

Abstract
Background
Wilms tumor, aniridia, genitourinary anomalies and mental retardation (WAGR) syndrome is a rare genetic disorder caused by heterozygous deletions of WT1 and PAX6 at chromosome 11p13. Deletion of BDNF is known eto be associated with hyperphagia and obesity in both humans and animal models; however, neuroendocrine and epigenetic profiles of individuals with WAGR syndrome remain to be determined.
Case presentation
We report a 5-year-old girl with the typical phenotype of WAGR syndrome. She showed profound delays in physical growth, motor and cognitive development without signs of obesity. Array comparative genome hybridization (CGH) revealed that she carried a 14.4 Mb deletion at 11p14.3p12, encompassing the WT1, PAX6 and BDNF genes. She experienced recurrent hypoglycemic episodes at 5 years of age. Insulin tolerance and hormonal loading tests showed normal hypothalamic responses to the hypoglycemic condition and other stimulations. Methylation analysis for freshly prepared DNA from peripheral lymphocytes using the pyro-sequencing-based system showed normal patterns of methylation at known imprinting control regions.
Conclusions
Children with WAGR syndrome may manifest profound delay in postnatal growth through unknown mechanisms. Epigenetic factors and growth-associated genes in WAGR syndrome remain to be characterized.
Electronic supplementary material
The online version of this article (10.1186/s12881-017-0477-5) contains supplementary material, which is available to authorized users.
Keywords: Wilms tumor, Aniridia, Genitourinary anomalies and mental retardation (WAGR) syndrome; Epigenetics; Neuroendocrine function; Methylation
Background
Wilms tumor, aniridia, genitourinary anomalies and mental retardation (WAGR) syndrome is a rare genetic disorder caused by the chromosomal defect at 11p13.3 [1]. Genetically, segmental deletions encompassing the WT1 and PAX6 genes are known to cause the syndromic phenotype, while an accompanying phenotype of obesity is linked to the extensive deletion involving the brain-derived neurotrophic factor (BDNF) locus [2]. Thus, patients with the WAGR and obesity (O) phenotypes are designated as having WAGRO syndrome [2].
Imprinting-associated disorders commonly present with abnormal physical growth and autism. Prader-Willi syndrome is an example of such diseases that are phenotypically characterized by muscular hypotonia in infancy, short stature and hyperphagic behavior with cognitive impairment later in childhood [3]. Although autistic and hyperphagic behaviors are known to overlap between WAGRO and Prader-Willi syndromes, no information is currently available for epigenetic impacts of BDNF insufficiency on gene expression in the developing brain of humans and animals. This study was thus directed to test whether WAGRO syndrome might have deregulated conditions in neuroendocrine or methylation profiles.
Case presentation
A 1-year and 10-month-old girl was referred to our department for assessment of growth delay and absence of speech. She was born at the 41st week of gestational age with birth weight of 2950 g. Congenital aniridia, cataracts and macular hypoplasia were noticed at birth. Genitourinary apparatus was normal in appearance.
When she was first referred to our department at 1 year and 10 months of age, her height (74.4 cm, −2.8SD) and weight (8.5 kg, −3.3SD) were evaluated to be small for age. At this age, her motor skills and cognitive development were considered as low as 10 months of age. She developed bilateral renal tumors at 2 years and 1 month of age, for which surgical resection was conducted. Pathological examination confirmed the diagnosis of Wilms tumor. The renal tumors responded favorably to standard regimens of chemotherapy. She was well nourished without recurrence of tumors for over 4 years after the chemotherapy. Thus, the clinical features of this case fit to the typical phenotypes of WAGR syndrome, as previously reported [1, 2, 4].
The G-band test revealed the karyotype of 46,XX,del(11)(p11.2p14) (Fig 1a). An array-comparative genome hybridization analysis (Clinical Microarray ver. 8.1 at Baylor MGL), and found the 14.5-Mb deletion at 11p14.3p12 (Fig 1a). The minimal segment of deletion were defined as arr[GRCh37] 11p14.3p12(24,792,569_39,222,929)×1. According to the GRCh37/hg19 assembly, the deleted region was located at the chromosomal band, 11p14.3p12. The deleted interval involved 50 genes in the adjacent loci, including PAX6 (chr11:31,806,340–31,839,509) and WT1 (chr11:32,409,322–32,457,081). The heterozygous deletions of WT1 and BDNF were validated with FISH and qPCR analyses (Fig. 1b, c; see Additional file 1). We further excluded that this patient had a variant coding sequence of BDNF at the non-deleted allele (data not shown). Together, these data indicate that the heterozygous deletion of BDNF, but not a hypomorphic mutation, such as Val66Met variant [5], caused the developmental delay in this patient. Because we were not allowed to analyze the parental chromosomes, it remained to be determined whether the deletion occurred de novo or as the consequence of chromosomal defects with lower penetrance, such as balanced translocations.
Fig. 1.
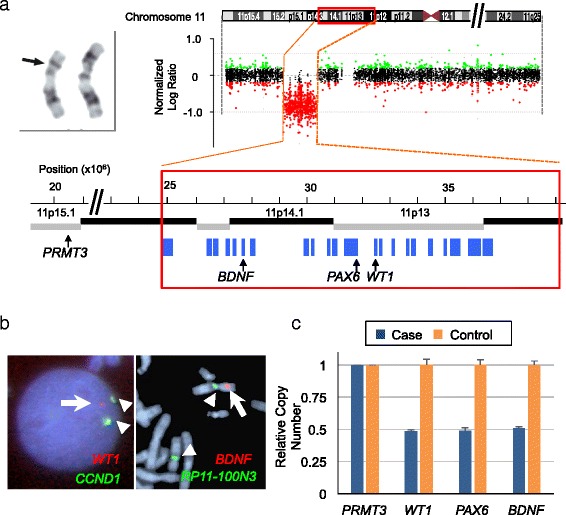
Genetic analyses of the present cases. a Array-CGH data for the present case. The left-top panel depicts the abnormal G-band pattern (arrow) of chromosome 11 in the peripheral leukocytes. On the right top, the log2 values of relative copy numbers are plotted against the base position (x10E6) at chromosome 11. Color codes represent gain (green) or loss (red) of the probed regions. The horizontal dashed lines indicate the thresholds of duplication (0.5) and heterozygous deletion (−1.0). The lower-panel shows a magnified view of the deleted region. G-band locus (thick, black and gray lines) and protein-coding genes (blue boxes) are mapped on the coordinates available at the UCSC genome browser (GRCh37/hg19). Chromosomal locations of the three WAGR syndrome-associated genes (BDNF, PAX6 and WT1) are denoted under the diagram. b The FISH image. The green (arrowheads) and red (arrows) signals show CCND1 (control) and WT1 loci, respectively. Note that one copy of the WT1 (arrow) is missing while the two copies of CCND1 signals (arrowheads) are unaffected. c Quantitative PCR data for this case and a healthy control. PRMT3 (chr11:20,409,076–20,530,879) serves as an internal control. Bar plots show the relative copy numbers of WT1, PAX6 and BDNF (mean ± SD, n = 4) to that of PRMT3 for each individual
At 5 years of age, she reached 91.3 cm (−3.5 SD) of height and 12.45 kg (−2.2 SD) of body weight. She had recurrent episodes of ketogenic hypoglycemia on starvation. To assess whether she may have neuro-endocrine dysfunction, we conducted a series of hormonal loading tests. Insulin tolerance test (0.1 U/kg) revealed that her blood glucose rapidly declined to 35 mg/dl after infusion, while growth hormone (GH), adrenocorticotropic hormone (ACTH) and cortisol were all normally increased in response to the hypoglycemic condition at 15–30 min (Additional file 1: Figure S2A). Combined infusion of 500 μg/kg L-arginine, 1.5 μg/kg CRH and 10 μg/kg TRH induced normal patterns of release in corresponding hormones (Additional file 1: Figure S2B). Oral glucose tolerance test with 1.75 g/kg of glucose resulted in normally fluctuated blood glucose and insulin levels, while lactate and pyruvate did not show abnormal patterns of accumulation (Additional file 1: Figure S2C). These data reassured that she had no obvious predisposing causes for hypoglycemia. She was likely intolerant of starved conditions owing to immature energy homeostasis as frequently observed in preschool children, but not as a consequence of WAGR-relevant phenotype.
This case had typical growth and neurodevelopmental profiles as WAGR syndrome with large chromosomal deletions (Table 1) [1]. We therefore considered it valuable to characterize her epigenetic profiles, which might disclose one of molecular mechanisms for delays in postnatal growth and development in WAGR syndrome. Recent studies indicated that chromosomal defects in the WAGR-critical region encompassed the LGR4 gene [6]. The heterozygous deletion of LGR4 was reported to cause the secondary effects on the epigenetic status through disturbing the expression of chromatin modifier proteins. Towards this end, we investigated whether children with WAGR syndrome might bear aberrant methylation profiles on previously known imprinting regions. However, this case had normal methylation patterns at known loci for imprinting-associated disorders when using lymphocytes (Additional file 1: Table S1) [7]. CARE guidelines were followed in reporting this case.
Table 1.
Phenotypic spectrum of WAGR syndrome with 10 Mb or larger deletions
| No | Case ID | Age | Sex | Reference (Author, Year) | Cytoband | Size (Mb) | Genome Assembly |
Start point | End point | Height | Body weight |
Neurological Signsa |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | F24a | 16 | F | Xu S, 2008 [4] | 11p12p15.1 | 23.0 | GRCh 35 (hg17) | 20,040,419 | 43,071,919 | NA | NA | ID, OCD |
| 2 | F8a | 11 | F | Xu S, 2008 [4] | 11p14p12.3 | 19.1 | GRCh 35 (hg17) | 25,086,501 | 44,212,278 | NA | NA | ID, OCD, ASD |
| 3 | NIH10 | 6 | M | Xu S, 2008 [4] | 11p11.2p14.1 | 18.2 | GRCh 35 (hg17) | 27,656,700 | 45,858,752 | NA | NA | DD |
| 4 | F17a | 36 | F | Xu S, 2008 [4] | 11p14p12.3 | 15.6 | GRCh 35 (hg17) | 25,086,501 | 40,651,423 | NA | NA | ID, ASD, ADHD, OCD, Anxiety |
| 5 | NIH3 | 4 | M | Xu S, 2008 [4] | 11p14p12.3 | 15.3 | GRCh 35 (hg17) | 24,202,909 | 39,529,029 | NA | NA | DD |
| 6 | F10a | 7 | F | Xu S, 2008 [4] | 11p11.2p13 | 14.9 | GRCh 35 (hg17) | 31,284,414 | 46,194,871 | NA | NA | ID, DD, OCD, Anxiety |
| 7 | F20a | 15 | M | Xu S, 2008 [4] | 11p14p12.3 | 14.6 | GRCh 35 (hg17) | 25,844,571 | 40,482,022 | NA | NA | ID, DD, ASD, ADHD, OCD, Anxiety |
| 8 | YS 013 | 5 | F | [This study] | 11p14.3p12 | 14.4 | GRCh37 (hg19) | 24,792,569 | 39,222,929 | 91.3 cm (−3.5 SD) |
12.45 kg (−2.2 SD) |
DD, ASD, Hyperphagia |
| 9 | NIH9 | 7 | F | Xu S, 2008 [4] | 11p13p15.1 | 14.4 | GRCh 35 (hg17 | 20,759,560 | 35,124,532 | NA | NA | ID, ASD |
| 10 | F7a | 13 | F | Xu S, 2008 [4] | 11p14p12.3 | 14.3 | GRCh 35 (hg17 | 24,542,321 | 38,824,714 | NA | NA | ASD, DD, ADHD |
| 11 | NIH6 | 17 | M | Xu S, 2008 [4] | 11p14p12.2 | 14.2 | GRCh 35 (hg17 | 26,005,134 | 40,174,102 | NA | NA | ID, ASD, ADHD, Anxiety |
| 12 | NIH2 | 19 | M | Xu S, 2008 [4] | 11p13p15.1 | 13.5 | GRCh 35 (hg17 | 20,135,621 | 33,614,001 | NA | NA | ID, ASD |
| 13 | F3a | 26 | F | Xu S, 2008 [4] | 11p12p13 | 12.9 | GRCh 35 (hg17 | 31,284,414 | 44,212,278 | NA | NA | ID, DD, Seizure, Depression |
| 14 | F21a | 12 | F | Xu S, 2008 [4] | 11p14p12.3 | 12.5 | GRCh 35 (hg17) | 25,336,593 | 37,873,278 | NA | NA | ADHD, OCD, Anxiety |
| 15 | F14a | 12 | F | Xu S, 2008 [4] | 11p14p12.1 | 12.2 | GRCh 35 (hg17) | 28,001,853 | 40,239,548 | NA | NA | ID, ASD, OCD, Anxiety, ADHD |
| 16 | F1a | 11 | M | Xu S, 2008 [4] | 11p13p14.3 | 11.4 | GRCh 35 (hg17) | 24,848,855 | 36,266,146 | NA | NA | ID, ASD, Anxiety, OCD |
| 17 | NIH11 | 4 | M | Xu S, 2008 [4] | 11p14p12.2 | 10.7 | GRCh 35 (hg17) | 26,690,778 | 37,341,623 | NA | NA | ID, ASD |
| 18 | F23a | 16 | M | Xu S, 2008 [4] | 11p14p12.1 | 10.6 | GRCh 35 (hg17) | 27,675,634 | 38,235,380 | NA | NA | ID, ASD, DD, OCD |
| 19 | Patient 2 | 2 | F | Yamamoto T, 2013 [13] |
11p13p12 | 10.5 | GRCh 35 (hg17) | 32,990,627 | 43,492,580 | 75.6 cm (−3.5SD) |
11.9 kg (+0.8SD) |
DD |
| 20 | NIH1 | 26 | M | Xu S, 2008 [4] | 11p14p12.1 | 10.2 | GRCh 35 (hg17) | 27,692,635 | 37,916,281 | NA | NA | ID, ASD |
| 21 | F19a | 5 | M | Xu S, 2008 [4] | 11p14p12.2 | 10.1 | GRCh 35 (hg17) | 26,699,475 | 36,757,882 | NA | NA | ID, DD, ASD |
a Abbreviations: ID intellectual disability, OCD obsessive compulsive disorder, ASD, autism spectrum disorder, DD, developmental delay, ADHD, attention deficit hyperactive disorder
Discussion and conclusions
We described a girl with typical phenotypes of WAGR syndrome. By defining the deleted interval in the short arm of chromosome 11, we confirmed that she had heterozygous deletions of WT1, PAX6 and BDNF1. The BDNF gene encodes brain-derived neurotrophic factor, one of the most potent activators of neurogenesis and synaptogenesis in the central nervous system [8]. Precise mechanisms for developing the obesity phenotype with BDNF deletion remain to be elusive; however, hypothalamic dysfunctions due to hypomorphic mutation of BDNF have been proposed in both humans and animal models [9, 10]. Although our case did not show hyperphagic behaviors during this study period, her metabolic status may be deranged to drive this phenotype in later stages.
BDNF and its cognate receptor, tyrosine receptor kinase B (TrkB), coordinate glucose metabolism and energy homeostasis in the hypothalamus [11, 12]. Because we were unable to detect hyper- or hypo-functioning responses to hormonal loading tests in this case, she was likely to experience hypoglycemic episodes on starvation without specific causes in the neuro-endocrine system. However, one could still argue that such hypothalamic dysfunctions might be evident only when she develops obesity. Thus, serial observation of physical growth and its correlation with hyperphagic and autistic behaviors in childhood through adolescence will be necessary to clarify the neural bases for obese phenotype of WAGR syndrome.
We further verified that she had unaltered methylation profiles at the CpG sites of previously known imprinting genes. Although it still remains unknown why the segmental deletion at 11p15 lead to short stature in WAGR syndrome in early childhood, these data provide a clue for directing future epigenetic study on WAGR syndrome. Consecutive evidence in future studies will warrant these data and provide insight into genotype-phenotype correlations for children with WAGR syndrome.
Acknowledgments
We thank staffs at Baylor MGL for array-CGH analysis; Yoshiaki Kinoshita, Tomoaki Taguchi (Department of Pediatric Surgery, Kyushu University) for surgical treatments; Tatsuro Tajiri (Kyoto Prefectural University of Medicine) for diagnostic approaches.
Funding
This study was supported by JSPS KAKENHI Grant Number 17K16268 (Y. Matsushita), 15 K0962 (Y.S.) and 17 K16301 (M.T.), a Health and Labour Sciences Research Grant on Evidence-based Early Diagnosis and Treatment Strategies for Neuroimmunological Diseases from the Ministry of Health, Labour and Welfare of Japan (Y.S.), Life Science Foundation of Japan, Takeda Science Foundation, The Mother and Child Health Foundation, The Japan Epilepsy Research Foundation (Y.S.).
Abbreviations
- a-CGH
Array-based Comparative Genome Hybridization
- BDNF
Brain-derived neurotrophic factor
- CNV
Copy Number Variation
- qPCR
Quantitative Polymerase Chain Reaction
- WAGR
Wilms tumor, aniridia, genitourinary anomalies and mental retardation
Additional file
For cytogenetics, quantitative PCR for copy number and gene expression analyses, methylation analysis, and ethics statement. Figure S1. Neuro-endocrine profile of the present case. Table S1. Methylation profile of the present case. Additional file References 1–3. (PDF 652 kb)
Authors’ contributions
YS, TH and SO directed this study; YT, YM performed CNV analyses and SA supported experiments; KI and KO conducted and evaluated the neuro-endocrine profiles; YK managed chemotherapy; MK performed and supervised methylation analyses; MT, SA, YI, MS and HT assessed neuro-developmental findings; CAS mapped CNVs; YT, SO and YS wrote the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
This study was approved by the institutional review board at Kyushu University. The parents of the present case were informed to participate in this study with written consents.
Consent for publication
Written informed consent was obtained from the parents for publication of the clinical details and any images in this report. A copy of the written consent is available for review by the Editor of this journal.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12881-017-0477-5) contains supplementary material, which is available to authorized users.
Contributor Information
Yui Takada, Email: yuiyui113jp@yahoo.co.jp.
Yasunari Sakai, Phone: +81-92-642-5421, Email: ysakai22q13@gmail.com.
Yuki Matsushita, Email: matuyuki@pediatr.med.kyushu-u.ac.jp.
Kazuhiro Ohkubo, Email: kohkubo@pediatr.med.kyushu-u.ac.jp.
Yuhki Koga, Email: yuuki-k@pediatr.med.kyushu-u.ac.jp.
Satoshi Akamine, Email: akamine@pediatr.med.kyushu-u.ac.jp.
Michiko Torio, Email: torio-m@pediatr.med.kyushu-u.ac.jp.
Yoshito Ishizaki, Email: ishizaki@pediatr.med.kyushu-u.ac.jp.
Masafumi Sanefuji, Email: sanefuji39@gmail.com.
Hiroyuki Torisu, Email: htorys@gmail.com.
Chad A. Shaw, Email: cashaw@bcm.edu
Masayo Kagami, Email: kagami-ms@ncchd.go.jp.
Toshiro Hara, Email: harat@pediatr.med.kyushu-u.ac.jp.
Shouichi Ohga, Email: ohgas@pediatr.med.kyushu-u.ac.jp.
References
- 1.Fantes JA, Bickmore WA, Fletcher JM, Ballesta F, Hanson IM, van Heyningen V. Submicroscopic deletions at the WAGR locus, revealed by nonradioactive in situ hybridization. Am J Hum Genet. 1992;51(6):1286–1294. [PMC free article] [PubMed] [Google Scholar]
- 2.Han JC, Liu QR, Jones M, Levinn RL, Menzie CM, Jefferson-George KS, Adler-Wailes DC, Sanford EL, Lacbawan FL, Uhl GR, et al. Brain-derived neurotrophic factor and obesity in the WAGR syndrome. N Engl J Med. 2008;359(9):918–927. doi: 10.1056/NEJMoa0801119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Diagnostic testing for Prader-Willi and Angleman syndromes: Report of the ASHG/ACMG Test and Technology Transfer Committee. Am J Hum Genet. 1996;58(5):1085–8. [PMC free article] [PubMed]
- 4.Xu S, Han JC, Morales A, Menzie CM, Williams K, Fan YS. Characterization of 11p14-p12 deletion in WAGR syndrome by array CGH for identifying genes contributing to mental retardation and autism. Cytogenet Genome Res. 2008;122(2):181–187. doi: 10.1159/000172086. [DOI] [PubMed] [Google Scholar]
- 5.Egan MF, Kojima M, Callicott JH, Goldberg TE, Kolachana BS, Bertolino A, Zaitsev E, Gold B, Goldman D, Dean M, et al. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell. 2003;112(2):257–269. doi: 10.1016/S0092-8674(03)00035-7. [DOI] [PubMed] [Google Scholar]
- 6.Yi T, Weng J, Siwko S, Luo J, Li D, Liu M. LGR4/GPR48 inactivation leads to aniridia-genitourinary anomalies-mental retardation syndrome defects. J Biol Chem. 2014;289(13):8767–8780. doi: 10.1074/jbc.M113.530816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fuke T, Mizuno S, Nagai T, Hasegawa T, Horikawa R, Miyoshi Y, Muroya K, Kondoh T, Numakura C, Sato S, et al. Molecular and clinical studies in 138 Japanese patients with silver-Russell syndrome. PLoS One. 2013;8(3):e60105. doi: 10.1371/journal.pone.0060105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Park H, Poo MM. Neurotrophin regulation of neural circuit development and function. Nat Rev Neurosci. 2013;14(1):7–23. doi: 10.1038/nrn3379. [DOI] [PubMed] [Google Scholar]
- 9.Gray J, Yeo GS, Cox JJ, Morton J, Adlam AL, Keogh JM, Yanovski JA, El Gharbawy A, Han JC, Tung YC, et al. Hyperphagia, severe obesity, impaired cognitive function, and hyperactivity associated with functional loss of one copy of the brain-derived neurotrophic factor (BDNF) gene. Diabetes. 2006;55(12):3366–3371. doi: 10.2337/db06-0550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lyons WE, Mamounas LA, Ricaurte GA, Coppola V, Reid SW, Bora SH, Wihler C, Koliatsos VE, Tessarollo L. Brain-derived neurotrophic factor-deficient mice develop aggressiveness and hyperphagia in conjunction with brain serotonergic abnormalities. Proc Natl Acad Sci U S A. 1999;96(26):15239–15244. doi: 10.1073/pnas.96.26.15239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu B, Goulding EH, Zang K, Cepoi D, Cone RD, Jones KR, Tecott LH, Reichardt LF. Brain-derived neurotrophic factor regulates energy balance downstream of melanocortin-4 receptor. Nat Neurosci. 2003;6(7):736–742. doi: 10.1038/nn1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yeo GS, Connie Hung CC, Rochford J, Keogh J, Gray J, Sivaramakrishnan S, O'Rahilly S, Farooqi IS. A de novo mutation affecting human TrkB associated with severe obesity and developmental delay. Nat Neurosci. 2004;7(11):1187–1189. doi: 10.1038/nn1336. [DOI] [PubMed] [Google Scholar]
- 13.Yamamoto T, Togawa M, Shimada S, Sangu N, Shimojima K, Okamoto N. Narrowing of the responsible region for severe developmental delay and autistic behaviors in WAGR syndrome down to 1.6 Mb including PAX6, WT1, and PRRG4. Am J Med Genet. 2014;164A(3):634–638. [DOI] [PubMed]