

Abstract
Background
The relationship between specific genome alterations and hepatocellular carcinoma (HCC) cancer stem cells (CSCs) remains unclear. In this study, we evaluated the relationship between somatic mutations and epithelial cell adhesion molecule positive (EpCAM+) CSCs.
Methods
Two patient-derived HCC samples (HCC1 and HCC2) were sorted by EpCAM expression and analyzed by whole exome sequence. We measured PCDH18 expression level in eight HCC cell lines as well as HCC1 and HCC2 by real-time quantitative RT-PCR. We validated the identified gene mutations in 57 paired of HCC and matched non-cancerous liver tissues by Sanger sequence.
Results
Whole exome sequencing on the sorted EpCAM+ and EpCAM− HCC1 and HCC2 cells revealed 19,263 nonsynonymous mutations in the cording region. We selected mutations that potentially impair the function of the encoded protein. Ultimately, 60 mutations including 13 novel nonsense and frameshift mutations were identified. Among them, PCDH18 mutation was more frequently detected in sorted EpCAM+ cells than in EpCAM− cells in HCC1 by whole exome sequences. However, we could not confirm the difference of PCDH18 mutation frequency between sorted EpCAM+ and EpCAM− cells by Sanger sequencing, indicating that PCDH18 mutation could not explain intracellular heterogeneity. In contrast, we found novel PCDH18 mutations, including c.2556_2557delTG, c.1474C>G, c.2337A>G, and c.2976G>T, were detected in HCC1 and 3/57 (5.3%) additional HCC surgical specimens. All four HCCs with PCDH18 mutations were EpCAM-positive, suggesting that PCDH18 somatic mutations might explain the intertumor heterogeneity of HCCs in terms of the expression status of EpCAM. Furthermore, EpCAM-positive cell lines (Huh1, Huh7, HepG2, and Hep3B) had lower PCDH18 expression than EpCAM-negative cell lines (PLC/PRL/5, HLE, HLF, and SK-Hep-1), and PCDH18 knockdown in HCC2 cells slightly enhanced cell proliferation.
Conclusions
Our data suggest that PCDH18 is functionally suppressed in a subset of EpCAM-positive HCCs through somatic mutations, and may play a role in the development of EpCAM-positive HCCs.
Electronic supplementary material
The online version of this article (doi:10.1186/s12935-017-0467-x) contains supplementary material, which is available to authorized users.
Keywords: Hepatocellular carcinoma, EpCAM, PCDH18, Whole exome sequencing, Cancer stem cell
Background
Hepatocellular carcinoma (HCC) is a leading cause of cancer death worldwide [1, 2], and it is usually associated with specific risk factors including hepatitis B or C virus infection, high alcohol intake, hemochromatosis, and nonalcoholic fatty liver disease [3]. A typical cancer can harbor thousands of somatic mutations, of which 10–100 might occur in the coding region of genes [4–8]. With the advent of next generation sequencing, recent studies have shown that the HCC genome can contain various somatic mutations, intrachromosomal rearrangements, gene fusions, and focal copy number alterations [9–11]. These studies have also indicated that genes related to two pathways, the p53 and Wnt/β-catenin signaling pathways, are most frequently mutated in HCC. Furthermore, whole genome analysis of the HCC genome has indicated that one of the most frequent mutations identified in HCC is TERT promoter region mutation [12].
HCC is a heterogeneous disease in terms of its morphology, biological behavior, response to treatment, and clinical outcome. Traditionally, this heterogeneity has been explained by cancer cell clonal evolution, and the step-wise acquisition of genetic mutations [13]. However, recent evidence has suggested that HCC may conform to the cancer stem cell (CSC) hypothesis; this hypothesis proposes that a subset of cells with stem cell-like features divide asymmetrically to generate a heterogeneous cell population, and these stem cell-like cells play a fundamental role in tumor maintenance, chemoresistance, and metastasis [14].
In HCC, several CSC markers, including CD133, CD90, CD44, CD24, and CD13 have been identified [15–19]. We have previously demonstrated that HCC subtypes can be defined by the expression of the hepatic stem/progenitor cell markers epithelial cell adhesion molecule (EpCAM) and α-fetoprotein, and that these subtypes correlate with distinct gene expression signatures and patient prognoses [20, 21]. Our previous data also suggested that EpCAM is a marker of liver CSCs, and might be used to enrich a highly tumorigenic and chemoresistant cell population.
In the present study, we sorted EpCAM+ and EpCAM− cell populations from fresh HCC specimens and performed whole exome sequencing on the sorted cell populations, to identify the somatic mutations that may explain the intratumor heterogeneity of cells (EpCAM+ CSCs and EpCAM− non-CSCs in the same tumor). We further evaluated the identified somatic mutations in independent 57 HCC tissues and EpCAM expression status, to identify the somatic mutations that may explain the intertumor heterogeneity of HCCs (EpCAM-positive and -negative HCCs). Our aim was to examine whether EpCAM expression is associated with specific genetic mutations in EpCAM+ CSCs (intratumor heterogeneity) or EpCAM-positive HCCs (intertumor heterogeneity), and to determine whether HCC conforms to the clonal evolution or CSC model.
Methods
Cell culture
HuH1, HuH7, HLE, HLF, Hep3B, HEP-G2, SK-Hep-1, and PLC/PRL/5 human liver cancer cell lines were obtained from the Japanese Collection of Research Bioresources (JCRB; Osaka, Japan) or American Type Culture Collection (ATCC; Manassas, VA). Cells were routinely cultured in DMEM supplemented with 10% FBS. Two fresh HCC specimens (HCC1 and HCC2) were obtained and were used for xenotransplantation and to prepare single-cell suspensions for analysis. Primary HCC tissues were dissected and digested in 1 mg/mL type 4 collagenase (Sigma-Aldrich Japan K.K., Tokyo, Japan) solution at 37 °C for 15–30 min. Contaminated red blood cells were lysed with ammonium chloride solution (STEMCELL Technologies, Vancouver, BC, Canada) on ice for 5 min.
Fluorescence activated cell sorting (FACS)
Cultured cells were trypsinized, washed, and resuspended in Hank’s Balanced Salt Solution (Lonza, Basel, Switzerland) supplemented with 1% HEPES and 2% FBS. Cells were then incubated with antibodies on ice for 30 min. Labeled cells were analyzed by FACS using a FACSCalibur (BD Biosciences, San Jose, CA). The antibodies used were: a FITC-conjugated anti-EpCAM monoclonal antibody (Clone Ber-EP4; DAKO, Carpinteria, CA); an APC-conjugated anti-CD326 (EpCAM) antibody (Miltenyi Biotec K.K., Tokyo, Japan); an APC-conjugated anti-CD90 monoclonal antibody (Clone 5E10; eBioscience, San Diego, CA); an APC-conjugated anti-CD133/2 antibody (Clone 293C3; Miltenyi Biotec K.K.); an APC-conjugated anti-CD44 mouse monoclonal antibody (eBioscience); an APC-conjugated anti-CD13 antibody (eBioscience); and a PE-conjugated anti-CD24 antibody (Miltenyi Biotec K.K.).
Cell sorting
Cells were trypsinized, washed, and resuspended in Hank’s Balanced Salt Solution supplemented with 1% HEPES and 2% FBS. Cells were then incubated with an APC-conjugated anti-CD326 (EpCAM) on ice for 30 min, and EpCAM positive and negative cells were isolated using a BD FACSAria II cell sorting system (BD Biosciences). In addition, EpCAM+ and EpCAM− cells were also sorted for functional studies using an autoMACS pro cell separator and CD326 (EpCAM) microbeads (Miltenyi Biotec K.K.).
Immunohistochemistry (IHC) analyses
HCC tissue samples were obtained from patients who had undergone radical resection at the Center for Liver Diseases in Kanazawa University Hospital, Kanazawa, Japan. All patients provided informed consent and the tissue acquisition procedures were approved by the Ethics Committee of Kanazawa University. In total, 57 formalin-fixed paraffin-embedded HCC samples, resected between 2008 and 2011, were used for the IHC analyses. IHC was performed using EnVision+ Kits (DAKO, Carpinteria, CA), according to the manufacturer’s instructions. An anti-EpCAM monoclonal antibody (VU-1D9; Oncogene Research Products, San Diego, CA) was used to detect EpCAM expression.
Spheroid formation assays
For spheroid formation assays, single cell suspensions from HCC1 and HCC2 were generated using FACS and 1.5 × 104 HCC1 cells or 1.0 × 104 HCC2 cells were seeded in 6-well Ultra-Low Attachment Microplates (Corning, Corning, NY). The number of spheroids was determined 21 days after seeding.
Tumorigenicity in NOD/SCID mice
The protocol for animal procedures was approved by the Kanazawa University Animal Care and Use Committee. Cells were suspended in 200 µL of 1:1 DMEM:Matrigel (BD Biosciences) and were subcutaneously injected into 6-week-old NOD/SCID mice (NOD/NCrCRl-Prkdcscid) purchased from Charles River Laboratories, Inc. (Wilmington, MA). The size and incidence of subcutaneous tumors was recorded. For histological evaluation, tumors were formalin-fixed and paraffin-embedded prior to storage.
Quantitative reverse transcription-PCR analysis
Total RNA was extracted using High Pure RNA Isolation Kit (Roche Diagnostics K.K., Tokyo, Japan) according to the manufacturer’s instructions. The expression of selected genes was determined in triplicate using the 7900 Sequence Detection System (Applied Biosystems, Foster City, CA). Each sample was normalized relative to 18s rRNA expression. The probes used were PCDH18, Hs01556217_m1; CYP2R1, Hs01379776_m1; TECTA, Hs00193706_m1; ITGB8, Hs00174456_m1; CSMD1, Hs00899130_m1; PER1, Hs01092603_m1; ALKBH3, Hs00286731_m1; OSCP1, Hs00376771_m1; and 18s rRNA, Hs99999901_s1 (Applied Biosystems).
RNA interference
Small interfering RNAs (siRNAs) specific to PCDH18 (#1, HSS122980: #2, HSS122982) and a negative control (12935200) siRNA were designed and synthesized by Invitrogen (Invitrogen, Carlsbad, CA). A total of 2 × 105 cells were seeded into 6-well plates 24 h before transfection. Transfection was performed using Lipofectamine RNAiMAX Transfection Reagent (Invitrogen), according to the manufacturer’s instructions. A total of 20, 40, 60, and 100 pmol/L of siRNAs was used for each transfection in SK-Hep-1, HCC2, HLE, and HLF, respectively.
DNA extraction and whole exome sequencing
DNA extraction was performed using the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany). The SureSelect Human All Exon V4 Kit (Agilent Technologies, Santa Clara, CA) was used for whole exome capture, and the HiSeq 2000 Sequencing System (Illumina Inc., San Diego, CA) was used for massive parallel sequencing. The sequence reads were mapped against the University of California, Santa Cruz hg19 Genome Browser (http://hgdownload.cse.ucsc.edu/goldenPath/hg19/chromosomes/). Sequence variations, including single nucleotide polymorphisms and insertion/deletions were detected using the Genome Analysis Toolkit software (GATK; Broad Institute, Cambridge, MA). All of the whole exome sequencing and analysis was performed at Riken Genesis (Riken Genesis, Tokyo, Japan). To predict the effect of nonsynonymous single nucleotide substitutions on protein structure, function, and phenotype, we used tools available online, such as SIFT (http://sift.jcvi.org/) [22] and Polyphen2 (http://genetics.bwh.harvard.edu/pph2/) [23].
DNA extraction and Sanger sequencing
DNA extraction was performed using the QIAamp DNA Mini Kit (Qiagen). PCR Primers were designed by Primer-BLAST (http://www.ncbi.nlm.nih.gov/tools/primer-blast/). Primers are listed in Tables 1 and 2. PCR amplifications were performed using Takara Taq Hot Start Version (Takara, Shiga, Japan), or PrimeSTAR GXL DNA Polymerase (Takara) using a standard application protocol and the manufacturer’s instructions. PCR cleanup was performed using the QIAquick PCR Purification Kit (Qiagen). The cleaned PCR products were sequenced using a BigDyeTerminator v3.1 cycle sequencing KIT (Applied Biosystems). Sequenced products were run on the Life Technologies 3130xl Genetic Analyzer (Applied Biosystems). Electropherograms were visualized and analyzed using Sequence Scanner v2.0 software (Applied Biosystems).
Table 1.
Primers used for PCR amplification
| Gene | Exon number | Primer sequence |
|---|---|---|
| PCDH18 | Exon 1 | Forward 5′-TAATCTGGGAAGCAAGGGGAC-3′ |
| Reverse 5′-ACGACCAAACAAACGCAAGG-3′ | ||
| Exon 2 | Forward 5′-CACTGTCTCCTGCCTCACTG-3′ | |
| Reverse 5′-ATAGTTGGTAGCTGGCTGCG-3′ | ||
| Exon 3 | Forward 5′-GGCTGTATCGGATGAGGTGG-3′ | |
| Reverse 5′-CCAGCAGGTCTCTCAGCTTC-3′ | ||
| Exon 4 | Forward 5′-CAGTGGCTAGTTTCTACACGAC3′ | |
| Reverse 5′-TCACACCTAGTTCTTCCCACG-3′ |
Table 2.
Primers used for Sanger sequencing
| Gene | Target | Primer sequence |
|---|---|---|
| PCDH18 | Exon 1 #1 | 5′-GCTAAAGTGTGCATCTTTGCTAC-3′ |
| Exon 1 #2 | 5′-CAGCAACACTGCACAAATTGC-3′ | |
| Exon 1 #3 | 5′-CTTCGGGCTTCCTCCATCTC-3′ | |
| Exon 1 #4 | 5′-TCAGCCCAGAATCCTTGTCC-3′ | |
| Exon 1 #5 | 5′-TCTGAGGCAGTGAGCTGAAG-3′ | |
| Exon 2 | 5′-CACTGTCTCCTGCCTCACTG-3′ | |
| Exon 3 | 5′-GGCTGTATCGGATGAGGTGG-3′ | |
| Exon 4 #1 | 5′-CACACTTGCATTGTGTACATACG-3′ | |
| Exon 4 #2 | 5′-GAAGGCGGTAAGAGACGCTG-3′ |
Cell proliferation assays
For cell proliferation assays, single cell suspensions of 2 × 103 cells were seeded in 96-well plates, and cell density was evaluated 48 h after seeding using the Cell Counting Kit-8 (Dojindo Laboratories, Kumamoto, Japan) according to the manufacturer’s instructions.
Statistical analysis
Different test groups were compared using GraphPad Prism software 6.0 (GraphPad Software, San Diego, CA).
Results
EpCAM+ cells show CSC features in primary HCCs
For this study, we obtained two patient-derived hepatitis C virus-associated HCC samples (HCC1 and HCC2). FACS analysis indicated that both HCC1 and HCC2 contained subsets of EpCAM+ cells. HCC1 and HCC2 also contained subsets of cells expressing CD133, CD90, CD44, CD24, and CD13 (Fig. 1a). We enriched EpCAM+ and EpCAM− cell populations from HCC1 and HCC2 with > 80% purity using magnetic-activated cell sorting and > 95% purity using FACS. When the sorted EpCAM+ HCC cells were cultured for a week, the EpCAM+ fraction slightly decreased from 90.3 to 87.1% in HCC1 and 86.6 to 75.3% in HCC2 (Fig. 1b); this is consistent with the ability of EpCAM+ cells to divide asymmetrically in vitro and to generate EpCAM− cells. In sorted EpCAM+ cells in vivo xenografts, EpCAM+ cells also divided asymmetrically and produced tumors with a mixture of both EpCAM+ and EpCAM− cells (Fig. 1c). EpCAM+ cells also showed strong spheroid forming capacity compared with EpCAM− cells in vitro (Fig. 1d, e), and could efficiently initiate large tumors in NOD/SCID mice (Fig. 1f). All of these results suggest that the EpCAM+ cells are CSCs and HCC1 and HCC2 cells conform to the CSC hypothesis.
Fig. 1.
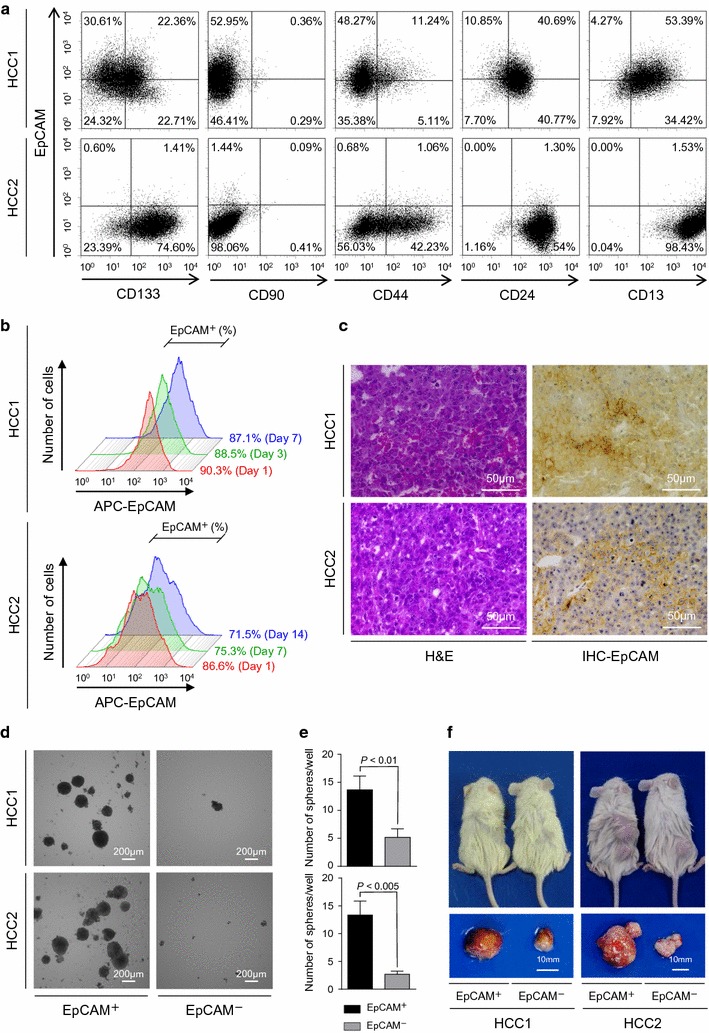
Hepatic stem cell marker expression in HCC1 and HCC2 cells. a Flow cytometry of HCC1 and HCC2 cells using fluorescently-labeled antibodies against EpCAM, CD133, CD90, CD44, CD24, and CD13. b Flow cytometry of EpCAM+ cells using an anti-EpCAM antibody. Figure shows EpCAM+ HCC1 cells on days 1, 3, and 7 after cell sorting and EpCAM+ HCC2 cells on days 1, 7, and 14 after cell sorting. c Histological analysis of EpCAM+ HCC1 and HCC2 xenografts. The figure shows hematoxylin and eosin (H&E) staining and anti-EpCAM immunohistochemistry (IHC) staining of the tumors. d Representative phase-contrast images of sorted EpCAM+ and EpCAM− HCC1 and HCC2 cell spheroids. e EpCAM+ and EpCAM− HCC1 and HCC2 spheroid formation. Experiments were performed in triplicate. Bars indicate the mean ± standard deviation. f Tumorigenic potential of EpCAM+ cells. Representative photomicrographs of NOD/SCID mice (upper panel) and subcutaneous tumors (lower panel) from EpCAM+ and EpCAM− HCC1 and HCC2 cell xenografts
Genomic features of patient-derived HCC1 and HCC2 cells
We performed whole exome analysis on the sorted EpCAM+ CSCs and EpCAM− non-CSCs in HCC1 and HCC2 cells, obtained from the two primary HCCs, and the patient’s matched peripheral blood mononuclear cells (PBMCs). In total, 19,263 nonsynonymous mutations were identified in the coding region. To isolate the potential somatic mutations that were only present in the EpCAM+ CSCs or EpCAM− non-CSCs, and were not present in the matched PBMCs, we performed nucleotide variants analysis by filtering the sequence data according to its qualities, sequence depth, and the available single nucleotide polymorphism database (Fig. 2a). We further selected genetic mutations that potentially impair the function of the encoded protein. Through this, we identified a known TP53 hotspot mutation in HCC2, c.844C>T; the c.844C>T mutation is reported to a induce p.R282W phenotype [24] and occurs in the second most frequently altered pathway in HCC [10]. In addition, we also identified, and confirmed by Sanger sequencing, 60 potential somatic mutations that affect 56 different genes in HCC1 and HCC2 (see Additional file 1). A novel TP53 missense mutation, c.767C>T, which would induce p.T256I, was identified in HCC1 (Fig. 2b). In HCC1, we predominantly detected mutations in genes associated with the chromatin remodeling pathway (ARID1A and ARID2) and NF-κB/MAPK signaling pathway (NTRK3, TLR5, and AR). In contrast, genes associated with the cell cycle G1/S checkpoint (CDKN1B and CDKN2A) and the vitamin D signaling pathway (CYP2R1 and CYP24A1) were predominantly detected in HCC2. These mutations might reflect frequently mutated pathways, such as TP53 in EpCAM-positive HCCs [25].
Fig. 2.
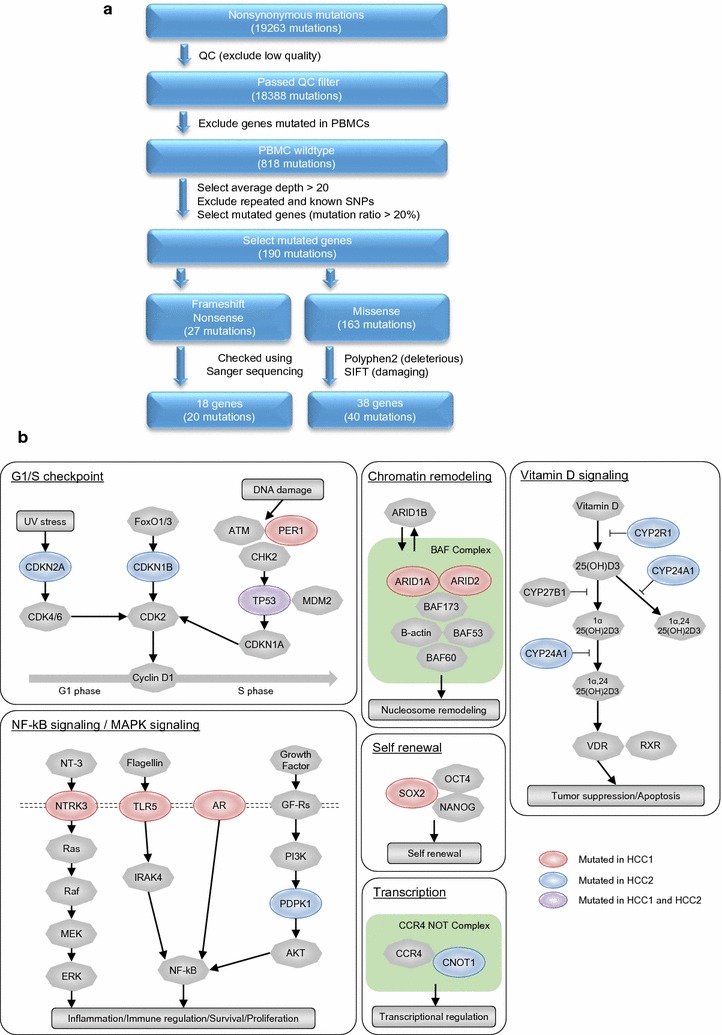
Whole exome sequencing analysis of HCC1 and HCC2. a Flowchart of the whole exome sequencing process. Nonsynonymous mutations were extracted from HCC1 and HCC2 cells. Boxes refer to major bioinformatic processes. Variants were filtered for their coding localization, annotation in dbSNP or 1000 genomes, and somatic and functional impairment. b The major pathways in which genetic mutations were detected in HCC1 and HCC2 cells. Somatic mutations detected in HCC1 or HCC2 are shown in red or blue, respectively. Mutations detected in both HCC1 and HCC2 are shown in purple
EpCAM+ CSCs and EpCAM− non-CSCs had similar somatic mutation patterns
We proceeded to select frameshift and nonsense mutations that have not previously been published to try to identify potential driver genes that alter protein function in HCC1 and HCC2. We identified 13 novel somatic mutations in HCC1 and HCC2 by Sanger sequencing (Table 3 and Fig. 3a). The number of point mutations as well as their function reported previously were assessed according to the previous publications and are available in Additional file 2 [26–40]. We tried to identify any mutations that were more frequently detected in either the sorted EpCAM+ CSCs or EpCAM− non-CSCs in HCC.
Table 3.
Previously unpublished frameshift and nonsense mutations
| Gene | Position | Mutation type | Mutant allele (%) in EpCAM+ CSCs fraction | Mutant allele (%) in EpCAM− non-CSCs fraction | Difference (%) EpCAM+ (%) − EpCAM− (%) |
Sample |
|---|---|---|---|---|---|---|
| PCDH18 | c.2556_2557delTG | Frameshift | 67.7 | 45.9 | 21.8 | HCC 1 |
| KIAA2026 | c.1411delC | Frameshift | 34.4 | 19.6 | 14.8 | HCC 1 |
| ALKBH3 | c.182delA | Frameshift | 68.4 | 54.5 | 13.9 | HCC 1 |
| CCDC168 | c.13741G>A | Nonsense | 33.2 | 28.9 | 4.3 | HCC 2 |
| CYP2R1 | c.881_882insG | Frameshift | 38.7 | 38.4 | 0.3 | HCC 2 |
| PER1 | c.709G>A | Nonsense | 100 | 100 | 0 | HCC 1 |
| OSCP1 | c.401_402insC | Frameshift | 94.3 | 95.2 | − 1 | HCC 2 |
| ITGB8 | c.391delG | Frameshift | 74.2 | 80 | − 5.8 | HCC 2 |
| INTS12 | c.914_917delGAAG | Frameshift | 25.5 | 33 | − 7.5 | HCC 1 |
| TECTA | c.5455A>T | Nonsense | 60.1 | 68.2 | − 8 | HCC 2 |
| MKI67 | c.2101C>A | Nonsense | 23.6 | 33 | − 9.4 | HCC 1 |
| TRIP4 | c1693_1694delAA | Frameshift | 28.9 | 38.5 | − 9.6 | HCC 2 |
| CSMD1 | c6711_6712delTG | Frameshift | 86 | 100 | − 14 | HCC 2 |
Fig. 3.
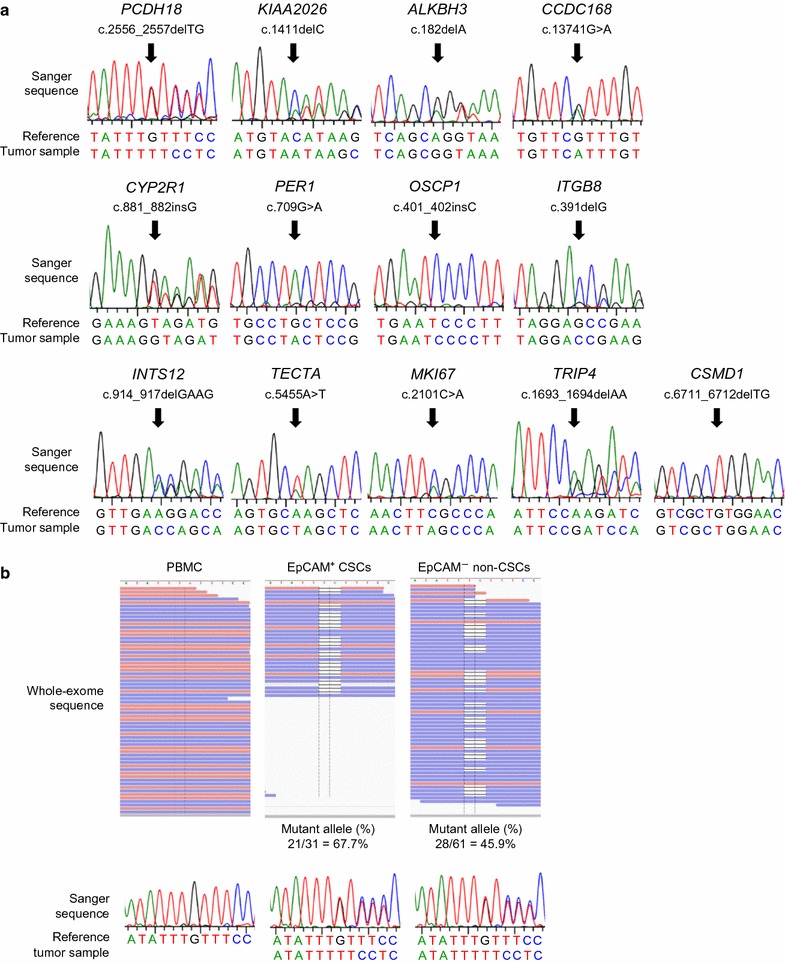
Validation of genetic mutations. a Electropherograms of the mutated sequences identified. b Frequency of PCDH18 mutation in PBMCs, EpCAM+ CSCs, and EpCAM− non-CSCs. Mutations were evaluated by whole exome sequencing (upper panel) and Sanger sequencing (lower panel)
Among them, PCDH18 was the most candidate gene frequently mutated in sorted EpCAM+ CSCs than in EpCAM− non-CSCs. However, this difference was too subtle and not clarified by conventional Sanger sequencing (Fig. 3b).
Furthermore, we could not detect other somatic mutations specific to sorted EpCAM+ CSCs or EpCAM− non-CSCs by whole exome sequencing. This indicates that the CSCs and non-CSCs showed similar somatic gene mutation patterns and frequencies at least in HCC1 and HCC2 that follow the CSC hypothesis.
PCDH18 mutations or loss of expression may be associated with the proliferation in EpCAM-positive HCC
Having identified 13 novel mutated genes in the HCC1 and HCC2 cells, we then evaluated their expression to determine whether gene expression is altered by the somatic mutations. We were able to successfully evaluate the expression of 8/13 of the genes in HCC1 cells, HCC2 cells, and eight standard HCC cell lines (Huh1, Huh7, HepG2, Hep3B, PLC/PRF/5, HLE, HLF, and SK-Hep-1; Fig. 4). Interestingly, the level of PCDH18 gene expression was significantly lower in EpCAM-positive HCC cells (HCC1, HCC2, Huh1, Huh7, HepG2, and Hep3B) compared with EpCAM-negative HCC cells (PLC/PRF/5, HLE, HLF, and SK-Hep-1; P = 0.038; Fig. 4). Although we did not detect PCDH18 mutations in the EpCAM-positive HCC cell lines, our data suggest that functional suppression of PCDH18, through somatic mutation or other mechanisms, may underlie the EpCAM-positive HCC cancer phenotype.
Fig. 4.
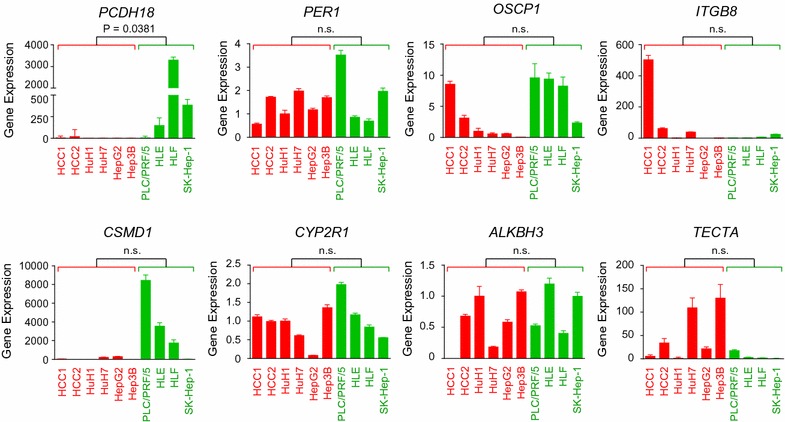
Alteration of gene expression in HCCs. Quantitative reverse transcription-PCR (qRT-PCR) analysis of HCC1 cells, HCC2 cells, and eight HCC cell lines. EpCAM-positive and EpCAM-negative HCC cells are indicated by red and green bars, respectively
Having determined that the level of PCDH18 gene expression was significantly lower in EpCAM-positive HCC cells, we proceeded to evaluate the frequency of PCDH18 mutation in 57 additional surgically resected HCC samples. Compared with matched non-tumor liver tissues, 3/57 (5.3%) HCCs harbored PCDH18 somatic mutations in the tumor tissues, which was validated by Sanger sequencing. We found the novel PCDH18 mutations including c.1474C>G (p.P492A), c.2337A>G (p.S780P), and c.2976G>T (p.N992K) missense mutations, and a c.2556_2557delTG frameshift mutation in HCC (Fig. 5a). We then evaluated EpCAM expression in the PCDH18-mutated HCC cases using IHC analysis (Fig. 5b). In all 57 of the HCC cases, 19/57 (33.3%) HCCs expressed EpCAM, and 3/19 (15.8%) harbored PCDH18 somatic mutations. In contrast, 38/57 (66.7%) HCCs did not express EpCAM, and using Sanger sequencing, no PCDH18 somatic mutations were detected in these EpCAM-negative HCCs; this difference was statistically significant (P = 0.033).
Fig. 5.
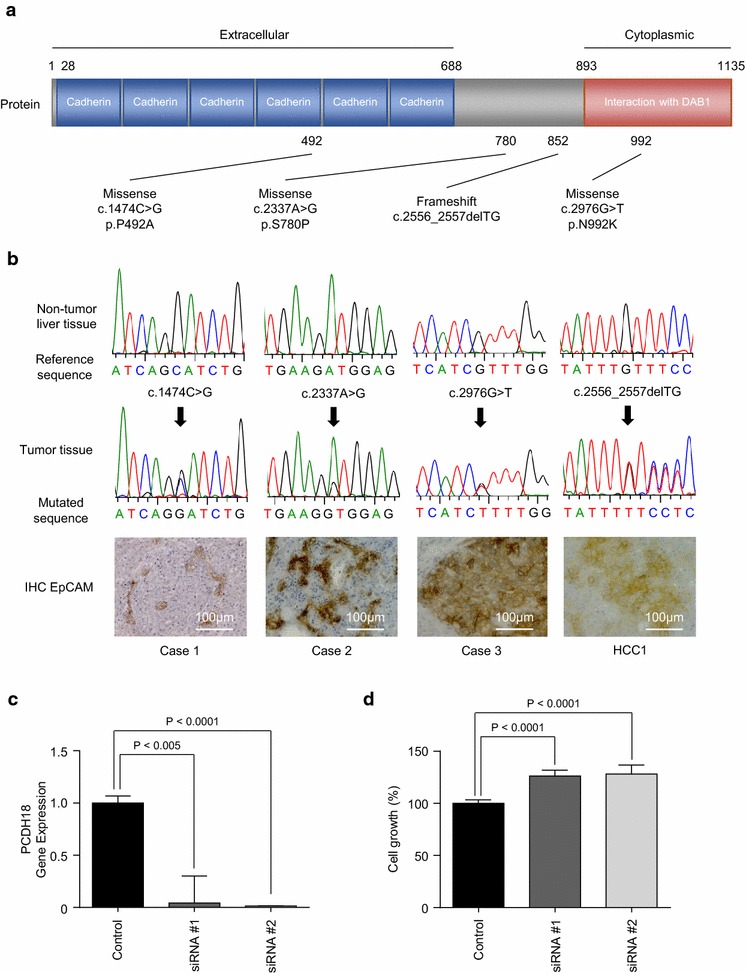
PCDH18 mutations in HCC. a Somatic mutation spectra in 57 HCCs. Nonsynonymous mutations are shown. Functional domains are indicated by colored boxes. b Electropherograms of PCDH18 mutated sequences in HCCs (upper). IHC analysis of HCCs (lower). c qRT-PCR analysis of PCDH18 in HCC2 cells transfected with control or PCDH18-specific siRNAs. d Cell proliferation assay of HCC2 cells transfected with control or PCDH18-specific siRNAs
To evaluate the functional role of PCDH18 in HCC, we transiently knocked down PCDH18 using small interfering RNAs (siRNAs; PCDH18 siRNA#1 and PCDH18 siRNA#2) in HCC2 cells. HCC2 cells were used because PCDH18 expression was only detectable in EpCAM-positive HCC2 cells, and could not be detected in the other EpCAM-positive HCC cells. PCDH18 expression was successfully suppressed using PCDH18-specific siRNAs compared with control siRNAs (Fig. 5c). Compared with the control, PCDH18 knockdown slightly enhanced cell proliferation in HCC2 cells (Fig. 5d). In contrast, knockdown of PCDH18 in EpCAM-negative HLE, HLF, and SK-Hep-1 cells resulted in the inhibition of cell proliferation (Fig. 6a–e). Taken together, our data suggests the different roles of PCDH18 on cell proliferation between EpCAM-positive and EpCAM-negative HCCs.
Fig. 6.
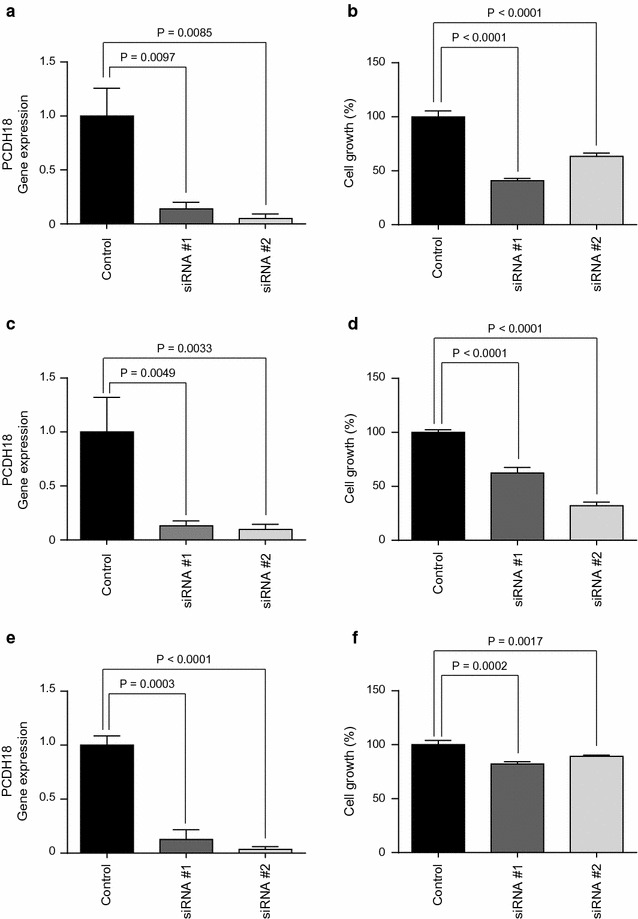
Cell proliferation assay of EpCAM-negative HCC cells. a qRT-PCR analysis of PCDH18 in HLE cells transfected with control or PCDH18-specific siRNAs. b Cell proliferation assay of HLE cells transfected with control or PCDH18-specific siRNAs. c qRT-PCR analysis of PCDH18 in HLF cells transfected with control or PCDH18-specific siRNAs. d Cell proliferation assay of HLF cells transfected with control or PCDH18-specific siRNAs. e qRT-PCR analysis of PCDH18 in SK-Hep-1 cells transfected with control or PCDH18-specific siRNAs. f Cell proliferation assay of SK-Hep-1 cells transfected with control or PCDH18-specific siRNAs
Discussion
It remains to be determined whether intratumor heterogeneity originates from the clonal evolution of tumor cells, with the step-wise acquisition of genetic changes (clonal evolution model), or a balance of self-renewal and differentiation by CSCs, which could potentially be regulated by the microenvironmental niche (CSC model). It is also possible that both models are true to a greater or lesser extent [41–44]. We have postulated that CSCs have a greater potential to acquire genetic mutations than non-CSCs because they are reported to be more resistant to chemo/radiation therapy, and are highly tumorigenic and metastatic. However, in the current study, we found that our two primary HCC cells that follow the CSC model had similar somatic mutation patterns in EpCAM+ CSCs and EpCAM− non-CSCs. This suggests that at certain points and conditions in the process of tumorigenesis, CSCs and non-CSCs are genetically similar, and that differences in their tumorigenic/metastatic ability may be conferred by signaling pathways rather than genetic alterations. However, because our data only reflect the exome status of two HCC nodules following the CSC hypothesis, it is possible that HCC CSCs acquire more genetic mutations at different organ sites or after chemo/radiation treatments that may confer a treatment-resistant phenotype. Further studies are required to evaluate the relationship between cancer cell evolution, CSCs, and treatment resistance.
Although we did not detect unique mutations that were enriched in EpCAM+ CSCs in our two primary HCC samples, we did identify a number of novel somatic mutations. One of these somatic mutations, PCDH18 (HCC1), was detected in 3/57 HCCs, and was significantly associated with EpCAM-positive HCC. The total PCDH18 mutation frequency was 5.3%, but in EpCAM-positive HCCs the PCDH18 mutation frequency was 15.8%. Furthermore, although we did not detect PCDH18 mutations in HCC cell lines, we did find that PCDH18 gene expression was suppressed in EpCAM-positive HCC cell lines compared with EpCAM-negative cell lines. These data suggest that a functional loss of PCDH18, by genetic mutation or other mechanisms such as epigenetic gene silencing, may be associated with the generation of EpCAM-positive HCCs. Indeed, PCDH18 knockdown in EpCAM-positive HCC2 cells resulted in a slightly enhanced rate of proliferation, indicating that the requirement for PCDH18 expression may have been bypassed in EpCAM-positive HCCs; in EpCAM-negative HCC cell lines, PCDH18 knockdown instead inhibited cell proliferation. Taken together, these data suggest that PCDH18 may play different roles in EpCAM-positive and EpCAM-negative HCCs.
Protocadherins (PCDHs) are members of the nonclassic subfamily of calcium-dependent cell–cell adhesion molecules, which is part of the cadherin superfamily [45]. The cadherin superfamily is classified into classical cadherins, desmosomal cadherins, and PCDHs. The PCDH family is largely divided into two groups based on their genomic structure: clustered PCDHs and non-clustered PCDHs. Non-clustered PCDHs are further classified into three subgroups: δ1, δ2, and ε.
PCDH18 belongs to the δ2-PCDH subgroup. Other δ2-PCDHs include PCDH8, PCDH10, PCDH17, and PCDH19 [46]. PCDH18 is reported to be expressed in the central nervous system and pharyngeal arches of zebrafish embryos [47], and plays a role in cell adhesion, behavior, and migration in zebrafish development [48]. Although the function of PCDH18 in humans is unclear, some studies have shown that PCDH18 deletion may be associated with altered brain development, intellectual disability, and multiple malformations with pulmonary hypertension [26–28, 49, 50]. Significantly, several δ2-PCDH members have been reported to function as tumor suppressor genes. For example, PCDH8 is genetically or epigenetically silenced in breast cancer [51] and mantle cell lymphoma [52]. The PCDH10 and PCDH17 promoter regions are reported to be hypermethylated in uterine cervical cancer [53], head and neck cancer, and some gastrointestinal cancers [54–56]. And our study has shown that a loss of PCDH18 gene expression may be related to the development of EpCAM-positive HCCs. In contrast, our data indicated the requirement of PCDH18 expression in EpCAM-negative HCC cell lines. We previously demonstrated that EpCAM+ CSCs show epithelial cell feature with highly tumorigenic capacity with activation of Wnt signaling, whereas CD90+ CSCs show mesenchymal cell feature with highly metastatic capacity with activation of c-Kit signaling. Furthermore, CD90+ CSCs were detected in all EpCAM-negative HCC cell lines [57]. Therefore, it is plausible that PCDH18 may have a role on maintenance of mesenchymal features of CD90+ CSCs. The different role of PCDH18 gene in different cellular contexts needs to be further evaluated in the future.
Conclusions
PCDH18 is mutated and might be functionally abrogated in a subset of EpCAM-positive HCCs. These data illustrated the potential role of PCDH18 as tumor suppressor, warranting the further exploration of its function in EpCAM-positive HCCs.
Additional files
Additional file 1. Validated nonsynonymous somatic mutations.
Additional file 2. The number of point mutations in 300 liver cancers and the function of 13 novel mutated genes.
Authors’ contributions
TH, TY, MH, and SK contributed to the study design. TH, TY, and HO contributed to data acquisition and analysis. KN, YH, YN, TH, YA, MY, NO, HS, and HT discussed the interpretation of the data. TH and TY wrote the manuscript. All authors read and approved the final manuscript.
Acknowledgements
We would like to thank Ms. Masayo Baba and Mika Ohwada for excellent technical assistance.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
All data used for this study are available from the corresponding author upon reasonable request.
Ethics approval and consent to participate
All patients provided written informed consent. The tissue acquisition procedures were approved by the Ethics Committee of Kanazawa University. The protocol for animal procedures was approved by the Kanazawa University Animal Care and Use Committee.
Funding
This study was supported by a Grant from the Japanese Ministry of Education, Culture, Sports, Science and Technology (23590967).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- HCC
hepatocellular carcinoma
- CSC
cancer stem cell
- EpCAM
epithelial cell adhesion molecule
- PCDH18
protocadherin 18
- FACS
fluorescence activated cell sorting
- qRT-PCR
quantitative reverse transcription-polymerase chain reaction
- siRNA
small interfering RNA
- IHC
immunohistochemistry
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1186/s12935-017-0467-x) contains supplementary material, which is available to authorized users.
Contributor Information
Takehiro Hayashi, Email: hayassy@live.jp.
Taro Yamashita, Phone: +81-76-265-2042, Email: taroy@m-kanazawa.jp.
Hikari Okada, Email: okada0922@gmail.com.
Kouki Nio, Email: niokouki2000@yahoo.co.jp.
Yasumasa Hara, Email: h_yasumasa173@yahoo.co.jp.
Yoshimoto Nomura, Email: y.nomura0331@gmail.com.
Tomoyuki Hayashi, Email: doctorugbyknzwtym@yahoo.co.jp.
Yoshiro Asahina, Email: yoshiro_11@yahoo.co.jp.
Mariko Yoshida, Email: marikoyoshida721@yahoo.co.jp.
Naoki Oishi, Email: naoooishi@gmail.com.
Hajime Sunagozaka, Email: suna@m-kanazawa.jp.
Hajime Takatori, Email: hajimetakatori@gmail.com.
Masao Honda, Email: mhonda@m-kanazawa.jp.
Shuichi Kaneko, Email: skaneko@m-kanazawa.jp.
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Bruix J, Gores GJ, Mazzaferro V. Hepatocellular carcinoma: clinical frontiers and perspectives. Gut. 2014;63(5):844–855. doi: 10.1136/gutjnl-2013-306627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132(7):2557–2576. doi: 10.1053/j.gastro.2007.04.061. [DOI] [PubMed] [Google Scholar]
- 4.Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, Davies H, Teague J, Butler A, Stevens C, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446(7132):153–158. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ley TJ, Mardis ER, Ding L, Fulton B, McLellan MD, Chen K, Dooling D, Dunford-Shore BH, McGrath S, Hickenbotham M, et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature. 2008;456(7218):66–72. doi: 10.1038/nature07485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shah SP, Morin RD, Khattra J, Prentice L, Pugh T, Burleigh A, Delaney A, Gelmon K, Guliany R, Senz J, et al. Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature. 2009;461(7265):809–813. doi: 10.1038/nature08489. [DOI] [PubMed] [Google Scholar]
- 7.Pleasance ED, Stephens PJ, O’Meara S, McBride DJ, Meynert A, Jones D, Lin ML, Beare D, Lau KW, Greenman C, et al. A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature. 2010;463(7278):184–190. doi: 10.1038/nature08629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ding L, Ellis MJ, Li S, Larson DE, Chen K, Wallis JW, Harris CC, McLellan MD, Fulton RS, Fulton LL, et al. Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature. 2010;464(7291):999–1005. doi: 10.1038/nature08989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Totoki Y, Tatsuno K, Yamamoto S, Arai Y, Hosoda F, Ishikawa S, Tsutsumi S, Sonoda K, Totsuka H, Shirakihara T, et al. High-resolution characterization of a hepatocellular carcinoma genome. Nat Genet. 2011;43(5):464–469. doi: 10.1038/ng.804. [DOI] [PubMed] [Google Scholar]
- 10.Guichard C, Amaddeo G, Imbeaud S, Ladeiro Y, Pelletier L, Maad IB, Calderaro J, Bioulac-Sage P, Letexier M, Degos F, et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet. 2012;44(6):694–698. doi: 10.1038/ng.2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang J, Deng Q, Wang Q, Li KY, Dai JH, Li N, Zhu ZD, Zhou B, Liu XY, Liu RF, et al. Exome sequencing of hepatitis B virus-associated hepatocellular carcinoma. Nat Genet. 2012;44(10):1117–1121. doi: 10.1038/ng.2391. [DOI] [PubMed] [Google Scholar]
- 12.Nault JC, Mallet M, Pilati C, Calderaro J, Bioulac-Sage P, Laurent C, Laurent A, Cherqui D, Balabaud C, Zucman-Rossi J. High frequency of telomerase reverse-transcriptase promoter somatic mutations in hepatocellular carcinoma and preneoplastic lesions. Nat Commun. 2013;4:2218. doi: 10.1038/ncomms3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Visvader JE. Cells of origin in cancer. Nature. 2011;469(7330):314–322. doi: 10.1038/nature09781. [DOI] [PubMed] [Google Scholar]
- 14.Yamashita T, Wang XW. Cancer stem cells in the development of liver cancer. J Clin Investig. 2013;123(5):1911–1918. doi: 10.1172/JCI66024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ma S, Tang KH, Chan YP, Lee TK, Kwan PS, Castilho A, Ng I, Man K, Wong N, To KF, et al. miR-130b Promotes CD133(+) liver tumor-initiating cell growth and self-renewal via tumor protein 53-induced nuclear protein 1. Cell Stem Cell. 2010;7(6):694–707. doi: 10.1016/j.stem.2010.11.010. [DOI] [PubMed] [Google Scholar]
- 16.Yang ZF, Ho DW, Ng MN, Lau CK, Yu WC, Ngai P, Chu PW, Lam CT, Poon RT, Fan ST. Significance of CD90+ cancer stem cells in human liver cancer. Cancer Cell. 2008;13(2):153–166. doi: 10.1016/j.ccr.2008.01.013. [DOI] [PubMed] [Google Scholar]
- 17.Zhu Z, Hao X, Yan M, Yao M, Ge C, Gu J, Li J. Cancer stem/progenitor cells are highly enriched in CD133+CD44+ population in hepatocellular carcinoma. Int J Cancer. 2010;126(9):2067–2078. doi: 10.1002/ijc.24868. [DOI] [PubMed] [Google Scholar]
- 18.Lee TK, Castilho A, Cheung VC, Tang KH, Ma S, Ng IO. CD24(+) liver tumor-initiating cells drive self-renewal and tumor initiation through STAT3-mediated NANOG regulation. Cell Stem Cell. 2011;9(1):50–63. doi: 10.1016/j.stem.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 19.Haraguchi N, Ishii H, Mimori K, Tanaka F, Ohkuma M, Kim HM, Akita H, Takiuchi D, Hatano H, Nagano H, et al. CD13 is a therapeutic target in human liver cancer stem cells. J Clin Invest. 2010;120(9):3326–3339. doi: 10.1172/JCI42550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamashita T, Forgues M, Wang W, Kim JW, Ye Q, Jia H, Budhu A, Zanetti KA, Chen Y, Qin LX, et al. EpCAM and alpha-fetoprotein expression defines novel prognostic subtypes of hepatocellular carcinoma. Cancer Res. 2008;68(5):1451–1461. doi: 10.1158/0008-5472.CAN-07-6013. [DOI] [PubMed] [Google Scholar]
- 21.Yamashita T, Ji J, Budhu A, Forgues M, Yang W, Wang HY, Jia H, Ye Q, Qin LX, Wauthier E, et al. EpCAM-positive hepatocellular carcinoma cells are tumor-initiating cells with stem/progenitor cell features. Gastroenterology. 2009;136(3):1012–1024. doi: 10.1053/j.gastro.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001;11(5):863–874. doi: 10.1101/gr.176601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Muller PA, Vousden KH. p53 mutations in cancer. Nat Cell Biol. 2013;15(1):2–8. doi: 10.1038/ncb2641. [DOI] [PubMed] [Google Scholar]
- 25.Woo HG, Wang XW, Budhu A, Kim YH, Kwon SM, Tang ZY, Sun Z, Harris CC, Thorgeirsson SS. Association of TP53 mutations with stem cell-like gene expression and survival of patients with hepatocellular carcinoma. Gastroenterology. 2011;140(3):1063–1070. doi: 10.1053/j.gastro.2010.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kasnauskiene J, Ciuladaite Z, Preiksaitiene E, Matuleviciene A, Alexandrou A, Koumbaris G, Sismani C, Pepalyte I, Patsalis PC, Kucinskas V. A single gene deletion on 4q28.3: PCDH18—a new candidate gene for intellectual disability? Eur J Med Genet. 2012;55(4):274–277. doi: 10.1016/j.ejmg.2012.02.010. [DOI] [PubMed] [Google Scholar]
- 27.Iruretagoyena JI, Davis W, Bird C, Olsen J, Radue R, Teo Broman A, Kendziorski C, Splinter BonDurant S, Golos T, Bird I, et al. Differential changes in gene expression in human brain during late first trimester and early second trimester of pregnancy. Prenat Diagn. 2014;34(5):431–437. doi: 10.1002/pd.4322. [DOI] [PubMed] [Google Scholar]
- 28.Duga B, Czako M, Komlosi K, Hadzsiev K, Torok K, Sumegi K, Kisfali P, Kosztolanyi G, Melegh B. Deletion of 4q28.3-31.23 in the background of multiple malformations with pulmonary hypertension. Mol Cytogenet. 2014;7:36. doi: 10.1186/1755-8166-7-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beharry AA, Lacoste S, O’Connor TR, Kool ET. Fluorescence monitoring of the oxidative repair of DNA alkylation damage by ALKBH3, a prostate cancer marker. J Am Chem Soc. 2016;138(11):3647–3650. doi: 10.1021/jacs.6b00986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dango S, Mosammaparast N, Sowa ME, Xiong LJ, Wu F, Park K, Rubin M, Gygi S, Harper JW, Shi Y. DNA unwinding by ASCC3 helicase is coupled to ALKBH3-dependent DNA alkylation repair and cancer cell proliferation. Mol Cell. 2011;44(3):373–384. doi: 10.1016/j.molcel.2011.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Christakos S, Dhawan P, Verstuyf A, Verlinden L, Carmeliet G. Vitamin D: metabolism, molecular mechanism of action, and pleiotropic effects. Physiol Rev. 2016;96(1):365–408. doi: 10.1152/physrev.00014.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sahar S, Sassone-Corsi P. Metabolism and cancer: the circadian clock connection. Nat Rev Cancer. 2009;9(12):886–896. doi: 10.1038/nrc2747. [DOI] [PubMed] [Google Scholar]
- 33.Yi M, Yang J, Li W, Li X, Xiong W, McCarthy JB, Li G, Xiang B. The NOR1/OSCP1 proteins in cancer: from epigenetic silencing to functional characterization of a novel tumor suppressor. J Cancer. 2017;8(4):626–635. doi: 10.7150/jca.17579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pozzi A, Zent R. TGF-beta sequestration by mesangial cell integrin alphavbeta8: a novel mechanism of glomerular endothelial cell regulation. Am J Pathol. 2011;178(2):485–489. doi: 10.1016/j.ajpath.2010.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kheirallah AK, de Moor CH, Faiz A, Sayers I, Hall IP. Lung function associated gene Integrator Complex subunit 12 regulates protein synthesis pathways. BMC Genom. 2017;18(1):248. doi: 10.1186/s12864-017-3628-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Asgharzade S, Tabatabaiefar MA, Modarressi MH, Ghahremani MH, Reiisi S, Tahmasebi P, Abdollahnejad F, Chaleshtori MH. A novel TECTA mutation causes ARNSHL. Int J Pediatr Otorhinolaryngol. 2017;92:88–93. doi: 10.1016/j.ijporl.2016.11.010. [DOI] [PubMed] [Google Scholar]
- 37.Cuylen S, Blaukopf C, Politi AZ, Muller-Reichert T, Neumann B, Poser I, Ellenberg J, Hyman AA, Gerlich DW. Ki-67 acts as a biological surfactant to disperse mitotic chromosomes. Nature. 2016;535(7611):308–312. doi: 10.1038/nature18610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ruiz A, Heilmann S, Becker T, Hernandez I, Wagner H, Thelen M, Mauleon A, Rosende-Roca M, Bellenguez C, Bis JC, et al. Follow-up of loci from the International Genomics of Alzheimer’s Disease Project identifies TRIP4 as a novel susceptibility gene. Transl Psychiatry. 2014;4:e358. doi: 10.1038/tp.2014.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Escudero-Esparza A, Bartoschek M, Gialeli C, Okroj M, Owen S, Jirstrom K, Orimo A, Jiang WG, Pietras K, Blom AM. Complement inhibitor CSMD1 acts as tumor suppressor in human breast cancer. Oncotarget. 2016;7(47):76920–76933. doi: 10.18632/oncotarget.12729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fujimoto A, Furuta M, Totoki Y, Tsunoda T, Kato M, Shiraishi Y, Tanaka H, Taniguchi H, Kawakami Y, Ueno M, et al. Whole-genome mutational landscape and characterization of noncoding and structural mutations in liver cancer. Nat Genet. 2016;48(5):500–509. doi: 10.1038/ng.3547. [DOI] [PubMed] [Google Scholar]
- 41.Gerlinger M, Horswell S, Larkin J, Rowan AJ, Salm MP, Varela I, Fisher R, McGranahan N, Matthews N, Santos CR, et al. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat Genet. 2014;46(3):225–233. doi: 10.1038/ng.2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366(10):883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li L, Cole J, Margolin DA. Cancer stem cell and stromal microenvironment. Ochsner J. 2013;13(1):109–118. [PMC free article] [PubMed] [Google Scholar]
- 44.Marquardt JU, Factor VM, Thorgeirsson SS. Epigenetic regulation of cancer stem cells in liver cancer: current concepts and clinical implications. J Hepatol. 2010;53(3):568–577. doi: 10.1016/j.jhep.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wolverton T, Lalande M. Identification and characterization of three members of a novel subclass of protocadherins. Genomics. 2001;76(1–3):66–72. doi: 10.1006/geno.2001.6592. [DOI] [PubMed] [Google Scholar]
- 46.Kim S-Y, Yasuda S, Tanaka H, Yamagata K, Kim H. Non-clustered protocadherin. Cell Adhes Migr. 2011;5(2):97–105. doi: 10.4161/cam.5.2.14374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kubota F, Murakami T, Tajika Y, Yorifuji H. Expression of protocadherin 18 in the CNS and pharyngeal arches of zebrafish embryos. Int J Dev Biol. 2008;52(4):397–405. doi: 10.1387/ijdb.072424fk. [DOI] [PubMed] [Google Scholar]
- 48.Aamar E, Dawid IB. Protocadherin-18a has a role in cell adhesion, behavior and migration in zebrafish development. Dev Biol. 2008;318(2):335–346. doi: 10.1016/j.ydbio.2008.03.040. [DOI] [PubMed] [Google Scholar]
- 49.Yue W, Wang T, Zachariah E, Lin Y, Yang CS, Xu Q, DiPaola RS, Tan XL. Transcriptomic analysis of pancreatic cancer cells in response to metformin and aspirin: an implication of synergy. Sci Rep. 2015;5:13390. doi: 10.1038/srep13390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gao L, Dang X, Huang L, Zhu L, Fang M, Zhang J, Xu X, Zhu L, Li T, Zhao L, et al. Search for the potential “second-hit” mechanism underlying the onset of familial hemophagocytic lymphohistiocytosis type 2 by whole-exome sequencing analysis. Transl Res. 2016;170:26–39. doi: 10.1016/j.trsl.2015.12.004. [DOI] [PubMed] [Google Scholar]
- 51.Yu JS, Koujak S, Nagase S, Li CM, Su T, Wang X, Keniry M, Memeo L, Rojtman A, Mansukhani M, et al. PCDH8, the human homolog of PAPC, is a candidate tumor suppressor of breast cancer. Oncogene. 2008;27(34):4657–4665. doi: 10.1038/onc.2008.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Leshchenko VV, Kuo PY, Shaknovich R, Yang DT, Gellen T, Petrich A, Yu Y, Remache Y, Weniger MA, Rafiq S, et al. Genomewide DNA methylation analysis reveals novel targets for drug development in mantle cell lymphoma. Blood. 2010;116(7):1025–1034. doi: 10.1182/blood-2009-12-257485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang KH, Liu HW, Lin SR, Ding DC, Chu TY. Field methylation silencing of the protocadherin 10 gene in cervical carcinogenesis as a potential specific diagnostic test from cervical scrapings. Cancer Sci. 2009;100(11):2175–2180. doi: 10.1111/j.1349-7006.2009.01285.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yu B, Yang H, Zhang C, Wu Q, Shao Y, Zhang J, Guan M, Wan J, Zhang W. High-resolution melting analysis of PCDH10 methylation levels in gastric, colorectal and pancreatic cancers. Neoplasma. 2010;57(3):247–252. doi: 10.4149/neo_2010_03_247. [DOI] [PubMed] [Google Scholar]
- 55.Ying J, Li H, Seng TJ, Langford C, Srivastava G, Tsao SW, Putti T, Murray P, Chan AT, Tao Q. Functional epigenetics identifies a protocadherin PCDH10 as a candidate tumor suppressor for nasopharyngeal, esophageal and multiple other carcinomas with frequent methylation. Oncogene. 2006;25(7):1070–1080. doi: 10.1038/sj.onc.1209154. [DOI] [PubMed] [Google Scholar]
- 56.Haruki S, Imoto I, Kozaki K, Matsui T, Kawachi H, Komatsu S, Muramatsu T, Shimada Y, Kawano T, Inazawa J. Frequent silencing of protocadherin 17, a candidate tumour suppressor for esophageal squamous cell carcinoma. Carcinogenesis. 2010;31(6):1027–1036. doi: 10.1093/carcin/bgq053. [DOI] [PubMed] [Google Scholar]
- 57.Yamashita T, Honda M, Nakamoto Y, Baba M, Nio K, Hara Y, Zeng SS, Hayashi T, Kondo M, Takatori H, et al. Discrete nature of EpCAM+ and CD90+ cancer stem cells in human hepatocellular carcinoma. Hepatology. 2013;57(4):1484–1497. doi: 10.1002/hep.26168. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. Validated nonsynonymous somatic mutations.
Additional file 2. The number of point mutations in 300 liver cancers and the function of 13 novel mutated genes.
Data Availability Statement
All data used for this study are available from the corresponding author upon reasonable request.