

Abstract
Background
Platelet-derived growth factor receptor A is altered by amplification and/or mutation in diffuse intrinsic pontine glioma (DIPG). We explored in vitro on new DIPG models the efficacy of dasatinib, a multi-tyrosine kinase inhibitor targeting this receptor.
Methods
Gene expression profiles were generated from 41 DIPGs biopsied at diagnosis and compared with the signature associated with sensitivity/resistance to dasatinib. A panel of 12 new DIPG cell lines were established from biopsy at diagnosis, serially passaged, and characterized by gene expression analyses. Effects of dasatinib (1–10 μM) on proliferation, invasion, and cytotoxicity were determined on 4 of these cell lines using live-cell imaging and flow cytometry assays. Downstream signaling and receptor tyrosine kinases (RTKs) were assessed by western blot and phospho-RTK array. The effect of the combination with the c-Met inhibitor cabozantinib was studied on cellular growth and invasion analyzed by the Chou–Talaly method.
Results
DIPG primary tumors and cell lines exhibited the gene expression signature of sensitivity to dasatinib. Dasatinib reduced proliferation (half-maximal inhibitory concentration = 10–100 nM) and invasion (30%–60% reduction) at 100 nM in 4/4 cultures and induced apoptosis in 1 of 4 DIPG cell lines. Activity of downstream effectors of dasatinib targets including activin receptor 1 was strongly reduced. Since multiple RTKs were activated simultaneously in DIPG cell lines, including c-Met, which can be also amplified in DIPG, the benefit of the combination of dasatinib with cabozantinib was explored for its synergistic effects on proliferation and migration/invasion in these cell lines.
Conclusion
Dasatinib exhibits antitumor effects in vitro that could be increased by the combination with another RTK inhibitor targeting c-Met.
Keywords: ACVR1, brainstem, preclinical model, PDGFRA, Src
Diffuse intrinsic pontine glioma (DIPG) is one of the most frequent malignant brain tumors in children. Its prognosis is dismal, with almost all patients dying before the second year after diagnosis.1 Due to its infiltrative nature, surgery cannot be performed without severe functional damages. Radiotherapy is the only effective treatment, albeit only transiently, while the addition of chemotherapy did not increase survival significantly.2
Diagnosis is usually made by radiology without biopsy collection. Lack of tumor tissue has limited our knowledge about the biology of this tumor. However, autopsy programs and systematic diagnostic biopsies performed by some groups allowed the conduct of specific high-throughput genomic studies.3
The most striking features of these neoplasms are first their ability to grow fast thanks to the overexpression of various growth factors4 and second their propensity to infiltrate the brain tissue2; symptoms are often present only a few weeks before diagnosis, while this interval is usually over 3 months for the other brain tumors, and during the course of the disease, the tumor tends to spread extensively in the cerebellum and the deep gray nuclei.
Platelet-derived growth factor receptor alpha (PDGFRA) signaling plays a key role in pediatric gliomagenesis5 and represents one of the few drugable targets in DIPG so far. PDGFRA is the most commonly amplified receptor tyrosine kinase (RTK) gene in DIPG,6–10 and activating mutations occur in 10% of DIPGs.9–11 Previous study by gene expression analysis revealed the existence of a subgroup of DIPGs characterized by the gene expression signature related to PDGFRA amplification irrespective of the presence of its genomic alteration, amplification, or mutation.9 In DIPG, the second most frequently amplified/mutated gene is MET.4,9,12 These alterations in one of the major pathways of invasion are in accordance with the highly infiltrative nature of this tumor.
We established new adherent DIPG cell lines from stereotactic biopsies performed at diagnosis to explore agents currently in development for their potential role in the treatment of DIPG. Dasatinib is an oral inhibitor of multiple targets, including PDGFRA and B, c-Kit, and Src.13 Cabozantinib is a potent, ATP-competitive inhibitor of c-Met, vascular endothelial growth factor receptor 2 (VEGFR2), and RET (“rearranged during transfection”).14 Both drugs are approved for other malignancies and are currently being tested individually in clinical trials in adults with glioblastoma.
We show here in vitro on newly developed DIPG models the efficacy on cell growth and invasion of dasatinib alone and its synergistic effect in combination with cabozantinib.
Materials and Methods
Proximity Ligation Assay
Sections were deparaffinized and rehydrated, and antigen retrieval was performed in a pressure cooker (5 min at 110°C, Decloaker chamber, Biocare Medical) using Diva decloaker as antigen retrieval (DV2004MX, Biocare Medical) followed by washes in Hot Rinse (HTR1001M, Biocare Medical). After wash in Tris-buffered saline (TBS)–Tween 20 (0.05%), staining was continued according to manufacturer’s instructions (Sigma Aldrich), with the exception that blocking solution of 20% goat serum in TBS-Tween 20 (0.05%) was used instead. Primary antibodies were anti-PDGFRA at 1:100 dilution (#3164, Cell Signaling Technology) and anti–phospho-tyrosine at 1:2000 (#9411, Cell Signaling Technology) in 20% goat serum in TBS-Tween 20 (0.05%). All washes were done in TBS-Tween 20 (0.05%). Staining was performed using proximity ligation assay (PLA) probes donkey anti-mouse minus (DUO92004, Sigma Aldrich) and donkey anti-rabbit plus (DUO92002, Sigma Aldrich) and the brightfield detection kit (DUO92012, Sigma Aldrich). Samples were then mounted using Pertex (Histolab).
Tumor and Acid Nucleic Extraction
Tumor samples and clinical information were collected as described earlier.9 Tumor biopsies were snap frozen in liquid nitrogen in the operating room to ensure preservation of high-quality RNA until extraction performed using the DNA/RNA Mini Kit (Qiagen).
Gene Expression Analyses
RNA microarray hybridization was carried out by the Functional Genomics Platform of the Integrated Research Cancer Institute in Villejuif using the Agilent SurePrint G3 Hmn GE 8*60 K Whole Human Gene Expression microarray (http://www.agilent.com). The microarray data related to this paper were compliant with the Minimum Information About a Microarray Experiment, and the raw data will be submitted to the Array Express data repository at the European Bioinformatics Institute (http://www.ebi.ac.uk/arrayexpress/) upon publication.
Bioinformatics Analysis
Raw gene expression data using normal brainstem (BSR) as reference were transferred into R software for statistical analysis. Gene Set Enrichment Analysis (GSEA)15 was performed with the preranked tool on the gene list ranked by increasing false discovery rate (FDR) adjusted P-values, for each contrast of interest, with default parameter values. A nominal FDR of <0.25 was considered statistically significant for GSEA. We ran GSEA with a t-test option as a metric parameter.
Establishment of DIPG Cell Lines and Cell Culture
Tumor pieces were collected into Dulbecco's modified Eagle's medium (DMEM; PAA Laboratories). Biopsies were cut into 1-mm3 pieces and placed into either DMEM for immediate processing or into freezing medium (90% serum, 10% dimethyl sulfoxide [DMSO]) prior to being progressively cooled to −80°C. Cell dissociation was performed mechanically by passage through increasingly finer needles (19G to 26G). Single cells were seeded in AmnioMAX C-100 complete medium (Gibco) and maintained at 37°C in a 5% CO2 humidified atmosphere. The cells were further cultured until appearance of adherent cells and colony formation and then weekly passaged.
The adult glioblastoma cell line T98G was obtained from American Type Culture Collection and the pediatric cell line SF188 was kindly provided by Dr Chris Jones (of the Institute of Cancer Research, Sutton, UK). Both were maintained as monolayers in DMEM-GlutaMAX plus 10% fetal bovine serum in 5% CO2.
Drugs
Dasatinib (Sprycel, BMS-354825) and cabozantinib (Cometriq, XL184) were provided by Bristol-Myers Squibb and Exelixis, respectively, and were prepared as a 20 mmol/L stock solution in DMSO.
Growth Inhibition
Cells (2000–3000/well) were plated in a 96-well plate. After 24 h, the cells were exposed to dasatinib at various concentrations for 72 h. Growth curves were constructed by imaging plates using the IncuCyte system (Essen Instruments), where the growth curves were built from confluence measurements acquired during automated kinetic imaging. Values of half-maximal inhibitory concentration (IC50) were determined using sigmoidal dose-response (variable slope) statistics and normalized in GraphPad Prism.
For the assessment of combination effects, cells were treated with increasing concentrations of drugs either alone or concurrently at their equipotent molar ratio, and combination indices were calculated by the Chou–Talaly method16 and CalcuSyn software (Biosoft).
Cytotoxicity
Cells were plated in triplicate in a 96-well plate to achieve optimal confluence of 25%–30% before treatment. Cells were then treated with 100 nmol/L dasatinib or vehicle (DMSO) diluted with complete medium containing Yoyo-1 (Life Technologies) at a final concentration of 0.1 μM. Images automatically were collected by the IncuCyte system every 2–3 h in both contrast phase and fluorescence. Quantitative measurement of nuclear staining was determined by object counting with image analysis IncuCyte software.
Apoptosis Assay
Subconfluent cells were treated with 50 and 100 nmol/L dasatinib for 72 h. Cells were then harvested and stained with annexin V and propidium iodide according to the protocol indicated by the Annexin V–FITC Apoptosis Detection Kit from Calbiochem and analyzed on an AccuriC6 flow cytometer and software (BD Bioscience).
Cell Cycle Analysis
Cells were plated at 60%–70% confluent and treated with 100 nmol/L dasatinib the following day. Cells were harvested, washed in phosphate buffered saline (PBS), and suspended in 70% ethanol at –20°C before staining with propidium iodide solution containing RNase A. DNA content was analyzed on an AccuriC6 flow cytometer and data were analyzed using FlowJo software (Tree Star).
Invasion/Migration Assays
Cells were grown to confluence on a 96-well Essen Bioscience ImageLoke plate previously coated with BD Matrigel. Using an IncuCyte wound maker, scratch wounds were made simultaneously in all culture wells. Cells growing as a monolayer were washed with PBS and overlaid with BD Matrigel; subsequently, complete medium containing 100 nmol/L dasatinib or DMSO alone was added. Wound images were automatically acquired and analyzed by integrated metric calculated through confluence algorithm. Migration protocol included the same procedure without coating and overlay with BD Matrigel.
Western Blot
After washing in cold PBS, cells were treated with the indicated dose of dasatinib or vehicle (DMSO) and harvested in Tris NaCl EDTA NP40 containing anti-protease (Complete Mini, Roche Diagnostics) and anti-phosphatases (Sigma Aldrich). Lysates were spun in a centrifuge at 14 000 rpm for 5 min and the supernatant was collected. Protein extracts were resolved on 4%–15% sodium dodecyl sulfate–polyacrylamide electrophoresis gels and transferred to nitrocellulose membrane (Trans-Blot Turbo system, Bio-Rad), following the manufacturer's instructions. Membranes were incubated at 4°C overnight with the following antibodies: p-PDGFRA, PDGFRA, p-Src family kinase, Src, phosphorylated mitogen-activated protein kinase kinase (MEK), MEK, phosphorylated extracellular signal-regulated kinase (ERK), ERK, p-Smad1/5/8, and actin (Cell Signaling) diluted (1:1000) in 5% bovine serum albumin in TBS-Tween 20. Blots were incubated with secondary goat anti-rabbit horseradish peroxidase–conjugated antibodies and enhanced by chemiluminescence reagent. The signals were captured and analyzed using a ChemiDoc MP Imaging System (Bio-Rad).
Phosphorylated Receptor Tyrosine Kinase Array
A human phospho-RTK array (R&D Systems) was used to detect simultaneously the phosphorylation status of RTKs (n = 49) in DIPG cells cultured in AmnioMAX C-100 basal medium supplemented in AmnioMAX C-100. Membranes were incubated with whole-cell lysates (1 mg) overnight and used according to the manufacturer's protocol.
Results
PDGFRA Is Activated in DIPG Tumor Samples Independently From Genomic Alterations
PDGFRA expression and phosphorylation status were analyzed for formalin-fixed paraffin-embedded tumor tissues from 13 DIPG patients at diagnosis (Supplementary Table S1). Most of these samples showed a strong expression of PDGFRA with associated phosphorylation. Although activation, expression, and gene copy number of PDGFRA may be related in some cases (Fig. 1A, upper-left panel), there was no significant correlation because some samples harbored overexpression/phosphorylation without amplification and/or mutation (Fig. 1A, upper-right panel).
Fig. 1.
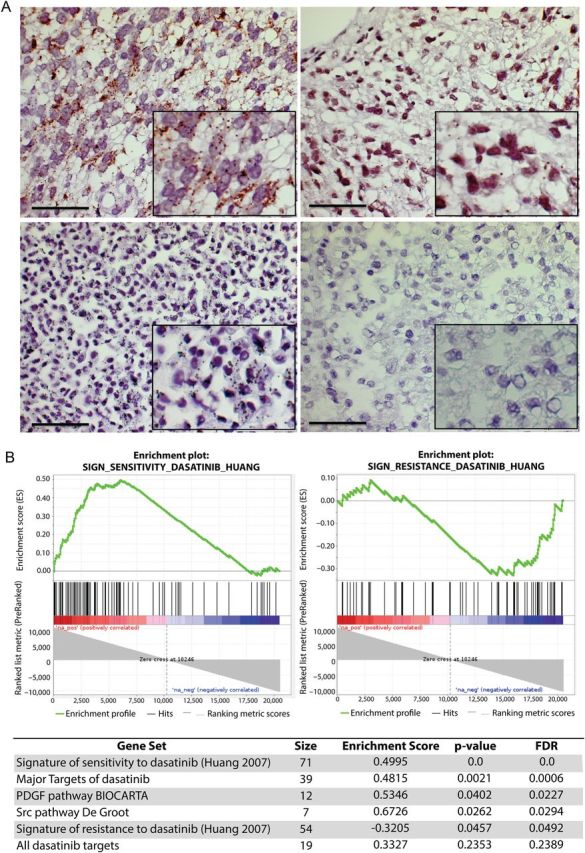
Frequent activation of PDGFRA and prediction of sensitivity to dasatinib in DIPG patients. (A) Phospho-PDGFRA proximity ligation assay. Upper panels represent tumors from DIPG patients (#4 and #11, described in the Supplementary Table S1). Lower panels represent positive and negative controls. Positive immunochemical staining appears as cytoplasmic brown dots. Scale bar = 50 µM. (B) GSEA plot comparing DIPG gene expression profile with the signature described for sensitivity and resistance to dasatinib (upper panels) and summary of other significant gene sets (FDR ≤ 0.25) related to dasatinib targets (lower panel).
Prediction of Sensitivity to Dasatinib in DIPG Patients
Using GSEA, the expression profiles of 41 DIPG samples in contrast with BSR were compared with the gene expression signature associated with sensitivity or resistance to dasatinib.17 This signature of sensitivity to dasatinib, determined in breast cancer cell lines and validated in lung cancer cell lines, was significantly enriched in the gene expression profile of DIPG (enrichment score = 0.4995; nominal P = .0; FDR q = 0.0) (Fig. 1B, upper left panel). Conversely the gene signature related to resistance to dasatinib was significantly downregulated in these samples (enrichment score = −0.32; nominal P = .04574; FDR q = 0.0492) (Fig. 1B, upper right panel). Because dasatinib is a broad-spectrum inhibitor, we looked for the expression of its target genes (Supplementary Table S2). We found an enrichment of the gene sets of the major targets (IC50 <100 nM) or all known targets of dasatinib (n = 64) and also for the Src pathway described by De Groot.18
Isolation and Establishment of DIPG Cell Lines
Over a period of 10 months, 12 patients underwent stereotactic biopsies in the Neurosurgery Department of Necker–Enfants Malades Hospital in Paris. Only samples from patients at diagnosis without prior radiotherapy or chemotherapy were selected. In most instances, 1 biopsy was transferred immediately to the laboratory to establish cell cultures, 1 or 2 biopsies were used for histological diagnosis and immunohistochemistry, and the remaining biopsies were snap frozen. Single cell suspensions were plated after dissociation of the tumor, and we observed the emergence of growing cells systematically. All of these cultures were weekly passaged and maintained during at least 13 weeks.
We performed comparative genomic hybridization analysis in 9 DIPG tumors, from which the panel of DIPG cultures were established and in these cultures per se at early passage (passages 2–8). While we observed the recurrent gain of chromosome 1q (6/9), no tumor exhibited PDGFRA amplification. Broadly, DIPG culture showed a flattening of the copy number profile compared with the primary tumors. However, the presence of the H3K27M mutation in 9 of the 12 DIPG cultures demonstrated reasonable similarity of these cell lines with the corresponding primary tumors. Among the 12 cultures, 4 gave rise to cell lines that could be passaged without undergoing senescence. Interestingly, NEM157 and NEM168 had disruptive TP53 mutations. Clinical data and biological characteristics relating to these 4 DIPG cell lines are summarized in Table 1. Both PDGFRA and Src were expressed and activated at different levels depending on the DIPG cell line (Fig. 4A). GSEA further demonstrated an enrichment of the dasatinib sensitivity signature (enrichment score = 0; nominal P = .0; FDR q = 0.0; Supplementary Fig. S1).
Table 1.
Clinical and experimental characteristics of DIPG cell lines
| NEM157 | NEM163 | NEM165 | NEM168 | |
|---|---|---|---|---|
| Clinical characteristics | ||||
| Age at diagnosis, y | 5.7 | 5.7 | 3.3 | 10.6 |
| Sex | F | M | M | F |
| Histological diagnosis | Oligoastrocytoma | Oligoastrocytoma | Oligoastrocytoma | Astrocytoma |
| Grade | II | II | III | III |
| Time to progression, mo | 10.3 | 8.5 | 16.9 | 7.0 |
| Survival, mo | 12.9 | 10.6 | 23.9 | 8.4 |
| In vitro characteristics | ||||
| Doubling time | 30 h | 36 h | 32 h | 34 h |
| PDGFRA activation | + | ++ | ++ | +++ |
| Src activation | + | ++ | ++ | + |
| Histone Mutation | K27M H3F3A | K27M H3F3A | K27M H3F3A | K27M H3F3A |
| P53 status | Δ337–340 R273C | Wild type | Wild type | Δ156 |
Fig. 4.
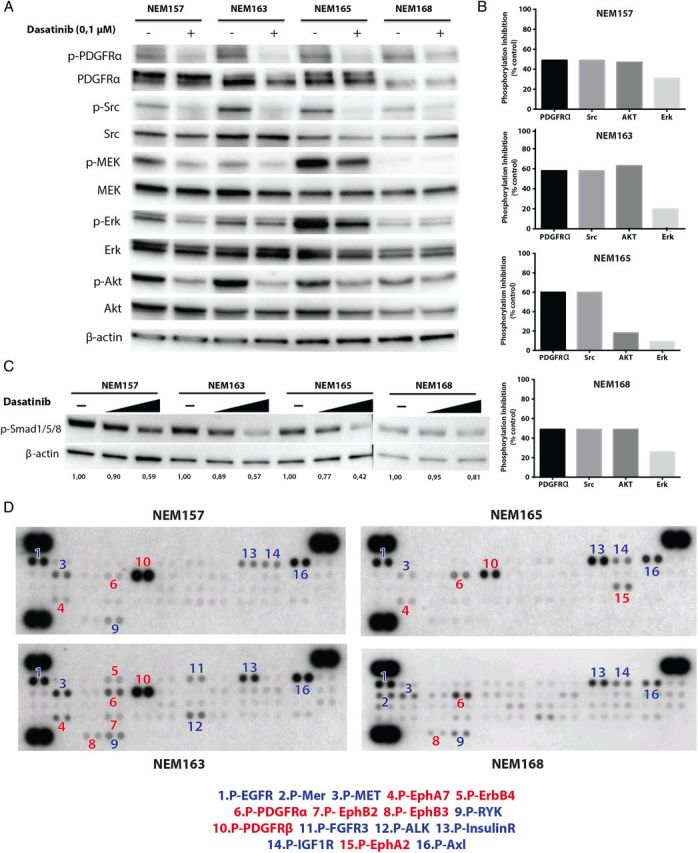
Targets downstream of dasatinib and potential escape mechanisms. (A) Effect of dasatinib on target activation and downstream signaling was assessed by western blot. Cells were treated with the indicated doses of dasatinib or vehicle for 1 h, in complete medium. Cells were lysed in Tris NaCl EDTA NP40 buffer, and equal protein (30 μg) was analyzed by western blotting on 4%–15% sodium dodecyl sulfate–polyacrylamide electrophoresis gels with the indicated antibodies. (B) Phosphorylation inhibition induced by dasatinib was calculated by densitometric analysis for each DIPG cell line. (C) Effect of dasatinib (1 μM and 10 μM) on Smad1/5/8 signaling. (D) Multiple RTKs were activated simultaneously in glioma cell lines. Whole-cell extracts from the DIPG cell lines were incubated on RTK antibody arrays, and phosphorylation status was determined by subsequent incubation with anti-phosphotyrosine horseradish peroxidase. Each RTK is spotted in duplicate: the pairs of dots in each corner are positive controls. Each pair of positive RTK dots is denoted by a numeral, with the identity of the corresponding RTKs listed below the arrays, in red and blue for dasatinib target or not, respectively. EGFR, epidermal growth factor receptor; FGFR3, fibroblast growth factor receptor 3; ALK, anaplastic lymphoma receptor tyrosine kinase; IGF1R, insulin-like growth factor 1 receptor.
Dasatinib Inhibits Cell Growth at Submicromolar Concentration
Values of IC50 were calculated after treatment for 72 h. Assay by MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium), which measures cell viability, was simultaneously performed at this endpoint, and we observed a good correlation of both methods (Supplementary Fig. S2). All of the DIPG cell lines tested showed clear sensitivity to dasatinib, with lower IC50 values than the well-described dasatinib sensitive adult glioma cell line T98G and pediatric supratentorial glioma cell line SF188 (Fig. 2A). Representative data of the ranking of sensitivity are shown in Fig. 2B.
Fig. 2.
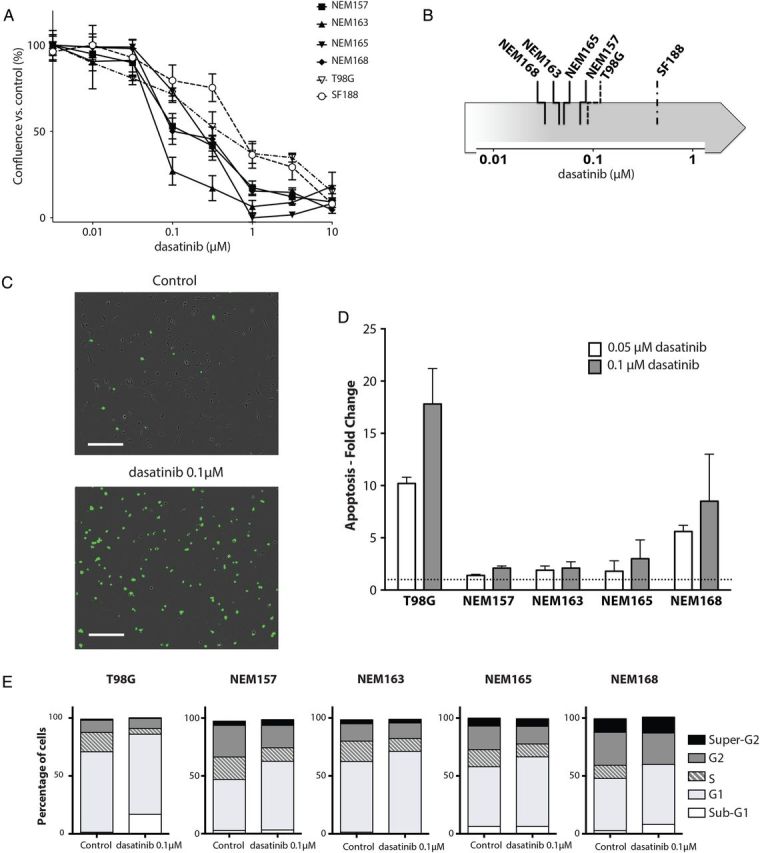
Effect of dasatinib on DIPG cell line in vitro. (A) DIPG cell lines (NEM), T98G, and SF188 were treated with the indicated doses of dasatinib or vehicle for 3 days and growth was measured using the confluence algorithm. Results are the mean percent inhibition compared with control cells ± SEM of 3 experiments carried out in triplicate. (B) Representative classification of dasatinib sensitivity by IC50 determination for each cell line. (C) Contrast phase fluorescent images of NEM168 12 h posttreatment. Nuclear staining indicates loss of membrane integrity. Scale bar = 300 µM. (D) Cells were plated in complete medium with or without the indicated dose of dasatinib or vehicle for 72 h, stained with annexin V and propidium iodide, and analyzed by flow cytometer, as described in Materials and Methods. Apoptosis detected in the control cells was set to 1 and is represented by the dashed line. Results are the mean ± SD of duplicate representatives of 3 experiments carried out. (E) Distribution of cells in sub-G1, G1, S, G2, and super-G2 phases was determined after propidium iodide staining and by using the Dean-Jet-Fox algorithm in FlowJo software.
Dasatinib Induces G1 Arrest or Cell Death in DIPG Cell Lines
Because of the significant antiproliferative effect of dasatinib on cell growth, we therefore investigated its effect on cell cycle and cell death. Cytotoxicity assay showed massive cytotoxicity in NEM168-treated cells (Fig. 2C), whereas it was less pronounced in the other DIPG cell lines (data not shown).
We next assessed whether dasatinib induced apoptosis. DIPG and T98G cell lines were treated with 50 or 100 nM dasatinib in complete medium for 72 h. Cells were stained with both annexin V–fluorescein isothiocyanate and propidium iodide and analyzed by flow cytometry. Induction of apoptosis was calculated as fold change of annexin V–positive cells compared with the control (Fig. 2D). We observed a significant induction of apoptosis in NEM168 and, as expected, in the well-described sensitive T98G cell line. By contrast, no apoptosis was found in the other DIPG lines. We next performed cell cycle analyses following dasatinib treatment of 100 nM and observed a block at the G1 phase of the cycle in all DIPG cell lines (Fig. 2E). This was also associated with an increase in the sub-G1 fraction in NEM168 and T98G corresponding to the apoptosis observed above. Together, these data show that dasatinib impairs cellular progression through the G1 phase or leads to apoptosis in these DIPG cell lines.
Autophagy was not observed using western blotting for light chain 3B cleavage (data not shown).
Dasatinib Decreases Migration and Invasion of DIPG Cells
Relative wound density was calculated after 12 h (ie, before any effect on cell growth could be observed; Table 1) to study migration and invasion regardless of proliferation by relying on the measurement of the spatial cell density in the wound area relative to spatial cell density outside of the wound area at every time point with self-normalization by the initial wound density (Fig. 3A).
Fig. 3.

Effect of dasatinib on cell migration and cell invasion. Cells were seeded in 96-well plates and incubated until subconfluent. A wound was scratched across each well (WoundMaker, Essen BioScience), and BD Matrigel was then added (or not for migration assay) with treatment. Wound closure was automatically imaged each 6 h and calculated as a percentage of wound confluence that the cell gained. (A) Quantitative wound-repair analysis at 12 h after dasatinib treatment (0.1 μM) for migration and invasion assay, respectively. (B) Contrast-phase images of NEM168 invasion at 24 h. Red and blue lines correspond to the frontline of the scratch wound at 0 h and 24 h, respectively. Scale bar = 300 µM.
All untreated cell lines migrated and invaded in this assay with similar rate with complete wound closure in 24–36 h. Cell migration and invasion were significantly inhibited by dasatinib at 12 h.
Effects of Dasatinib Treatment on Targets and Downstream Signaling
PDGFRA and Src activities were significantly downregulated for all DIPG cell lines in response to dasatinib treatment for 1 h in all 4 DIPG cell lines, as shown on western blot (Fig. 4B). We found a significant inhibition of Akt (50%–60% reduction) in NEM157, NEM163, and NEM168, but this effect was lower in NEM165 (20% reduction). Moreover, dasatinib induced partial inhibition of the MEK/ERK pathway (10%–30% reduction).
Activin receptor 1 (ACVR1) is a known secondary target of dasatinib and was recently unveiled as being mutated in DIPG.12,19–21 ACVR1 is responsible for the activation of the bone morphogenic protein–dependent transforming growth factor β pathway by increased phosphorylation of Smad1/5/8. In our cell lines, ACVR1 was not mutated but overexpressed compared with BSR (log2 ratios between 2.14 and 2.71). We showed here that a significant inhibition of phospho-Smad1/5/8 could be obtained at high doses of dasatinib (1 μM and 10 μM) (Fig. 4C).
Multiple Receptor Tyrosine Kinases Are Concomitantly Activated in DIPG Cell Lines
To define other coactivated RTKs in these DIPG cell lines, we used an antibody array that allows simultaneous assessment of phosphorylation status of 49 RTKs (Fig. 4D). Profiling of the phosphorylation status of 49 RTKs showed prominent coactivation of multiple phosphorylated RTKs, including targets of dasatinib: PDGFRA/B (4/4) and ephrin receptors (EphA2, EphB2, and EphB3) (4/4), as well as other RTKs that could potentially be responsible for escape mechanisms to the inhibition of one RTK. The other recurrent coactivated RTKs not targeted by dasatinib were: epidermal growth factor receptor (4/4), mesenchymal epithelial transition factor (MET) (4/4), insulin receptor (4/4), AXL (4/4), insulin-like growth factor 1 receptor (3/4), RYK (3/4), anaplastic lymphoma receptor tyrosine kinase (2/4), and fibroblast growth factor receptor (1/4). These results indicate that various RTKs are activated in the DIPG cell lines and contribute to maintain the activation of several downstream signaling pathways. This suggests that targeting multiple tyrosine kinases might be effective in DIPG cells.
Antiproliferative Effect of Dasatinib Is Synergized by Cabozantinib In vitro
MET amplification or mutation has been found in 5%–10% of DIPG primary tumors, and this genomic aberration is frequently found concomitantly with the PDGFRA amplification.4,9,12 Transcriptomic data exhibited an overexpression of hepatocyte growth factor and MET compared with BSR in the 41 DIPGs and also in these 4 DIPG cell lines, suggesting autocrine activation of the MET pathway, as also suggested by the phospho-RTK array (Fig. 4D).
According to these genomic data, c-Met phosphorylation, and evidence that hepatocyte growth factor contributes to resistance to RTK inhibitors,22 we analyzed the MET pathway as a therapeutic target alone or in combination with dasatinib. Treatment of the cells with the MET inhibitor cabozantinib as a single agent did not show strong antitumor effects, with IC50 values higher than micromolar concentration (data not shown).
We then tested the combination of both dasatinib and cabozantinib to assess synergy on cell growth in vitro. Synergistic effects of the combination on cell proliferation were observed in 3 of the 4 DIPG cell lines. The combination indices (CIs) at IC50 of dasatinib and a submicromolar dose of cabozantinib (0.75 μM) were synergistic in NEM157, NEM163, and NEM165 (CI = 0.53, CI = 0.28, and CI = 0.45, respectively) and additive for NEM165 (CI = 0.81) (Fig. 5A).
Fig. 5.
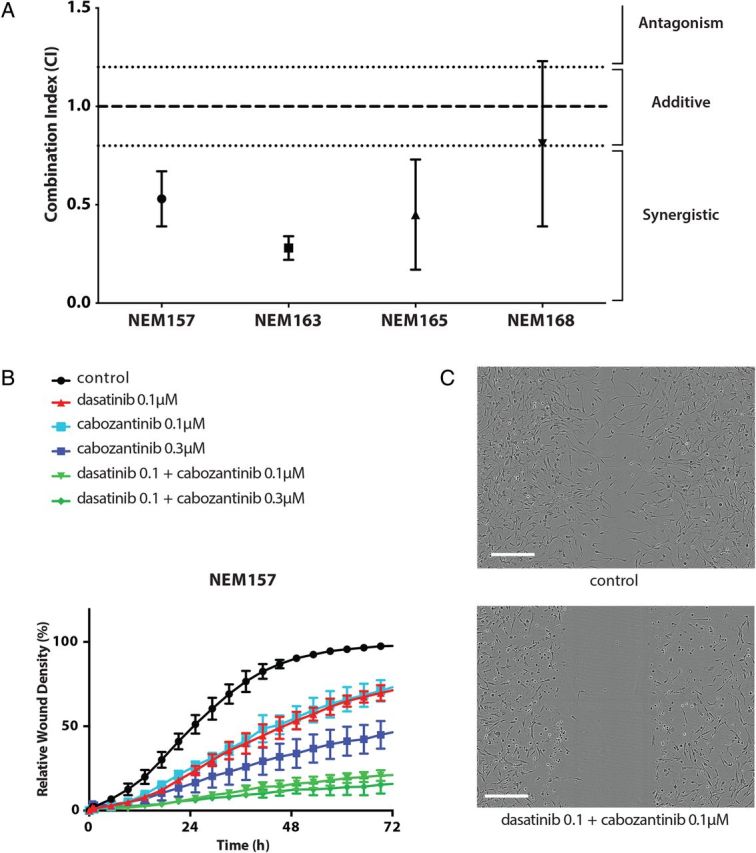
Synergistic effect of dasatinib and cabozantinib combination in vitro. (A) Synergistic effect at median effect of dasatinib and submicromolar dose of c-Met inhibitor (cabozantinib) (0.75 μM) on cell growth in vitro. Cells were treated with increasing concentrations of drugs either alone or concurrently at their equipotent molar ratio and combination indices (CIs) calculated by the method of Chou and Talaly (CI calculates synergism <0.8; additivity between >0.8 and <1.2; antagonism >1.2. All values are given as mean ± SD of at least 3 independent experiments). (B) Time course of relative wound density of the high invasive NEM157 cell line. (C) Contrast-phase images of NEM157 invasion at 24 h in control and combined condition (upper and lower panel, respectively). Scale bar = 300 µM.
Dasatinib Combined With Cabozantinib Inhibits Cell Invasion
Given the importance of the MET pathway in the invasive phenotype of glioma cells,23,24 we studied the effect of cabozantinib on invasion with the scratch wound assay. We chose the most invasive cell line, NEM157, which is also the least dasatinib sensitive cell line. We found an anti-invasive effect of cabozantinib alone at submicromolar concentration (Fig. 5B). Combination of both targeted inhibitors showed enhanced activity compared with each agent alone and abrogated invasion (Fig. 5B and C; Supplementary material, Movie S1).
Discussion
We have shown by PLA that DIPG samples exhibit frequent PDGFRA activation irrespective of their genomic status. These observations are consistent with the “PDGFRA signature” found in some DIPGs irrespective of the presence of a PDGFRA amplification.9 Accordingly, the broad activation of PDGFRA in DIPG would expand the usefulness to target this receptor in patients. Dasatinib appeared to be the most suitable inhibitor, since it has a stronger activity in vitro than imatinib, which had only limited activity in DIPG patients.25,26 Moreover, gene expression profiling performed in 41 DIPG samples would predict dasatinib sensitivity (Fig. 1B), and this signature of dasatinib sensitivity was also expressed in these DIPG cell lines (Supplementary Fig. S1). Dasatinib showed in vitro efficacy on this unique DIPG cell panel with antiproliferative effect for all, anti-invasive effect for most, and cytotoxic effect for one. Interestingly the cell line exhibiting apoptosis when treated with dasatinib was the only one without pathway activation of mitogen-activated protein kinase, suggesting that constitutional activation of this pathway in the other cell lines could explain the absence of cell death upon treatment. The doses tested here were achievable clinically given the maximal concentration reached in serum and measured in cerebrospinal fluid,27 but higher doses may prove to be useful when delivered directly to the tumor by alternative drug delivery strategies such as convection enhanced delivery, coadministration of an inhibitor of P-glycoprotein,28 or nanoformulations for increased delivery in the brain.29 Dasatinib has been tested in patients together with vandetanib in a recently published phase 1 trial for DIPG27; the maximal tolerated dose of dasatinib in combination with vandetanib could not be escalated to the levels achievable when given in monotherapy, but the authors concluded that dasatinib was an interesting agent to develop further, which is now supported with our preclinical data.
Given that DIPGs show activation of multiple RTKs, inhibiting concomitantly these other pathways by using other small inhibitor molecules is worth investigating. Moreover, multiple coamplification may be a relatively common phenomenon in high-grade glioma,30 and in this respect DIPG tumors show simultaneous amplification of PDGFRA and MET.4,9
Our interest then focused on the MET pathway, which we think is a relevant target in DIPG due to its activation in our cell lines shown by RTK arrays and by western blot and due to its known role in the invasive phenotype in glioma.23 We explored here the effect of cabozantinib, which is a tyrosine kinase inhibitor of c-Met, RET, and VEGFR2. Navis and colleagues31 have shown its antitumoral activity in an invasive glioma model, where the phenotype was driven by c-Met. Clinical activity of this compound has been reported in a phase 2 trial for recurrent glioblastoma.32 Among the DIPG cell lines of the present study, none exhibited either a mutation or an amplification of MET. However, we observed a synergistic antiproliferative effect for 3 of the 4 and additive activity for the fourth. The benefit of c-Met inhibition in combination with dasatinib was particularly strong in DIPG cell lines less sensitive to dasatinib.
Our results should be reproduced in DIPG tumor stem cells to ensure that the cancer-stem-cell fraction of the tumor is also targeted by these agents. Culture conditions (such as the type of medium or the oxygen pressure) can also influence the response to the drugs and should be taken into account when extrapolating the results. Moreover, these results should be reproduced in vivo when orthotopic models become available. An engineered mouse PDGF-induced model is available33 but may not recapitulate the patient tumors, especially with respect to multi-RTK activation, and therefore may not be the most relevant model to test these combinations. Xenograft models from primary tumors that mimic more closely DIPG are still needed. The synergy of dasatinib and cabozantinib suggests the potential of combination trials in patients with DIPG. Such a combination trial with dasatinib and crizotinib is currently recruiting (NCT01644773). An important future task will be to define markers to identify the patients most suitable for combination treatment or dasatinib single treatment. The fact that an aberrant PDGFRA pathway is not necessarily linked to amplification and/or mutation raises the question of the best test method to detect the involvement of this receptor in DIPG. PLA might be the suitable solution to explore this pathway activation. However, it should also be considered that part of the efficacy may come through the inhibition of other targets such as the non-receptor membrane-associated Src tyrosine kinase34–36 or ACVR1 that can be activated in these tumors as shown here and in recent publications.12,19–21
In summary, these results demonstrate synergistic antiproliferative and anti-invasive effects of dasatinib with a c-Met inhibitor in DIPG cell line models in vitro, which warrant further translational efforts. These 2 agents and their biosimilars are ready for clinical evaluation in children with DIPG for which there is currently no valid therapeutic option.
Funding
This work was supported by Bristol-Myers Squibb for drug supply and a grant (to J.G.), by Etoile de Martin (to N.T.), by Lemos Family and Friends (to J.G. and G.C.), and by Le Défi de Fortunée (to J.G.).
Supplementary Material
Acknowledgments
The authors would like to thank the sponsors of the study: the associations “L'Etoile de Martin” (N.T.) and “Le Défi de Fortunée” (J.G.) and the Lemos Family and Friends (J.G. and G.C.). The authors thank the Tumor Bank of Necker Hospital.
Conflict of interest statement . Bristol-Myers Squibb partially funded this study and also provided one of the study drugs.
References
- 1. Hargrave D, Bartels U, Bouffet E. Diffuse brainstem glioma in children: critical review of clinical trials. Lancet Oncol. 2006;7(3):241–248. [DOI] [PubMed] [Google Scholar]
- 2. Warren KE. Diffuse intrinsic pontine glioma: poised for progress. Front Oncol. 2012;2:205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sturm D, Witt H, Hovestadt V, et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell. 2012;22(4):425–437. [DOI] [PubMed] [Google Scholar]
- 4. Paugh BS, Broniscer A, Qu C, et al. Genome-wide analyses identify recurrent amplifications of receptor tyrosine kinases and cell-cycle regulatory genes in diffuse intrinsic pontine glioma. J Clin Oncol. 2011;29(30):3999–4006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. MacDonald TJ. Pediatric glioma: role of platelet derived growth factor receptor. In: Hayat MA, ed., Pediatric Cancer, Vol. 2:Teratoid/Rhabdoid, Brain Tumors, and Glioma. New York: Springer; 2012:259–267. [Google Scholar]
- 6. Barrow J, Adamowicz-Brice M, Cartmill M, et al. Homozygous loss of ADAM3A revealed by genome-wide analysis of pediatric high-grade glioma and diffuse intrinsic pontine gliomas. Neuro Oncol. 2011;13(2):212–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zarghooni M, Bartels U, Lee E, et al. Whole-genome profiling of pediatric diffuse intrinsic pontine gliomas highlights platelet-derived growth factor receptor alpha and poly (ADP-ribose) polymerase as potential therapeutic targets. J Clin Oncol. 2010;28(8):1337–1344. [DOI] [PubMed] [Google Scholar]
- 8. Paugh BS, Qu C, Jones C, et al. Integrated molecular genetic profiling of pediatric high-grade gliomas reveals key differences with the adult disease. J Clin Oncol. 2010;28(18):3061–3068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Puget S, Philippe C, Bax DA, et al. Mesenchymal transition and PDGFRA amplification/mutation are key distinct oncogenic events in pediatric diffuse intrinsic pontine gliomas. PLoS One. 2012;7(2):e30313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Paugh BS, Zhu X, Qu C, et al. Novel oncogenic PDGFRA mutations in pediatric high-grade gliomas. Cancer Res. 2013;73(20):6219–6229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Balss J, Meyer J, Mueller W, et al. Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol. 2008;116(6):597–602. [DOI] [PubMed] [Google Scholar]
- 12. Taylor KR, Mackay A, Truffaux N, et al. Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nat Genet. 2014;46(5):457–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Karaman MW, Herrgard S, Treiber DK, et al. A quantitative analysis of kinase inhibitor selectivity. Nat Biotech. 2008;26(1):127–132. [DOI] [PubMed] [Google Scholar]
- 14. Yakes FM, Chen J, Tan J, et al. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol Cancer Ther. 2011;10(12):2298–2308. [DOI] [PubMed] [Google Scholar]
- 15. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chou TC, Talaly P. A simple generalized equation for the analysis of multiple inhibitions of Michaelis-Menten kinetic systems. J Biol Chem. 1977;252(18):6438–6442. [PubMed] [Google Scholar]
- 17. Huang F, Reeves K, Han X, et al. Identification of candidate molecular markers predicting sensitivity in solid tumors to dasatinib: rationale for patient selection. Cancer Res. 2007;67(5):2226–2238. [DOI] [PubMed] [Google Scholar]
- 18. Ahluwalia MS, Groot JD, Liu W, et al. Targeting SRC in glioblastoma tumors and brain metastases: rationale and preclinical studies. Cancer Lett. 2010;298(2):139–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Buczkowicz P, Hoeman C, Rakopoulos P, et al. Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat Genet. 2014;46(5):451–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wu G, Diaz AK, Paugh BS, et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet. 2014;46(5):444–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fontebasso AM, Papillon-Cavanagh S, Schwartzentruber J, et al. Recurrent somatic mutations in ACVR1 in pediatric midline high-grade astrocytoma. Nat Genet. 2014;46(5):462–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wilson TR, Fridlyand J, Yan Y, et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature. 2012;487(7408):505–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kong D-S, Song S-Y, Kim D-H, et al. Prognostic significance of c-Met expression in glioblastomas. Cancer. 2009;115(1):140–148. [DOI] [PubMed] [Google Scholar]
- 24. Abounader R, Laterra J. Scatter factor/hepatocyte growth factor in brain tumor growth and angiogenesis. Neuro Oncol. 2005;7(4):436–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pollack IF, Jakacki RI, Blaney SM, et al. Phase I trial of imatinib in children with newly diagnosed brainstem and recurrent malignant gliomas: a Pediatric Brain Tumor Consortium report. Neuro Oncol. 2007;9(2):145–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Geoerger B, Morland B, Ndiaye A, et al. Target-driven exploratory study of imatinib mesylate in children with solid malignancies by the Innovative Therapies for Children with Cancer (ITCC) European Consortium. Eur J Cancer. 2009;45(13):2342–2351. [DOI] [PubMed] [Google Scholar]
- 27. Broniscer A, Baker SD, Wetmore C, et al. Phase I trial, pharmacokinetics, and pharmacodynamics of vandetanib and dasatinib in children with newly diagnosed diffuse intrinsic pontine glioma. Clin Cancer Res. 2013;19(11):3050–3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lagas JS, van Waterschoot RAB, van Tilburg VACJ, et al. Brain accumulation of dasatinib is restricted by P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) and can be enhanced by elacridar treatment. Clin Cancer Res. 2009;15(7):2344–2351. [DOI] [PubMed] [Google Scholar]
- 29. Benezra M, Hambardzumyan D, Penate-Medina O, et al. Fluorine-labeled dasatinib nanoformulations as targeted molecular imaging probes in a PDGFB-driven murine glioblastoma model. Neoplasia. 2012;1412:1132–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Little SE, Popov S, Jury A, et al. Receptor tyrosine kinase genes amplified in glioblastoma exhibit a mutual exclusivity in variable proportions reflective of individual tumor heterogeneity. Cancer Res. 2012;72(7):1614–1620. [DOI] [PubMed] [Google Scholar]
- 31. Navis AC, Bourgonje A, Wesseling P, et al. Effects of dual targeting of tumor cells and stroma in human glioblastoma xenografts with a tyrosine kinase inhibitor against c-MET and VEGFR2. PLoS One. 2013;8(3):e58262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wen PY, Macdonald DR, Reardon DA, et al. Updated response assessment criteria for high-grade gliomas: response assessment in neuro-oncology working group. J Clin Oncol. 2010;28(11):1963–1972. [DOI] [PubMed] [Google Scholar]
- 33. Becher OJ, Hambardzumyan D, Walker TR, et al. Preclinical evaluation of radiation and perifosine in a genetically and histologically accurate model of brainstem glioma. Cancer Res. 2010;70(6):2548–2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Groot J, Milano V. Improving the prognosis for patients with glioblastoma: the rationale for targeting Src. J Neurooncol. 2009;95(2):151–163. [DOI] [PubMed] [Google Scholar]
- 35. Du J, Bernasconi P, Clauser KR, et al. Bead-based profiling of tyrosine kinase phosphorylation identifies SRC as a potential target for glioblastoma therapy. Nat Biotech. 2009;27(1):77–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sikkema AH, Diks SH, Dunnen WFAd, et al. Kinome profiling in pediatric brain tumors as a new approach for target discovery. Cancer Res. 2009;69(14):5987–5995. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.