

Abstract
TP53 mutations have been associated with anaplasia in Wilms tumour, which conveys a high risk for relapse and fatal outcome. Nevertheless, TP53 alterations have been reported in no more than 60% of anaplastic tumours, and recent data have suggested their presence in tumours that do not fulfil the criteria for anaplasia, questioning the clinical utility of TP53 analysis. Therefore, we characterized the TP53 status in 84 fatal cases of Wilms tumour, irrespective of histological subtype. We identified TP53 alterations in at least 90% of fatal cases of anaplastic Wilms tumour, and even more when diffuse anaplasia was present, indicating a very strong if not absolute coupling between anaplasia and deregulation of p53 function. Unfortunately, TP53 mutations do not provide additional predictive value in anaplastic tumours since the same mutation rate was found in a cohort of non‐fatal anaplastic tumours. When classified according to tumour stage, patients with stage I diffuse anaplastic tumours still had a high chance of survival (87%), but this rate dropped to 26% for stages II–IV. Thus, volume of anaplasia or possible spread may turn out to be critical parameters. Importantly, among non‐anaplastic fatal tumours, 26% had TP53 alterations, indicating that TP53 screening may identify additional cases at risk. Several of these non‐anaplastic tumours fulfilled some criteria for anaplasia, for example nuclear unrest, suggesting that such partial phenotypes should be under special scrutiny to enhance detection of high‐risk tumours via TP53 screening. A major drawback is that these alterations are secondary changes that occur only later in tumour development, leading to striking intratumour heterogeneity that requires multiple biopsies and analysis guided by histological criteria. In conclusion, we found a very close correlation between histological signs of anaplasia and TP53 alterations. The latter may precede development of anaplasia and thereby provide diagnostic value pointing towards aggressive disease.
Keywords: Wilms tumour, nephroblastoma, anaplasia, TP53, tumour heterogeneity
Introduction
Wilms tumour (WT) or nephroblastoma is the most common paediatric renal tumour, affecting about 1/10 000 children before the age of 15 years, with a median age of approximately 3.5 years 1. With current treatment strategies, overall survival rate is about 90%, but is strongly dependent on the histological subtype and tumour stage 2. In Europe, patients are treated according to International Society of Paediatric Oncology (SIOP) protocols 3. These usually include preoperative chemotherapy, followed by surgery, adjusted postoperative chemotherapy and in some cases radiotherapy, stratified by tumour stage and histology.
Wilms tumours are histologically diverse, with variable contributions of blastemal, stromal, and epithelial elements. While stromal or epithelial subtype, mixed (triphasic) and regressive tumours are classified as intermediate risk, blastemal subtype, and those containing diffuse anaplastic elements are assigned to the high‐risk group after preoperative chemotherapy. While patients with low and intermediate risk tumours have 88% 5‐year event‐free and 92% overall survival, event‐free survival for patients with anaplastic WT is only 42% and overall survival about 48%. Patients with blastemal predominant tumours show 58% event‐free survival and overall survival of 84%, respectively 2, 4, 5.
Anaplastic Wilms tumours account for 5–8% of all WTs 6, 7, 8 and are equally associated with adverse outcome in both the SIOP as well as the North American COG histological classification system, which is based on upfront surgery. Diffuse anaplasia especially carries a strongly elevated risk for relapse and fatal outcome, while focal anaplasia, on the other hand, is classified as intermediate risk, but it can be speculated whether this represents a quantitative or qualitative mechanistic difference 9.
The anaplastic variant of Wilms tumour is known to be associated with TP53 mutations 10. Nevertheless, this correlation of histology and mutation does not include all anaplastic tumours. Rather, evidence for TP53 mutation has been found in only 48–55% of anaplastic cases, with another 11% having suspicious loss of chromosome 17p 11, 12. On the other hand, we have found TP53 mutations in tumours with sometimes inconclusive or even missing features of anaplasia 13, but this has not been studied systematically in non‐anaplastic WT. Furthermore, it is unclear whether TP53 alterations or histologically defined anaplasia will be the more relevant parameter for risk stratification.
Therefore, we characterized the TP53 status in a larger cohort of fatal Wilms tumour cases irrespective of histological subtype. In addition, anaplastic tumours of surviving patients were analyzed to evaluate the prognostic relevance of TP53 mutations.
Material and methods
Patients and samples
All cases were obtained from the German SIOP93–01/GPOH and SIOP2001/GPOH studies. All subjects (or their parents) provided written consent for tumour banking and future research use according to national regulations including ethical approval (Ethikkommission der Ärztekammer des Saarlandes. Germany; No.: 23.4.93/Ls and No.: 136/01). For fatal cases, only those classified with ‘death of disease’ were included. Surviving patients had at least 2 years of follow up. The histotype was based on reference and/or panel pathology.
Genomic DNA of tumour and corresponding normal kidney tissue was isolated from either formalin‐fixed, paraffin‐embedded (FFPE) or snap frozen material stored at −70 °C using QIAamp and AllPrep kits (QIAGEN, Hilden, Germany), or from white blood cells as described previously 14.
Histological analysis of FFPE material
For FISH analysis and immunohistochemistry, we constructed tissue microarrays containing two 1.5 mm punches of tumour tissue per case. In cases with diffuse or focal anaplasia, one anaplastic region and – if present – a non‐anaplastic tumour region were punched out. For mutation screening, anaplastic and non‐anaplastic tumour regions were macrodissected.
DNA sequencing
Exons 4–10 of the TP53 gene were amplified individually from genomic tumour DNA with primers described in supplementary material, Table S1. For FFPE material, nested PCR with 30 plus 35 cycles was employed to improve yield. Amplified fragments were sequenced using primers indicated in supplementary material, Table S1 by Sanger sequencing (GATC, Konstanz, Germany).
The web‐tools Provean 15 (http://provean.jcvi.org), Sift 16 (http://sift.jcvi.org), and PolyPhen‐2 17 (http://genetics.bwh.harvard.edu/pph2) were used to predict effects of missense mutations. The TP53 reference sequence was taken from the TP53 database (R18).
Copy number variation analysis
Copy number variations (CNVs) of chromosome 17p were analyzed in DNA from frozen tumour material by multiplex ligation‐dependent probe amplification (MLPA) using the P380‐X2 kit (MRC‐Holland, Amsterdam, the Netherlands), and data were analyzed with the Coffalyzer software (MRC‐Holland).
For FFPE material, CNVs were analyzed by FISH using 3 µm thick tissue microarray sections according to standard procedures 18. The SPEC TP53/CEN17 Dual Color Probe kit (ZytoVision, Bremerhaven, Germany) containing a specific probe for TP53 and the reference probe CEN17 was used to score deletions of TP53. Fifty to hundred non‐overlapping nuclei were counted. Deletions were defined as TP53 copy number loss in more than 50% of counted tumour cells. Monosomy was assumed if more than 50% of nuclei showed only one centromere and one TP53 signal 19.
Immunohistochemistry
Immunohistochemistry (IHC) analysis of p53 expression was performed on 2 µm sections mounted on glass slides in a Leica Bond‐Max automated immunostainer (Leica Microsystems Inc., Bannockburn, Ireland). Heat‐induced epitope retrieval was performed with high‐pH buffer (Leica) for 20 min. The p53 specific antibody DO7 (Leica Biosystems, Wetzlar, Germany) was diluted 1:100 and incubated for 30 min, followed by the Leica polymer (15 min). Diaminobenzidine was used as the chromogen with subsequent light haematoxylin counterstain.
Results
TP53 alteration in fatal WT cases
A set of 86 tumour samples from 84 unselected fatal cases (including two bilateral tumours) was collected from the German GPOH/SIOP 93–01 and 2001 studies. In all instances, the patients had died from progressive disease on average 2.2 (0.4–8.5) years after initial diagnosis. The set contained 47 high risk (30 diffuse anaplasia [DA], 17 blastemal) and 39 low/intermediate risk tumours (1 completely necrotic, 1 epithelial, 2 stromal, 15 mixed type, 16 regressive, 2 focal anaplasia [FA], 2 primary operated blastemal predominant). About 70% of patients had higher stage disease (13 stage I, 12 stage II, 23 stage III, 32 stage IV, and 2 stage V; 2 unknown). Clinical data for all cases are listed in Table 1. All diagnoses were confirmed by reference pathologists. Tumour DNA was extracted from paraffin sections (57 cases), frozen material (46 cases) or both (17 cases, with generally concordant results).
Table 1.
WT samples analyzed for TP53 alterations
| ID | Single nucleotide variant (SNV) | Het/hom | Copy number | IHC | Histology † | Stage local | Stage global | Sex | Age (month) | Relapse | Metastasis | Death ‡ |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 267fs*1 | hom | 1 | 0% | R | 1 | 1 | m | 34.0 | y | n | y |
| 2 § | R175H, R342* | het | 2 | 70% | T | 1 | 1 | f | 195.9 | n | y | y |
| 3 § | L194R | hom | 1 | 5% | T, nu | 1 | 1 | m | 206.7 | y | y | y |
| 4 | splice in3 | hom | 2 | 0% | T, nu | 3 | 3 | m | 230.1 | y | y | y |
| 5 | R282W | hom | 1 | 80% | FA | 1 | 5 | f | 69.2 | n | y | y |
| 6 | R175H | het | 1 | 80% | FA | 3 | 3 | m | 35.4 | y | y | y |
| 7 | 241fs*16 | het | 1 | 0% | pr. B | 2 | 2 | f | 127.6 | y | y | y |
| 8 | R273H | het | 2 | 0% | B | 1 | 1 | m | 71.4 | y | y | y |
| 9 | R342P | het | 2 | na | B | 3 | 5 | m | 59.0 | n | y | y |
| 10 | splice in9 | hom | 2 | 5% | B | 2 | 5 | f | 15.0 | y | y | y |
| 11 | G226S | hom | 1 | 3% | B | 2 | 2 | f | 84.6 | n | y | y |
| 12 | G245S | het | 2 | 40% | B | 2 | 2 | m | 109.1 | y | y | y |
| 13 | R248W | hom | 2 | na | B | 3 | 4 | f | 89.0 | y | y | y |
| 14 | R196* | hom | na | 0% | DA | f | y | y | ||||
| 15 | V197M | het | 2 | 70% | DA | 2 | 2 | f | 51.3 | y | y | y |
| 16 | R273H | hom | na | 0% | DA | 3 | 3 | f | 44.0 | y | y | y |
| 17 | R158H, G226D, G245S, R280K | het/hom | na | 80% | DA | 3 | 3 | f | 90.3 | n | y | y |
| 18 | T231I | hom | na | 80% | DA | 1 | 1 | m | 67.7 | y | y | y |
| 19 | 292fs*11 | het | 2 | 80% | DA | 3 | 3 | f | 72.6 | y | y | y |
| 20 | S127F | het | 1 | 80% | DA | 3 | 3 | m | 79.6 | y | y | y |
| 21 | K319E | hom | 1 | 90% | DA | 1 | 1 | m | 21.1 | y | y | y |
| 22 | R175H, R342* | het | na | 25% ‐ 100% | DA | 3 | 4 | f | 73.0 | n | y | y |
| 23 § | 346fs*4 & 336fs*4 ¶ | hom | 1 | 80% | DA | 3 | 5 | f | 58.8 | y | y | y |
| 24 | R273H | het | 2 | 40% | DA | 3 | 3 | f | 127.4 | n | y | y |
| 25 | R175_C176 del | hom | 1 | 70% | DA | 2 | 2 | m | 52.9 | y | n | y |
| 26 | splice in9 | het | 1 | 0% | DA | 3 | 4 | m | 77.1 | n | y | y |
| 27** | L252_I254 del | hom | 2 | 95% | DA | 3 | 3 | m | 27.5 | y | y | y |
| 28 †† | R267W | hom | 2 | 80% | DA | 2 | 2 | f | 61.3 | y | y | y |
| 29 | R273H | hom | 1 | 80% | DA | 3 | 3 | f | 39.8 | y | y | y |
| 30 | G105V | hom | 2 | 10% | DA | 2 | 4 | m | 292.6 | n | y | y |
| 31 § | splice in9 | het | 1 | 0% | DA | 3 | 4 | f | 94.3 | y | y | y |
| 32 | G245D | hom | 1 | 70% | DA | 3 | 4 | m | 62.6 | n | y | y |
| 33 | R273C | hom | 1 | 95% | DA | 3 | 4 | m | 59.7 | n | y | y |
| 34 | R273H | hom | 1 | 100% | DA | 3 | 3 | m | 65.6 | n | y | y |
| 35 | N131 del | hom | 1 | 70% | DA | 3 | 4 | f | 135.9 | y | n | y |
| 36 | 335fs*1 | hom | 2 | 70% | DA | 3 | 4 | f | 98.1 | n | y | y |
| 37 | C238R | hom | 1 | 40% | DA | 3 | 5 | m | 70.8 | n | y | n ‡‡ |
| 38 | R273C | hom | 2 | 100% | DA | 2 | m | y | y | |||
| 39 | P151S, H179R | het | 1 | 95% | DA | 2 | 2 | f | 46.8 | n | y | y |
| 40 | normal | 1 | na | R | 1 | 4 | m | 41.1 | y | y | y | |
| 41 | normal | 1 | 0% | pr. B | 1 | 1 | f | 46.2 | y | n | y | |
| 42 | normal | 1 | 0% | DA | 3 | 3 | m | 71.2 | n | y | y | |
| 43 §§ | deletion | 0 | 0% | DA | 3 | 4 | f | 66.7 | n | y | y | |
| 44 | normal | na | 90% | B | 2 | 2 | m | 39.4 | n | y | y | |
| 45 | normal | 2 | 20%, foc. 70% | DA | 3 | 4 | m | 53.5 | n | y | y | |
| 46 | normal | 2 | 15% | N | 3 | 4 | f | 102.8 | y | y | y | |
| 47 | normal | 2 | 0% | E | 3 | 3 | m | 80.2 | y | y | y | |
| 48 | normal | 2 | na | S | 3 | 3 | f | 54.0 | y | y | y | |
| 49 § | normal | 2 | 0% | S | 1 | 1 | f | 62.8 | n | y | y | |
| 50 | normal | na | 0% | T | 3 | 3 | f | 23.6 | y | y | y | |
| 51 | normal | na | na | T | 2 | 4 | f | 69.4 | y | y | y | |
| 52 | normal | 2 | 3% | T | 1 | 1 | m | 53.1 | y | y | y | |
| 53 | normal | na | 3% | T | 3 | 3 | f | 38.3 | n | y | y | |
| 54 | normal | na | 0% | T | 3 | 3 | m | 77.6 | y | n | y | |
| 55 | normal | na | 3% | T | 3 | 4 | f | 71.5 | y | n | y | |
| 56 | normal | 2 | 10% | T | 3 | 4 | f | 57.8 | y | y | y | |
| 57 | normal | 2 | 0% | T | 3 | 4 | m | 11.5 | n | y | y | |
| 58 | normal | 2 | 0% | T | 1 | 4 | f | 39.9 | n | y | y | |
| 59 | normal | 2 | na | T | 2 | 2 | m | 87.0 | n | y | y | |
| 60 | normal | na | na | T | 3 | 4 | m | 57.5 | n | y | y | |
| 61l | normal | 2 | na | T | 1 | 5 | m | 56.2 | n | y | y | |
| 61r | normal | 2 | na | R | 1 | 5 | m | 56.2 | y | y | y | |
| 62 | normal | na | na | R | 2 | 2 | m | 31.3 | y | y | y | |
| 63 | normal | na | na | R | 3 | 4 | f | 74.6 | n | y | y | |
| 64 | normal | 2 | na | R | 2 | 4 | m | 38.3 | y | y | y | |
| 65 †† | G360A (SNP) | 2 | 0% | R | 3 | 4 | m | 125.6 | y | n | y | |
| 66 | normal | na | 3% | R | 1 | 1 | f | 66.9 | n | y | y | |
| 67 | normal | 2 | na | R | 3 | 4 | f | 175.4 | n | y | y | |
| 68 | normal | 2 | na | R | 1 | 4 | f | 26.9 | n | y | y | |
| 69 | normal | 2 | na | R | 1 | 1 | m | 55.3 | y | n | y | |
| 70 | normal | 2 | na | R | 1 | 4 | m | 83.4 | n | n | y | |
| 71 | normal | 2 | na | R | 3 | 4 | m | 126.5 | n | y | y | |
| 72 ¶¶ | normal | 2 | 2% | R | 2 | 2 | m | 36.7 | y | y | y | |
| 73 | normal | 2 | na | R | 3 | 4 | f | 60.3 | n | y | y | |
| 74r | normal | 2 | na | R | 1 | 5 | m | 46.7 | y | y | y | |
| 74l | normal | 2 | na | B | 3 | 5 | m | 46.7 | y | y | y | |
| 75 | normal | na | 30% | B | 3 | 3 | f | 61.1 | y | y | y | |
| 76 | normal | 2 | 1% | B | 3 | 3 | m | 123.4 | n | y | y | |
| 77 | normal | na | na | B | 1 | 4 | m | 46.6 | y | y | y | |
| 78 | normal | na | na | B | 3 | 3 | f | 77.4 | y | y | y | |
| 79 | normal | na | na | B | 1 | 1 | m | 50.4 | y | y | y | |
| 80 | normal | 2 | na | B | 3 | 5 | f | 22.3 | y | y | y | |
| 81 | normal | 2 | 10% | B | 1 | 1 | m | 121.0 | y | y | y | |
| 82 | normal | na | 3% | B | 3 | 3 | f | 27.1 | y | y | y | |
| 83 | normal | 2 | 25% | B | 3 | 4 | m | 127.6 | y | y | y | |
| 84 | normal | 2 | 0% | DA | 3 | 3 | m | 6.0 | y | n | y | |
| Survivingcases | ||||||||||||
| 85 | P278R | het | 2 | 70% | DA | 1 | 1 | m | 90.5 | n | n | n |
| 86 | splice in8 | hom | 2 | 2% | DA | 1 | 1 | f | 54.4 | n | n | n (3y) |
| 87 | G245S | het | 1 | 80% | DA | 1 | 1 | f | 61.6 | n | n | n (3y) |
| 88 | splice in8 & G245D ¶ | het | 2 | 1% | DA | 2 | 4 | f | 160.2 | n | n | n (3y) |
| 89 | 342fs*2 | hom | 1 | 80% | DA | 2 | 2 | m | n | n | n | |
| 90 | R175H | hom | 2 | 75% | DA | 3 | 3 | f | 149.2 | n | y | n (2y) |
| 91 | R342P | hom | 1 | 80% | DA | 1 | 1 | m | 86.4 | n | n | n (2.5y) |
| 92 | R273H | het | 1 | 70% | DA | 3 | 3 | f | 48.7 | n | n | n (2y) |
| 93 | P278S | hom | 1 | 90% | DA | 4 | f | 61.3 | n | y | n (3y) | |
| 94 | splice in6 | het | 2 | 10%, foc. 80% | DA | 1 | 1 | m | 10.2 | n | n | n (3.5y) |
| 95 | R175H | hom | 1 | 80% | DA | 3 | 3 | m | 66.3 | n | n | n |
| 96 | R175H & F338dupl ¶ | hom | 1 | 100% | DA | 1 | 4 | f | 36.4 | n | n | n |
| 97 | Q192* | hom | 1 | 0% | DA | 1 | 1 | f | 47.5 | n | n | n |
| 98 | R342P | hom | 2 | 90% | DA | 1 | 1 | f | 61.7 | n | y | n |
| 99 | R175H | hom | 1 | 90% | DA | 1 | 1 | m | 69.5 | y | y | n |
| 100 | R342* | hom | 1 | 40% | DA | 3 | 3 | m | 193.2 | n | n | n |
| 101 | R342P | hom | 1 | 90% | DA | 1 | 1 | m | 71.7 | n | n | n |
| 102 | 240fs*1 | hom | 1 | 0% | DA | 3 | 4 | m | 59.4 | n | n | n |
| 103 | G334_R337 del | hom | 1 | 80% | DA | 1 | 1 | m | 35.0 | n | n | n |
| 104 | R213* | hom | 1 | 0% | FA | 1 | 1 | m | 24.0 | n | n | n (3.5y) |
| 105 | R158L | het | 1 | 80% | FA | 2 | 4 | f | 64.3 | n | n | n (3y) |
| 106 | 342fs*2 | hom | 1 | 95% | FA | 2 | 4 | f | 71.1 | n | n | n |
| 107 | R175H | hom | 1 | 90% | FA | 1 | 1 | f | 33.6 | n | n | n |
| 108 | P309L | hom | 1 | 0% | FA | 1 | 4 | f | 27.4 | n | n | n |
| 109 | S362N | hom | 2 | 0% | FA | 2 | 2 | f | 44.4 | n | n | n |
| 110 | normal | 1 | 0% | DA | 1 | 1 | f | 35.4 | n | n | n | |
| 111 | normal | 2 | 90% | DA | 1 | 1 | m | 35.5 | n | n | n | |
| 112 | normal | 2 | 70% | FA | 3 | 4 | m | 32.5 | n | n | n (3y) | |
| 113 | normal | 2 | 2% | DA | 1 | 1 | f | 60.5 | n | n | n (2y) | |
| 114 | normal | 2 | 0% | FA | 1 | 1 | m | 153.4 | n | n | n | |
| 115 | normal | 2 | 25% | FA | 3 | 4 | f | 66.8 | n | n | n (3.5y) | |
†R, regressive; T, triphasic; nu, nuclear unrest; FA, focal anaplasia; pr. B, primary operated blastemal predominant; B, blastemal; DA, diffuse anaplasia.
‡Years of follow‐up are given for surviving patients (if < 4 years).
§Mutation found in tumour and relapse.
¶Different mutations in different regions.
**Familial WT.
††Germline mutation.
‡‡Not reported, but therapy cancelled with progressive disease (bilateral, lung and liver metastases).
§§Previously diagnosed with adrenal tumour and rhabdomyosarcoma.
¶¶Mutation in relapse only.
The TP53 gene was analyzed by sequencing of exons 4–10 including splice junctions that encompass 98.55% of reported mutations 20. TP53 mutations were found in 46% (39/84) of fatal cases. These included missense, nonsense, and splice mutations as well as small indels (Table 1). To screen for other types of alterations, we searched for TP53 CNVs by MLPA or FISH. In 18/66 cases, we detected copy number loss: one tumour with homozygous TP53 loss, 14 tumours with heterozygous loss and mutation of the other allele, while three cases had a heterozygous loss with no evidence for alteration of the second copy.
Since TP53 mutations lead to either complete absence of the protein or abnormal stabilization of a functionally impaired protein, we assessed the expression of p53 by immunohistochemistry. Most mutations led to clear positive staining on corresponding sections, while cases with deletions and most protein truncations did not produce discernible signals. Two tumours showed p53 positive IHC (up to 90% of cells) while copy number analysis and TP53 sequencing remained inconspicuous, suggesting an alteration of this pathway by other means.
Overall, our analyses of TP53 sequence, copy number and protein expression revealed 45/86 tumours with TP53 alterations, predominantly in high risk DA (29/30) and blastemal tumours (7/17). However, we also detected mutations in triphasic (3/15), regressive (2/16), and primary operated blastemal predominant (2/2) tumours as well as in 2/2 tumours with FA (Figure 1). No mutations were detected in necrotic (1), epithelial (1), or stromal type (2) tumours. Among the non‐DA tumours 15/56 (26%) had TP53 alterations with 9/39 (23.1%) from the low/intermediate risk group scoring positive. Among the 30 fatal DA tumours, there were 26 tumours with sequence alterations, two with heterozygous TP53 loss and one with p53 stabilization of unknown cause. Only one DA tumour was unaffected based on our testing.
Figure 1.
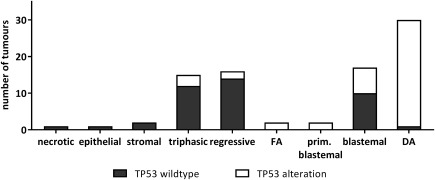
TP53 alterations in WT samples from patients with fatal disease outcome. Number of cases is given for each histotype. Black boxes represent tumour samples with normal TP53 sequence, copy number, and IHC staining, while tumours with any TP53 alteration (SNV, CNV, or IHC) are depicted by white boxes. FA, focal anaplasia; DA, diffuse anaplasia.
There was no difference in TP53 status with respect to sex, tumour stage, or presence of metastases at diagnosis. All deceased patients died from recurrent disease. Patients with TP53‐mutant tumours (median age 67.5 months, range 15–288) tended to be older at diagnosis compared to those with wildtype TP53 (median age 56.8 months, range 11–173), but this did not reach statistical significance (Figure 2). It may be due to the distribution of histological subtypes as 64.4% of the mutant group had DA, which is known to preferentially occur at an older age 9.
Figure 2.
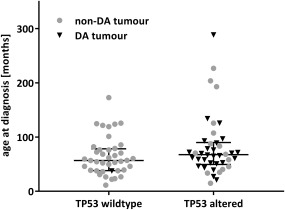
Age at diagnosis of patients with fatal disease. Median and interquartile range are indicated.
TP53 in anaplastic WT from survivors
The strong link between anaplasia and TP53 alterations in the cohort of fatal cases raised the question whether these are equally frequent in non‐fatal anaplastic Wilms tumours. We therefore analyzed samples from 31 surviving patients diagnosed with FA (9 cases) or DA Wilms tumour (22 cases) to elucidate the predictive impact of TP53 alterations in anaplastic Wilms tumours. The cohort contained 16 stage I, 2 stage II, 4 stage III, and 9 stage IV patients with >2 years of follow‐up (Table 1). Pathology review ensured the presence of anaplastic cells specifically in the tumour sections analyzed.
Single nucleotide variants (SNVs) or small indels were detected in 25 tumours. Two additional cases showed p53 immunostaining despite wildtype TP53 sequence, while one case had lost one TP53 copy based on FISH (Table 1). Overall, TP53 was affected in 90% (28/31) of non‐fatal anaplastic WT. Only two FA and one DA tumour lacked alterations detectable by our tests.
Thus, among the high risk cohorts of DA‐WT there was no difference in the frequency of TP53 alterations between surviving and fatal cases (21/22 in survivor versus 29/30 in fatal groups). However, tumour stage appeared to be crucial for survival (Mann‐Whitney test p = 0.002): the majority of surviving patients with DA‐WT had stage I disease, while most deceased patients were diagnosed with stage II, III, or IV (Figure 3). For FA‐WT, there were more survivors, but numbers were too small to draw conclusions.
Figure 3.
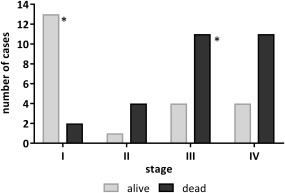
Stage, survival, and TP53 status of diffuse anaplasia Wilms tumours. Tumour stage is indicated for 22 surviving (grey) and 28 fatal (black) cases of DA‐WT. The two TP53 wildtype tumours are indicated by an asterisk.
Intratumour heterogeneity
The possibility of intra‐tumour heterogeneity is well known in Wilms tumours. While many genetic events – presumed to be initiation lesions – are found uniformly in all biopsies, alterations in WTX or 1q gain are quite heterogeneous upon multiple sampling 21, 22. To assess if TP53 alterations are early events affecting all tumour cells or occur in tumour subclones only at later time points, we analyzed multiple specimens (median 3) from 23 mutant tumours (Table 2). The presence of anaplastic cells was re‐examined by reference pathology.
Table 2.
Analysis of multiple WT specimens
| ID | Histology | DNA source | Localization | TP53 SNV | IHC | CNV |
|---|---|---|---|---|---|---|
| 3 a | Mixed with nuclear unrest | FFPE | Primary tumour | L194R hom | 1 | |
| 3 b | Necrotic | cryo | Relapse | L194R het | 1 | |
| 3 c | Unknown | FFPE | Relapse | wt | 1 | |
| 23 a | PLNR | FFPE | Right kidney | wt | 0% | |
| 23 b | PLNR | FFPE | Right kidney | wt | ||
| 23 c | Anaplasia | FFPE | Left kidney | 346fs*4 hom | 70% | 1 |
| 23 d | Anaplasia | FFPE | Left kidney | 346fs*4 hom | 40% | |
| 23 e | Anaplasia | FFPE | Left kidney | 346fs*4 het | 60% | |
| 23 f | Anaplasia | FFPE | Left kidney | 346fs*4 hom | 70% | 1 |
| 23 g | Anaplasia | FFPE | Left kidney | 346fs*4 hom | 40% | |
| 23 h | PLNR | FFPE | Left kidney | wt | 2 | |
| 23 i | Necrotic, 2% viable tumour | FFPE | Left kidney | 336fs*4 het | ||
| 23 j | Necrotic, 1% viable tumour | FFPE | Left kidney | 336fs*4 het | 90% | |
| 23 k | Unclear | FFPE | Lymph node | wt | ||
| 23 l | Anaplasia, 5% tumour | FFPE | Left kidney, relapse | 346fs*4 het | 60% | |
| 23 m | Unclear | FFPE | Left kidney, relapse | wt | ||
| 23 n | PLNR | FFPE | Left kidney, relapse | wt | ||
| 23 o | 25% tumour, anaplasia | FFPE | Left kidney, relapse | wt | ||
| 23 p | 2% tumour | FFPE | Left kidney, relapse | 336fs*4 het | 90% | |
| 23 q | PLNR | FFPE | Right kidney, relapse | wt | ||
| 23 r | PLNR | FFPE | Right kidney, relapse | wt | ||
| 23 s | PLNR | FFPE | Right kidney, relapse | wt | ||
| 23 t | PLNR | FFPE | Right kidney, relapse | wt | ||
| 23 u | Anaplasia | FFPE | Lymph node | 336fs*4 hom | 90% | 2 |
| 23 v | Anaplasia | FFPE | Lymph node | 336fs*4 het | 90% | 2 |
| 29 a | Anaplasia | FFPE | R273H hom | |||
| 29 b | Anaplasia | cryo | R273H 80% | 1 | ||
| 34 a | Macrodissected anaplasia | FFPE | R273H hom | 1 | ||
| 34 b | Unknown | cryo | R273H hom | 1 | ||
| 35 a | Macrodissected anaplasia | FFPE | del N131 hom | 1 | ||
| 35 b | Anaplasia | cryo | del N131 hom | 1 | ||
| 35 c | Anaplasia | cryo | del N131 het | 1 | ||
| 35 d | Nuclear unrest | cryo | del N131 het | |||
| 37 a | Macrodissected anaplasia | FFPE | C238R 80% | 1 | ||
| 37 b | Anaplasia | cryo | wt | 1 | ||
| 37 c | Stroma + regression, no anaplasia | cryo | wt | 1% | 2 | |
| 38 a | Macrodissected non‐anaplasia | FFPE | wt | |||
| 38 b | Macrodissected anaplasia | FFPE | R273C hom | |||
| 85 a | Macrodissected anaplasia | FFPE | P278R | 70% | ||
| 85 b | Not done | cryo | P278R het | 2 | ||
| 86 a | Macrodissected anaplasia | FFPE | splice ex9 hom | |||
| 86 b | 100% regression | cryo | splice het 20% | 2 | ||
| 86 c | Anaplasia | cryo | splice het 60% | 2 | ||
| 87 a | Macrodissected anaplasia | FFPE | G245S het | |||
| 87 b | Anaplasia | cryo | G245S hom | 80% | 1 | |
| 87 c | 98% regression, 2% tumour, no anaplasia | cryo | G245S het | 80% | 1 | |
| 88 a | Macrodissected anaplasia | FFPE | splice ex9, het | |||
| 88 b | Anaplasia | cryo | G245D, het | 2 | ||
| 88 c | 100% regression | cryo | wt | 2 | ||
| 93 a | Macrodissected non‐anaplasia | FFPE | wt | |||
| 93 b | Macrodissected anaplasia | FFPE | P278S hom | |||
| 93 c | Not done | cryo | P278S hom | 1 | ||
| 94 a | Macrodissected anaplasia | FFPE | splice ex7 het | |||
| 94 b | Macrodissected non‐anaplasia | FFPE | wt | |||
| 95 a | Macrodissected anaplasia | FFPE | Primary tumour | R175H, hom | ||
| 95 b | Macrodissected non‐anaplasia | FFPE | Primary tumour | wt | ||
| 95 c | Anaplasia | cryo | Primary tumour | F338dupl, hom | 100% | 1 |
| 95 d | Anaplasia | cryo | Primary tumour | F338dupl, hom | 100% | |
| 95 e | Anaplasia | cryo | Primary tumour | F338dupl, hom | 100% | 1 |
| 95 f | Anaplasia | cryo | Relapse | F338dupl, hom | 100% | 1 |
| 95 g | Anaplasia | cryo | Relapse | F338dupl, hom | 100% | |
| 95 h | Anaplasia | cryo | Relapse | F338dupl, hom | 100% | 1 |
| 96 a | Macrodissected anaplasia | FFPE | R175H | 80% | ||
| 96 b | Macrodissected anaplasia | FFPE | R175H het | |||
| 96 c | Macrodissected non‐anaplasia | FFPE | wt | |||
| 99 a | No anaplasia | cryo | wt | nd | 2 | |
| 99 b | Anaplasia | cryo | R175H hom | 90% | 1 | |
| 102 a | Anaplasia | cryo | fsS240*1 hom | 90% | 1 | |
| 102 b | Anaplasia | cryo | fsS240*1 hom | 90% | 1 | |
| 103 a | Anaplasia | cryo | G334_R337 del | 90% | 1 | |
| 103 b | No anaplasia | cryo | wt | 0% | ||
| 104 a | Macrodissected anaplasia | FFPE | hom R213* | 1 | ||
| 104 b | Anaplasia | cryo | hom R213* | 1 | ||
| 104 c | Stroma (myogenic), no anaplasia | cryo | wt | 2 | ||
| 105 a | Macrodissected anaplasia | FFPE | R158L het | 1 | ||
| 105 b | Blastema, no anaplasia | cryo | R158L het | 2 | ||
| 105 c | 99% regression | cryo | wt | 2 | ||
| 106 a | Macrodissected anaplasia | FFPE | fsR342*2 hom | |||
| 106 b | 99% regression, no anaplasia | cryo | wt | |||
| 106 c | 99% regression, no anaplasia | cryo | wt | |||
| 108 a | Macrodissected non‐anaplasia | FFPE | wt | |||
| 108 b | Macrodissected anaplasia | FFPE | P309L hom | 1 | ||
| 108 c | Blastema | cryo | wt | 1 | ||
| 108 d | Blastema | cryo | wt | 1 | ||
| 108 e | Blastema | cryo | wt | 2 | ||
| 109 a | Macrodissected anaplasia | FFPE | S362N hom | |||
| 109 b | Blastema | cryo | wt |
In five tumours, all regions tested showed anaplasia and consistently exhibited TP53 alterations. Areas with and without anaplastic cells were found in 18 patients. In 13 of these tumours, TP53 alterations were present in anaplastic regions only. In contrast, in four tumours even regions without signs of anaplasia contained TP53 mutations and, in one patient, some of the anaplastic regions appeared to lack TP53 alterations. Six of eighteen tumours were subjected to macrodissection of paired anaplastic and non‐anaplastic regions from the same histological sections. In these macrodissected specimens, TP53 mutations were clearly restricted to anaplastic regions.
One patient with variable presence of TP53 alterations presented with a DA tumour in the left kidney and contralateral perilobar nephrogenic rests (PLNR) followed by a relapse with multiple lymph node metastases. Mutations (exon 10 indel) were absent from the PLNR, but present in anaplastic specimens and in metastases with anaplasia (Figure 4). Interestingly, two distinct frameshift mutations (E336fs*4 and E346fs*4) were found in different regions of the anaplastic tumour, indicating late and independent development of TP53 alterations.
Figure 4.
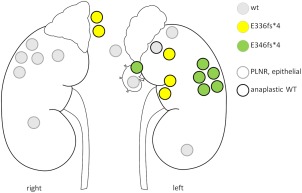
Analysis of TP53 in multiple WT specimens from a single patient. The patient was diagnosed with perilobar nephrogenic rests (PLNR, grey border) in the right kidney and anaplastic WT (black border) in the left kidney. Two different frameshift mutations (green versus yellow) within exon 10 were found in anaplastic lesions, while PLNR harboured wildtype TP53 (grey).
We could analyze primary and relapse/metastasis material of seven patients. Both carried the same TP53 mutation in five patients and in one patient only relapse material carried mutant TP53. In one case, all samples lacked TP53 alterations. Thus, TP53 alterations were found primarily, but not exclusively, in anaplastic areas.
Significance of p53 alterations
In the two cohorts of 117 tumours in total, we identified 73 cases with alterations affecting the TP53 gene or protein (Figure 5A). In 64 tumours, TP53 mutations, either SNVs or indels, were detected. CNVs as ascertained by MLPA in frozen material or by FISH in FFPE sections could be analyzed in 97 of the tumours. Loss affecting one TP53 copy was seen more often in TP53‐mutant tumours (37/59) than in samples without such mutations (5/38). The latter may still harbour alterations in flanking exons or introns that are not detected by our approach. The same may apply to the four p53‐IHC positive samples that lacked sequence or CNV alterations. There was only a single tumour sample with a homozygous loss of TP53.
Figure 5.
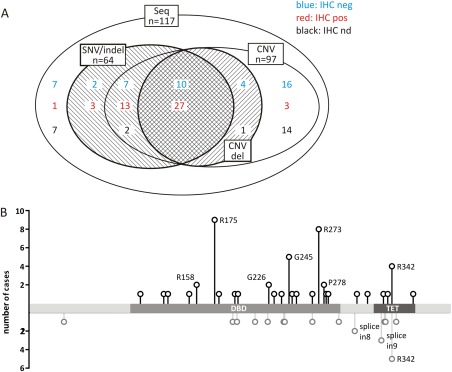
Types of TP53 alteration in 117 tumours. (A) Alterations based on sequencing (SNV/indel) and copy number analysis (CNV del) are shown with respect to immunohistochemical detection (blue versus red). Shaded areas represent corresponding TP53 alterations. (B) Distribution of TP53 point mutations found in Wilms tumour: black lollypop symbols in the upper panel represent missensemutations and in‐frame deletions of single amino acids; grey lollypop symbols in the lower panel represent non‐sense, frameshift, and splice mutations. Mutations occurring twice or more are labelled. DNA binding domain and tetramerization domain are indicated.
TP53 sequence alterations included 50 missense, 6 nonsense, 9 frameshift, and 7 splice site mutations. Four homozygous splice mutant tumours were tested at the RNA level: three showed transcription of the mutant variant carrying intron sequences, while in the fourth only wildtype mRNA could be detected, perhaps due to nonsense‐mediated decay and amplification of wild type transcripts from non‐tumour cells. Four tumours presented with multiple mutations [R158H(hom) + G226D(het) + G245S(het) + R280K(hom), two compound heterozygotes R175H + R342* (het, CNV 2) and P151S + H179R (het, CNV1)].
Assessment of TP53 missense alterations using the prediction tools PolyPhen‐2, Provean, Sift, and the TP53 database showed that the vast majority are expected to disrupt normal TP53 function (Table 3). In only two cases was there no clear‐cut evidence for abnormal TP53 function, but both were restricted to macrodissected anaplastic regions: P309L is predicted as deleterious only by SIFT and S362N is described as neutral by all tools. Both are missing from the TP53 database, while all other missense mutations were recorded.
Table 3.
Functional effects of TP53 sequence alterations
| TP53 mutation (protein) | Mutation annotation (cDNA) | Provean | Sift | PolyPhen2 | Counts in TP53 database somatic/germline |
|---|---|---|---|---|---|
| p.G105V | c.314G>T | D | D | D | 5/0 |
| p.S127F | c.380C>T | D | D | D | 26/0 |
| p.N131 del | c.391_393del3 | D | n.a. | n.a. | 12/0 |
| p.P151S | c.451C>T | D | D | D | 102/4 |
| p.R158H | c.473G>A | D | D | D | 114/19 |
| p.R158L | c.473G>T | D | D | D | 102/1 |
| p.R175_C176 del | c.523_528del6 | D | n.a. | n.a. | 1/0 |
| p.R175H | c.524G>A | D | D | D | 1216/75 |
| p.H179R | c.536A>G | D | D | D | 174/0 |
| p.Q192* | c.574C>T | stop | stop | stop | 112/0 |
| p.L194R | c.581T>G | D | D | D | 69/0 |
| p.R196* | c.586C>T | stop | stop | stop | 240/18 |
| p.V197M | c.589G>A | N | D | D | 11/1 |
| p.R213* | c.637C>T | stop | stop | stop | 321/27 |
| p.G226S | c.676G>A | D | D | D | 3/0 |
| p.G226D | c.677G>A | D | D | D | 4/0 |
| p.T231I | c.692C>T | D | D | D | 4/0 |
| p.C238R | c.712T>C | D | D | D | 23/0 |
| p.G245S | c.733G>A | D | D | D | 456/40 |
| p.G245D | c.734G>A | D | D | D | 162/8 |
| p.R248W | c.742C>T | D | D | D | 739/88 |
| p.L252_I254 del | c.754_762del9 | D | n.a. | n.a. | 6/0 |
| p.R267W | c.799C>T | D | D | D | 33/1 |
| p.R273C | c.817C>T | D | D | D | 707/35 |
| p.R273H | c.818G>A | D | D | D | 858/63 |
| p.P278R | c.833C>G | D | D | D | 44/0 |
| p.P278S | c.832C>T | D | D | D | 91/1 |
| p.R280K | c.839G>A | D | D | D | 75/1 |
| p.R282W | c.844C>T | D | D | D | 581/47 |
| p.P309L | c.926C>T | N | D | N | 0/0 |
| p.K319E | c.955A>G | N | D | D | 4/0 |
| p.G334_R337 del | c.1000_1011del12 | D | n.a. | n.a. | 0/0 |
| p.F338 dupl | c.1014_1015ins3 | D | n.a. | n.a. | 0/0 |
| p.R342* | c.1024C>T | stop | stop | stop | 93/16 |
| p.R342P | c.1025G>C | N | N | D | 6/10 |
| p.S362N | c.1085G>A | N | N | N | 0/0 |
| p.fs240*1 | c.717_718ins4 | stop † | |||
| p.fs241*16 | c.723_736del14 | stop † | |||
| p.fs267*1 | c.800_806del7 | stop † | |||
| p.fs292*11 | c.876_877del2 | stop † | |||
| p.fs335*1 | c.1004_1005ins1 | stop † | |||
| p.fs336*4 | c.1005_1021del17 | stop † | |||
| p.fs342*2 | c.1024_1024del1 | stop † | |||
| p.fs346*4 | c.1036_1037ins13 | stop † | |||
| splice in 3 | c.97–1G>T | splice defect † | 0/0 | ||
| splice in 6 | c.673–1G>C | splice defect † | 2/0 | ||
| splice in 8 | c.919 + 1G>A | splice defect † | 13/7 | ||
| splice in 8 | c.920–1G>A | splice defect † | 16/0 | ||
| splice in 9 | c.993 + 1G>T | splice defect † | 17/0 | ||
| splice in 9 | c.994–1G>C | splice defect † | 2/6 | ||
†Frameshift and splice site alterations were not analyzed by the prediction tools used.
D, deleterious; N, neutral; n.a., not analyzable by the particular prediction tool.
The distribution of mutations along the TP53 coding region was similar to the one described in the TP53 database (Figure 5B). The most frequent sites were R175H, G245S/D, R273C/H, and R342P. Seventy percent of mutations affect the DNA binding domain (DBD); another 28% specifically disturb the multimerization domain.
Control tissue was tested for 52 of 64 mutant cases. Only one patient carried a germline mutation (R267W) that may cause Li‐Fraumeni syndrome (patient was previously diagnosed with neurofibromatosis type I).
Effects on p53 stabilization
Deregulation of TP53 function in mutant tumours was corroborated by immunostaining of FFPE or frozen sections of all TP53‐mutant tumours (for two samples no additional material was available). Furthermore, 31 non‐mutant tumours were stained to check for other p53 stabilizing effects. Four of these tumours showed p53 stabilization despite the presence of a wildtype sequence.
The effects of TP53 missense mutations were position dependent. The four most common (R175H, G245S/D, R273C/H, R342P) and several other deleterious missense mutations led to p53 accumulation, as did the in‐frame deletions N131, R175_C176, L252_I254, and G334_R337. On the other hand, tumours with G105V, L194R, and G226S and, unexpectedly, two of the R273H cases showed almost no p53 positive cells. The tumours with predicted neutral mutations (P309L, S362N) likewise lacked p53 staining. The splice mutations led to very weak or absent p53 staining and the effect of stop mutations was position dependent: p53 staining was missing for stops up to amino acid 268, while stabilization was seen with stop mutations at 303 and beyond.
IHC staining of tumours with the frequent stabilizing TP53 mutations revealed a striking specificity: strong p53 signals were detected in regions with anaplasia, while corresponding non‐anaplastic regions with wildtype TP53 sequence were p53 negative (Figure 6). This further supports the strong link between mutation and anaplasia as seen in macrodissected samples.
Figure 6.
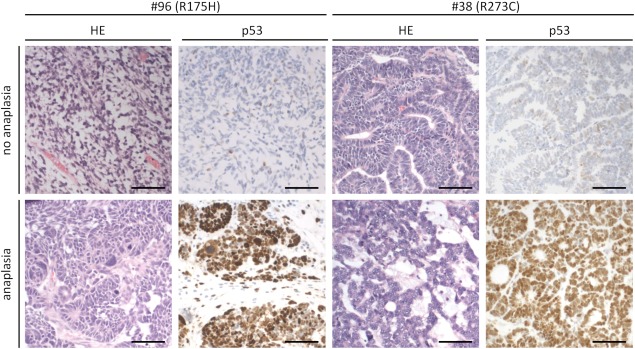
Immunohistochemical p53 staining of anaplastic and non‐anaplastic regions of two tumours (#96 and #38) with common TP53 mutations (R175H and R273C respectively). Bar: 100 µm.
Discussion
Even though TP53 is frequently mutated in human cancer, mutations have rarely been reported in Wilms tumour in general, but they are considered pathognomonic for the infrequent anaplastic subtype. In a previous screen of blastemal type tumours, we had identified a significant fraction carrying TP53 alterations, but several of these cases did not fulfil diagnostic criteria for anaplasia upon re‐examination 13. Nevertheless, these TP53 alterations were tightly linked to aneuploidy and chromothripsis. Given the very high lethality in this subset, we decided to analyze a larger cohort of tumours with fatal outcome to determine the prognostic relevance of TP53 status in WT and to reevaluate the restriction of TP53 alterations to the anaplastic subtype.
TP53 mutation rate and heterogeneity
Our data show that the vast majority of DA tumours (87%, 45/52) carry SNVs and indels of TP53, independent of later survival. A high mutation rate was also found in tumours with only focal anaplasia (73%, 8/11). This is much higher than mutation rates of about 50% reported in prior studies 11, 12. There is likely no ascertainment bias due to our initial focus on tumours with lethal outcome, since we found similarly high mutation rates in fatal as well as long‐term surviving anaplastic cases.
Testing of just a single sample of bulk tumour tissue may be an important difference in earlier analyses. We likely increased the sensitivity by multiple sampling in many cases and through dissection of anaplastic regions. Our data clearly indicate that neighbouring non‐anaplastic tumour tissue frequently lacks TP53 mutations. This is not based only on sequence analysis, since immunohistochemical staining confirmed that TP53 alterations may be restricted to small anaplastic regions that can easily be missed when single biopsies or bulk tumour material are analyzed.
The striking heterogeneity is consistent with data from the recent study on TP53 mutations in DA‐WT 11, where mutations were revealed only by deep sequencing in 2/22 cases. This also fits with a previous report on pre‐chemotherapy biopsies describing frequent failure to detect anaplastic features, while anaplasia was more readily diagnosed in post‐chemotherapy nephrectomy specimens 23.
The value of multiple biopsies in Wilms tumours has been demonstrated by Cresswell et al 21: while alterations of 11p15 tended to be present in all biopsies from a given tumour, occurrence of 1q gain was quite heterogeneous, similar to our data on WTX mutations 22. Thus, TP53 alterations clearly belong to the class of late, progression type mutations affecting just a fraction of tumour cells that may only be revealed by multiple sampling.
TP53 deletions
Many TP53 mutations act in a dominant‐negative manner, with heterozygous mutations being fully effective 24, 25. Only a small number of tumours in our cohort harboured two or more TP53 mutations, the phase of which is not known. In addition, we detected 17p loss in about 63% (37/59) of mutant tumours, even if this may be secondary to p53‐dependent genome instability. Thus, inactivation of both copies is frequent, but apparently not a prerequisite for oncogenic action. These numbers almost match the rates of 17p loss in 20/25 and 42/53 anaplastic TP53 mutant tumours reported previously 11, 12. The difference may be due to reduced sensitivity in our testing of FFPE material, since the reference FISH probe used (CEN17) may lead to an underestimation of TP53 loss if the entire chromosome is lost.
IHC‐only p53 alterations
Besides sequence alteration and copy number loss of TP53, disruption of the p53‐pathway may occur via other affected pathway members. In our cohort, four tumours exhibited stabilized p53 by IHC despite a wildtype DNA sequence, suggesting the presence of rare mutations in flanking genomic regions or pathway activation by other means. Indeed, the analysis of seven TP53 wildtype anaplastic Wilms tumours by Ooms et al 12 suggested that the p53 pathway can be activated in the absence of mutations in p53‐pathway genes. This clearly demonstrates that despite the strong correlation of histology and genetics, only one of the methods may yield positive results in difficult cases.
TP53 and Wilms tumour stratification
As patients with DA tumours carry a high risk for relapse and fatal outcome, the p53 status is a prime candidate to further improve clinical risk stratification. In our cohort of anaplastic Wilms tumours, the rate of TP53 alterations was very high in both groups (96% in fatal versus 90% in surviving group) and thus not predictive of adverse events or death. Tumour stage was the only relevant prognostic factor, which has already been established previously 4. In contrast, mutant TP53 correlated with increased recurrence and risk of death, irrespective of tumour stage in the study by Maschietto et al 11. Ooms et al 12 reported no difference in outcome for stage I/II DA‐WT patients, but p53 abnormalities correlated with worse overall survival for patients with stage III/IV disease. It is difficult to reconcile these data, but differences in detection rates with the possible unmasking of anaplasia after pre‐operative chemotherapy in our samples may add to this discrepancy.
The strong influence of stage on patient outcome suggests that TP53‐mutant tumours are more resistant to current chemotherapy and only complete surgical resection is able to control them. Any growth into neighbouring tissues or vessel ingression, even without clinically detectable remnants or spread, increases the risk of relapse and progression. Given the histological heterogeneity, it may be worthwhile in future to assess if the estimated volume of the anaplastic compartment of a tumour may help to better predict outcome, similar to the predictive value of blastemal volume for event‐free survival 26.
Histological criteria and the continuum of TP53 phenotypes
While anaplastic tumours were predominantly affected, TP53 alterations were also observed in 16/52 fatal non‐anaplastic Wilms tumours, preferentially in blastemal type WT (7/17, 41%). Thus, it would be worthwhile to test whether p53 status is informative in this second group of high risk Wilms tumours. In a prior study, we detected TP53 alterations in 7/58 blastemal WT by exome sequencing and all of these patients died due to disease 13, while neither TP53 mutation nor 17p loss were reported in the exome analysis of 77 North American favourable histology WT 27. These findings point to blastemal type tumours as primary targets for testing.
A highly relevant question relates to histological coverage and criteria. The presence of partial features of anaplasia in some of the blastemal tumours in our prior study suggests a continuum of TP53‐induced histological changes that may be more difficult to assess if present in small quantity or only partially penetrant. Non‐anaplastic tumours may contain p53 mutant subclones that do not meet all criteria of anaplasia, but can subsequently develop a full anaplastic phenotype. This is supported by the detection of nuclear unrest in two triphasic tumours with TP53 alterations. Nuclear unrest, defined by enlarged, hyperchromatic nuclei without multipolar mitotic figures, is more common in stage I/II disease than in stage III/IV 28. Interestingly, these patients had intermediate event‐free survival between favourable histology and anaplastic WT, but overall survival was equal for the nuclear unrest and favourable histology groups. This step‐wise evolution of anaplasia from TP53 mutant, but phenotypically inconspicuous, cells is in line with p53 positive staining being not just limited to anaplastic cells, but also present in a larger surrounding area without anaplasia. Thus, TP53 screening could enhance the sensitivity of high‐risk subtype detection.
Future diagnostic approaches
The strong correlation of TP53 alterations and anaplasia as the prime adverse predictor in Wilms tumours strengthens their causal mechanistic link. The presence of TP53 mutations in blastemal type and even in intermediate risk histology tumours supports the addition of TP53 screening to the diagnostic workup, but the striking heterogeneity calls for multiple sampling of tumours. It also classifies TP53 alterations as progression markers indicating the development of a more malignant and difficult to treat subpopulation of tumour cells. With falling costs of sequencing, there may be a rationale to consider liquid biopsies, which hold the promise to provide a global picture of an entire tumour, including mutations present in only a fraction of cells. The genetic instability in p53‐impaired tumour cells may lead to higher rates of cell death with concomitant release of cell‐free DNA into the circulation, favouring their detection.
Author contributions statement
JW, CV, IL, RF, NG, MG: conceived the study design; JW, MG: wrote the paper. All authors were involved in data collection, data analysis, data interpretation, and had final approval of the submitted version.
Supporting information
SUPPLEMENTARY MATERIAL ONLINE
Table S1. Primers for amplification and sequencing of TP53 exons 4–10
Acknowledgements
This work was funded by the Deutsche Forschungsgemeinschaft (grant GE 539/13–1) and Deutsche Krebshilfe (grant 50–2709‐Gr2). The authors thank all members of the GPOH Wilms tumour study group for their efforts in collecting precious samples and data. The authors thank Sabrina Bausenwein for technical support. Publication was funded by the German Research Foundation (DFG) and the University of Wuerzburg in the funding programme Open Access Publishing.
No conflicts of interest were declared.
References
- 1. Breslow N, Olshan A, Beckwith JB, et al Epidemiology of Wilms tumor. Med Pediatr Oncol 1993; 21 : 172–181. [DOI] [PubMed] [Google Scholar]
- 2. Dome JS, Graf N, Geller JI, et al Advances in Wilms tumor treatment and biology: progress through international collaboration. J Clin Oncol 2015; 33 : 2999–3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Vujanic GM, Sandstedt B. The pathology of Wilms' tumour (nephroblastoma): the International Society of Paediatric Oncology approach. J Clin Pathol 2010; 63 : 102–109. [DOI] [PubMed] [Google Scholar]
- 4. Weirich A, Ludwig R, Graf N, et al Survival in nephroblastoma treated according to the trial and study SIOP‐9/GPOH with respect to relapse and morbidity. Ann Oncol 2004; 15 : 808–820. [DOI] [PubMed] [Google Scholar]
- 5. Reinhard H, Semler O, Burger D, et al Results of the SIOP 93–01/GPOH trial and study for the treatment of patients with unilateral nonmetastatic Wilms tumor. Klin Padiatr 2004; 216 : 132–140. [DOI] [PubMed] [Google Scholar]
- 6. Dome JS, Cotton CA, Perlman EJ, et al Treatment of anaplastic histology Wilms' tumor: results from the fifth National Wilms' Tumor Study. J Clin Oncol 2006; 24 : 2352–2358. [DOI] [PubMed] [Google Scholar]
- 7. Popov SD, Sebire NJ, Vujanic GM. Wilms' tumour ‐ histology and differential diagnosis In Wilms Tumor, van den Heuvel‐Eibrink MM. (Ed). Codon Publications: Brisbane (AU), 2016. [PubMed] [Google Scholar]
- 8. Reinhard H, Schmidt A, Furtwangler R, et al Outcome of relapses of nephroblastoma in patients registered in the SIOP/GPOH trials and studies. Oncol Rep 2008; 20 : 463–467. [PubMed] [Google Scholar]
- 9. Vujanic GM, Harms D, Sandstedt B, et al New definitions of focal and diffuse anaplasia in Wilms tumor: the International Society of Paediatric Oncology (SIOP) experience. Med Pediatr Oncol 1999; 32 : 317–323. [DOI] [PubMed] [Google Scholar]
- 10. Bardeesy N, Falkoff D, Petruzzi MJ, et al Anaplastic Wilms' tumour, a subtype displaying poor prognosis, harbours p53 gene mutations. Nat Genet 1994; 7 : 91–97. [DOI] [PubMed] [Google Scholar]
- 11. Maschietto M, Williams RD, Chagtai T, et al TP53 mutational status is a potential marker for risk stratification in Wilms tumour with diffuse anaplasia. PLoS One 2014; 9 : e109924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ooms AH, Gadd S, Gerhard DS, et al Significance of TP53 mutation in Wilms tumors with diffuse anaplasia: a report from the children's oncology group. Clin Cancer Res 2016; 22 : 5582–5591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wegert J, Ishaque N, Vardapour R, et al Mutations in the SIX1/2 pathway and the DROSHA/DGCR8 miRNA microprocessor complex underlie high‐risk blastemal type Wilms tumors. Cancer Cell 2015; 27 : 298–311. [DOI] [PubMed] [Google Scholar]
- 14. Wittmann S, Zirn B, Alkassar M, et al Loss of 11q and 16q in Wilms tumors is associated with anaplasia, tumor recurrence, and poor prognosis. Genes Chromosomes Cancer 2007; 46 : 163–170. [DOI] [PubMed] [Google Scholar]
- 15. Choi Y, Sims GE, Murphy S, et al Predicting the functional effect of amino acid substitutions and indels. PLoS One 2012; 7 : e46688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non‐synonymous variants on protein function using the SIFT algorithm. Nat Protoc 2009; 4 : 1073–1081. [DOI] [PubMed] [Google Scholar]
- 17. Adzhubei IA, Schmidt S, Peshkin L, et al A method and server for predicting damaging missense mutations. Nat Methods 2010; 7 : 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vokuhl C, Oyen F, Haberle B, et al Small cell undifferentiated (SCUD) hepatoblastomas: all malignant rhabdoid tumors? Genes Chromosomes Cancer 2016; 55 : 925–931. [DOI] [PubMed] [Google Scholar]
- 19. Ambros PF, Ambros IM, SIOP Europe Neuroblastoma Pathology B , et al Pathology and biology guidelines for resectable and unresectable neuroblastic tumors and bone marrow examination guidelines. Med Pediatr Oncol 2001; 37 : 492–504. [DOI] [PubMed] [Google Scholar]
- 20. Leroy B, Ballinger ML, Baran‐Marszak F, et al Recommended guidelines for validation, quality control, and reporting of TP53 variants in clinical practice. Cancer Res 2017; 77 : 1250–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cresswell GD, Apps JR, Chagtai T, et al Intra‐tumor genetic heterogeneity in Wilms tumor: clonal evolution and clinical implications. EBioMedicine 2016; 9 : 120–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wegert J, Wittmann S, Leuschner I, et al WTX inactivation is a frequent, but late event in Wilms tumors without apparent clinical impact. Genes Chromosomes Cancer 2009; 48 : 1102–1111. [DOI] [PubMed] [Google Scholar]
- 23. Vujanic GM, Kelsey A, Mitchell C, et al The role of biopsy in the diagnosis of renal tumors of childhood: results of the UKCCSG Wilms tumor study 3. Med Pediatr Oncol 2003; 40 : 18–22. [DOI] [PubMed] [Google Scholar]
- 24. Marutani M, Tonoki H, Tada M, et al Dominant‐negative mutations of the tumor suppressor p53 relating to early onset of glioblastoma multiforme. Cancer Res 1999; 59 : 4765–4769. [PubMed] [Google Scholar]
- 25. Oren M, Rotter V. Mutant p53 gain‐of‐function in cancer. Cold Spring Harb Perspect Biol 2010; 2 : a001107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dome JS, Perlman EJ, Graf N. Risk stratification for wilms tumor: current approach and future directions. Am Soc Clin Oncol Educ Book 2014, 215–223. [DOI] [PubMed] [Google Scholar]
- 27. Walz AL, Ooms A, Gadd S, et al Recurrent DGCR8, DROSHA, and SIX homeodomain mutations in favorable histology Wilms tumors. Cancer Cell 2015; 27 : 286–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hill DA, Shear TD, Liu T, et al Clinical and biologic significance of nuclear unrest in Wilms tumor. Cancer 2003; 97 : 2318–2326. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SUPPLEMENTARY MATERIAL ONLINE
Table S1. Primers for amplification and sequencing of TP53 exons 4–10