undergoes rapamycin-induced heterodimerization with FKBP-12 (Fang et al., 2001). How PA binding to the FRB domain affects mTORC1 kinase activity remains unclear. PLD1 was shown to localize to endosomes and lysosomes in a nutrient-regulated manner (Dall’Armi et al., 2010, Yoon et al., 2011). Thus, it was proposed that local production of PA at the lysosomal surface may synergize with other nutrient inputs, primarily amino acids, in activating mTORC1 locally. However, a caveat to this model is the observation that PLD1−/− mice are viable and do not display a phenotype consistent with reduced mTORC1 activation (Elvers et al., 2010).
Signaling phospholipids known as phosphoinositides play established roles in mTORC1 regulation. Binding of insulin to the insulin receptor at the plasma membrane triggers the activation of type-I PI3K, which phosphorylates the 3-OH position in the inositol ring of PI(4,5)P2 to generate PI(3,4,5)P3. Production of PI(3,4,5)P3 on the inner leaflet of the plasma membrane is a key event for activation of Akt, which then phosphorylates and suppresses TSC, resulting in Rheb activation and triggering of mTORC1 kinase function at the lysosome (reviewed in (Saxton and Sabatini, 2017)). Additional phosphoinositides found on endomembranes have been linked to mTORC1 regulation, including phosphatidylinositol 3-phosphate (PI3P) (the product of the type-III PI3-Kinase, the human orthologue of yeast Vps34) and phosphatidylinositol 3,4-bisphosphate [PI(3,4)P2], generated from phosphorylation of PI4P by class II PI3K β (PI3KC2β) (Gulati et al., 2008, Marat et al., 2017).
Recently, it was found that cholesterol activates mTORC1 in a cell-autonomous manner. In particular, cholesterol delivered to the lysosome by endocytosed LDL particles induced the recruitment of mTORC1 to the lysosomal surface (Castellano et al., 2017). Combining in vitro and in cell experiments, it was shown that lysosomal cholesterol levels affect the activation status of the Rag GTPases. SLC38A9, the putative lysosomal solute carrier that is involved in mTORC1 activation by arginine, harbors putative cholesterol-interacting motifs in its transmembrane helix 8, known as cholesterol recognition amino acid consensus (CRAC) and the inverse motif, known as CARC (Fantini and Barrantes, 2013). These motifs were shown to mediate the specific interaction of SLC38A9 with cholesterol, and were required for mTORC1 activation by this ligand (Castellano et al., 2017, Derler et al., 2016). A further finding from this study was that the NPC1 protein was also essential for mTORC1 regulation by cholesterol, suggesting that dysregulated mTORC1 could contribute to the metabolic derangement observed in Niemann-Pick type C disease. How this pathway for cell-autonomous cholesterol sensing plays out in a whole organism context, and whether it may take up tissue-specific and organ-specific roles remains to be determined.
In summary, our understanding of how information on cellular lipid availability feeds back on mTORC1 is in its infancy. Further studies are needed to elucidate the biochemical mechanisms as well as the physiological context of lipid sensing by mTORC1. The evidence gathered so far sets the stage for exciting future work spanning reconstituted systems, functional studies in cells and genetic and metabolic analysis in mice, which will shed light on fundamental rules of lipid homeostasis.
Disruption of lysosomal lipid homeostasis in disease and aging
Loss of the degradative capacity of the lysosome, which occurs over time or as a consequence of gene mutations, is increasingly viewed as a driving force in metabolic derangement and neurodegeneration that characterize aging and certain inherited disorders. Loss of hydrolytic or lysosomal transport ability causes aberrant accumulation of macromolecules within the lumen, leading to broad functional impairment of trafficking, signaling, and quality control pathways (reviewed in (Ballabio and Gieselmann, 2009)). A majority of lysosomal storage disorders involve the aberrant accumulation of lipid species including sphingolipids (Niemann-Pick types A and B, Gaucher, Sandhoff, Krabbe, Fabry), mucolipids (collectively known as mucolipidoses), and sterols (Niemann Pick type C, Wolman, and cholesterol ester storage disease). Accumulation of these lipid species impairs the overall degradative capacity of the lysosome and alters its physiology leading to aberrant size, trafficking, and attenuated autophagy (Ballabio and Gieselmann, 2009, Cox and Cachon-Gonzalez, 2012, Platt et al., 2012). These functional deficiencies impact multiple cell types, particularly neurons resulting in the neurodegenerative phenotypes associated with a majority of storage diseases.
Often accumulation of a primary lipid species occurs with concomitant buildup of secondary lipids. For example, in Niemann-Pick type C, cholesterol build up occurs along with oxysterols, sphingomyelin, gangliosides GM3 and GM2, lysobisphosphatidic acid (LBPA), and sphingosine (Kobayashi et al., 1999, Lloyd-Evans et al., 2008). Collectively, these effects lead to further functional impairment, which goes beyond the lysosome. In cells lacking NPC1, mitochondria also show increased cholesterol content, possibly through the sterol transfer activity of MLN64 (Balboa et al., 2017, Kennedy et al., 2014). In turn, mitochondrial cholesterol accumulation led to an increase in glycolytic flux and mitochondrial oxidative stress, linking aberrant lipid composition and transfer to metabolic alterations. More generally, there is evidence that lysosomal storage disorders, particularly lipid storage disorders, result in severe mitochondrial dysfunction including their altered morphology, calcium signaling, redox imbalance, and altered TCA cycle flux (for comprehensive review see (Plotegher and Duchen, 2017)).
A major consequence of lipid accumulation within the lysosome is the disruption of autophagy (Settembre et al., 2008). For example, increased levels of cholesterol in Niemann-Pick type C is thought to disrupt the function of Rab GTPases that control delivery of hydrolases to the lysosome, potentially reducing its degradative capacity (Ganley and Pfeffer, 2006). NPC lysosomes display aberrant trafficking, due at least in part to the increased association with the minus end directed microtubule motor dynein-dynactin (Rocha et al., 2009). Moreover, alterations in membrane composition of NPC lysosomes may impair the function of the soluble N-ethylmaleimide-sensitive factor protein (SNAP) receptors (SNAREs), which are key components of vesicle fusion machinery resulting in defective fusion ability of lysosomes with autophagosomes (Fraldi et al., 2010, Sarkar et al., 2013).
Loss of lysosomal function is emerging as a driving force in age-related degeneration of cells and tissues (Colacurcio and Nixon, 2016, Hughes and Gottschling, 2012). A time dependent decrease in autophagic flux progressively impairs cellular homeostasis, as evidenced by accumulation of aggregated proteins and extensive mitochondrial damage observed in brain tissue from healthy aged subjects. Electron micrographs from aged humans as well as mice show that a significant percentage of lysosomes appear to be filled with insoluble lipid precipitates known as lipofuscins. These are the insoluble residues left by multiple rounds of fusion and degradation, and are thought to impair the catabolic capacity of the lysosome they reside in. The accumulation of lipofuscin filled lysosomes is especially evident in non-dividing cells such as neurons, which are critically dependent on the autophagic-lysosomal system for their function and survival (Hara et al., 2006).
Interestingly, a class of lipid storage disorders known as neuronal ceroid lipofuscinoses (NCL) display massive accumulation of lipofuscin-like material inside of lysosomes at an early age and are characterized by severe neurodegeneration (Carcel-Trullols et al., 2015). Therefore, some of the pathophysiological mechanisms that underlie lysosomal storage disorders might occur during normal aging albeit at a slower rate. Multiple lines of evidence from organisms spanning from yeast to higher mammals strongly suggests that maintenance of lysosomal function is key for lifespan and healthy aging, or healthspan. In addition to maintenance of lipid homeostasis, there is evidence that lipid processing and trafficking within the lysosome may generate signals that control lifespan in the roundworm C. elegans (Folick et al., 2015).
Although many storage diseases are linked to impairment of lysosomal function, enhanced lysosomal activity may also promote disease progression. For example, in the case of KRAS driven cancers, generation of nutrients through lysosomal processing may confer a selective advantage under conditions of nutrient scarcity (Perera and Zoncu, 2016). Recently, it has been shown that this enhanced lysosomal activity relies on specific transcriptional programs. For example, in pancreatic ductal adenocarcinoma (PDA), the MiT/TFE transcription factors evade regulation by mTORC1 and become constitutively localized to the nucleus where they drive enhanced lysosomal biogenesis (Perera et al., 2015). This adaptation enables scavenging of energy rich nutrients, including lipids, which are delivered to the lysosome through macropinocytosis and autophagy, driving cellular growth and survival (Commisso et al., 2013, Mancias and Kimmelman, 2011). Interestingly, concomitant upregulation of ACAT1 in pancreatic cancer may enable storage of LDL-derived cholesterol in lipid droplets and promote tumor growth and metastasis (Li et al., 2016a). This mechanism may be important not only for cancer growth, but also in the context of resistance to therapy. In mouse models where mutant KRAS can be inducibly ablated the appearance of resistant cells relies on enhanced lysosomal function, lipid droplet accumulation, and lipid catabolism (Viale et al., 2014). Thus, a deeper understanding of lysosomal function, both in normal and disease states, may inform new approaches for preventing and treating cancer and age-related conditions.
Concluding Remarks
Lysosomes promote lipid catabolism and transport, both of which are critical in maintaining cellular lipid homeostasis. Recent work has established that the lysosome is a hub for nutrient sensing and signaling, and it is likely that the lipid sensing and trafficking functions of the lysosome are intimately linked. In particular, organelle contact sites may mediate the transfer of information between subcellular compartments, each with specialized roles such as storage of lipids in lipid droplets or beta-oxidation of lipids in mitochondria and peroxisomes. Organelle crosstalk via membrane contact sites is a rapidly progressing area of research with many outstanding questions, and future work may shed light on the role of various lipid species in supporting the function of the organelle in which they reside.
In the case of lysosomes, mTORC1 acts as a sensor of lysosomal lipids, although it remains to be seen whether other species beside cholesterol and phosphatidic acid can impact mTORC1 signaling. Interestingly, mTORC2, a distinct protein complex that shares the same core kinase subunit with mTORC1, senses sphingolipid levels at the plasma membrane, and plays a key role in maintaining sphingolipid homeostasis in response to a variety of stressors (Berchtold et al., 2012, Muir et al., 2014). Importantly, aberrant mTORC1 signaling may contribute to the pathogenesis of lysosomal disorders characterized by abnormal lipid storage, and thus targeting mTORC1 may prove beneficial in these disease settings. Clarifying the role of mTORC1 in maintaining lipid homeostasis may increase our understanding of processes where lipids are key building blocks, such as cell growth, proliferation and differentiation processes that require expansion of membranes, as well as energy storage. Furthermore, it will be interesting to compare how these lipid-sensing and lipid-trafficking networks acquire specialized functions in different tissues, according to specific metabolic roles (see Outstanding Questions).
Outstanding questions.
How do organelle contact sites regulate the lipid composition of organelle membranes?
How are contacts between different organelles regulated and do they serve functions other than lipid transfer?
What is the mechanism of cholesterol export from the lysosome?
Do other lipids besides cholesterol mediate mTORC1 activation at the lysosome?
Fig. 1. Cellular routes for cholesterol trafficking.

Low-density lipoprotein receptors (LDLR) on the plasma membrane bind LDL particles and are delivered into early endosomes (EE) via clathrin-mediated endocytosis (1). As EE acidify and mature into late endosomes (LE), LDL dissociates from the LDLR and the LDLR is sorted into the endocytic recycling compartment (ERC) and returned to the plasma membrane. LDL continues along the endocytic pathway and ultimately reaches the lysosome (2), where the LDL is broken down (3). Lysosomal acid lipase (LipA) hydrolyzes cholesteryl esters to release free cholesterol. Cholesterol is shuttled through the lysosomal lumen by NPC2, and subsequently transferred to the luminal N-terminal domain of the polytopic transmembrane protein NPC1 (4). The mechanisms of cholesterol export from the lysosome are still unknown, although they are likely to occur through several parallel routes that may involve the formation of membrane contact sites (shown by bidirectional dashed lines) between the lysosome and acceptor membranes (5). Endogenous cholesterol synthesis at the endoplasmic reticulum (ER) requires multiple biosynthetic enzymes including the rate-limiting enzyme HMG-CoA reductase. Under conditions of cholesterol abundance, ER cholesterol can be esterified by Acetyl-CoA Acetyltransferase 1 (ACAT1) and stored in lipid droplets (6).
Fig. 2. Lipid transfer proteins mediate lipid exchange between membranes.

Sterols and phospholipids are transferred between two adjacent organelle membranes via lipid transfer proteins. ER anchored VAP serves as the interacting partner for various lipid transfer proteins including OSBP on the Golgi as well as ORP1L and STARD3 on the LE/LY. ORP1L and STARD3 are involved in cholesterol transfer from the LE/LY to the ER and from the ER to the LE/LY, respectively. ORP5 is a VAP-independent ER associated protein that interacts with NPC1 and is thought to act as a cholesterol acceptor, thus facilitating lysosomal cholesterol export. At the Golgi, OSBP transports PI4P down its concentration gradient to the ER, where the PI4P specific phosphatase Sac1 hydrolyzes it to PI to maintain low levels of PI4P at the ER membrane. After depositing the PI4P at the ER, OSBP binds cholesterol and transfers it to the Golgi, thus completing the cycle and establishing a net transfer of cholesterol in the ER-to-Golgi direction.
Fig. 3. mTORC1 activation promotes lipid biogenesis and suppresses lipid catabolism.

In the presence of nutrients, mTORC1 translocates from the cytoplasm to the surface of the lysosome where it becomes activated and phosphorylates downstream substrates thereby activating (green box) or inhibiting them (red box). S6-kinase 1 (S6K1) is an mTORC1 substrate that promotes proteolytic processing of the transcription factors SREBP1/2. Inactive, full-length SREBP is retained at the ER membrane by its association with SCAP and INSIG in the presence of cholesterol. Under low cholesterol conditions in the ER, SCAP undergoes a conformational change releasing SREBP and promoting its COPII vesicle mediated trafficking to the Golgi. At the Golgi, two proteases, S1P and S2P, cleave SREBP to release the soluble, N-terminal fragment, which translocates to the nucleus and binds to sterol response elements (SRE) upstream of promoters for genes involved in sterol uptake and de novo lipid biosynthesis. Furthermore, mTORC1 phosphorylation blocks the nuclear entry of lipin1, a phosphatidic acid phosphatase that acts as an inhibitor of nuclear SREBP activity. Similarly, phosphorylation of TFEB prevents its translocation to the nucleus, repressing transcriptional activation of CLEAR genes involved in β-oxidation of fatty acids, lipid catabolism, and lysosomal biogenesis. mTORC1 is thought to promote adipocyte differentiation by releasing a translational block mediated by 4E-BP1. Finally, mTORC1 inhibits autophagic degradation of lipid droplets through phosphorylation of ULK1 kinase.
Trends.
The lysosome regulates lipid metabolism through transcriptional as well as post-translational mechanisms.
Inter-organelle contacts allow exchange of lipids across adjacent membranes in order to dynamically regulate lipid composition.
Lysosomes are a hub for different nutrient classes including amino acids and lipids, which are integrated upstream of mTORC1.
Acknowledgments
We thank Mathieu Laplante and Carmine Settembre for critical reading of the manuscript. This work was supported by the NIH Director’s New Innovator Award (1DP2CA195761-01), the Pew-Stewart Scholarship for Cancer Research, the Damon Runyon-Rachleff Innovation Award, the Edward Mallinckrodt, Jr. Foundation Grant and the Packer Wentz Endowment to R.Z., and a National Science Foundation Graduate Research Fellowship (DGE 1106400) to A.M.T.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Alpy F, Rousseau A, Schwab Y, Legueux F, Stoll I, Wendling C, … Tomasetto C. STARD3 or STARD3NL and VAP form a novel molecular tether between late endosomes and the ER. Journal of cell science. 2013;126:5500–5512. doi: 10.1242/jcs.139295. [DOI] [PubMed] [Google Scholar]
- 2.Alpy F, Tomasetto C. START ships lipids across interorganelle space. Biochimie. 2014;96:85–95. doi: 10.1016/j.biochi.2013.09.015. [DOI] [PubMed] [Google Scholar]
- 3.Balboa E, Castro J, Pinochet MJ, Cancino GI, Matias N, Jose Saez P, … Zanlungo S. MLN64 induces mitochondrial dysfunction associated with increased mitochondrial cholesterol content. Redox Biol. 2017;12:274–284. doi: 10.1016/j.redox.2017.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ballabio A, Gieselmann V. Lysosomal disorders: from storage to cellular damage. Biochim Biophys Acta. 2009;1793:684–696. doi: 10.1016/j.bbamcr.2008.12.001. [DOI] [PubMed] [Google Scholar]
- 5.Bar-Peled L, Chantranupong L, Cherniack AD, Chen WW, Ottina KA, Grabiner BC, … Sabatini DM. A Tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science. 2013;340:1100–1106. doi: 10.1126/science.1232044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bar-Peled L, Schweitzer LD, Zoncu R, Sabatini DM. Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell. 2012;150:1196–1208. doi: 10.1016/j.cell.2012.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barbosa AD, Siniossoglou S. Function of lipid droplet-organelle interactions in lipid homeostasis. Biochim Biophys Acta. 2017 doi: 10.1016/j.bbamcr.2017.04.001. [DOI] [PubMed] [Google Scholar]
- 8.Ben-Sahra I, Howell JJ, Asara JM, Manning BD. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science. 2013;339:1323–1328. doi: 10.1126/science.1228792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ben-Sahra I, Hoxhaj G, Ricoult SJ, Asara JM, Manning BD. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science. 2016;351:728–733. doi: 10.1126/science.aad0489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Berchtold D, Piccolis M, Chiaruttini N, Riezman I, Riezman H, Roux A, … Loewith R. Plasma membrane stress induces relocalization of Slm proteins and activation of TORC2 to promote sphingolipid synthesis. Nature cell biology. 2012;14:542–547. doi: 10.1038/ncb2480. [DOI] [PubMed] [Google Scholar]
- 11.Binda M, Peli-Gulli MP, Bonfils G, Panchaud N, Urban J, Sturgill TW, … De Virgilio C. The Vam6 GEF controls TORC1 by activating the EGO complex. Mol Cell. 2009;35:563–573. doi: 10.1016/j.molcel.2009.06.033. [DOI] [PubMed] [Google Scholar]
- 12.Brown AJ, Sun L, Feramisco JD, Brown MS, Goldstein JL. Cholesterol addition to ER membranes alters conformation of SCAP, the SREBP escort protein that regulates cholesterol metabolism. Mol Cell. 2002;10:237–245. doi: 10.1016/s1097-2765(02)00591-9. [DOI] [PubMed] [Google Scholar]
- 13.Carcel-Trullols J, Kovacs AD, Pearce DA. Cell biology of the NCL proteins: What they do and don’t do. Biochim Biophys Acta. 2015;1852:2242–2255. doi: 10.1016/j.bbadis.2015.04.027. [DOI] [PubMed] [Google Scholar]
- 14.Caron A, Richard D, Laplante M. The Roles of mTOR Complexes in Lipid Metabolism. Annu Rev Nutr. 2015;35:321–348. doi: 10.1146/annurev-nutr-071714-034355. [DOI] [PubMed] [Google Scholar]
- 15.Castellano BM, Thelen AM, Moldavski O, Feltes M, van der Welle RE, Mydock-McGrane L, … Zoncu R. Lysosomal cholesterol activates mTORC1 via an SLC38A9-Niemann-Pick C1 signaling complex. Science. 2017;355:1306–1311. doi: 10.1126/science.aag1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang TY, Chang CC, Ohgami N, Yamauchi Y. Cholesterol sensing, trafficking, and esterification. Annual review of cell and developmental biology. 2006;22:129–157. doi: 10.1146/annurev.cellbio.22.010305.104656. [DOI] [PubMed] [Google Scholar]
- 17.Chantranupong L, Scaria SM, Saxton RA, Gygi MP, Shen K, Wyant GA, … Sabatini DM. The CASTOR Proteins Are Arginine Sensors for the mTORC1 Pathway. Cell. 2016;165:153–164. doi: 10.1016/j.cell.2016.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Charman M, Kennedy BE, Osborne N, Karten B. MLN64 mediates egress of cholesterol from endosomes to mitochondria in the absence of functional Niemann-Pick Type C1 protein. J Lipid Res. 2010;51:1023–1034. doi: 10.1194/jlr.M002345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chu BB, Liao YC, Qi W, Xie C, Du X, Wang J, … Song BL. Cholesterol transport through lysosome-peroxisome membrane contacts. Cell. 2015;161:291–306. doi: 10.1016/j.cell.2015.02.019. [DOI] [PubMed] [Google Scholar]
- 20.Colacurcio DJ, Nixon RA. Disorders of lysosomal acidification-The emerging role of v-ATPase in aging and neurodegenerative disease. Ageing Res Rev. 2016;32:75–88. doi: 10.1016/j.arr.2016.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Commisso C, Davidson SM, Soydaner-Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S, … Bar-Sagi D. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature. 2013;497:633–637. doi: 10.1038/nature12138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cox TM, Cachon-Gonzalez MB. The cellular pathology of lysosomal diseases. The Journal of pathology. 2012;226:241–254. doi: 10.1002/path.3021. [DOI] [PubMed] [Google Scholar]
- 23.Dall’Armi C, Hurtado-Lorenzo A, Tian H, Morel E, Nezu A, Chan RB, … Di Paolo G. The phospholipase D1 pathway modulates macroautophagy. Nature communications. 2010;1:142. doi: 10.1038/ncomms1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Daniele T, Schiaffino MV. Lipid transfer and metabolism across the endolysosomal-mitochondrial boundary. Biochim Biophys Acta. 2016;1861:880–894. doi: 10.1016/j.bbalip.2016.02.001. [DOI] [PubMed] [Google Scholar]
- 25.Das A, Brown MS, Anderson DD, Goldstein JL, Radhakrishnan A. Three pools of plasma membrane cholesterol and their relation to cholesterol homeostasis. Elife. 2014:3. doi: 10.7554/eLife.02882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deffieu MS, Pfeffer SR. Niemann-Pick type C 1 function requires lumenal domain residues that mediate cholesterol-dependent NPC2 binding. Proc Natl Acad Sci U S A. 2011;108:18932–18936. doi: 10.1073/pnas.1110439108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Derler I, Jardin I, Stathopulos PB, Muik M, Fahrner M, Zayats V, … Romanin C. Cholesterol modulates Orai1 channel function. Science signaling. 2016;9:ra10. doi: 10.1126/scisignal.aad7808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Di Malta C, Siciliano D, Calcagni A, Monfregola J, Punzi S, Pastore N, … Ballabio A. Transcriptional activation of RagD GTPase controls mTORC1 and promotes cancer growth. Science. 2017;356:1188–1192. doi: 10.1126/science.aag2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dibble CC, Manning BD. Signal integration by mTORC1 coordinates nutrient input with biosynthetic output. Nature cell biology. 2013;15:555–564. doi: 10.1038/ncb2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Du X, Kumar J, Ferguson C, Schulz TA, Ong YS, Hong W, … Yang H. A role for oxysterol-binding protein-related protein 5 in endosomal cholesterol trafficking. J Cell Biol. 2011;192:121–135. doi: 10.1083/jcb.201004142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Duvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, … Manning BD. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell. 2010;39:171–183. doi: 10.1016/j.molcel.2010.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Egan DF, Chun MG, Vamos M, Zou H, Rong J, Miller CJ, … Shaw RJ. Small Molecule Inhibition of the Autophagy Kinase ULK1 and Identification of ULK1 Substrates. Mol Cell. 2015;59:285–297. doi: 10.1016/j.molcel.2015.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Elvers M, Stegner D, Hagedorn I, Kleinschnitz C, Braun A, Kuijpers ME, … Nieswandt B. Impaired alpha(IIb)beta(3) integrin activation and shear-dependent thrombus formation in mice lacking phospholipase D1. Science signaling. 2010;3:ra1. doi: 10.1126/scisignal.2000551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Espenshade PJ, Hughes AL. Regulation of sterol synthesis in eukaryotes. Annual review of genetics. 2007;41:401–427. doi: 10.1146/annurev.genet.41.110306.130315. [DOI] [PubMed] [Google Scholar]
- 35.Fang Y, Vilella-Bach M, Bachmann R, Flanigan A, Chen J. Phosphatidic acid-mediated mitogenic activation of mTOR signaling. Science. 2001;294:1942–1945. doi: 10.1126/science.1066015. [DOI] [PubMed] [Google Scholar]
- 36.Fantini J, Barrantes FJ. How cholesterol interacts with membrane proteins: an exploration of cholesterol-binding sites including CRAC, CARC, and tilted domains. Frontiers in physiology. 2013;4:31. doi: 10.3389/fphys.2013.00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Folick A, Oakley HD, Yu Y, Armstrong EH, Kumari M, Sanor L, … Wang MC. Aging. Lysosomal signaling molecules regulate longevity in Caenorhabditis elegans. Science. 2015;347:83–86. doi: 10.1126/science.1258857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Foster DA, Salloum D, Menon D, Frias MA. Phospholipase D and the maintenance of phosphatidic acid levels for regulation of mammalian target of rapamycin (mTOR) J Biol Chem. 2014;289:22583–22588. doi: 10.1074/jbc.R114.566091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fraldi A, Annunziata F, Lombardi A, Kaiser HJ, Medina DL, Spampanato C, … Ballabio A. Lysosomal fusion and SNARE function are impaired by cholesterol accumulation in lysosomal storage disorders. Embo J. 2010;29:3607–3620. doi: 10.1038/emboj.2010.237. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 40.Ganley IG, Pfeffer SR. Cholesterol accumulation sequesters Rab9 and disrupts late endosome function in NPC1-deficient cells. J Biol Chem. 2006;281:17890–17899. doi: 10.1074/jbc.M601679200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goldstein JL, Brown MS. A century of cholesterol and coronaries: from plaques to genes to statins. Cell. 2015;161:161–172. doi: 10.1016/j.cell.2015.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gong X, Qian H, Zhou X, Wu J, Wan T, Cao P, … Yan N. Structural Insights into the Niemann-Pick C1 (NPC1)-Mediated Cholesterol Transfer and Ebola Infection. Cell. 2016;165:1467–1478. doi: 10.1016/j.cell.2016.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gulati P, Gaspers LD, Dann SG, Joaquin M, Nobukuni T, Natt F, … Thomas G. Amino acids activate mTOR complex 1 via Ca2+/CaM signaling to hVps34. Cell Metab. 2008;7:456–465. doi: 10.1016/j.cmet.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, … Mizushima N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 45.He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annual review of genetics. 2009;43:67–93. doi: 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hughes AL, Gottschling DE. An early age increase in vacuolar pH limits mitochondrial function and lifespan in yeast. Nature. 2012;492:261–265. doi: 10.1038/nature11654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hurley JH, Schulman BA. Atomistic autophagy: the structures of cellular self-digestion. Cell. 2014;157:300–311. doi: 10.1016/j.cell.2014.01.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ikonen E. Cellular cholesterol trafficking and compartmentalization. Nat Rev Mol Cell Biol. 2008;9:125–138. doi: 10.1038/nrm2336. [DOI] [PubMed] [Google Scholar]
- 49.Im YJ, Raychaudhuri S, Prinz WA, Hurley JH. Structural mechanism for sterol sensing and transport by OSBP-related proteins. Nature. 2005;437:154–158. doi: 10.1038/nature03923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Infante RE, Wang ML, Radhakrishnan A, Kwon HJ, Brown MS, Goldstein JL. NPC2 facilitates bidirectional transfer of cholesterol between NPC1 and lipid bilayers, a step in cholesterol egress from lysosomes. Proc Natl Acad Sci U S A. 2008;105:15287–15292. doi: 10.1073/pnas.0807328105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Inoki K, Li Y, Xu T, Guan KL. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003;17:1829–1834. doi: 10.1101/gad.1110003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jung J, Genau HM, Behrends C. Amino Acid-Dependent mTORC1 Regulation by the Lysosomal Membrane Protein SLC38A9. Molecular and cellular biology. 2015;35:2479–2494. doi: 10.1128/MCB.00125-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kennedy BE, Madreiter CT, Vishnu N, Malli R, Graier WF, Karten B. Adaptations of energy metabolism associated with increased levels of mitochondrial cholesterol in Niemann-Pick type C1-deficient cells. J Biol Chem. 2014;289:16278–16289. doi: 10.1074/jbc.M114.559914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Khamzina L, Veilleux A, Bergeron S, Marette A. Increased activation of the mammalian target of rapamycin pathway in liver and skeletal muscle of obese rats: possible involvement in obesity-linked insulin resistance. Endocrinology. 2005;146:1473–1481. doi: 10.1210/en.2004-0921. [DOI] [PubMed] [Google Scholar]
- 55.Kim E, Goraksha-Hicks P, Li L, Neufeld TP, Guan KL. Regulation of TORC1 by Rag GTPases in nutrient response. Nature cell biology. 2008;10:935–945. doi: 10.1038/ncb1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim JE, Chen J. regulation of peroxisome proliferator-activated receptor-gamma activity by mammalian target of rapamycin and amino acids in adipogenesis. Diabetes. 2004;53:2748–2756. doi: 10.2337/diabetes.53.11.2748. [DOI] [PubMed] [Google Scholar]
- 57.Kobayashi T, Beuchat MH, Lindsay M, Frias S, Palmiter RD, Sakuraba H, … Gruenberg J. Late endosomal membranes rich in lysobisphosphatidic acid regulate cholesterol transport. Nature cell biology. 1999;1:113–118. doi: 10.1038/10084. [DOI] [PubMed] [Google Scholar]
- 58.Kornmann B, Currie E, Collins SR, Schuldiner M, Nunnari J, Weissman JS, Walter P. An ER-mitochondria tethering complex revealed by a synthetic biology screen. Science. 2009;325:477–481. doi: 10.1126/science.1175088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Korsheninnikova E, van der Zon GC, Voshol PJ, Janssen GM, Havekes LM, Grefhorst A, … Maassen JA. Sustained activation of the mammalian target of rapamycin nutrient sensing pathway is associated with hepatic insulin resistance, but not with steatosis, in mice. Diabetologia. 2006;49:3049–3057. doi: 10.1007/s00125-006-0439-5. [DOI] [PubMed] [Google Scholar]
- 60.Kwon HJ, Abi-Mosleh L, Wang ML, Deisenhofer J, Goldstein JL, Brown MS, Infante RE. Structure of N-terminal domain of NPC1 reveals distinct subdomains for binding and transfer of cholesterol. Cell. 2009;137:1213–1224. doi: 10.1016/j.cell.2009.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lahiri S, Toulmay A, Prinz WA. Membrane contact sites, gateways for lipid homeostasis. Curr Opin Cell Biol. 2015;33:82–87. doi: 10.1016/j.ceb.2014.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Laplante M, Horvat S, Festuccia WT, Birsoy K, Prevorsek Z, Efeyan A, Sabatini DM. DEPTOR cell-autonomously promotes adipogenesis, and its expression is associated with obesity. Cell Metab. 2012;16:202–212. doi: 10.1016/j.cmet.2012.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li J, Gu D, Lee SS, Song B, Bandyopadhyay S, Chen S, … Cheng JX. Abrogating cholesterol esterification suppresses growth and metastasis of pancreatic cancer. Oncogene. 2016a;35:6378–6388. doi: 10.1038/onc.2016.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li J, Pfeffer SR. Lysosomal membrane glycoproteins bind cholesterol and contribute to lysosomal cholesterol export. Elife. 2016:5. doi: 10.7554/eLife.21635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li X, Saha P, Li J, Blobel G, Pfeffer SR. Clues to the mechanism of cholesterol transfer from the structure of NPC1 middle lumenal domain bound to NPC2. Proc Natl Acad Sci U S A. 2016b;113:10079–10084. doi: 10.1073/pnas.1611956113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lin MG, Hurley JH. Structure and function of the ULK1 complex in autophagy. Curr Opin Cell Biol. 2016;39:61–68. doi: 10.1016/j.ceb.2016.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lloyd-Evans E, Morgan AJ, He X, Smith DA, Elliot-Smith E, Sillence DJ, … Platt FM. Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nature medicine. 2008;14:1247–1255. doi: 10.1038/nm.1876. [DOI] [PubMed] [Google Scholar]
- 68.Long X, Lin Y, Ortiz-Vega S, Yonezawa K, Avruch J. Rheb binds and regulates the mTOR kinase. Current biology : CB. 2005;15:702–713. doi: 10.1016/j.cub.2005.02.053. [DOI] [PubMed] [Google Scholar]
- 69.Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol. 2009;10:307–318. doi: 10.1038/nrm2672. [DOI] [PubMed] [Google Scholar]
- 70.Maeda K, Anand K, Chiapparino A, Kumar A, Poletto M, Kaksonen M, Gavin AC. Interactome map uncovers phosphatidylserine transport by oxysterol-binding proteins. Nature. 2013;501:257–261. doi: 10.1038/nature12430. [DOI] [PubMed] [Google Scholar]
- 71.Mancias JD, Kimmelman AC. Targeting autophagy addiction in cancer. Oncotarget. 2011;2:1302–1306. doi: 10.18632/oncotarget.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Marat AL, Wallroth A, Lo WT, Muller R, Norata GD, Falasca M, … Haucke V. mTORC1 activity repression by late endosomal phosphatidylinositol 3,4-bisphosphate. Science. 2017;356:968–972. doi: 10.1126/science.aaf8310. [DOI] [PubMed] [Google Scholar]
- 73.Martin V, Carrillo G, Torroja C, Guerrero I. The sterol-sensing domain of Patched protein seems to control Smoothened activity through Patched vesicular trafficking. Current biology : CB. 2001;11:601–607. doi: 10.1016/s0960-9822(01)00178-6. [DOI] [PubMed] [Google Scholar]
- 74.Martina JA, Chen Y, Gucek M, Puertollano R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy. 2012;8:903–914. doi: 10.4161/auto.19653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Martina JA, Diab HI, Lishu L, Jeong AL, Patange S, Raben N, Puertollano R. The nutrient-responsive transcription factor TFE3 promotes autophagy, lysosomal biogenesis, and clearance of cellular debris. Science signaling. 2014;7:ra9. doi: 10.1126/scisignal.2004754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.McCauliff LA, Xu Z, Li R, Kodukula S, Ko DC, Scott MP, … Storch J. Multiple Surface Regions on the Niemann-Pick C2 Protein Facilitate Intracellular Cholesterol Transport. J Biol Chem. 2015;290:27321–27331. doi: 10.1074/jbc.M115.667469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Menon S, Dibble CC, Talbott G, Hoxhaj G, Valvezan AJ, Takahashi H, … Manning BD. Spatial control of the TSC complex integrates insulin and nutrient regulation of mTORC1 at the lysosome. Cell. 2014;156:771–785. doi: 10.1016/j.cell.2013.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mesmin B, Antonny B, Drin G. Insights into the mechanisms of sterol transport between organelles. Cellular and molecular life sciences : CMLS. 2013a;70:3405–3421. doi: 10.1007/s00018-012-1247-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mesmin B, Bigay J, Moser von Filseck J, Lacas-Gervais S, Drin G, Antonny B. A four-step cycle driven by PI(4)P hydrolysis directs sterol/PI(4)P exchange by the ER-Golgi tether OSBP. Cell. 2013b;155:830–843. doi: 10.1016/j.cell.2013.09.056. [DOI] [PubMed] [Google Scholar]
- 80.Millard EE, Gale SE, Dudley N, Zhang J, Schaffer JE, Ory DS. The sterol-sensing domain of the Niemann-Pick C1 (NPC1) protein regulates trafficking of low density lipoprotein cholesterol. J Biol Chem. 2005;280:28581–28590. doi: 10.1074/jbc.M414024200. [DOI] [PubMed] [Google Scholar]
- 81.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mobius W, Ohno-Iwashita Y, van Donselaar EG, Oorschot VM, Shimada Y, Fujimoto T, … Slot JW. Immunoelectron microscopic localization of cholesterol using biotinylated and non-cytolytic perfringolysin O. J Histochem Cytochem. 2002;50:43–55. doi: 10.1177/002215540205000105. [DOI] [PubMed] [Google Scholar]
- 83.Muir A, Ramachandran S, Roelants FM, Timmons G, Thorner J. TORC2-dependent protein kinase Ypk1 phosphorylates ceramide synthase to stimulate synthesis of complex sphingolipids. Elife. 2014:3. doi: 10.7554/eLife.03779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Murley A, Sarsam RD, Toulmay A, Yamada J, Prinz WA, Nunnari J. Ltc1 is an ER-localized sterol transporter and a component of ER-mitochondria and ER-vacuole contacts. J Cell Biol. 2015;209:539–548. doi: 10.1083/jcb.201502033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nguyen TB, Louie SM, Daniele JR, Tran Q, Dillin A, Zoncu R, … Olzmann JA. DGAT1-Dependent Lipid Droplet Biogenesis Protects Mitochondrial Function during Starvation-Induced Autophagy. Developmental cell. 2017;42:9–21. e25. doi: 10.1016/j.devcel.2017.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.O’Rourke EJ, Ruvkun G. MXL-3 and HLH-30 transcriptionally link lipolysis and autophagy to nutrient availability. Nature cell biology. 2013;15:668–676. doi: 10.1038/ncb2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Owen JL, Zhang Y, Bae SH, Farooqi MS, Liang G, Hammer RE, … Brown MS. Insulin stimulation of SREBP-1c processing in transgenic rat hepatocytes requires p70 S6-kinase. Proc Natl Acad Sci U S A. 2012;109:16184–16189. doi: 10.1073/pnas.1213343109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Panchaud N, Peli-Gulli MP, De Virgilio C. Amino acid deprivation inhibits TORC1 through a GTPase-activating protein complex for the Rag family GTPase Gtr1. Science signaling. 2013;6:ra42. doi: 10.1126/scisignal.2004112. [DOI] [PubMed] [Google Scholar]
- 89.Peli-Gulli MP, Sardu A, Panchaud N, Raucci S, De Virgilio C. Amino Acids Stimulate TORC1 through Lst4-Lst7, a GTPase-Activating Protein Complex for the Rag Family GTPase Gtr2. Cell reports. 2015;13:1–7. doi: 10.1016/j.celrep.2015.08.059. [DOI] [PubMed] [Google Scholar]
- 90.Peng M, Yin N, Li MO. SZT2 dictates GATOR control of mTORC1 signalling. Nature. 2017;543:433–437. doi: 10.1038/nature21378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Perera RM, Stoykova S, Nicolay BN, Ross KN, Fitamant J, Boukhali M, … Bardeesy N. Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature. 2015 doi: 10.1038/nature14587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Perera RM, Zoncu R. The Lysosome as a Regulatory Hub. Annual review of cell and developmental biology. 2016;32:223–253. doi: 10.1146/annurev-cellbio-111315-125125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Peterson TR, Sengupta SS, Harris TE, Carmack AE, Kang SA, Balderas E, … Sabatini DM. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell. 2011;146:408–420. doi: 10.1016/j.cell.2011.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Petit CS, Roczniak-Ferguson A, Ferguson SM. Recruitment of folliculin to lysosomes supports the amino acid-dependent activation of Rag GTPases. J Cell Biol. 2013;202:1107–1122. doi: 10.1083/jcb.201307084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Phillips MJ, Voeltz GK. Structure and function of ER membrane contact sites with other organelles. Nat Rev Mol Cell Biol. 2016;17:69–82. doi: 10.1038/nrm.2015.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Platt FM, Boland B, van der Spoel AC. The cell biology of disease: lysosomal storage disorders: the cellular impact of lysosomal dysfunction. J Cell Biol. 2012;199:723–734. doi: 10.1083/jcb.201208152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Plotegher N, Duchen MR. Mitochondrial Dysfunction and Neurodegeneration in Lysosomal Storage Disorders. Trends Mol Med. 2017;23:116–134. doi: 10.1016/j.molmed.2016.12.003. [DOI] [PubMed] [Google Scholar]
- 98.Polak P, Cybulski N, Feige JN, Auwerx J, Ruegg MA, Hall MN. Adipose-specific knockout of raptor results in lean mice with enhanced mitochondrial respiration. Cell Metab. 2008;8:399–410. doi: 10.1016/j.cmet.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 99.Porstmann T, Santos CR, Griffiths B, Cully M, Wu M, Leevers S, … Schulze A. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 2008;8:224–236. doi: 10.1016/j.cmet.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Powis K, Zhang T, Panchaud N, Wang R, De Virgilio C, Ding J. Crystal structure of the Ego1-Ego2-Ego3 complex and its role in promoting Rag GTPase-dependent TORC1 signaling. Cell Res. 2015;25:1043–1059. doi: 10.1038/cr.2015.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Radhakrishnan A, Goldstein JL, McDonald JG, Brown MS. Switch-like control of SREBP-2 transport triggered by small changes in ER cholesterol: a delicate balance. Cell Metab. 2008;8:512–521. doi: 10.1016/j.cmet.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Radhakrishnan A, Sun LP, Kwon HJ, Brown MS, Goldstein JL. Direct binding of cholesterol to the purified membrane region of SCAP: mechanism for a sterol-sensing domain. Mol Cell. 2004;15:259–268. doi: 10.1016/j.molcel.2004.06.019. [DOI] [PubMed] [Google Scholar]
- 103.Rambold AS, Cohen S, Lippincott-Schwartz J. Fatty acid trafficking in starved cells: regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Developmental cell. 2015;32:678–692. doi: 10.1016/j.devcel.2015.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Rebsamen M, Pochini L, Stasyk T, de Araujo ME, Galluccio M, Kandasamy RK, … Superti-Furga G. SLC38A9 is a component of the lysosomal amino acid sensing machinery that controls mTORC1. Nature. 2015;519:477–481. doi: 10.1038/nature14107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Reinisch KM, De Camilli P. SMP-domain proteins at membrane contact sites: Structure and function. Biochim Biophys Acta. 2016;1861:924–927. doi: 10.1016/j.bbalip.2015.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ricoult SJ, Manning BD. The multifaceted role of mTORC1 in the control of lipid metabolism. EMBO Rep. 2013;14:242–251. doi: 10.1038/embor.2013.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ridgway ND, Dawson PA, Ho YK, Brown MS, Goldstein JL. Translocation of oxysterol binding protein to Golgi apparatus triggered by ligand binding. J Cell Biol. 1992;116:307–319. doi: 10.1083/jcb.116.2.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Rocha N, Kuijl C, van der Kant R, Janssen L, Houben D, Janssen H, … Neefjes J. Cholesterol sensor ORP1L contacts the ER protein VAP to control Rab7-RILP-p150 Glued and late endosome positioning. J Cell Biol. 2009;185:1209–1225. doi: 10.1083/jcb.200811005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Roczniak-Ferguson A, Petit CS, Froehlich F, Qian S, Ky J, Angarola B, … Ferguson SM. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Science signaling. 2012;5:ra42. doi: 10.1126/scisignal.2002790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Saftig P, Klumperman J. Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat Rev Mol Cell Biol. 2009;10:623–635. doi: 10.1038/nrm2745. [DOI] [PubMed] [Google Scholar]
- 111.Sakai J, Rawson RB, Espenshade PJ, Cheng D, Seegmiller AC, Goldstein JL, Brown MS. Molecular identification of the sterol-regulated luminal protease that cleaves SREBPs and controls lipid composition of animal cells. Mol Cell. 1998;2:505–514. doi: 10.1016/s1097-2765(00)80150-1. [DOI] [PubMed] [Google Scholar]
- 112.Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. 2010;141:290–303. doi: 10.1016/j.cell.2010.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, Sabatini DM. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science. 2008;320:1496–1501. doi: 10.1126/science.1157535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, … Ballabio A. A gene network regulating lysosomal biogenesis and function. Science. 2009;325:473–477. doi: 10.1126/science.1174447. [DOI] [PubMed] [Google Scholar]
- 115.Sarkar S, Carroll B, Buganim Y, Maetzel D, Ng AH, Cassady JP, … Jaenisch R. Impaired autophagy in the lipid-storage disorder Niemann-Pick type C1 disease. Cell reports. 2013;5:1302–1315. doi: 10.1016/j.celrep.2013.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Saxton RA, Chantranupong L, Knockenhauer KE, Schwartz TU, Sabatini DM. Mechanism of arginine sensing by CASTOR1 upstream of mTORC1. Nature. 2016a;536:229–233. doi: 10.1038/nature19079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Saxton RA, Knockenhauer KE, Wolfson RL, Chantranupong L, Pacold ME, Wang T, … Sabatini DM. Structural basis for leucine sensing by the Sestrin2-mTORC1 pathway. Science. 2016b;351:53–58. doi: 10.1126/science.aad2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Saxton RA, Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell. 2017;169:361–371. doi: 10.1016/j.cell.2017.03.035. [DOI] [PubMed] [Google Scholar]
- 119.Schauder CM, Wu X, Saheki Y, Narayanaswamy P, Torta F, Wenk MR, … Reinisch KM. Structure of a lipid-bound extended synaptotagmin indicates a role in lipid transfer. Nature. 2014;510:552–555. doi: 10.1038/nature13269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Schoneberg J, Lee IH, Iwasa JH, Hurley JH. Reverse-topology membrane scission by the ESCRT proteins. Nat Rev Mol Cell Biol. 2017;18:5–17. doi: 10.1038/nrm.2016.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Schulze RJ, Weller SG, Schroeder B, Krueger EW, Chi S, Casey CA, McNiven MA. Lipid droplet breakdown requires dynamin 2 for vesiculation of autolysosomal tubules in hepatocytes. J Cell Biol. 2013;203:315–326. doi: 10.1083/jcb.201306140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Seo AY, Lau PW, Feliciano D, Sengupta P, Gros MAL, Cinquin B, … Lippincott-Schwartz J. AMPK and vacuole-associated Atg14p orchestrate mu-lipophagy for energy production and long-term survival under glucose starvation. Elife. 2017:6. doi: 10.7554/eLife.21690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Settembre C, Ballabio A. Lysosome: regulator of lipid degradation pathways. Trends in cell biology. 2014;24:743–750. doi: 10.1016/j.tcb.2014.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Settembre C, De Cegli R, Mansueto G, Saha PK, Vetrini F, Visvikis O, … Ballabio A. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nature cell biology. 2013a;15:647–658. doi: 10.1038/ncb2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, … Ballabio A. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332:1429–1433. doi: 10.1126/science.1204592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Settembre C, Fraldi A, Jahreiss L, Spampanato C, Venturi C, Medina D, … Ballabio A. A block of autophagy in lysosomal storage disorders. Hum Mol Genet. 2008;17:119–129. doi: 10.1093/hmg/ddm289. [DOI] [PubMed] [Google Scholar]
- 127.Settembre C, Fraldi A, Medina DL, Ballabio A. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol. 2013b;14:283–296. doi: 10.1038/nrm3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, … Ballabio A. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. Embo J. 2012;31:1095–1108. doi: 10.1038/emboj.2012.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Sever N, Song BL, Yabe D, Goldstein JL, Brown MS, DeBose-Boyd RA. Insig-dependent ubiquitination and degradation of mammalian 3-hydroxy-3-methylglutaryl-CoA reductase stimulated by sterols and geranylgeraniol. J Biol Chem. 2003;278:52479–52490. doi: 10.1074/jbc.M310053200. [DOI] [PubMed] [Google Scholar]
- 130.Singh R, Cuervo AM. Autophagy in the cellular energetic balance. Cell Metab. 2011;13:495–504. doi: 10.1016/j.cmet.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, … Czaja MJ. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–1135. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Smulan LJ, Ding W, Freinkman E, Gujja S, Edwards YJ, Walker AK. Cholesterol-Independent SREBP-1 Maturation Is Linked to ARF1 Inactivation. Cell reports. 2016;16:9–18. doi: 10.1016/j.celrep.2016.05.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Sudhof TC, Russell DW, Brown MS, Goldstein JL. 42 bp element from LDL receptor gene confers end-product repression by sterols when inserted into viral TK promoter. Cell. 1987;48:1061–1069. doi: 10.1016/0092-8674(87)90713-6. [DOI] [PubMed] [Google Scholar]
- 134.Sun Y, Fang Y, Yoon MS, Zhang C, Roccio M, Zwartkruis FJ, … Chen J. Phospholipase D1 is an effector of Rheb in the mTOR pathway. Proc Natl Acad Sci U S A. 2008;105:8286–8291. doi: 10.1073/pnas.0712268105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Tee AR, Manning BD, Roux PP, Cantley LC, Blenis J. Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Current biology : CB. 2003;13:1259–1268. doi: 10.1016/s0960-9822(03)00506-2. [DOI] [PubMed] [Google Scholar]
- 136.Thiam AR, Farese RV, Jr, Walther TC. The biophysics and cell biology of lipid droplets. Nat Rev Mol Cell Biol. 2013;14:775–786. doi: 10.1038/nrm3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Thoreen CC, Chantranupong L, Keys HR, Wang T, Gray NS, Sabatini DM. A unifying model for mTORC1-mediated regulation of mRNA translation. Nature. 2012;485:109–113. doi: 10.1038/nature11083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Tong J, Yang H, Yang H, Eom SH, Im YJ. Structure of Osh3 reveals a conserved mode of phosphoinositide binding in oxysterol-binding proteins. Structure. 2013;21:1203–1213. doi: 10.1016/j.str.2013.05.007. [DOI] [PubMed] [Google Scholar]
- 139.Toulmay A, Prinz WA. Direct imaging reveals stable, micrometer-scale lipid domains that segregate proteins in live cells. J Cell Biol. 2013;202:35–44. doi: 10.1083/jcb.201301039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Tsujishita Y, Hurley JH. Structure and lipid transport mechanism of a StAR-related domain. Nat Struct Biol. 2000;7:408–414. doi: 10.1038/75192. [DOI] [PubMed] [Google Scholar]
- 141.Tsun ZY, Bar-Peled L, Chantranupong L, Zoncu R, Wang T, Kim C, … Sabatini DM. The Folliculin Tumor Suppressor Is a GAP for the RagC/D GTPases That Signal Amino Acid Levels to mTORC1. Mol Cell. 2013 doi: 10.1016/j.molcel.2013.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Um SH, Frigerio F, Watanabe M, Picard F, Joaquin M, Sticker M, … Thomas G. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature. 2004;431:200–205. doi: 10.1038/nature02866. [DOI] [PubMed] [Google Scholar]
- 143.van Meer G, Voelker DR, Feigenson GW. Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol. 2008;9:112–124. doi: 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Viale A, Pettazzoni P, Lyssiotis CA, Ying H, Sanchez N, Marchesini M, … Draetta GF. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature. 2014;514:628–632. doi: 10.1038/nature13611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Walker AK, Jacobs RL, Watts JL, Rottiers V, Jiang K, Finnegan DM, … Naar AM. A conserved SREBP-1/phosphatidylcholine feedback circuit regulates lipogenesis in metazoans. Cell. 2011;147:840–852. doi: 10.1016/j.cell.2011.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Wang CY, Stapleton DS, Schueler KL, Rabaglia ME, Oler AT, Keller MP, … Attie AD. Tsc2, a positional candidate gene underlying a quantitative trait locus for hepatic steatosis. J Lipid Res. 2012;53:1493–1501. doi: 10.1194/jlr.M025239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Wang S, Tsun ZY, Wolfson RL, Shen K, Wyant GA, Plovanich ME, … Sabatini DM. Metabolism. Lysosomal amino acid transporter SLC38A9 signals arginine sufficiency to mTORC1. Science. 2015;347:188–194. doi: 10.1126/science.1257132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Wilhelm LP, Wendling C, Vedie B, Kobayashi T, Chenard MP, Tomasetto C, … Alpy F. STARD3 mediates endoplasmic reticulum-to-endosome cholesterol transport at membrane contact sites. Embo J. 2017;36:1412–1433. doi: 10.15252/embj.201695917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Wolfson RL, Chantranupong L, Saxton RA, Shen K, Scaria SM, Cantor JR, Sabatini DM. Sestrin2 is a leucine sensor for the mTORC1 pathway. Science. 2016;351:43–48. doi: 10.1126/science.aab2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Wolfson RL, Chantranupong L, Wyant GA, Gu X, Orozco JM, Shen K, … Sabatini DM. KICSTOR recruits GATOR1 to the lysosome and is necessary for nutrients to regulate mTORC1. Nature. 2017;543:438–442. doi: 10.1038/nature21423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Xu S, Benoff B, Liou HL, Lobel P, Stock AM. Structural basis of sterol binding by NPC2, a lysosomal protein deficient in Niemann-Pick type C2 disease. J Biol Chem. 2007;282:23525–23531. doi: 10.1074/jbc.M703848200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Yabe D, Xia ZP, Adams CM, Rawson RB. Three mutations in sterol-sensing domain of SCAP block interaction with insig and render SREBP cleavage insensitive to sterols. Proc Natl Acad Sci U S A. 2002;99:16672–16677. doi: 10.1073/pnas.262669399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Yang T, Espenshade PJ, Wright ME, Yabe D, Gong Y, Aebersold R, … Brown MS. Crucial step in cholesterol homeostasis: sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell. 2002;110:489–500. doi: 10.1016/s0092-8674(02)00872-3. [DOI] [PubMed] [Google Scholar]
- 154.Yecies JL, Zhang HH, Menon S, Liu S, Yecies D, Lipovsky AI, … Manning BD. Akt stimulates hepatic SREBP1c and lipogenesis through parallel mTORC1-dependent and independent pathways. Cell Metab. 2011;14:21–32. doi: 10.1016/j.cmet.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Yoon MS, Du G, Backer JM, Frohman MA, Chen J. Class III PI-3-kinase activates phospholipase D in an amino acid-sensing mTORC1 pathway. J Cell Biol. 2011;195:435–447. doi: 10.1083/jcb.201107033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Zhang HH, Huang J, Duvel K, Boback B, Wu S, Squillace RM, … Manning BD. Insulin stimulates adipogenesis through the Akt-TSC2-mTORC1 pathway. PLoS One. 2009;4:e6189. doi: 10.1371/journal.pone.0006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Zhao K, Ridgway ND. Oxysterol-Binding Protein-Related Protein 1L Regulates Cholesterol Egress from the Endo-Lysosomal System. Cell reports. 2017;19:1807–1818. doi: 10.1016/j.celrep.2017.05.028. [DOI] [PubMed] [Google Scholar]
- 158.Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science. 2011;334:678–683. doi: 10.1126/science.1207056. [DOI] [PMC free article] [PubMed] [Google Scholar]