

Abstract
In recent years, whole-genome sequencing (WGS) has been perceived as a technology with the potential to revolutionise clinical microbiology. Herein, we reviewed the literature on the use of WGS for the most commonly encountered pathogens in clinical microbiology laboratories: Escherichia coli and other Enterobacteriaceae, Staphylococcus aureus and coagulase-negative staphylococci, streptococci and enterococci, mycobacteria and Chlamydia trachomatis. For each pathogen group, we focused on five different aspects: the genome characteristics, the most common genomic approaches and the clinical uses of WGS for (i) typing and outbreak analysis, (ii) virulence investigation and (iii) in silico antimicrobial susceptibility testing. Of all the clinical usages, the most frequent and straightforward usage was to type bacteria and to trace outbreaks back. A next step toward standardisation was made thanks to the development of several new genome-wide multi-locus sequence typing systems based on WGS data. Although virulence characterisation could help in various particular clinical settings, it was done mainly to describe outbreak strains. An increasing number of studies compared genotypic to phenotypic antibiotic susceptibility testing, with mostly promising results. However, routine implementation will preferentially be done in the workflow of particular pathogens, such as mycobacteria, rather than as a broadly applicable generic tool. Overall, concrete uses of WGS in routine clinical microbiology or infection control laboratories were done, but the next big challenges will be the standardisation and validation of the procedures and bioinformatics pipelines in order to reach clinical standards.
Keywords: Genomics, Clinical microbiology, Next-generation sequencing, Whole-genome sequencing
Introduction
Over the last decade, whole-genome sequencing (WGS) has been identified as one of the most promising techniques in clinical microbiology [1, 2]. Since the first bacterial genomes sequenced in 1995 [3, 4], it has come a long way and genome sequencing is now broadly implemented in research laboratories thanks to the rise of high-throughput sequencing [5]. Although its use in clinical microbiology increases, WGS is differentially implemented depending on the pathogen or the intended uses. Generally, clinical microbiology aims to provide a rapid detection and identification of a microorganism, for bacteria, combined or not with antimicrobial susceptibility testing (AST). Recent improvements of sequencing technologies with higher speed and output-to-cost ratios render WGS applicable for many aspects of clinical microbiology, including infectious disease control and epidemiology of pathogens [6, 7].
Even if WGS can be applied to all microorganisms (viruses, bacteria, parasites or fungi), this review focuses on clinical bacteriology. Very good review articles focusing on sequencing technologies or quality control have been published [5, 8, 9]. Herein, we aim to review the applications of WGS in clinical bacteriology focusing on the recent advances in terms of genomic approaches, applications for typing and outbreak, and in silico virulence-associated genes detection and antimicrobial susceptibility prediction for the most common pathogens encountered in blood cultures in our clinical microbiology laboratory [10], as well as for several intracellular bacteria of particular interest (Table 1). For antimicrobial susceptibility prediction based on genomic data, our review is aligned with the in-depth report of the European Committee on Antimicrobial Susceptibility Testing (EUCAST) by Ellington et al. that reviewed the literature on WGS prediction of phenotypic AST from genotypes [11]. We hope that our review will be useful for the clinical microbiologist wishing to obtain an update on the broad applications of WGS for very common pathogens.
Table 1.
Items investigated in this review for each pathogen
| Items | Pathogens |
|---|---|
| Genome characteristics | Escherichia coli and other Enterobacteriaceae |
| Genomic approach | Staphylococcus aureus and coagulase-negative staphylococci |
| Typing and outbreak | Streptococci and enterococci |
| Virulence | Pseudomonas aeruginosa and Acinetobacter baumannii |
| Antimicrobial susceptibility | Mycobacterium tuberculosis complex and other mycobacteria |
| Chlamydia trachomatis |
Escherichia coli and other Enterobacteriaceae
Genome characteristics
Escherichia coli is one of the most studied organisms in the world. Its genome size ranges from 4.6 Mb to 5.9 Mb for a median GC content of 50.6%, with 4200 to 5500 genes [12]. Overall, Enterobacteriaceae are characterised by a large variable genome with various intra-family horizontal gene transfer (HGT) or recombination, sometimes increased by the host’s medical conditions [13].
Genomic approach
So far, WGS was applied mainly on extracted DNA from cultivated bacterial isolates. However, metagenomic shotgun amplification allowed the identification of foodborne pathogens directly from food samples [14–16]. Interestingly, Loman et al. used metagenomic shotgun amplification to investigate an outbreak of Shiga toxin-producing E. coli, but sensitivity remained low (67%) compared to cultures [17]. Hasman et al. performed WGS directly on clinical urine samples and successfully identified E. coli, and complete congruence with the regular microbiology work-up was observed [18].
Typing and outbreak
Escherichia coli strains have been historically grouped into serotypes, biotypes, pathotypes and sequence types [12]. Serotypes (O and H antigens), pathotypes and sequence types [like multi-locus sequence typing (MLST) based on 7–8 housekeeping genes] can be inferred from WGS data [12, 19–21]. Moreover, WGS allows discrimination up to the single nucleotide polymorphisms (SNPs) level for real-time or retrospective investigation of outbreaks of E. coli [22–25], Salmonella enterica [26–31] or Klebsiella spp. [32–35]. Although variants detection allows the most sensitive discrimination between isolates based on DNA sequences, it is limited by the need for a reference genome or whole-genome alignment [36]. Moreover, they lack standardisation and usually do not allow straightforward comparison between studies [20]. New sequence typing methods, such as ribosomal MLST (rMLST, 53 loci) [37], core-genome MLST (cgMLST, >500 loci) or whole-genome MLST (wgMLST, all loci) have arisen since the era of WGS and allow typing up to the strain or clone levels [20]. The use of wgMLST was recently demonstrated by typing extended-spectrum beta-lactamase-producing Enterobacteriaceae [38]. These recent typing tools are available on EnteroBase (https://enterobase.warwick.ac.uk), an online database gathering metadata and genotypes inferred from genome assemblies for four gamma-proteobacteria (Escherichia/Shigella, Salmonella, Yersinia and Moraxella). Moreover, EnteroBase integrates a tool for Salmonella in silico serotyping developed by Yoshida et al. [39]. For K. pneumoniae, a cgMLST scheme was developed to type hypervirulent and multi-resistant strains [40]. Although there is controversy about differentiating the genus Shigella from Escherichia due to its genome similarities with enteroinvasive E. coli [41], a k-mer analysis coupled to MLST from inferred WGS data seems to be an effective discriminative approach [42].
Virulence
Robins-Browne et al. raised the question of the relevance of pathotypes for intestinal pathogenic E. coli (IPEC) in the era of WGS [12]. Although pathotypes remain the subtyping system that is the most clinically relevant, WGS is able to: (i) predict pathotypes with accuracy (Table 2) and (ii) overcome the limitations of this classification, for instance with the emergence of strains with new pathogenic features, such as the enteroaggregative Shiga toxin-producing E. coli [12, 43]. Contrary to the obligate pathogen IPEC, extraintestinal pathogenic E. coli (ExPEC) are opportunistic pathogens and infections arise from the commensal microbiota. Therefore, an identification based on the presence/absence of virulence-associated genes in ExPEC genomes is not straightforward since host medical predispositions also play a major role in the pathogenesis, despite the description of many virulence-associated genes [44]. For K. pneumoniae, several plasmidic and chromosomal genes have been identified as virulence genes associated with community-acquired pyogenic liver abscesses [45, 46]. WGS can identify hypervirulent clones in a rapid manner, which can be of great use to prevent a clonal spread [40, 45, 47].
Table 2.
Virulence-associated genetic determinants of the main Escherichia coli pathotypes
| Gene/genomic region/plasmid | Functional role | Comments |
|---|---|---|
| LEE PAI | Genomic island containing eae (encoding gene for an adhesin) as well as effectors and structural proteins associated with a type III secretion system | EPEC defining region |
| pINV | Encodes for a type III secretion system and for effectors allowing intracellular survival | EIEC/Shigella defining plasmid |
| est, elt | Heat-stable (ST) and heat-labile (LT) enterotoxins | ETEC defining genes |
| stx1, stx2 | Shiga toxins (verotoxins) 1 and 2 | EHEC defining genes |
| aggR, aatA, aaiC | Transcriptional regulator, transporter protein and secreted protein | Associated with EAEC phenotype |
Adapted from Robins-Browne et al. [12]
LEE PAI, Locus of enterocyte effacement pathogenicity island; EPEC, enteropathogenic E. coli; EIEC, enteroinvasive E. coli; ETEC, enterotoxigenic E. coli; EHEC, enterohaemorrhagic E. coli; EAEC, enteroaggregative E. coli
Antimicrobial susceptibility
Overall, several studies reported more than 95% concordance between genotypic and phenotypic antimicrobial resistances for Enterobacteriaceae, such as E. coli and K. pneumoniae [48–50]. However, in a significant proportion of carbapenem-resistant K. pneumoniae and E. cloacae isolates, no carbapenemase could be detected, showing the presence of other resistance mechanisms [51]. Indeed, particular resistance mechanisms, such as modification in the membrane permeability or up-regulation of efflux pumps, will be harder to predict, and further studies are required to improve accuracy among heterogeneous datasets [11]. Furthermore, important limitations with short-read technologies remain for plasmid assemblies due to the inability of assemblers to deal with repeats [11]. They can be overcome using long-read sequencing to improve their detection [51–54] but the cost remains too high for most clinical laboratories. Finally, the particular case of Salmonella spp. needs to be further assessed due to the limited number of studies [11].
Staphylococcus aureus and coagulase-negative staphylococci
Genome characteristics
Staphylococcus aureus has a genome size that ranges from 2.6 to 3.1 Mb, with a median GC content of 32.8%. Coagulase-negative staphylococci (CoNS) have similar genome features to S. aureus. Mobile genetic elements represent 15–20% of the S. aureus genome, emphasising the important transfer of virulence factors and/or antimicrobial resistances that can happen between strains [55] or even between species [56–58].
Genomic approach
The most common approach for S. aureus is WGS applied on extracted DNA from cultivated bacterial isolates. To our knowledge, no study reported culture-independent genome sequencing. Besides S. aureus, there are a limited number of studies on WGS application for CoNS in a clinical setting.
Typing and outbreak
In terms of discriminatory power, WGS and SNP-based methods overcome all previous methods used for typing, such as pulsed-field gel electrophoresis (PFGE), 7-loci MLST and spa typing [59]. To ensure backward compatibility with traditional genotyping, spa types could be inferred from genome assemblies with 97% [60] and 99.1% [61] accuracy, although spa typing is based on the number and order of repeats, which can theoretically impair reliable genome assemblies from short reads. For SCCmec—a mobile genetic element carrying the methicillin resistance gene in S. aureus [62] that shows a great diversity and a high rate of recombination—typing can also be done using WGS and has the advantage to allow the detection of new types or subtypes, although multiplex polymerase chain reaction (PCR) and DNA microarray remain widely used [63]. During outbreak investigations, many studies could rule in or out a direct transmission of closely related isolates using SNP-based approaches [64–67]. As for Enterobacteriaceae, rMLST, cgMLST, wgMLST or even pan-genome MLST show high discriminatory power and, if used more often, could be of great use for standardisation and inter-study comparisons [20, 68, 69].
Virulence
Staphylococcus aureus is a highly adapted pathogen and a number of its genes are related to virulence. WGS provides the possibility to screen the genomes for specific genes of interest, such as Panton–Valentine leucocidin (PVL) or superantigens encoding genes (Table 3), involved in severe clinical presentations, such as necrotising pneumonia or staphylococcal toxic shock syndrome [73]. Commercial multiplex PCRs or DNA microarrays are available and can already screen for some antibiotic resistance genes or particular virulence factors in a culture-independent manner. Their clinical utility remains controversial, although some authors recommend the adjunction of a clindamycin regimen for PVL+ necrotising pneumonia [73]. Thus, in the context of patient care, the use of WGS for virulence investigation remains limited if not done in a shorter time-to-result. Most of the CoNS virulence-associated genes known are genes related to biofilm or adherence to surface [74]. However, the pro-inflammatory and cytolytic phenol-soluble modulin (PSM) combined with the methicillin resistance island could play a critical role in CoNS sepsis pathogenesis [71].
Table 3.
Main Staphylococcus aureus toxins encoded on the accessory genome
| Gene/genomic region/plasmid | Functional role | Comments |
|---|---|---|
| PVL locus (lukF-PV, lukS-PV) | Pore-forming toxin targeting polymorphonuclear leucocytes | Phage-encoded toxin associated with necrotising pneumonia or severe skin and soft tissue formation |
| lukD/E, lukG/H | Pore-forming toxins targeting polymorphonuclear leucocytes | Located on a pathogenicity island (LukDE), they act synergistically with PVL |
| psm-mec locus | Cytolytic capacity, biofilm formation, methicillin resistance, cell spreading and expression of other virulence factors [70] | This locus may also be found in CoNS and could play a major role in CoNS sepsis [71] |
| eta, etb, etd | Exfoliative toxin A, B and D | Toxins involved in the pathogenesis of bullous impetigo and staphylococcal scaled-skin syndrome |
| se(a-e), se(g-j), se(r-t), sel(k-q), sel(u-w), tsst-1 | Staphylococcal enterotoxins and enterotoxin-like toxins | Superantigens associated with S. aureus food poisoning and toxic shock syndrome |
Adapted from Grumann et al. [72]
Antimicrobial susceptibility
Several studies report a high efficiency for in silico antimicrobial susceptibility testing [64, 75–78]. Mykrobe predictor, an online tool allowing a rapid discrimination between S. aureus and other staphylococci, predicts antimicrobial susceptibility with high sensitivity (99.1%) and specificity (99.6%) [79]. Moreover, the predictions are made from raw sequences and can be achieved in less than 3 min, thanks to a de Bruijn-based method. However, limitations for the antimicrobial susceptibility prediction remain (i) because of gaps in the knowledge and the important number of mechanisms of resistance existing for particular antibiotics such as aminoglycosides or glycopeptides [80, 81], as well as (ii) due to genetic instability with the loss of some mobile genetic elements such as erm(C) or the SCCmec cassette while passaging the isolate [11]. On the other hand, for mupirocin, mismatches between genotypic predictions and AST could be explained by laboratory variations. Indeed, those predicted resistant genotypes concerned isolates with a diameter of inhibition of 29 mm, whereas epidemiological cut-off (ECOFF) for the wild type is more than 30 mm for mupirocin. Therefore, it implies that the mupirocin zone diameter ECOFF needs to be revised [11]. For CoNS, studies comparing genotypic to phenotypic correlation remain limited.
Streptococci and enterococci
Genome characteristics
The median lengths are 1.8 Mb and 2.1 Mb for Streptococcus pyogenes and S. pneumoniae, respectively. Enterococci of medical importance, such as Enterococcus faecalis and E. faecium, have larger genomes, ranging from 2.6 to 3.4 Mb. The GC content for these two genera varies from 35% to 40%. Overall, streptococci and enterococci display high genome plasticity. HGT and homologous recombination can drive serotype modifications, as well as the spread of virulence factors and antibiotic resistance genes [82–84].
Genomic approach
Regular WGS from bacterial culture is the standard. To our knowledge, no study reports a culture-independent WGS approach for streptococci detection. Hasman et al. could successfully identify E. faecalis by WGS directly from urine samples [18]. In addition, the E. faecalis complete genome sequence could be obtained directly by a metagenomic approach from stool samples by Morowitz et al. [85].
Typing and outbreak
Molecular typing of S. pyogenes is classically done with the M-protein encoding gene (emm), as well as with the 7-loci MLST [86, 87]. However, for outbreak investigation, studies have shown the added value of WGS thanks to its high discriminatory power compared to other typing techniques [88–91]. Streptococcus pneumoniae serotypes are wildly used and important for epidemiological studies and vaccine development [92]. Interestingly, MLST is highly congruent with strain serotypes [93] and can be easily inferred from WGS data. Serotype prediction from WGS reads is possible thanks to PneumoCaT, a recently developed automated pipeline [94]. It holds the advantage of recognising particular cases of mixed serotypes or in the presence of new subtypes, possibly masked by regular methods. For enterococci, 7-loci MLST and SNP-based approaches are often used for epidemiological studies or outbreak investigations [95–101]. A cgMLST scheme for E. faecium was recently published by de Been et al. and reaches the same resolution as SNP-based approaches, which could facilitate standardisation and comparisons between laboratories [102].
Virulence
Genomes of streptococci hold many genes related to virulence (Table 4) [103, 105]. However, in addition to the presence or absence of virulence-related genes, mutations in regulators, such as two-component systems, are often involved in increased virulence. Due to the complexity of the paths regulating virulence in streptococci, WGS data could benefit from being combined with RNA sequencing and in vivo study for outbreak investigations [89]. However, we hypothesise that having pipelines and databases of virulence-associated genes and mutations in regulators of virulence would be useful for public health surveillance or to prevent further complications of particular clinical presentations, for example by adding clindamycin to patients at risk of developing toxic shock syndrome for S. pyogenes based on the strain genotype.
Table 4.
Main Streptococcus pyogenes virulence factors
| Gene/genomic region/plasmid | Functional role | Comments |
|---|---|---|
| hasA, hasB, hasC | Hyaluronic acid capsule synthesis | Prevention of phagocytosis |
| emm | Antiphagocytic protein (M protein) | Sequence used for typing S. pyogenes isolates |
| spyCEP | Interleukin-8 protease | Inhibition of PMN leucocytes diapedesis |
| sda1 | Streptodornase D (extracellular DNase) | Degradation of PMN DNA nets |
| sagA, sagB, sagC, slo | Streptolysin S and O | Lysis of red blood cells, epithelial cells, macrophages and PMN |
| speA, speC, speH, speI, speJ, speL/M, ssa, SMEZ | Superantigens | Involved in the pathogenesis of toxic shock syndrome or scarlet fever |
| speB | Cysteine protease | Tissue invasion and dissemination |
| fbaA, sclA | Adhesins |
Adapted from Cole et al. [103] and Reglinski and Sriskandan [104]
Many low-/non-virulent isolates hold virulence factors in their genome but their expression is under strong down-regulation. Mutations in two-component systems, such as covR/S or other regulators, have been associated with a dramatic up-regulation of most of those virulence-associated genes
PMN, Polymorphonuclear.
Antimicrobial susceptibility
Many studies focus on antimicrobial resistance and rely to some extent on genomic data [11, 106, 107]. For instance, Howden et al. used WGS to investigate the transmission in hospitalised patients of vancomycin-resistant E. faecium (VREfm), which is, in fact, mainly driven by de novo generation and not only by nosocomial transmission as previously thought [108]. To extend the example of VRE, gene clusters involved in vancomycin resistance in enterococci such as vanA and vanB can be routinely screened using multiplex PCRs with a good correlation with phenotypic AST [109, 110]. By extension, WGS could be used to screen and detect all known van gene clusters. However, to our knowledge, no large studies compared WGS-based genotypic AST to phenotypic AST for streptococci or enterococci, despite the increasing knowledge on the genomic basis of antimicrobial resistances and the rise of multidrug-resistant streptococci and enterococci.
Pseudomonas aeruginosa and Acinetobacter baumannii
Genome characteristics
The P. aeruginosa genome size ranges from 6.1 to 7.5 Mb, with a median GC content of 66.2%. For A. baumannii, its genome size is shorter and varies from 3.7 to 4.3 Mb, with a median GC content of 39%. HGT and genome-wide homologous recombination plays a major role in these two successful and often multidrug-resistant opportunistic pathogens [111–114]. Plasmid-mediated antibiotic resistances play a major role in the transmission of antimicrobial resistances between isolates and species, which may be hard to assess based only on short reads sequencing, as discussed already for Enterobacteriaceae.
Genomic approach
Most studies that investigated outbreaks used a regular culture-based approach for WGS. Nevertheless, culture-independent shotgun WGS was performed to investigate the composition of the microbiota of sputa sampled from patients with cystic fibrosis, without broad-range 16S rRNA PCR to avoid bias [115].
Typing and outbreak
Recent studies showed the added value of WGS for outbreak investigation retrospectively or prospectively compared to other typing techniques for P. aeruginosa [116–121] and A. baumannii [122–126]. Thrane et al. made public a web tool (https://cge.cbs.dtu.dk/services/PAst-1.0/) for in silico determination of the P. aeruginosa serotype, which can be useful to detect or characterise outbreak clones [120]. A real-time WGS investigation of an outbreak in a neonatal intensive care unit was performed and could be used to trace back the index patient and the source of the outbreak [127]. Although it has not been used for P. aeruginosa so far, cgMLST was recently carried out for typing A. baumannii and successfully differentiated a clonal spread among other isolates [128].
Virulence
WGS allowed indubitably a better understanding of acute or chronic P. aeruginosa and A. baumannii infections, and helps the development of new therapeutic approaches [129, 130]. However, besides its use for research or outbreak strain characterisation, a clinical application for the detection of virulence determinants to individualise treatments is currently too preliminary.
Antimicrobial susceptibility
A large study comparing phenotypic and genotypic AST for P. aeruginosa reports 91% sensitivity and 94% specificity for both meropenem- and levofloxacin-resistant phenotypes prediction [131]. However, for amikacin, only 60% of non-susceptible isolates based on AST were congruent with the genomic findings. In contrast, Wright et al. observed high concordance with AST for predicted aminoglycoside and carbapenem susceptibility using 75 isolates of A. baumannii [132]. ARG-ANNOT (Antibiotic Resistance Gene-ANNOTation), a downloadable tool for the detection of antimicrobial resistances, was validated using 174 isolates of A. baumannii with 100% sensitivity and 100% specificity for the genes analysed, even when querying partial sequences [133]. Although good sensitivity/specificity may be reached based on the presence or absence of genes or point mutations in antibiotic target genes, major challenges remain in the prediction of chromosomal alterations, resulting in the modification of expression of genes, such as efflux pumps or intrinsic beta-lactamases [11]. More studies starting from strain collections remain to be done to compare phenotypic and genotypic methods for AST.
Mycobacterium tuberculosis complex and other mycobacteria
Genome characteristics
Mycobacterium tuberculosis complex (MTBC) has a clonal, monomorphic genome of approximately 4.3 to 4.4 Mb. HGT or recombination do not occur in MTBC, whereas it is an important driving force for evolution in other mycobacteria (M. canetti or non-tuberculosis mycobacteria, NTM) [134]. Thus, antimicrobial resistances can only occur from SNPs or insertion–deletion events in MTBC.
Genomic approach
Although many genomic studies have been performed on classical mycobacterial culture, very concrete implementations were attempted in high-income countries [135, 136]. By performing WGS on positive MGIT, a complete report including species identification, in silico AST and calculation of genetic distance to detect outbreaks could be sent a median of 21 days faster than the final reference laboratory report [135]. Moreover, costs were 7% cheaper than the regular workflow for mycobacteria. Public Health England reports to be close to a broad implementation of WGS for the routine diagnosis of mycobacterial infections [137]. Finally, culture-independent WGS was performed directly on sputa. One study performed a proof of concept [138] and the other reported a high-quality sequencing for 20 out of 24 samples and highly concordant genotypic–phenotypic AST [139]. The time-to-AST was 14 days shorter than with other WGS workflows using MGIT. In addition, two sequenced samples did not grow in regular culture, emphasising the added value of WGS performed directly on clinical samples [139].
Typing and outbreak
Recent studies showed a higher resolution of WGS compared to other molecular typing techniques [140–143], such as restriction fragment length polymorphism (RFLP) [144], spoligotyping [145] or variable-number tandem repeats of mycobacterial interspersed repetitive units (MIRU-VNTR) [146]. Although, spoligotypes and MIRU-VNTR types can be determined from WGS, it is not a straightforward approach due to the repeats in the regions of interest, thus rendering assemblies difficult to make from short reads [147]. For public health, WGS was used to trace back outbreaks with high resolution, giving the possibility to identify clonal transmission between patients [148–150]. However, as discussed before, SNP-based approaches lack standardisation and inter-laboratory reproducibility. To tackle this issue, a cgMLST scheme was recently designed for MTBC [151].
Virulence
Lessons from M. tuberculosis genomics allowed the identification of a large number of virulence genes, such as catalases, superoxide dismutase, as well as effectors of the type VII secretion system (ESAT-6, CFP10, recently renamed EsxA and EsxB) [152, 153]. However, the relevance to search for specific virulence genes is limited since MTBC populations are mainly clonal and assessment of virulence based on lineages holds more promise. There are seven lineages of MTBC of human health relevance [147]. Lineages 2 (particularly the modern Beijing sublineage) and 4 are the most widespread and are more virulent than lineages 1 and 6, with more severe clinical presentations, more transmissibility and less immunogenicity [154–157]. Given their restrictive geographic distribution, lineages 3, 5 and 7 are also likely to be less virulent [156]. Thus, knowing lineage informs on virulence and is of public health interest. In addition, automatic web tools can type and assign lineage to a strain from WGS raw data very quickly [158, 159].
Antimicrobial susceptibility
Recent large studies compared AST with the detection of variants associated to antimicrobial resistances [160–162]. Moreover, several web-based automated tools, taking raw reads as input, are available [79, 158, 159, 163, 164]. Although sensitivity and specificity were high with the dataset used in these studies, the EUCAST study group identified several limitations [11]. (a) Low sensitivity for hetero-resistance is reported for molecular techniques [165] and coverage needs to be increased to overcome that, which, currently, would increase the cost and, thus, may not be suitable for a clinical microbiology laboratory setting. Moreover, most of the current pipelines are not designed to detect insertion–deletion events [166]. (b) Systematic errors may arise from poorly defined cut-offs for phenotypic AST that are used as standard for the validation of in silico AST. (c) Finally, genetic basis for antimicrobial resistance is not completely understood, particularly for non-essential genes involved in antimicrobial resistance, which means that WGS can mainly rule in rather than rule out antimicrobial resistance [11]. However, it is clear that WGS can improve the mycobacterial AST workflow and patient care by reducing dramatically the time to an effective antimicrobial regiment, despite it being unlikely that laboratories will dispense completely with phenotypic AST in the near future [11].
Chlamydia trachomatis
Genome characteristics
Chlamydia trachomatis has a small genome size, as a consequence of the adaptation to its intracellular habitat [167], of 1.0 Mb to 1.1 Mb, with a median GC content of 41.2%. Although there are evidences for HGT and especially for homologous recombination, these mechanisms seem to play a smaller role than point mutations for driving the evolution of C. trachomatis [168].
Genomic approach
Culture-dependant approaches are time- and resource-consuming, due to the intracellular lifestyle of C. trachomatis. To tackle this issue, several studies successfully performed WGS directly on clinical samples by using various techniques: (i) immunomagnetic separation for targeted bacterial enrichment with multiple displacement amplification, (ii) capture RNA bait set, (iii) whole-genome amplification before WGS and (iv) multiplexed microdroplet PCR enrichment technology [169–172]. A limitation for the clinical use of the first technique could be the lysis buffer, which is present in some commercial devices, and may prevent the binding of antibodies to the major outer membrane protein (MOMP) [173].
Typing and outbreak
Chlamydia trachomatis was historically classified by MOMP-based serology. Serovars are clinically important because they determine the tissue tropism of the infection (serovars A–C, ocular; D–K, urogenital and ocular; L1–L3, lymph nodes) [168]. In recent years, PCR of the ompA, the gene encoding for the MOMP, was developed for typing but exhibited very low epidemiological resolution [174]. The multi-locus variable-number tandem repeat (VNTR) analysis (MLVA) system and various MLST schemes as well as the multi-locus typing DNA array were developed, which provide more reliable topologies [175–177]. WGS was shown to have a higher resolution than regular phylogenies based on MLST [178].
Virulence
Numerous genes and variants were associated with specific tissue tropism or pathogenic effect [168]. However, besides a straightforward use of WGS to build robust core-genome phylogenies and to infer serovar from ompA to predict tropism, there is currently not enough knowledge on specific virulence factors that could have a clinical value.
Antimicrobial susceptibility
Although treatment failures have been reported, they are not likely due to antimicrobial resistance, which will hopefully remain rare [179]. Thus, there is currently a limited need for in silico antimicrobial resistance predictions for C. trachomatis.
Discussion
For all the major pathogens investigated during this review, we can observe an increasing number of publically available genomes (Fig. 1). Along with this trend, our review shows the development of various WGS-based approaches, as well as attempts of their implementation in a clinical microbiology routine. Knowledge on the genomics of the pathogens is a prerequisite before any clinical use and important features need to be kept in mind for each microorganism. Although horizontal gene transfer or recombination events are very frequent in most pathogens, they do not occur in M. tuberculosis. This is critical because HGT and recombination have a large impact on the transmission of virulence factors, antimicrobial resistance genes and on serovar modifications. Concerning the genomic approaches, WGS is regularly performed on cultivated isolates, but an increasing number of studies report culture-independent WGS, which could speed up the clinical laboratory workflow, particularly to decrease the time to genotypic AST. A straightforward and broadly recognised use of WGS is for the investigation of outbreaks and is nowadays broadly implemented in clinical microbiology and infection control laboratories. Although SNP-based methods have shown great successes, new typing approaches such as rMLST or cgMLST schemes, which offer standardisation and comparability between laboratories, are available for an increasing number of organisms. Moreover, they were shown to be highly reproducible and accurate [180]. Mellmann et al. used cgMLST to monitor prospectively the transmission of methicillin-resistant S. aureus, VRE, multidrug-resistant E. coli and multidrug-resistant P. aeruginosa. This approach was efficient and cost-effective in the setting of a majority of multi-bed rooms and because of the possibility to reduce a systematic isolation recommended by German guidelines [181]. Diseases pathogenesis is extremely diverse and complex. For most pathogens, there is no straightforward approach to predict an isolate’s virulence based on its genotype. Indeed, host factors as well as modification of the expression of virulence-associated genes add another layer of complexity. However, WGS can provide a map of the virulome, which can sometimes be determining for a patient’s care, for instance, by precisely determining the E. coli pathotype. The EUCAST subcommittee reports that there is currently not enough evidence to support clinical decision-making based on genotypic AST [11]. However, for mycobacteria, WGS implementation for diagnosis, in silico AST and outbreak investigation was shown to be successful and cost-effective, with a rapid turnaround time, saving weeks or even months of cultures [135].
Fig. 1.
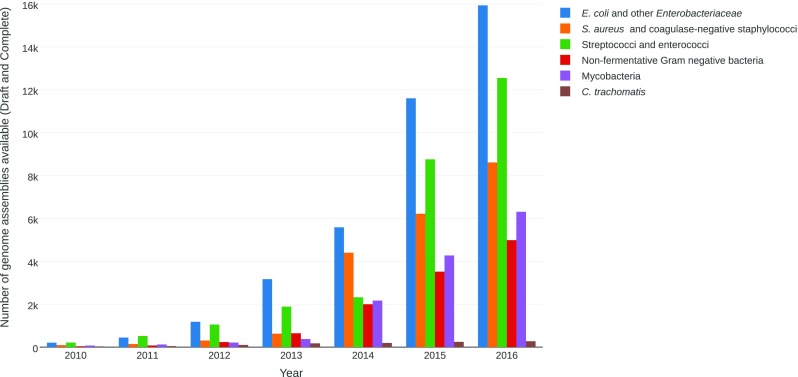
Number of genome assemblies available in the National Center for Biotechnology Information (NCBI) database per year
Finally, for an implementation in clinical microbiology, WGS-based methods will need standardised and validated (i) procedures, (ii) quality control and (iii) subsequent bioinformatics pipelines. Moreover, they will need to be in line with the clinical requirements for data protection.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
References
- 1.Didelot X, Bowden R, Wilson DJ, et al. Transforming clinical microbiology with bacterial genome sequencing. Nat Rev Genet. 2012;13:601–612. doi: 10.1038/nrg3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bertelli C, Greub G. Rapid bacterial genome sequencing: methods and applications in clinical microbiology. Clin Microbiol Infect. 2013;19:803–813. doi: 10.1111/1469-0691.12217. [DOI] [PubMed] [Google Scholar]
- 3.Fraser CM, Gocayne JD, White O, et al. The minimal gene complement of Mycoplasma genitalium. Science. 1995;270:397–404. doi: 10.1126/science.270.5235.397. [DOI] [PubMed] [Google Scholar]
- 4.Fleischmann RD, Adams MD, White O, et al. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science. 1995;269:496–512. doi: 10.1126/science.7542800. [DOI] [PubMed] [Google Scholar]
- 5.Metzker ML. Sequencing technologies—the next generation. Nat Rev Genet. 2010;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 6.Loman NJ, Constantinidou C, Chan JZM, et al. High-throughput bacterial genome sequencing: an embarrassment of choice, a world of opportunity. Nat Rev Microbiol. 2012;10:599–606. doi: 10.1038/nrmicro2850. [DOI] [PubMed] [Google Scholar]
- 7.Tang P, Croxen MA, Hasan MR, et al. Infection control in the new age of genomic epidemiology. Am J Infect Control. 2017;45:170–179. doi: 10.1016/j.ajic.2016.05.015. [DOI] [PubMed] [Google Scholar]
- 8.Wain J, Mavrogiorgou E. Next-generation sequencing in clinical microbiology. Expert Rev Mol Diagn. 2013;13:225–227. doi: 10.1586/erm.13.8. [DOI] [PubMed] [Google Scholar]
- 9.Gargis AS, Kalman L, Lubin IM. Assuring the quality of next-generation sequencing in clinical microbiology and public health laboratories. J Clin Microbiol. 2016;54:2857–2865. doi: 10.1128/JCM.00949-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Opota O, Croxatto A, Prod’hom G, et al. Blood culture-based diagnosis of bacteraemia: state of the art. Clin Microbiol Infect. 2015;21:313–322. doi: 10.1016/j.cmi.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 11.Ellington MJ, Ekelund O, Aarestrup FM, et al. The role of whole genome sequencing in antimicrobial susceptibility testing of bacteria: report from the EUCAST subcommittee. Clin Microbiol Infect. 2017;23:2–22. doi: 10.1016/j.cmi.2016.11.012. [DOI] [PubMed] [Google Scholar]
- 12.Robins-Browne RM, Holt KE, Ingle DJ, et al. Are Escherichia coli pathotypes still relevant in the era of whole-genome sequencing? Front Cell Infect Microbiol. 2016;6:141. doi: 10.3389/fcimb.2016.00141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stecher B, Denzler R, Maier L, et al. Gut inflammation can boost horizontal gene transfer between pathogenic and commensal Enterobacteriaceae. Proc Natl Acad Sci U S A. 2012;109:1269–1274. doi: 10.1073/pnas.1113246109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leonard SR, Mammel MK, Lacher DW, et al. Application of metagenomic sequencing to food safety: detection of Shiga toxin-producing Escherichia coli on fresh bagged spinach. Appl Environ Microbiol. 2015;81:8183–8191. doi: 10.1128/AEM.02601-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blagden T, Schneider W, Melcher U, et al. Adaptation and validation of E-probe diagnostic nucleic acid analysis for detection of Escherichia coli O157:H7 in metagenomic data from complex food matrices. J Food Prot. 2016;79:574–581. doi: 10.4315/0362-028X.JFP-15-440. [DOI] [PubMed] [Google Scholar]
- 16.Leonard SR, Mammel MK, Lacher DW, et al. Strain-level discrimination of Shiga toxin-producing Escherichia coli in spinach using metagenomic sequencing. PLoS One. 2016;11:e0167870. doi: 10.1371/journal.pone.0167870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Loman NJ, Constantinidou C, Christner M, et al. A culture-independent sequence-based metagenomics approach to the investigation of an outbreak of Shiga-toxigenic Escherichia coli O104:H4. JAMA. 2013;309:1502–1510. doi: 10.1001/jama.2013.3231. [DOI] [PubMed] [Google Scholar]
- 18.Hasman H, Saputra D, Sicheritz-Ponten T, et al. Rapid whole-genome sequencing for detection and characterization of microorganisms directly from clinical samples. J Clin Microbiol. 2014;52:139–146. doi: 10.1128/JCM.02452-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ingle DJ, Valcanis M, Kuzevski A, et al. In silico serotyping of E. coli from short read data identifies limited novel O-loci but extensive diversity of O:H serotype combinations within and between pathogenic lineages. Microb Genom. 2016;2:e000064. doi: 10.1099/mgen.0.000064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maiden MCJ, Jansen van Rensburg MJ, Bray JE, et al. MLST revisited: the gene-by-gene approach to bacterial genomics. Nat Rev Microbiol. 2013;11:728–736. doi: 10.1038/nrmicro3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clermont O, Gordon D, Denamur E. Guide to the various phylogenetic classification schemes for Escherichia coli and the correspondence among schemes. Microbiology. 2015;161:980–988. doi: 10.1099/mic.0.000063. [DOI] [PubMed] [Google Scholar]
- 22.Grad YH, Lipsitch M, Feldgarden M, et al. Genomic epidemiology of the Escherichia coli O104:H4 outbreaks in Europe, 2011. Proc Natl Acad Sci U S A. 2012;109:3065–3070. doi: 10.1073/pnas.1121491109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Joensen KG, Scheutz F, Lund O, et al. Real-time whole-genome sequencing for routine typing, surveillance, and outbreak detection of verotoxigenic Escherichia coli. J Clin Microbiol. 2014;52:1501–1510. doi: 10.1128/JCM.03617-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rusconi B, Sanjar F, Koenig SSK, et al. Whole genome sequencing for genomics-guided investigations of Escherichia coli O157:H7 outbreaks. Front Microbiol. 2016;7:985. doi: 10.3389/fmicb.2016.00985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mellmann A, Harmsen D, Cummings CA, et al. Prospective genomic characterization of the German enterohemorrhagic Escherichia coli O104:H4 outbreak by rapid next generation sequencing technology. PLoS One. 2011;6:e22751. doi: 10.1371/journal.pone.0022751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Quick J, Ashton P, Calus S, et al. Rapid draft sequencing and real-time nanopore sequencing in a hospital outbreak of Salmonella. Genome Biol. 2015;16:114. doi: 10.1186/s13059-015-0677-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Byrne L, Fisher I, Peters T, et al. A multi-country outbreak of Salmonella Newport gastroenteritis in Europe associated with watermelon from Brazil, confirmed by whole genome sequencing: October 2011 to January 2012. Euro Surveill. 2014;19:6–13. doi: 10.2807/1560-7917.ES2014.19.31.20866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.den Bakker HC, Allard MW, Bopp D, et al. Rapid whole-genome sequencing for surveillance of Salmonella enterica serovar enteritidis. Emerg Infect Dis. 2014;20:1306–1314. doi: 10.3201/eid2008.131399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Inns T, Lane C, Peters T et al (2015) A multi-country Salmonella enteritidis phage type 14b outbreak associated with eggs from a German producer: ‘near real-time’ application of whole genome sequencing and food chain investigations, United Kingdom, May to September 2014. Euro Surveill 20(16). pii: 21098 [DOI] [PubMed]
- 30.Octavia S, Wang Q, Tanaka MM, et al. Delineating community outbreaks of Salmonella enterica serovar Typhimurium by use of whole-genome sequencing: insights into genomic variability within an outbreak. J Clin Microbiol. 2015;53:1063–1071. doi: 10.1128/JCM.03235-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Phillips A, Sotomayor C, Wang Q, et al. Whole genome sequencing of Salmonella typhimurium illuminates distinct outbreaks caused by an endemic multi-locus variable number tandem repeat analysis type in Australia, 2014. BMC Microbiol. 2016;16:211. doi: 10.1186/s12866-016-0831-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Snitkin ES, Zelazny AM, Thomas PJ, et al. Tracking a hospital outbreak of carbapenem-resistant Klebsiella pneumoniae with whole-genome sequencing. Sci Transl Med. 2012;4:148ra116. doi: 10.1126/scitranslmed.3004129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kanamori H, Parobek CM, Juliano JJ et al (2017) A prolonged outbreak of KPC-3-producing Enterobacter cloacae and Klebsiella pneumoniae driven by multiple mechanisms of resistance transmission at a large academic burn center. Antimicrob Agents Chemother 61(2). pii: e01516-16. doi:10.1128/AAC.01516-16 [DOI] [PMC free article] [PubMed]
- 34.Zhou K, Lokate M, Deurenberg RH et al (2016) Use of whole-genome sequencing to trace, control and characterize the regional expansion of extended-spectrum β-lactamase producing ST15 Klebsiella pneumoniae. Sci Rep 6:20840. doi:10.1038/srep20840 [DOI] [PMC free article] [PubMed]
- 35.Jiang Y, Wei Z, Wang Y, et al. Tracking a hospital outbreak of KPC-producing ST11 Klebsiella pneumoniae with whole genome sequencing. Clin Microbiol Infect. 2015;21:1001–1007. doi: 10.1016/j.cmi.2015.07.001. [DOI] [PubMed] [Google Scholar]
- 36.Croucher NJ, Harris SR, Grad YH, et al. Bacterial genomes in epidemiology—present and future. Philos Trans R Soc Lond B Biol Sci. 2013;368:20120202. doi: 10.1098/rstb.2012.0202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jolley KA, Bliss CM, Bennett JS, et al. Ribosomal multilocus sequence typing: universal characterization of bacteria from domain to strain. Microbiology. 2012;158:1005–1015. doi: 10.1099/mic.0.055459-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kluytmans-van den Bergh MFQ, Rossen JWA, Bruijning-Verhagen PCJ, et al. Whole-genome multilocus sequence typing of extended-spectrum-beta-lactamase-producing Enterobacteriaceae. J Clin Microbiol. 2016;54:2919–2927. doi: 10.1128/JCM.01648-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yoshida CE, Kruczkiewicz P, Laing CR, et al. The Salmonella in silico typing resource (SISTR): an open web-accessible tool for rapidly typing and subtyping draft Salmonella genome assemblies. PLoS One. 2016;11:e0147101. doi: 10.1371/journal.pone.0147101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bialek-Davenet S, Criscuolo A, Ailloud F, et al. Genomic definition of hypervirulent and multidrug-resistant Klebsiella pneumoniae clonal groups. Emerg Infect Dis. 2014;20:1812–1820. doi: 10.3201/eid2011.140206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pettengill EA, Pettengill JB, Binet R. Phylogenetic analyses of Shigella and enteroinvasive Escherichia coli for the identification of molecular epidemiological markers: whole-genome comparative analysis does not support distinct genera designation. Front Microbiol. 2016;6:1573. doi: 10.3389/fmicb.2015.01573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chattaway MA, Schaefer U, Tewolde R, et al. Identification of Escherichia coli and Shigella species from whole-genome sequences. J Clin Microbiol. 2017;55:616–623. doi: 10.1128/JCM.01790-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Clements A, Young JC, Constantinou N, et al. Infection strategies of enteric pathogenic Escherichia coli. Gut Microbes. 2012;3:71–87. doi: 10.4161/gmic.19182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Köhler C-D, Dobrindt U. What defines extraintestinal pathogenic Escherichia coli? Int J Med Microbiol. 2011;301:642–647. doi: 10.1016/j.ijmm.2011.09.006. [DOI] [PubMed] [Google Scholar]
- 45.Struve C, Roe CC, Stegger M, et al. Mapping the evolution of hypervirulent Klebsiella pneumoniae. MBio. 2015;6:e00630. doi: 10.1128/mBio.00630-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ye M, Tu J, Jiang J, et al. Clinical and genomic analysis of liver abscess-causing Klebsiella pneumoniae identifies new liver abscess-associated virulence genes. Front Cell Infect Microbiol. 2016;6:165. doi: 10.3389/fcimb.2016.00165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Holt KE, Wertheim H, Zadoks RN, et al. Genomic analysis of diversity, population structure, virulence, and antimicrobial resistance in Klebsiella pneumoniae, an urgent threat to public health. Proc Natl Acad Sci U S A. 2015;112:E3574–E3581. doi: 10.1073/pnas.1501049112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stoesser N, Batty EM, Eyre DW, et al. Predicting antimicrobial susceptibilities for Escherichia coli and Klebsiella pneumoniae isolates using whole genomic sequence data. J Antimicrob Chemother. 2013;68:2234–2244. doi: 10.1093/jac/dkt180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zankari E, Hasman H, Kaas RS, et al. Genotyping using whole-genome sequencing is a realistic alternative to surveillance based on phenotypic antimicrobial susceptibility testing. J Antimicrob Chemother. 2013;68:771–777. doi: 10.1093/jac/dks496. [DOI] [PubMed] [Google Scholar]
- 50.Tyson GH, McDermott PF, Li C, et al. WGS accurately predicts antimicrobial resistance in Escherichia coli. J Antimicrob Chemother. 2015;70:2763–2769. doi: 10.1093/jac/dkv186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pecora ND, Li N, Allard M, et al. Genomically informed surveillance for carbapenem-resistant Enterobacteriaceae in a health care system. MBio. 2015;6:e01030-15. doi: 10.1128/mBio.01030-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hudson CM, Bent ZW, Meagher RJ, et al. Resistance determinants and mobile genetic elements of an NDM-1-encoding Klebsiella pneumoniae strain. PLoS One. 2014;9:e99209. doi: 10.1371/journal.pone.0099209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Doi Y, Hazen TH, Boitano M, et al. Whole-genome assembly of Klebsiella pneumoniae coproducing NDM-1 and OXA-232 carbapenemases using single-molecule, real-time sequencing. Antimicrob Agents Chemother. 2014;58:5947–5953. doi: 10.1128/AAC.03180-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Conlan S, Thomas PJ, Deming C, et al. Single-molecule sequencing to track plasmid diversity of hospital-associated carbapenemase-producing Enterobacteriaceae. Sci Transl Med. 2014;6:254ra126. doi: 10.1126/scitranslmed.3009845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lindsay JA. Genomic variation and evolution of Staphylococcus aureus. Int J Med Microbiol. 2010;300:98–103. doi: 10.1016/j.ijmm.2009.08.013. [DOI] [PubMed] [Google Scholar]
- 56.Bloemendaal ALA, Brouwer EC, Fluit AC. Methicillin resistance transfer from Staphylocccus epidermidis to methicillin-susceptible Staphylococcus aureus in a patient during antibiotic therapy. PLoS One. 2010;5:e11841. doi: 10.1371/journal.pone.0011841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhu W, Clark N, Patel JB. pSK41-like plasmid is necessary for Inc18-like vanA plasmid transfer from Enterococcus faecalis to Staphylococcus aureus in vitro. Antimicrob Agents Chemother. 2013;57:212–219. doi: 10.1128/AAC.01587-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhu W, Murray PR, Huskins WC, et al. Dissemination of an Enterococcus Inc18-like vanA plasmid associated with vancomycin-resistant Staphylococcus aureus. Antimicrob Agents Chemother. 2010;54:4314–4320. doi: 10.1128/AAC.00185-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Williamson DA, Heffernan H, Nimmo G. Contemporary genomic approaches in the diagnosis and typing of Staphylococcus aureus. Pathology. 2015;47:270–275. doi: 10.1097/PAT.0000000000000236. [DOI] [PubMed] [Google Scholar]
- 60.Bartels MD, Petersen A, Worning P, et al. Comparing whole-genome sequencing with Sanger sequencing for spa typing of methicillin-resistant Staphylococcus aureus. J Clin Microbiol. 2014;52:4305–4308. doi: 10.1128/JCM.01979-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bletz S, Mellmann A, Rothgänger J, et al. Ensuring backwards compatibility: traditional genotyping efforts in the era of whole genome sequencing. Clin Microbiol Infect. 2015;21:347.e1–347.e4. doi: 10.1016/j.cmi.2014.11.005. [DOI] [PubMed] [Google Scholar]
- 62.International Working Group on the Classification of Staphylococcal Cassette Chromosome Elements (IWG-SCC) Classification of staphylococcal cassette chromosome mec (SCCmec): guidelines for reporting novel SCCmec elements. Antimicrob Agents Chemother. 2009;53:4961–4967. doi: 10.1128/AAC.00579-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu J, Chen D, Peters BM, et al. Staphylococcal chromosomal cassettes mec (SCCmec): a mobile genetic element in methicillin-resistant Staphylococcus aureus. Microb Pathog. 2016;101:56–67. doi: 10.1016/j.micpath.2016.10.028. [DOI] [PubMed] [Google Scholar]
- 64.Köser CU, Holden MTG, Ellington MJ, et al. Rapid whole-genome sequencing for investigation of a neonatal MRSA outbreak. N Engl J Med. 2012;366:2267–2275. doi: 10.1056/NEJMoa1109910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Azarian T, Cook RL, Johnson JA, et al. Whole-genome sequencing for outbreak investigations of methicillin-resistant Staphylococcus aureus in the neonatal intensive care unit: time for routine practice? Infect Control Hosp Epidemiol. 2015;36:777–785. doi: 10.1017/ice.2015.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Layer F, Sanchini A, Strommenger B, et al. Molecular typing of toxic shock syndrome toxin-1- and enterotoxin A-producing methicillin-sensitive Staphylococcus aureus isolates from an outbreak in a neonatal intensive care unit. Int J Med Microbiol. 2015;305:790–798. doi: 10.1016/j.ijmm.2015.08.033. [DOI] [PubMed] [Google Scholar]
- 67.Jaton L, Pillonel T, Jaton K, et al. Common skin infection due to Panton–Valentine leucocidin-producing Staphylococcus aureus strains in asylum seekers from Eritrea: a genome-based investigation of a suspected outbreak. Clin Microbiol Infect. 2016;22:739.e5–739.e8. doi: 10.1016/j.cmi.2016.05.026. [DOI] [PubMed] [Google Scholar]
- 68.Leopold SR, Goering RV, Witten A, et al. Bacterial whole-genome sequencing revisited: portable, scalable, and standardized analysis for typing and detection of virulence and antibiotic resistance genes. J Clin Microbiol. 2014;52:2365–2370. doi: 10.1128/JCM.00262-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Roisin S, Gaudin C, De Mendonça R, et al. Pan-genome multilocus sequence typing and outbreak-specific reference-based single nucleotide polymorphism analysis to resolve two concurrent Staphylococcus aureus outbreaks in neonatal services. Clin Microbiol Infect. 2016;22:520–526. doi: 10.1016/j.cmi.2016.01.024. [DOI] [PubMed] [Google Scholar]
- 70.Qin L, McCausland JW, Cheung GYC, et al. PSM-mec—a virulence determinant that connects transcriptional regulation, virulence, and antibiotic resistance in staphylococci. Front Microbiol. 2016;7:1293. doi: 10.3389/fmicb.2016.01293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Qin L, Da F, Fisher EL, et al. Toxin mediates sepsis caused by methicillin-resistant Staphylococcus epidermidis. PLoS Pathog. 2017;13:e1006153. doi: 10.1371/journal.ppat.1006153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Grumann D, Nübel U, Bröker BM. Staphylococcus aureus toxins—their functions and genetics. Infect Genet Evol. 2014;21:583–592. doi: 10.1016/j.meegid.2013.03.013. [DOI] [PubMed] [Google Scholar]
- 73.Tong SYC, Davis JS, Eichenberger E, et al. Staphylococcus aureus infections: epidemiology, pathophysiology, clinical manifestations, and management. Clin Microbiol Rev. 2015;28:603–661. doi: 10.1128/CMR.00134-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Becker K, Heilmann C, Peters G. Coagulase-negative staphylococci. Clin Microbiol Rev. 2014;27:870–926. doi: 10.1128/CMR.00109-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Eyre DW, Golubchik T, Gordon NC, et al. A pilot study of rapid benchtop sequencing of Staphylococcus aureus and Clostridium difficile for outbreak detection and surveillance. BMJ Open. 2012;2:e001124. doi: 10.1136/bmjopen-2012-001124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Holden MTG, Hsu L-Y, Kurt K, et al. A genomic portrait of the emergence, evolution, and global spread of a methicillin-resistant Staphylococcus aureus pandemic. Genome Res. 2013;23:653–664. doi: 10.1101/gr.147710.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gordon NC, Price JR, Cole K, et al. Prediction of Staphylococcus aureus antimicrobial resistance by whole-genome sequencing. J Clin Microbiol. 2014;52:1182–1191. doi: 10.1128/JCM.03117-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lee GC, Long SW, Musser JM, et al. Comparative whole genome sequencing of community-associated methicillin-resistant Staphylococcus aureus sequence type 8 from primary care clinics in a Texas community. Pharmacotherapy. 2015;35:220–228. doi: 10.1002/phar.1536. [DOI] [PubMed] [Google Scholar]
- 79.Bradley P, Gordon NC, Walker TM, et al. Rapid antibiotic-resistance predictions from genome sequence data for Staphylococcus aureus and Mycobacterium tuberculosis. Nat Commun. 2015;6:10063. doi: 10.1038/ncomms10063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chen C-J, Huang Y-C, Chiu C-H. Multiple pathways of cross-resistance to glycopeptides and daptomycin in persistent MRSA bacteraemia. J Antimicrob Chemother. 2015;70:2965–2972. doi: 10.1093/jac/dkv225. [DOI] [PubMed] [Google Scholar]
- 81.McEvoy CRE, Tsuji B, Gao W, et al. Decreased vancomycin susceptibility in Staphylococcus aureus caused by IS256 tempering of WalKR expression. Antimicrob Agents Chemother. 2013;57:3240–3249. doi: 10.1128/AAC.00279-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Andam CP, Hanage WP. Mechanisms of genome evolution of Streptococcus. Infect Genet Evol. 2015;33:334–342. doi: 10.1016/j.meegid.2014.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hegstad K, Mikalsen T, Coque TM, et al. Mobile genetic elements and their contribution to the emergence of antimicrobial resistant Enterococcus faecalis and Enterococcus faecium. Clin Microbiol Infect. 2010;16:541–554. doi: 10.1111/j.1469-0691.2010.03226.x. [DOI] [PubMed] [Google Scholar]
- 84.Croucher NJ, Harris SR, Fraser C, et al. Rapid pneumococcal evolution in response to clinical interventions. Science. 2011;331:430–434. doi: 10.1126/science.1198545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Morowitz MJ, Denef VJ, Costello EK, et al. Strain-resolved community genomic analysis of gut microbial colonization in a premature infant. Proc Natl Acad Sci U S A. 2011;108:1128–1133. doi: 10.1073/pnas.1010992108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sanderson-Smith M, De Oliveira DM, Guglielmini J, et al. A systematic and functional classification of Streptococcus pyogenes that serves as a new tool for molecular typing and vaccine development. J Infect Dis. 2014;210:1325–1338. doi: 10.1093/infdis/jiu260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bessen DE, McShan WM, Nguyen SV, et al. Molecular epidemiology and genomics of group A Streptococcus. Infect Genet Evol. 2015;33:393–418. doi: 10.1016/j.meegid.2014.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ben Zakour NL, Venturini C, Beatson SA, et al. Analysis of a Streptococcus pyogenes puerperal sepsis cluster by use of whole-genome sequencing. J Clin Microbiol. 2012;50:2224–2228. doi: 10.1128/JCM.00675-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Olsen RJ, Fittipaldi N, Kachroo P, et al. Clinical laboratory response to a mock outbreak of invasive bacterial infections: a preparedness study. J Clin Microbiol. 2014;52:4210–4216. doi: 10.1128/JCM.02164-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Engelthaler DM, Valentine M, Bowers J, et al. Hypervirulent emm59 clone in invasive group A Streptococcus outbreak, southwestern United States. Emerg Infect Dis. 2016;22:734–738. doi: 10.3201/eid2204.151582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tagini F, Aubert B, Troillet N et al (2017) Importance of whole genome sequencing for the assessment of outbreaks in diagnostic laboratories: analysis of a case series of invasive Streptococcus pyogenes infections. Eur J Clin Microbiol Infect Dis. doi:10.1007/s10096-017-2905-z [DOI] [PMC free article] [PubMed]
- 92.Jauneikaite E, Tocheva AS, Jefferies JMC, et al. Current methods for capsular typing of Streptococcus pneumoniae. J Microbiol Methods. 2015;113:41–49. doi: 10.1016/j.mimet.2015.03.006. [DOI] [PubMed] [Google Scholar]
- 93.Harrison OB, Brueggemann AB, Caugant DA, et al. Molecular typing methods for outbreak detection and surveillance of invasive disease caused by Neisseria meningitidis, Haemophilus influenzae and Streptococcus pneumoniae, a review. Microbiology. 2011;157:2181–2195. doi: 10.1099/mic.0.050518-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kapatai G, Sheppard CL, Al-Shahib A, et al. Whole genome sequencing of Streptococcus pneumoniae: development, evaluation and verification of targets for serogroup and serotype prediction using an automated pipeline. PeerJ. 2016;4:e2477. doi: 10.7717/peerj.2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lister DM, Kotsanas D, Ballard SA, et al. Outbreak of vanB vancomycin-resistant Enterococcus faecium colonization in a neonatal service. Am J Infect Control. 2015;43:1061–1065. doi: 10.1016/j.ajic.2015.05.047. [DOI] [PubMed] [Google Scholar]
- 96.Sivertsen A, Billström H, Melefors Ö, et al. A multicentre hospital outbreak in Sweden caused by introduction of a vanB2 transposon into a stably maintained pRUM-plasmid in an Enterococcus faecium ST192 clone. PLoS One. 2014;9:e103274. doi: 10.1371/journal.pone.0103274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Schlebusch S, Price GR, Gallagher RL, et al. MALDI-TOF MS meets WGS in a VRE outbreak investigation. Eur J Clin Microbiol Infect Dis. 2017;36:495–499. doi: 10.1007/s10096-016-2824-4. [DOI] [PubMed] [Google Scholar]
- 98.Brodrick HJ, Raven KE, Harrison EM, et al. Whole-genome sequencing reveals transmission of vancomycin-resistant Enterococcus faecium in a healthcare network. Genome Med. 2016;8:4. doi: 10.1186/s13073-015-0259-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Raven KE, Reuter S, Reynolds R, et al. A decade of genomic history for healthcare-associated Enterococcus faecium in the United Kingdom and Ireland. Genome Res. 2016;26:1388–1396. doi: 10.1101/gr.204024.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Raven KE, Reuter S, Gouliouris T et al (2016) Genome-based characterization of hospital-adapted Enterococcus faecalis lineages. Nat Microbiol 1(3). pii: 15033. doi:10.1038/nmicrobiol.2015.33 [DOI] [PMC free article] [PubMed]
- 101.Santona A, Taviani E, Deligios M, et al. Vancomycin-resistant Enterococcus faecium high-resolution typing by core genome multilocus sequence typing. J Infect Dev Ctries. 2016;10:1159–1161. doi: 10.3855/jidc.9223. [DOI] [PubMed] [Google Scholar]
- 102.de Been M, Pinholt M, Top J, et al. Core genome multilocus sequence typing scheme for high-resolution typing of Enterococcus faecium. J Clin Microbiol. 2015;53:3788–3797. doi: 10.1128/JCM.01946-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cole JN, Barnett TC, Nizet V, et al. Molecular insight into invasive group A streptococcal disease. Nat Rev Microbiol. 2011;9:724–736. doi: 10.1038/nrmicro2648. [DOI] [PubMed] [Google Scholar]
- 104.Reglinski M, Sriskandan S (2014) The contribution of group A streptococcal virulence determinants to the pathogenesis of sepsis. Virulence 5:127–136. doi:10.4161/viru.26400 [DOI] [PMC free article] [PubMed]
- 105.Mitchell AM, Mitchell TJ (2010) Streptococcus pneumoniae: virulence factors and variation. Clin Microbiol Infect 16:411–418. doi:10.1111/j.1469-0691.2010.03183.x [DOI] [PubMed]
- 106.Cattoir V. Mechanisms of antibiotic resistance: Streptococcus pyogenes. In: Ferretti JJ, Stevens DL, Fischetti VA, editors. Streptococcus pyogenes: basic biology to clinical manifestations. Oklahoma City: University of Oklahoma Health Sciences Center; 2016. pp. 947–991. [Google Scholar]
- 107.Miller WR, Munita JM, Arias CA. Mechanisms of antibiotic resistance in enterococci. Expert Rev Anti Infect Ther. 2014;12:1221–1236. doi: 10.1586/14787210.2014.956092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Howden BP, Holt KE, Lam MMC, et al. Genomic insights to control the emergence of vancomycin-resistant enterococci. MBio. 2013;4:e00412-13. doi: 10.1128/mBio.00412-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Holzknecht BJ, Hansen DS, Nielsen L, et al. Screening for vancomycin-resistant enterococci with Xpert® vanA/vanB: diagnostic accuracy and impact on infection control decision making. New Microbes New Infect. 2017;16:54–59. doi: 10.1016/j.nmni.2016.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Huh HJ, Jang M-A, Seo JY, et al. Evaluation of the iNtRON VRE vanA/vanB real-time PCR assay for detection of vancomycin-resistant enterococci. Ann Lab Med. 2015;35:76–81. doi: 10.3343/alm.2015.35.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Klockgether J, Cramer N, Wiehlmann L, et al. Pseudomonas aeruginosa genomic structure and diversity. Front Microbiol. 2011;2:150. doi: 10.3389/fmicb.2011.00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Dettman JR, Rodrigue N, Kassen R. Genome-wide patterns of recombination in the opportunistic human pathogen Pseudomonas aeruginosa. Genome Biol Evol. 2014;7:18–34. doi: 10.1093/gbe/evu260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Antunes LCS, Visca P, Towner KJ. Acinetobacter baumannii: evolution of a global pathogen. Pathog Dis. 2014;71:292–301. doi: 10.1111/2049-632X.12125. [DOI] [PubMed] [Google Scholar]
- 114.Touchon M, Cury J, Yoon E-J, et al. The genomic diversification of the whole Acinetobacter genus: origins, mechanisms, and consequences. Genome Biol Evol. 2014;6:2866–2882. doi: 10.1093/gbe/evu225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hauser PM, Bernard T, Greub G, et al. Microbiota present in cystic fibrosis lungs as revealed by whole genome sequencing. PLoS One. 2014;9:e90934. doi: 10.1371/journal.pone.0090934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Snyder LA, Loman NJ, Faraj LA, et al. Epidemiological investigation of Pseudomonas aeruginosa isolates from a six-year-long hospital outbreak using high-throughput whole genome sequencing. Euro Surveill. 2013;18:20611. doi: 10.2807/1560-7917.ES2013.18.42.20611. [DOI] [PubMed] [Google Scholar]
- 117.Witney AA, Gould KA, Pope CF, et al. Genome sequencing and characterization of an extensively drug-resistant sequence type 111 serotype O12 hospital outbreak strain of Pseudomonas aeruginosa. Clin Microbiol Infect. 2014;20:O609–O618. doi: 10.1111/1469-0691.12528. [DOI] [PubMed] [Google Scholar]
- 118.Turton JF, Wright L, Underwood A, et al. High-resolution analysis by whole-genome sequencing of an international lineage (sequence type 111) of Pseudomonas aeruginosa associated with metallo-carbapenemases in the United Kingdom. J Clin Microbiol. 2015;53:2622–2631. doi: 10.1128/JCM.00505-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Willmann M, Bezdan D, Zapata L, et al. Analysis of a long-term outbreak of XDR Pseudomonas aeruginosa: a molecular epidemiological study. J Antimicrob Chemother. 2015;70:1322–1330. doi: 10.1093/jac/dku546. [DOI] [PubMed] [Google Scholar]
- 120.Thrane SW, Taylor VL, Lund O, et al. Application of whole-genome sequencing data for O-specific antigen analysis and in silico serotyping of Pseudomonas aeruginosa isolates. J Clin Microbiol. 2016;54:1782–1788. doi: 10.1128/JCM.00349-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Blanc DS, Gomes Magalhaes B, Abdelbary M, et al. Hand soap contamination by Pseudomonas aeruginosa in a tertiary care hospital: no evidence of impact on patients. J Hosp Infect. 2016;93:63–67. doi: 10.1016/j.jhin.2016.02.010. [DOI] [PubMed] [Google Scholar]
- 122.Lewis T, Loman NJ, Bingle L, et al. High-throughput whole-genome sequencing to dissect the epidemiology of Acinetobacter baumannii isolates from a hospital outbreak. J Hosp Infect. 2010;75:37–41. doi: 10.1016/j.jhin.2010.01.012. [DOI] [PubMed] [Google Scholar]
- 123.Halachev MR, Chan JZ-M, Constantinidou CI, et al. Genomic epidemiology of a protracted hospital outbreak caused by multidrug-resistant Acinetobacter baumannii in Birmingham, England. Genome Med. 2014;6:70. doi: 10.1186/s13073-014-0070-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Salipante SJ, SenGupta DJ, Cummings LA, et al. Application of whole-genome sequencing for bacterial strain typing in molecular epidemiology. J Clin Microbiol. 2015;53:1072–1079. doi: 10.1128/JCM.03385-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hammerum AM, Hansen F, Skov MN, et al. Investigation of a possible outbreak of carbapenem-resistant Acinetobacter baumannii in Odense, Denmark using PFGE, MLST and whole-genome-based SNPs. J Antimicrob Chemother. 2015;70:1965–1968. doi: 10.1093/jac/dkv072. [DOI] [PubMed] [Google Scholar]
- 126.Fitzpatrick MA, Ozer EA, Hauser AR. Utility of whole-genome sequencing in characterizing Acinetobacter epidemiology and analyzing hospital outbreaks. J Clin Microbiol. 2016;54:593–612. doi: 10.1128/JCM.01818-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Davis RJ, Jensen SO, Van Hal S, et al. Whole genome sequencing in real-time investigation and management of a Pseudomonas aeruginosa outbreak on a neonatal intensive care unit. Infect Control Hosp Epidemiol. 2015;36:1058–1064. doi: 10.1017/ice.2015.133. [DOI] [PubMed] [Google Scholar]
- 128.Willems S, Kampmeier S, Bletz S, et al. Whole-genome sequencing elucidates epidemiology of nosocomial clusters of Acinetobacter baumannii. J Clin Microbiol. 2016;54:2391–2394. doi: 10.1128/JCM.00721-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Fothergill JL, Winstanley C, James CE. Novel therapeutic strategies to counter Pseudomonas aeruginosa infections. Expert Rev Anti Infect Ther. 2012;10:219–235. doi: 10.1586/eri.11.168. [DOI] [PubMed] [Google Scholar]
- 130.Wang N, Ozer EA, Mandel MJ, et al. Genome-wide identification of Acinetobacter baumannii genes necessary for persistence in the lung. MBio. 2014;5:e01163-14. doi: 10.1128/mBio.01163-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kos VN, Déraspe M, McLaughlin RE, et al. The resistome of Pseudomonas aeruginosa in relationship to phenotypic susceptibility. Antimicrob Agents Chemother. 2015;59:427–436. doi: 10.1128/AAC.03954-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wright MS, Stockwell TB, Beck E, et al. SISPA-Seq for rapid whole genome surveys of bacterial isolates. Infect Genet Evol. 2015;32:191–198. doi: 10.1016/j.meegid.2015.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Gupta SK, Padmanabhan BR, Diene SM, et al. ARG-ANNOT, a new bioinformatic tool to discover antibiotic resistance genes in bacterial genomes. Antimicrob Agents Chemother. 2014;58:212–220. doi: 10.1128/AAC.01310-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Boritsch EC, Khanna V, Pawlik A, et al. Key experimental evidence of chromosomal DNA transfer among selected tuberculosis-causing mycobacteria. Proc Natl Acad Sci U S A. 2016;113:9876–9881. doi: 10.1073/pnas.1604921113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Pankhurst LJ, Del Ojo Elias C, Votintseva AA, et al. Rapid, comprehensive, and affordable mycobacterial diagnosis with whole-genome sequencing: a prospective study. Lancet Respir Med. 2016;4:49–58. doi: 10.1016/S2213-2600(15)00466-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Cirillo DM, Cabibbe AM, De Filippo MR, et al. Use of WGS in Mycobacterium tuberculosis routine diagnosis. Int J Mycobacteriol. 2016;5(Suppl 1):S252–S253. doi: 10.1016/j.ijmyco.2016.09.053. [DOI] [PubMed] [Google Scholar]
- 137.Public Health England (2016) Tuberculosis in England: 2016 report. Available online at: https://www.gov.uk/government/uploads/system/uploads/attachment_data/file/581238/TB_Annual_Report_2016_GTW2309_errata_v1.2.pdf. Accessed 21 Feb 2017
- 138.Doughty EL, Sergeant MJ, Adetifa I, et al. Culture-independent detection and characterisation of Mycobacterium tuberculosis and M. africanum in sputum samples using shotgun metagenomics on a benchtop sequencer. PeerJ. 2014;2:e585. doi: 10.7717/peerj.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Brown AC, Bryant JM, Einer-Jensen K, et al. Rapid whole-genome sequencing of Mycobacterium tuberculosis isolates directly from clinical samples. J Clin Microbiol. 2015;53:2230–2237. doi: 10.1128/JCM.00486-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Jamieson FB, Teatero S, Guthrie JL, et al. Whole-genome sequencing of the Mycobacterium tuberculosis Manila sublineage results in less clustering and better resolution than mycobacterial interspersed repetitive-unit–variable-number tandem-repeat (MIRU-VNTR) typing and spoligotyping. J Clin Microbiol. 2014;52:3795–3798. doi: 10.1128/JCM.01726-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Luo T, Yang C, Peng Y, et al. Whole-genome sequencing to detect recent transmission of Mycobacterium tuberculosis in settings with a high burden of tuberculosis. Tuberculosis (Edinb) 2014;94:434–440. doi: 10.1016/j.tube.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Mehaffy C, Guthrie JL, Alexander DC, et al. Marked microevolution of a unique Mycobacterium tuberculosis strain in 17 years of ongoing transmission in a high risk population. PLoS One. 2014;9:e112928. doi: 10.1371/journal.pone.0112928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Roetzer A, Diel R, Kohl TA, et al. Whole genome sequencing versus traditional genotyping for investigation of a Mycobacterium tuberculosis outbreak: a longitudinal molecular epidemiological study. PLoS Med. 2013;10:e1001387. doi: 10.1371/journal.pmed.1001387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Otal I, Martín C, Vincent-Lévy-Frebault V, et al. Restriction fragment length polymorphism analysis using IS6110 as an epidemiological marker in tuberculosis. J Clin Microbiol. 1991;29:1252–1254. doi: 10.1128/jcm.29.6.1252-1254.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kamerbeek J, Schouls L, Kolk A, et al. Simultaneous detection and strain differentiation of Mycobacterium tuberculosis for diagnosis and epidemiology. J Clin Microbiol. 1997;35:907–914. doi: 10.1128/jcm.35.4.907-914.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Supply P, Lesjean S, Savine E, et al. Automated high-throughput genotyping for study of global epidemiology of Mycobacterium tuberculosis based on mycobacterial interspersed repetitive units. J Clin Microbiol. 2001;39:3563–3571. doi: 10.1128/JCM.39.10.3563-3571.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Takiff HE, Feo O. Clinical value of whole-genome sequencing of Mycobacterium tuberculosis. Lancet Infect Dis. 2015;15:1077–1090. doi: 10.1016/S1473-3099(15)00071-7. [DOI] [PubMed] [Google Scholar]
- 148.Gardy JL, Johnston JC, Ho Sui SJ, et al. Whole-genome sequencing and social-network analysis of a tuberculosis outbreak. N Engl J Med. 2011;364:730–739. doi: 10.1056/NEJMoa1003176. [DOI] [PubMed] [Google Scholar]
- 149.Walker TM, Ip CL, Harrell RH, et al. Whole-genome sequencing to delineate Mycobacterium tuberculosis outbreaks: a retrospective observational study. Lancet Infect Dis. 2013;13:137–146. doi: 10.1016/S1473-3099(12)70277-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Stucki D, Ballif M, Bodmer T, et al. Tracking a tuberculosis outbreak over 21 years: strain-specific single-nucleotide polymorphism typing combined with targeted whole-genome sequencing. J Infect Dis. 2015;211:1306–1316. doi: 10.1093/infdis/jiu601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kohl TA, Diel R, Harmsen D, et al. Whole-genome-based Mycobacterium tuberculosis surveillance: a standardized, portable, and expandable approach. J Clin Microbiol. 2014;52:2479–2486. doi: 10.1128/JCM.00567-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Cole ST, Brosch R, Parkhill J, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 153.Forrellad MA, Klepp LI, Gioffré A, et al. Virulence factors of the Mycobacterium tuberculosis complex. Virulence. 2013;4:3–66. doi: 10.4161/viru.22329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Hanekom M, van der Spuy GD, Streicher E, et al. A recently evolved sublineage of the Mycobacterium tuberculosis Beijing strain family is associated with an increased ability to spread and cause disease. J Clin Microbiol. 2007;45:1483–1490. doi: 10.1128/JCM.02191-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Ribeiro SCM, Gomes LL, Amaral EP, et al. Mycobacterium tuberculosis strains of the modern sublineage of the Beijing family are more likely to display increased virulence than strains of the ancient sublineage. J Clin Microbiol. 2014;52:2615–2624. doi: 10.1128/JCM.00498-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Coscolla M, Gagneux S. Consequences of genomic diversity in Mycobacterium tuberculosis. Semin Immunol. 2014;26:431–444. doi: 10.1016/j.smim.2014.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Liu Q, Luo T, Dong X, et al. Genetic features of Mycobacterium tuberculosis modern Beijing sublineage. Emerg Microbes Infect. 2016;5:e14. doi: 10.1038/emi.2016.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Feuerriegel S, Schleusener V, Beckert P, et al. PhyResSE: a web tool delineating Mycobacterium tuberculosis antibiotic resistance and lineage from whole-genome sequencing data. J Clin Microbiol. 2015;53:1908–1914. doi: 10.1128/JCM.00025-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Iwai H, Kato-Miyazawa M, Kirikae T, et al. CASTB (the comprehensive analysis server for the Mycobacterium tuberculosis complex): a publicly accessible web server for epidemiological analyses, drug-resistance prediction and phylogenetic comparison of clinical isolates. Tuberculosis (Edinb) 2015;95:843–844. doi: 10.1016/j.tube.2015.09.002. [DOI] [PubMed] [Google Scholar]
- 160.Zhang H, Li D, Zhao L, et al. Genome sequencing of 161 Mycobacterium tuberculosis isolates from China identifies genes and intergenic regions associated with drug resistance. Nat Genet. 2013;45:1255–1260. doi: 10.1038/ng.2735. [DOI] [PubMed] [Google Scholar]
- 161.Köser CU, Bryant JM, Becq J, et al. Whole-genome sequencing for rapid susceptibility testing of M. tuberculosis. N Engl J Med. 2013;369:290–292. doi: 10.1056/NEJMc1215305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Walker TM, Kohl TA, Omar SV, et al. Whole-genome sequencing for prediction of Mycobacterium tuberculosis drug susceptibility and resistance: a retrospective cohort study. Lancet Infect Dis. 2015;15:1193–1202. doi: 10.1016/S1473-3099(15)00062-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Coll F, McNerney R, Preston MD, et al. Rapid determination of anti-tuberculosis drug resistance from whole-genome sequences. Genome Med. 2015;7:51. doi: 10.1186/s13073-015-0164-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Steiner A, Stucki D, Coscolla M, et al. KvarQ: targeted and direct variant calling from fastq reads of bacterial genomes. BMC Genomics. 2014;15:881. doi: 10.1186/1471-2164-15-881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Folkvardsen DB, Thomsen VØ, Rigouts L, et al. Rifampin heteroresistance in Mycobacterium tuberculosis cultures as detected by phenotypic and genotypic drug susceptibility test methods. J Clin Microbiol. 2013;51:4220–4222. doi: 10.1128/JCM.01602-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Loman NJ, Pallen MJ. Twenty years of bacterial genome sequencing. Nat Rev Microbiol. 2015;13:787–794. doi: 10.1038/nrmicro3565. [DOI] [PubMed] [Google Scholar]
- 167.Zomorodipour A, Andersson SG. Obligate intracellular parasites: Rickettsia prowazekii and Chlamydia trachomatis. FEBS Lett. 1999;452:11–15. doi: 10.1016/S0014-5793(99)00563-3. [DOI] [PubMed] [Google Scholar]
- 168.Nunes A, Borrego MJ, Gomes JP. Genomic features beyond Chlamydia trachomatis phenotypes: what do we think we know? Infect Genet Evol. 2013;16:392–400. doi: 10.1016/j.meegid.2013.03.018. [DOI] [PubMed] [Google Scholar]
- 169.Seth-Smith HMB, Harris SR, Skilton RJ, et al. Whole-genome sequences of Chlamydia trachomatis directly from clinical samples without culture. Genome Res. 2013;23:855–866. doi: 10.1101/gr.150037.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Christiansen MT, Brown AC, Kundu S, et al. Whole-genome enrichment and sequencing of Chlamydia trachomatis directly from clinical samples. BMC Infect Dis. 2014;14:591. doi: 10.1186/s12879-014-0591-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Andersson P, Klein M, Lilliebridge RA, et al. Sequences of multiple bacterial genomes and a Chlamydia trachomatis genotype from direct sequencing of DNA derived from a vaginal swab diagnostic specimen. Clin Microbiol Infect. 2013;19:E405–E408. doi: 10.1111/1469-0691.12237. [DOI] [PubMed] [Google Scholar]
- 172.Joseph SJ, Li B, Ghonasgi T, et al. Direct amplification, sequencing and profiling of Chlamydia trachomatis strains in single and mixed infection clinical samples. PLoS One. 2014;9:e99290. doi: 10.1371/journal.pone.0099290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.de Vries HJC, Schim van der Loeff MF, Bruisten SM. High-resolution typing of Chlamydia trachomatis: epidemiological and clinical uses. Curr Opin Infect Dis. 2015;28:61–71. doi: 10.1097/QCO.0000000000000129. [DOI] [PubMed] [Google Scholar]
- 174.Pedersen LN, Herrmann B, Møller JK. Typing Chlamydia trachomatis: from egg yolk to nanotechnology. FEMS Immunol Med Microbiol. 2009;55:120–130. doi: 10.1111/j.1574-695X.2008.00526.x. [DOI] [PubMed] [Google Scholar]
- 175.Klint M, Fuxelius H-H, Goldkuhl RR, et al. High-resolution genotyping of Chlamydia trachomatis strains by multilocus sequence analysis. J Clin Microbiol. 2007;45:1410–1414. doi: 10.1128/JCM.02301-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Pedersen LN, Pødenphant L, Møller JK. Highly discriminative genotyping of Chlamydia trachomatis using omp1 and a set of variable number tandem repeats. Clin Microbiol Infect. 2008;14:644–652. doi: 10.1111/j.1469-0691.2008.02011.x. [DOI] [PubMed] [Google Scholar]
- 177.Labiran C, Clarke IN, Cutcliffe LT, et al. Genotyping markers used for multi locus VNTR analysis with ompA (MLVA-ompA) and multi sequence typing (MST) retain stability in Chlamydia trachomatis. Front Cell Infect Microbiol. 2012;2:68. doi: 10.3389/fcimb.2012.00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Harris SR, Clarke IN, Seth-Smith HMB, et al. Whole-genome analysis of diverse Chlamydia trachomatis strains identifies phylogenetic relationships masked by current clinical typing. Nat Genet. 2012;44:413–419. doi: 10.1038/ng.2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Kong FYS, Hocking JS. Treatment challenges for urogenital and anorectal Chlamydia trachomatis. BMC Infect Dis. 2015;15:293. doi: 10.1186/s12879-015-1030-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Mellmann A, Andersen PS, Bletz S, et al. High interlaboratory reproducibility and accuracy of next-generation-sequencing-based bacterial genotyping in a ring trial. J Clin Microbiol. 2017;55:908–913. doi: 10.1128/JCM.02242-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Mellmann A, Bletz S, Böking T, et al. Real-time genome sequencing of resistant bacteria provides precision infection control in an institutional setting. J Clin Microbiol. 2016;54:2874–2881. doi: 10.1128/JCM.00790-16. [DOI] [PMC free article] [PubMed] [Google Scholar]