

Abstract
Background
New therapeutic targets are needed to eliminate cancer stem cells (CSCs). We hypothesize that direct comparison of paired CSCs and nonstem tumor cells (NSTCs) will facilitate identification of primary “driver” chromosomal aberrations that can serve as diagnostic markers and/or therapeutic targets.
Methods
We applied spectral karyotyping and G-banding to matched pairs of neurospheres (CSC-enriched cultures) and fetal bovine serum-based monolayer cultures (enriched with NSTCs) from 16 patient-derived orthotopic xenograft mouse models, including 9 medulloblastomas (MBs) and 7 high-grade gliomas (HGGs), followed by direct comparison of their numerical and structural abnormalities.
Results
Chromosomal aberrations were detected in neurospheres of all 16 models, and 82.0% numerical and 82.4% structural abnormalities were maintained in their matching monolayer cultures. Among the shared abnormalities, recurrent clonal changes were identified including gain of chromosomes 18 and 7 and loss of chromosome 10/10q (5/16 models), isochromosome 17q in 2 MBs, and a new breakpoint of 13q14 in 3 HGGs. Chromothripsis-like evidence was also observed in 3 HGG pairs. Additionally, we noted 20 numerical and 15 structural aberrations that were lost from the neurospheres and found 26 numerical and 23 structural aberrations that were only present in the NSTCs. Compared with MBs, the neurosphere karyotypes of HGG were more complex, with fewer chromosomal aberrations preserved in their matching NSTCs.
Conclusion
Self-renewing CSCs in MBs and pediatric HGGs harbor recurrent numerical and structural aberrations that were maintained in the matching monolayer cultures. These primary chromosomal changes may represent new markers for anti-CSC therapies.
Keywords: chromosomal aberration, cancer stem cell, neurosphere, orthotopic xenograft model
Brain tumors are the most common class of pediatric solid tumor. Despite advances in radiological diagnosis and multimodal therapies, brain tumors remain the leading cause of cancer-related death in children. The 5-year survival of high-grade gliomas (HGGs), including anaplastic astrocytoma (AA) and glioblastoma multiforme (GBM), is still only 20%–25%.1 Similarly, children with recurrent medulloblastoma (MB) fare dismally, with <10% long-term survival.2,3 Even in children who survive these diseases, many are left with severe neurocognitive and neuroendocrine sequelae due to therapy-related toxicities on their developing brain.
Recent isolation of cancer stem cells (CSCs) in various human cancers4–9 has led to a paradigm shift in our understanding of tumor biology and development of new therapies. Accumulating evidence suggests that CSCs are inherently resistant to conventional chemo- and radiation therapies and thus may represent the ultimate cause of tumor recurrence.10,11 One of the major impediments for CSC-specific therapies is that many key drug-resistant mechanisms and self-renewal molecular pathways active in CSCs are shared by normal stem cells. Therefore, targeting these pathways in CSCs carries a risk of damaging normal stem cells.
Chromosomal abnormalities often result in oncogenic gene fusion, amplification, or deletion. They represent a characteristic attribute of cancer cells and have long been recognized as biological and diagnostic markers as well as key therapeutic targets in several human cancers.12 However, similar to most solid tumors, cytogenetic aberrations in malignant brain tumors are usually very complex and often involve multiple chromosomes, thus making it difficult to identify the primary or driver abnormalities that play central roles in initiating and/or propagating human cancers. Since CSCs have been shown to possess the exclusive capability of self-renewal for driving tumor formation, we hypothesized that the chromosomal abnormalities critical to initial tumor formation or sustained maintenance of CSCs would be passed down to offspring nonstem tumor cells (NSTCs) and that a direct comparative analysis of chromosomal aberrations between matched CSCs and NSTCs would differentiate the primary abnormalities originated in CSCs from the secondary abnormalities acquired in NSTCs.
Limited availability of tumor tissues, coupled with the relatively rarity of CSCs (although the advanced cancers may have higher content of CSCs) remains a major challenge that impedes chromosomal analysis of CSCs in human solid tumors. We have recently shown that direct implantation of fresh surgical specimens of pediatric brain tumors into anatomically matched locations in mouse brains led to formation of orthotopic xenograft tumors that replicated the histopathological and invasive features of original patient tumors and preserved the CSC pool.13–16 Since orthotopic xenograft tumors can be serially sub-transplanted in vivo into mouse brains and cryopreserved as seed cells for subsequent expansion of a mouse model cohort, these xenograft tumors would serve as a valuable and reusable resource for the sustained supply of biologically accurate CSCs and NSTCs.
In this study, we applied cytogenetic analysis and spectral karyotyping (SKY) to tumor cells harvested from a panel of 16 patient tumor-derived orthotopic xenograft mouse models (9 MBs and 7 HGGs). Recognizing the limitations of cell surface markers in identifying CSCs, we utilized a neurosphere assay17 to functionally enrich self-renewing CSCs in vitro (hereafter referred as “CSC-enriched cultures”) and compared them with traditional primary cultured cells in fetal bovine serum (FBS)-based medium, which is shown to facilitate the growth of non-stem tumor cells (hereafter referred as “serum-treated cultures”).18 Our aims were to determine (i) if the self-renewing CSCs in the neurospheres harbor any chromosomal aberrations, (ii) if the primary cytogenetic changes identified in self-renewing CSCs in CSC-enriched cultures are maintained in NSTCs grown in serum-treated cultures, and (iii) if there are any recurrent chromosomal abnormalities that are shared among neurospheres of same or different tumor types.
Materials and Methods
Patient-derived Orthotopic Xenograft Mouse Models
All experiments were performed following a human protocol approved by the institutional review board and an animal protocol approved by the institutional animal care and use committee of Baylor College of Medicine. Signed informed consent was obtained from all patients or their legal guardians prior to sample acquisition. A total of 9 MB and 7 HGG orthotopic xenograft mouse models were included (Table 1). These models were established through direct implantation of fresh surgical specimens into matched locations in the brains of Rag2/severe combined immunodeficiency (SCID) mice.13–15,19 Briefly, Rag2/SCID mice were bred and maintained in a specific pathogen-free animal facility at Texas Children's Hospital. Mice of both sexes, aged 6–8 weeks, were anesthetized with sodium pentobarbital (50 mg/kg). Tumor cells (1 × 105) were suspended in 2 µL of culture medium and injected into cerebral hemisphere (1 mm to the right of the midline, 1.5 mm anterior to the lambdoid suture, and 3 mm deep) or cerebellar hemisphere (1 mm to the right of the midline, 1 mm posterior to the lambdoid suture, and 3 mm deep) via a 10-µL 26-gauge Hamilton Gastight 1701 syringe needle. The animals were monitored daily until they developed signs of neurological deficit or became moribund, at which time they were euthanized and their brains removed to harvest xenograft tumors.
Table 1.
Clinical and pathological features of the orthotopic xenograft mouse models
| No. | Model ID | Age | Sex | Final Diagnosis | Molecular Subtype |
|---|---|---|---|---|---|
| 1 | ICb-984MB | 7 years 10 months | Female | MB (anaplastic) | SHH |
| 2 | ICb-1299MB | 2 years 9 months | Female | MB (anaplastic) | C/D |
| 3 | ICb-1572MB | 14 years 9 months | Male | MB (large cell) | C |
| 4 | ICb-1338MB | 0 years 6 months | Male | MB (nodular) | SHH |
| 5 | ICb-1595MB | 1 years 3 months | Male | MB (anaplastic) | C |
| 6 | ICb-1078MB | 11 years 9 months | Male | MB (anaplastic) | D |
| 7 | ICb-1494MB | 5 years 2 months | Female | MB (anaplastic) | C |
| 8 | ICb-Z61109MB | 7 years | Male | MB (anaplastic) | (ND) |
| 9 | ICb-J1017MB | 9 years | Male | MB (anaplastic) | C |
| 10 | ICb-1227AA | 16 years 11 months | Female | AA | – |
| 11 | IC-1502GBM | 4 years 8 months | Female | GBM | – |
| 12 | IC-3704GBM | 12 years | Male | GBM | – |
| 13 | IC-3752GBM | 5 years | Female | GBM | – |
| 14 | IC-1128GBM | 8 years 7 months | Male | GBM | – |
| 15 | IC-2305GBM | 9 years | Male | GBM | – |
| 16 | IC-1406GBM | 5 years | Female | GBM | – |
Abbreviations: AA, anaplastic astrocytoma; GBM, glioblastoma multiforme; MB, medulloblastoma; ND, not done.
Fetal Bovine Serum-based Monolayer Culture of Xenograft Tumor Cells
Xenograft tumors from patient-derived orthotopic xenograft (PDOX) models were mechanically dissociated into single cells, as described previously,13 and incubated in Dulbecco's modified Eagle's medium supplemented with 10% FBS in a CO2 incubator at 37°C. The culture medium was changed every 3–4 days. Since variable length of time in culture can potentially introduce significant differences of cell growth, the seeded tumor cells were allow to attach and reach ∼80% confluence between 1–39 (14.6 ± 11.3) days (Fig. 2B) when the tumor cells were harvested for chromosome analysis following standard protocols.
Fig. 2.

Experimental outline. (A) Orthotopic xenograft mouse models were established by direct implantation of patient tumor cells into the matched locations in mouse brains (ie, glioblastomas to mouse cerebrum [as shown] and medulloblastomas to mouse cerebellum). (B) Paired cultures of tumor cells from the same xenograft tumors were initiated in serum-free medium to facilitate the growth of cancer stem cells (CSCs) into neurospheres and in traditional fetal bovine serum-based medium to propagate cells enriched in nonstem tumor cells (NSTCs). (C) Representative spectral karyotyping images of IC-1227AA showing the preservation of CSC chromosome aberrations in the NSTCs (exampled by the 2 linked squares) and the complex translocations that indicate chromothripsis involving chromosomes 2, 3, 8, and 10 (circled in white). (D) NSTCs showed 2 clones that appear to suggest a split from the major clone shown in the CSCs in case IC-1128 GBM. Aberrant chromosomes in neurospheres were color coded (ie, blue indicates those maintained in clone 1, red in clone 2, and green in both clones of NSTCs).
Neurosphere Assay (Cancer Stem Cell-enriched Cultures)
Single cell suspensions were freshly prepared from xenograft tumors and incubated in serum-free stem cell growth medium consisting of neurobasal media, N2 and B27 supplements (0.5× each; Invitrogen), human recombinant basic fibroblast growth factor (50 ng/mL), epidermal growth factor (40 ng/mL; R&D Systems),18 and penicillin G and streptomycin sulfate (1:100; Invitrogen). Formation of neurospheres (Fig. 2B, >50 cells) was monitored under a Nikon phase-contrast microscope. Neurospheres were harvested between 8 and 28 (15.5 ± 7.4) days for chromosome analysis.
Flow Cytometry
Tumor cells were labeled with monoclonal antibodies against human CD133, CD44 conjugated with APC, and CD49f conjugated with FITC (Milteny) at 4°C for 10 minutes per manufacturer's instructions.13–15 Cells were then washed and resuspended in Hanks′ balanced saline solution containing 5% FBS. Isotype control was included to set the baseline gate. Cells were analyzed using LSR II (Life Technologies), and data were graphed with Kaluza flow analysis software (Beckman Coulter, Inc., Brea, CA). Dead cells were excluded by propidium iodide staining.
Spectral Karyotyping and G-banding Chromosome Analysis
Cultured neurospheres and monolayer cells were trypsinized and harvested. G-banding was completed using standard cytogenetic procedures. Chromosomal results were interpreted according to the International System for Human Cytogenetic Nomenclature (ISCN 2005 and 2009). For SKY, a cocktail of human chromosome paints was obtained from Applied Spectral Imaging (ASI) . Hybridization and detection were carried out according to the manufacturer's protocol. For each case, a minimum of 10 metaphase cells was analyzed. Images were acquired with a SD300H Spectra cube (ASI) mounted on a Zeiss Axioplan II microscope using a custom-designed optical filter (SKY-1; Chroma Technology) and analyzed using SKY View 2.1.1 software (ASI). Breakpoints on the SKY-painted chromosomes were determined by comparison of corresponding DAPI and/or G-banding of the same chromosome. A breakpoint was considered recurrent if identified in 2 or more cases. Primary karyotyping data are available upon request.
Statistical Analysis
Comparison of the numbers of structural and numerical aberrations between MB and HGGs were performed with the Student t test, and the differences in sample distribution were analyzed with the Pearson' chi-square test. P values < .05 are considered significant.
Results
Differences of Cancer Stem Cell Contents Between Neurospheres and Fetal Bovine Serum-based Monolayer Cultures
CSC theory suggests that CSCs give rise to NSTCs, which populate the tumor mass.4,20–22 It is therefore our hypothesis that the key cytogenetic abnormalities of CSCs critical for sustained growth of tumor mass would be passed on to their offspring NSTCs. Although it would be best to compare CSCs directly with matching NSTCs, not all CSC markers have been identified in brain tumors. Additionally, there remain technical difficulties to isolating biologically accurate and quantitatively sufficient CSC cells, even using existing cell surface markers such as CD133, CD44 and CD49, because the positive tumor cells tend to be rare in many tumors (Fig. 1A and C).
Fig. 1.
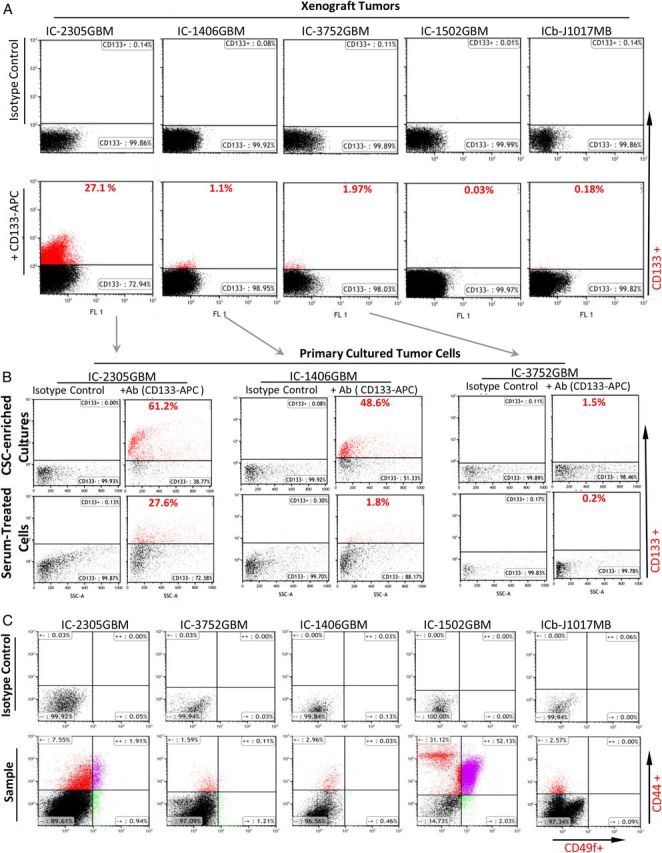
Flow cytometric analysis of cancer stem cell (CSC) content using CD133 as a representative marker of brain tumor CSCs. (A) Direct analysis of CD133+ content in patient-derived orthotopic xenograft (PDOX) tumors. (B) Comparison of CD133+ cell content between neurospheres and fetal bovine serum-based monolayer cultures in 3 glioblastoma models. Isotype control was used for each sample to set baseline. (C) Direct analysis of CD44+ and/or CD49f+ cells through double staining in 5 PDOX models.
While formation of neurospheres from single cells is a well-established assay for tfunctional evaluation of stem cell self-renewal capacity independent of cell surface markers,17,23 strategies for isolating and propagating pure NSTCs in vitro have not been well established primarily because not all brain tumor stem cell markers have been identified. To overcome this difficulty, we examined if primary cultured monolayer cells (the primary method in clinical cytogenetic laboratories) can be used as a reliable reference of NSTCs by comparing their contents of CD133+ cells with neurospheres. As shown in (Fig. 1B), the contents of CD133+ cells in FBS-based monolayer cells were much lower than those in the matching neurospheres and ranged from 3% in IC-1406GBM and 13.3% in IC-3752GBM to 45% in IC-2305GBM. These data confirmed that FBS-based monolayer cultures favored the growth of CD133− tumor cells, suggesting that direct comparison with self-renewing CSCs in neurosphere should provide important insights about the cytogenetic differences between CSCs and NSTCs (Fig. 2A–C).
Neurospheres in Cancer Stem Cell-enriched Cultures Displayed Both Numerical and Structural Chromosomal Aberrations
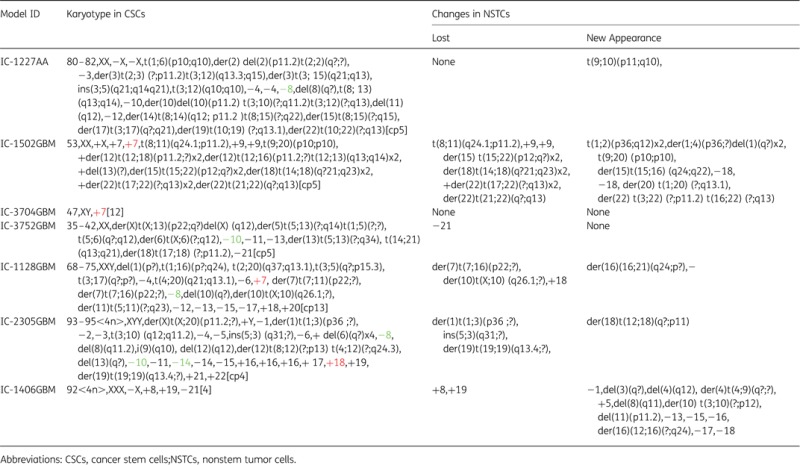
CSC theory suggests that human cancers are driven by rare populations of CSCs that possess exclusive self-renewal capabilities.4,20–22 Little, however, is known about the cytogenetic changes in CSCs. To examine if brain tumor CSCs contain chromosomal aberrations, we applied both SKY and G-banding in neurospheres that had been freshly grown in CSC-enriched cultures (Fig. 2B) derived from 16 PDOX mouse models that were shown to have preserved CD133+ stem cells.13,14 In all neurospheres, we observed chromosomal abnormalities (Fig. 2C). Numerical aberrations (gain or loss of whole chromosome) were detected in all tumors (100%), ranging from 1–14 per cell (6.6 ± 5.5) in MBs (Tables 2 and 4) and 1–21 per cell (7.4 ± 6.6) in HGGs (Tables 3 and 4). The difference between MBs and HGGs, however, was not significant (P = .77). Structural aberrations ranged from 0–8 per cell (2.7 ± 2.6) in MBs (Tables 2 and 4) and from 0–15 per cell (8.7 ± 6.5) in HGGs (Tables 3 and 4). The difference between MBs and HGGs was significant (P = .023). These data showed that numerical and structural aberrations were frequent in both MB and HGG self-renewing stem cells (CSCs), and that HGG neurospheres exhibited greater structural abnormalities.
Table 2.
Spectral karyotyping and G-banding of medulloblastoma xenograft tumors

|
Table 4.
Summary of numerical and structural aberrations observed in cancer stem cells and nonstem tumor cells
| Model ID | Numerical Aberrations |
Structural Aberrations |
||||||
|---|---|---|---|---|---|---|---|---|
| Total in CSC | Shared in NSTCs | Lost in NSTCs | New in NSTCs | Total in CSC | Shared in NSTCs | Lost in NSTCs | New in NSTCs | |
| ICb-984MB | 14 | 14 | 0 | 0 | 8 | 8 | 0 | 2 |
| ICb-1299MB | 2 | 2 | 0 | 1 | 0 | 0 | 0 | 0 |
| ICb-1572MB | 1 | 1 | 0 | 0 | 3 | 3 | 0 | 0 |
| ICb-1338MB | 1 | 1 | 0 | 0 | 1 | 1 | 0 | 0 |
| ICb-1595MB | 9 | 3 | 6 | 4 | 0 | 0 | 0 | 0 |
| ICb-1078MB | 11 | 6 | 5 | 8 | 4 | 3 | 1 | 0 |
| ICb-1494MB | 14 | 11 | 3 | 4 | 1 | 1 | 0 | 0 |
| ICb-61109MB | 2 | 2 | 0 | 0 | 5 | 5 | 0 | 0 |
| ICb-J1017MB | 5 | 5 | 0 | 0 | 2 | 2 | 0 | 3 |
| MB Total | 59 | 45 | 14 | 17 | 24 | 23 | 1 | 5 |
| (%)a | 76.3% | 23.7% | (28.8%) | 95.9% | 4.1% | (20.8%) | ||
| IC-1227AA | 8 | 8 | 0 | 0 | 15 | 15 | 0 | 1 |
| IC-1502GBM | 5 | 3 | 2 | 2 | 14 | 5 | 9 | 8 |
| IC-3704GBM | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| IC-3752GBM | 4 | 3 | 1 | 0 | 7 | 7 | 0 | 0 |
| IC-1128GBM | 9 | 8 | 1 | 0 | 11 | 9 | 2 | 1 |
| IC-2305GBM | 21 | 21 | 0 | 0 | 14 | 11 | 3 | 1 |
| IC-1406GBM | 4 | 2 | 2 | 7 | 0 | 0 | 0 | 7 |
| HGGb Total | 52 | 46 | 6 | 9 | 61 | 47 | 14 | 18 |
| (%) | 88.5% | 11.5% | (17.3%) | 77.1% | 22.9% | (29.5%) | ||
Abbreviations: AA, astrocytoma; CSCs, cancer stem cells; GBM, glioblastoma multiforme; NSTCs, nonstem tumor cells.
aPercentage of the total numerical or structural changes in CSCs.
bHGG includes both AA and GBM.
Table 3.
Spectral karyotyping and G-banding of malignant glioma xenograft tumors
|
Structural and Numerical Aberrations Found in Neurospheres Were Maintained in Primary Monolayer Cells in Serum-treated Cultures
When all chromosomal aberrations (both recurrent and sporadic) were taken into consideration, we found 3 MBs and one GBM in which the monolayer cultures maintained 100% of the abnormalities present in their corresponding neurospheres. For the remaining 6 MBs and 6 HGGs, partial retention of chromosomal aberrations was observed.
Overall, 82% of numerical changes and 82.4% of structural changes that were detected in neurospheres in CSC-enriched cultures were maintained in the monolayer cells in serum-treated cultures enriched with NSTCs. In MB neurospheres, 45 of 59 (76.3%) numerical changes (gain or loss of whole chromosome or the entire chromosome arm) (Tables 2 and 4) were retained in their matching monolayer cultures, whereas 46 of 52 (88.5) HGG neurospheres were retained (Tables 3 and 4). Of the 24 structural chromosomal aberrations found in MB neurospheres, 23 (95.9%) were detected in the monolayer cultures (Table 2), whereas 47 of the 61 (77.1%) HGG neurospheres with structural aberrations were preserved in their monolayer cultures enriched with NSTCs (Tables 3 and 4). Worthy of note is that complete preservation of neurosphere structural aberrations in their corresponding monolayer cultures was observed in 6 of 7 (85.7%) MBs (2 additional MB neurospheres did not have structural changes) and in 2 of 6 (33.3%) HGGs (Tables 2–4). Chi-square analysis showed that the distribution differences between HGG and MB samples were significant (P < .01), suggesting that HGG cells had higher genomic instability than MB cells.
Identification of Recurrent and Clonal Chromosomal Aberrations That Were Shared by Neurospheres and Their Matching Monolayer Cultures Enriched With Nonstem Tumor Cells
Recurrent (in more than one tumor) and clonal (in more than 2 cells per tumor) chromosomal aberrations have long been recognized as being more important than random changes. It is therefore reasonable to believe that the recurrent and/or clonal aberrations present in both neurospheres and monolayer cultures have the potential to be the drivers sustaining tumor growth.
A total of 25 recurrent clonal chromosomal aneuploidies (9 gains and 16 losses) were identified. Gains of chromosomes 7, 9, 16, 18, 19, 20, and 21 were shared by both MBs and HGGs, while gain of chromosome 22 was only observed in MBs. Among the 16 whole chromosomes that were lost, 9 chromosomes (chromosomes 1, 2, 8, 10/10q, 11, 13, 14, 17, and X) were observed in both MBs and HGGs, and 7 (chromosomes 3, 4, 6, 12, 15, 18 and 21) were observed only in HGGs (Fig. 3). Among these aneuploidies, gain of chromosome 18 was the most common aberration and was found in 5 of 9 pairs (55.5%) of MBs and 1 of 7 pairs (14.3%) of HGGs. Gain of chromosome 7, which has been previously reported to be a recurrent finding in GBMs,24,25 was found in 3 of 7 pairs (42.8%) of HGGs and 2 of 9 pairs (22.2%) of MBs. In addition, loss of chromosome 10 or 10q, which has also been previously reported as a recurrent cytogenetic finding in HGGs, was observed in 3 of 7 pairs (42.8%) of HGGs and 2 of 9 pairs (22.2%) of MBs. One MB or HGG had concurrent loss of chromosome 10 and gain of chromosome 18. Chromosomal gains of 14, 18, 19, 21 and 22 were more frequently found in MBs, whereas the losses of chromosome 3, 4, 6, 8, 12, 13, 18, and 21 in HGGs (Fig. 3).
Fig. 3.
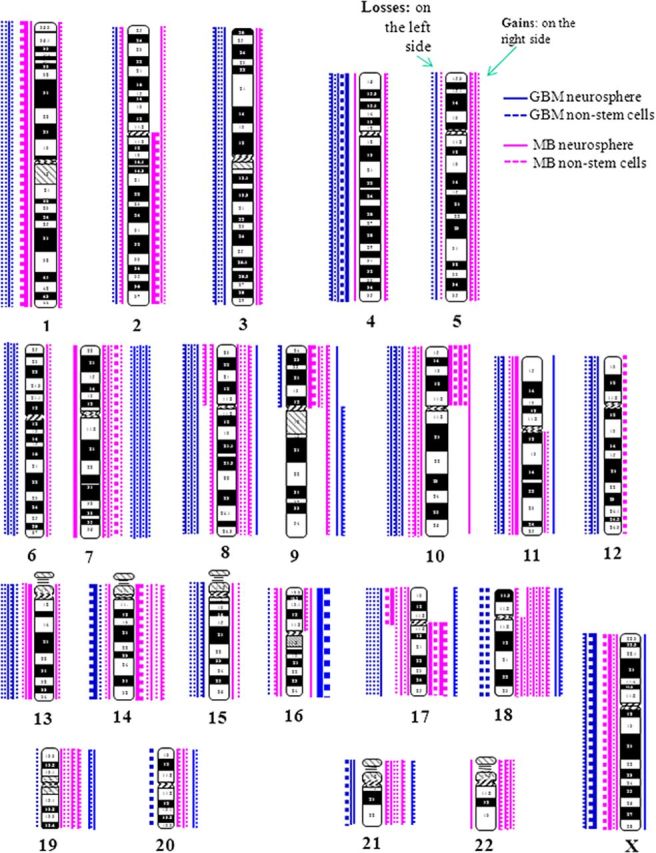
Ideogram showing chromosome aneuploidies or chromosomal arm gains/losses. Chromosomal losses were plotted at the left side and gains at the right side of each chromosome (blue for high-grade gliomas and pink for medulloblastomas). Neurospheres are represented by solid lines and monolayer cultured NSTCs by dotted lines.
From the 24 clonal structural aberrations identified in the neurospheres of 7 MBs, we found iso17q (10), which is one of the most common chromosomal aberrations described in MBs,26,27 in 2 models (ICb-984MB and ICb-1078MB). Although no similar recurrent structural aberrations were identified from the 61 total clonal structural aberrations in HGG neurospheres, we detected 3 structural aberrations in 3 different tumors (IC-1227AA, IC-1502GBM, and IC-3752GBM) that involved in a breakpoint at 13q14 (Table 3). This abnormality has not been previously described in pediatric HGGs.
New Clonal Chromosomal Abnormalities in Monolayer Cells in Serum-treated Cultures
Genetic instability is one of the hallmarks of cancer cells.28–30 It is possible that rapid division and proliferation of NTSC would introduce new structural and/or numerical abnormalities. Some of these abnormalities, particularly recurrent and new clonal aberrations, might be important for facilitating rapid proliferation of tumor mass. In this study, 26 new numerical and 23 structural aberrations were observed in monolayer cultures enriched with NSTCs. Among them, 17 new clonal numerical changes were detected in 4 MBs (4/9, 44.4%), and 9 were identified in 2 HGGs (2/7, 28.5%), respectively. Interestingly, recurrent losses of chromosomes 13, 17, and 18 were found only in HGGs (Tables 3 and 4).
New clonal structural aberrations were found in 2 MBs: ICb-984MB showing der(22)(q10)t(15;22)(q?;?) and ICb-J1017MB showing der(2)t(2;12), der(9)t(9;15) and del(12). In HGGs, we found new structural aberrations in 3 tumors: (i) IC-1128GBM showing der(16)(16;21)(q24;p?)(n = 1), IC-1502GBM showing t(1;2)(p36;q12)x2, der(1;4)(p36;?) del(1)(q?)x2, der(20)t(1;20)(?;q13.1), and der(22)t(3;22)(?;p11.2)t(16;22)(?;q13) (n = 6), and (iii) IC-1127AA showing t(9;10)(p11;q10) (n = 1). The lack of shared structural aberrations among these tumors is indicative of the random nature of these chromosomal abnormalities, even though the overall case number is small.
Loss of Clonal Chromosomal Aberrations Originally Present in the Neurospheres
We next examined if clonal aberrations of CSCs can be lost in the monolayer cultures enriched with NSTCs since they may represent alterations that are either associated with stem cell status, (eg, self-renewal and multipotent capacity) or are not critically required for the expansion of tumor mass. A total of 14 numerical changes of neurospheres were lost in 3 of the 9 MBs (Tables 2 and 4), and 6 numerical aberrations disappeared in the monolayer cultures in 4 of the 7 HGGs (Tables 3 and 4). Additionally, a single structural aberration disappeared in one of 9 MBs and 14 structural aberrations were lost in 3 of 7 HGGs. No aberrations disappeared in more than one tumor (ie, recurrent), suggesting that they may not play important roles in sustaining tumor mass expansion. The distribution differences of clonal chromosomal losses (chi-square analysis) and the percentage of loss per tumor (Student t test) between HGG and MB neurospheres were not significant (P > .05). One interesting phenomenon observed in IC-1128 GBM was that this model's monolayer cultures showed 2 different clones that appeared to have derived from the primary clone found in the neurospheres (Fig. 2D), although it is possible that these changes originated from 2 independent clones.
Both Neurospheres and Monolayer Cultures Harbor Chromothripsis-like Complex Chromosomal Rearrangements
Chromothripsis is a relatively new concept describing the complexity of genome abnormalities that can either involve a single chromosome or the entire genome.31,32 It has been reported in both germline and tumors by high resolution genome-wide analysis. Here, we defined “chromothripsis-like” as a single chromosome or chromosome arm involving 3 or more events including deletions, duplications, and unbalanced translocations at chromosomal level. Three HGGs (3/7, 42.9%) showed evidence of chromothripsis33 (Table 3 and Fig. 2C) including chromosomes 2, 3, 8, and 10 in IC-1227AA, chromosome 12 in IC-1502GBM, and chromosomes X and 5 in IC-3752GBM. In addition, we also detected double minutes (a structural change also proposed to result from a chromothripsis event34) in both neurospheres and monolayer cultures in ICb-1078MB. In cases ICb-J1017MB and IC-2305GBM, a chromothripsis-like event involving chromosome 12 was observed in the monolayer cells only. Altogether, these data indicated that chromothripsis-like events exist in CSC-enriched cultures and in monolayer cells grown in serum-treated cultures.
Discussion
Here we report, for the first time, the molecular cytogenetic analysis of self-renewing CSCs in CSC-enriched neurosphere cultures and the direct comparison with their matching FBS-based monolayer cultures enriched with NSTCs using a novel set of PDOX xenograft mouse models. Our data demonstrated that neurospheres in malignant brain tumors contain recurrent numerical and structural chromosomal aberrations and that the majority of these chromosomal abnormalities were preserved in their matching serum-treated monolayer cultures that were enriched with NSTCs. We also found multiple chromosomal aberrations that were either lost from the neurospheres or were present only in the monolayer cultures. Our data provided experimental evidence to support the CSC theory and offered a novel approach for differentiating primary and secondary chromosomal aberrations in human solid tumors.
Compared with hematological malignancies, few cytogenetic markers (including MBs and GBMs) have been identified in solid human cancers. This is primarily due to the extreme complexity of chromosomal aberrations. Although it has been widely recognized that many chromosome changes result from the genetic instabilities of cancer cells and are thus termed secondary changes, there has been little success in differentiating secondary and primary cytogenetic changes. Recent identification of CSCs has provided a new clue for pinpointing primary cytogenetic abnormalities in solid tumors. However, there are no phenotypic markers that can reliably isolate all CSC populations. Although several markers, such as CD133 and CD15, have been utilized for isolating and characterizing CSCs, there are still controversies about their specificity and relative abundance.35–37 Formation of neurospheres has been a reliable functional assay for examining in vitro self-renewal capabilities of both normal and cancerous stem cells.17,38 Our examination of structural and numerical chromosomal abnormalities of neurospheres in CSC-enriched cultures would therefore provide functional and unbiased analysis of chromosomal aberrations in brain tumor CSCs. It is noteworthy that cells in neurospheres are heterogeneous. Although each neurosphere is derived from single CSC cells, some of the daughter cells have undergone differentiation to become rapidly proliferating NSTCs. Because asymmetrical division is an intrinsic feature of stem cell proliferation and there are currently no assays that ensure complete symmetrical division—in which both daughter cells maintain stem cell status—the neurosphere assay remains the assay of choice for propagating brain tumor stem cells. Since we only incubated tumor cells for an average of 15.5 days, the single CSCs may need as few as 6 cell divisions to form neurospheres of 64 cells (assuming every cell in the neurosphere was actively proliferating). By analyzing the chromosomal changes in these “short-term” cultured neurospheres, we have a better chance for capturing at least some of the primary aberrations of the original and single CSCs.
Direct comparison between tumor cells grown in CSC-enriched cultures (eg, neurospheres) and those grown in nonstem-promoting conditions (eg, serum-treated cultures) at the single-cell level has an unique advantage: (i) it can identify primary clonal changes that were shared by neurospheres and monolayer cultures (enriched with NSTCs) and (ii) detect secondary changes that were present only in the monolayers (described as new aberrations in this study) or in the neurospheres (described as lost aberrations). Although the role of these secondary changes remains to be determined, we hypothesize that the recurrent primary aberrations (preserved clonal changes that occurred in at least 2 independent tumors) might have played critical roles in sustaining tumor growth. Our identification of 3 previously described chromosomal aberrations (ie, iso17q in MB,26,27,39 trisomy 7 in GBM,25,40 and loss of chromosome 10 and 10q in GBM41) were in fact recurrent abnormalities present in self-renewing CSCs provided solid initial data to support this hypothesis.
The advantage of our approach is further enhanced by the discovery of a new recurrent breakpoint at 13q14 in GBMs and the detection of new recurrent and clonal gains and losses of chromosomes from the relatively small tumor populations. Our finding of the high frequencies of gain for chromosome 18 in 55.5% of MBs and loss of chromosomes 3, 4, 6, and 12 in 42.8% of HGGs has provided important new clues that may lead to identification of new gene(s) or signaling pathways critical for the initiation and progression of malignant brain tumors. These findings are consistent with the model of CSCs in which NSTCs are derived from the seed cells (CSCs) and that further independent progression/evolution of these offspring cells would result in divergent karyotypes; however, it is also possible that CSCs themselves became heterogeneous during tumor development and that some NSTC cells only retained the abnormalities of more primitive CSCs that had fewer aberrations.
Chromothripsis is a relatively new concept depicting the complexity of a single chromosome or the entire genome.31,32,42 The majority of cases (12/16, 75%) in our study showed complex chromosomal aberrations. While the overall frequencies of chromothripsis in MBs and HGGs were in agreement with the data previously reported in MBs31 and gliomas,42 our findings further showed that this chromosomal catastrophe can happen both in neurospheres in CSC-enriched cultures and monolayer cells in serum-treated cultures enriched with NSTCs in pediatric HGGs, suggesting that chromothripsis may have played a role in these tumors.
With regard to a comparison between MB and HGG, our studies showed that the levels of divergence from normal karyotype between these 2 types of malignant brain tumors were not identical. The HGG cells in serum-treated cultures exhibited a higher degree of disparity from their neurospheres grown in CSC-enriched cultures than that of the MB cells (Tables 2 and 3). Biologically, these findings suggested that the degree of genomic instability in HGGs is higher than that of MBs. Clinically, these data suggested that therapies only effective against the NSTC populations in HGG cells will have a greater chance of failure against CSC population because of the vast differences in their genetic abnormalities; at the same time, new therapies may have to be developed for the recurrent tumors due to increased genetic drift during tumor progression and evolution.
Compared with the genome-wide single nucleotide polymorphism (SNP) microarray analysis and whole genome sequencing,42 the resolution of SKY is limited at ∼10–15 Mb levels. It is therefore possible that some cryptic structural aberrations (deletion or amplification) or copy-neutral loss of heterozygosity were missed and that the breakpoints may still need to be defined with additional methods such as fluorescence in-situ hybridization. However, our approach has some unique advantages that cannot be matched by the aforementioned technologies. Specifically, the molecular cytogenetic approach allowed us to examine independent and truly single tumor cells to identify subtle changes pertaining to specific or even smaller fraction of tumor cells. Combining the capability of molecular cytogenetics for analyzing genetic changes of single tumor cells with the power of high-resolution whole-genome analysis approaches (ie, OligoArray, comparative genomic hybridization, or SNP array), may facilitate identification of recurrent primary genetic changes critical for tumor initiation and progression at single gene or exon levels.
In summary, we report for the first time that neurospheres in CSC-enriched culture in malignant brain tumors contain recurrent structural and numerical chromosomal aberrations. We showed that the majority of these chromosomal abnormalities were shared by their matching monolayer cultures enriched with NSTCs, hence representing the primary chromosomal aberrations. At the same time, we identified chromosomal aberrations that were only present in the monolayer cells in serum-treated cultures (ie, the secondary changes). Additionally, we found that the cytogenetic features of monolayer cultures enriched with NSTCs in HGG tend to diverge more from their neurospheres than those in MBs. This finding not only revealed the increased genetic instability in HGGs, it also suggested that multiple therapeutic strategies will be needed to effectively target different subpopulations in HGGs.
Funding
This study is supported by Cancer Fighters of Houston and Clayton Foundation for Research (Li, XN).
Conflict of interest statement. None to be declared for all authors.
References
- 1. Cohen KJ, Gibbs IC, Fisher PG, et al. A phase I trial of arsenic trioxide chemoradiotherapy for infiltrating astrocytomas of childhood. Neuro Oncol. 2013;15(6):783–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ellison DW, Dalton J, Kocak M, et al. Medulloblastoma: clinicopathological correlates of SHH, WNT, and non-SHH/WNT molecular subgroups. Acta Neuropathol. 2011;121(3):381–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Northcott PA, Korshunov A, Witt H, et al. Medulloblastoma comprises four distinct molecular variants. J Clin Oncol. 2011;29(11):1408–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hemmati HD, Nakano I, Lazareff JA, et al. Cancerous stem cells can arise from pediatric brain tumors. Proc Natl Acad Sci USA. 2003;100(25):15178–15183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Singh SK, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature. 2004;432(7015):396–401. [DOI] [PubMed] [Google Scholar]
- 6. Hope KJ, Jin L, Dick JE. Acute myeloid leukemia originates from a hierarchy of leukemic stem cell classes that differ in self-renewal capacity. Nat Immunol. 2004;5(7):738–743. [DOI] [PubMed] [Google Scholar]
- 7. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3(7):730–737. [DOI] [PubMed] [Google Scholar]
- 8. Hudson DL. Epithelial stem cells in human prostate growth and disease. Prostate Cancer Prostatic Dis. 2004;7(3):188–194. [DOI] [PubMed] [Google Scholar]
- 9. Fan X, Eberhart CG. Medulloblastoma stem cells. J Clin Oncol. 2008;26(17):2821–2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005;5(4):275–284. [DOI] [PubMed] [Google Scholar]
- 11. Jones RJ, Matsui WH, Smith BD. Cancer stem cells: are we missing the target? J Natl Cancer Inst. 2004;96(8):583–585. [DOI] [PubMed] [Google Scholar]
- 12. Shaw AT, Yeap BY, Solomon BJ, et al. Effect of crizotinib on overall survival in patients with advanced non-small-cell lung cancer harbouring ALK gene rearrangement: a retrospective analysis. Lancet Oncol. 2011;12(11):1004–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shu Q, Wong KK, Su JM, et al. Direct orthotopic transplantation of fresh surgical specimen preserves CD133+ tumor cells in clinically relevant mouse models of medulloblastoma and glioma. Stem Cells. 2008;26(6):1414–1424. [DOI] [PubMed] [Google Scholar]
- 14. Yu L, Baxter PA, Voicu H, et al. A clinically relevant orthotopic xenograft model of ependymoma that maintains the genomic signature of the primary tumor and preserves cancer stem cells in vivo. Neuro Oncol. 2010;12(6):580–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liu Z, Zhao X, Mao H, et al. Intravenous injection of oncolytic picornavirus SVV-001 prolongs animal survival in a panel of primary tumor-based orthotopic xenograft mouse models of pediatric glioma. Neuro Oncol. 2013;15(9):1173–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu Z, Zhao X, Wang Y, et al. A patient tumor-derived orthotopic xenograft mouse model replicating the group 3 supratentorial primitive neuroectodermal tumor in children. Neuro Oncol. 2014;16(6):787–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chaichana K, Zamora-Berridi G, Camara-Quintana J, et al. Neurosphere assays: growth factors and hormone differences in tumor and nontumor studies. Stem Cells. 2006;24(12):2851–2857. [DOI] [PubMed] [Google Scholar]
- 18. Lee J, Kotliarova S, Kotliarov Y, et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell. 2006;9(5):391–403. [DOI] [PubMed] [Google Scholar]
- 19. Shu Q, Antalffy B, Su JM, et al. Valproic Acid prolongs survival time of severe combined immunodeficient mice bearing intracerebellar orthotopic medulloblastoma xenografts. Clin Cancer Res. 2006;12(15):4687–4694. [DOI] [PubMed] [Google Scholar]
- 20. Singh SK, Clarke ID, Terasaki M, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63(18):5821–5828. [PubMed] [Google Scholar]
- 21. Marx J. Cancer research. Mutant stem cells may seed cancer. Science. 2003;301(5638):1308–1310. [DOI] [PubMed] [Google Scholar]
- 22. Al Hajj M, Becker MW, Wicha M, et al. Therapeutic implications of cancer stem cells. Curr Opin Genet Dev. 2004;14(1):43–47. [DOI] [PubMed] [Google Scholar]
- 23. Wan F, Zhang S, Xie R, et al. The utility and limitations of neurosphere assay, CD133 immunophenotyping and side population assay in glioma stem cell research. Brain Pathol. 2010;205:877–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bigner SH, Mark J, Mahaley MS, et al. Patterns of the early, gross chromosomal changes in malignant human gliomas. Hereditas. 1984;101(1):103–113. [DOI] [PubMed] [Google Scholar]
- 25. Bigner SH, Humphrey PA, Wong AJ, et al. Characterization of the epidermal growth factor receptor in human glioma cell lines and xenografts. Cancer Res. 1990;50(24):8017–8022. [PubMed] [Google Scholar]
- 26. Biegel JA, Janss AJ, Raffel C, et al. Prognostic significance of chromosome 17p deletions in childhood primitive neuroectodermal tumors (medulloblastomas) of the central nervous system. Clin Cancer Res. 1997;3(3):473–478. [PubMed] [Google Scholar]
- 27. Cogen PH, Daneshvar L, Metzger AK, et al. Involvement of multiple chromosome 17p loci in medulloblastoma tumorigenesis. Am J Hum Genet. 1992;50(3):584–589. [PMC free article] [PubMed] [Google Scholar]
- 28. Beckman RA, Loeb LA. Genetic instability in cancer: theory and experiment. Semin Cancer Biol. 2005;15(6):423–435. [DOI] [PubMed] [Google Scholar]
- 29. Lagasse E. Cancer stem cells with genetic instability: the best vehicle with the best engine for cancer. Gene Ther. 2008;15(2):136–142. [DOI] [PubMed] [Google Scholar]
- 30. Damia G, D'Incalci M. Genetic instability influences drug response in cancer cells. Curr Drug Targets. 2010;11(10):1317–1324. [DOI] [PubMed] [Google Scholar]
- 31. Rausch T, Jones DT, Zapatka M, et al. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell. 2012;148(1–2):59–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Stephens PJ, Greenman CD, Fu B, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 2011;144(1):27–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Righolt C, Mai S. Shattered and stitched chromosomes-chromothripsis and chromoanasynthesis-manifestations of a new chromosome crisis? Genes Chromosomes Cancer. 2012;51(11):975–981. [DOI] [PubMed] [Google Scholar]
- 34. Holland AJ, Cleveland DW. Chromoanagenesis and cancer: mechanisms and consequences of localized, complex chromosomal rearrangements. Nat Med. 2012;18(11):1630–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang J, Sakariassen PO, Tsinkalovsky O, et al. CD133 negative glioma cells form tumors in nude rats and give rise to CD133 positive cells. Int J Cancer. 2008;122(4):761–768. [DOI] [PubMed] [Google Scholar]
- 36. Bidlingmaier S, Zhu X, Liu B. The utility and limitations of glycosylated human CD133 epitopes in defining cancer stem cells. J Mol Med. 2008;869:1025–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Prestegarden L, Enger PO. Cancer stem cells in the central nervous system--a critical review. Cancer Res. 2010;70(21):8255–8258. [DOI] [PubMed] [Google Scholar]
- 38. Laks DR, Masterman-Smith M, Visnyei K, et al. Neurosphere formation is an independent predictor of clinical outcome in malignant glioma. Stem Cells. 2009;27(4):980–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Burnett ME, White EC, Sih S, et al. Chromosome arm 17p deletion analysis reveals molecular genetic heterogeneity in supratentorial and infratentorial primitive neuroectodermal tumors of the central nervous system. Cancer Genet Cytogenet. 1997;97(1):25–31. [DOI] [PubMed] [Google Scholar]
- 40. Bigner SH, Mark J, Bullard DE, et al. Chromosomal evolution in malignant human gliomas starts with specific and usually numerical deviations. Cancer Genet Cytogenet. 1986;22(2):121–135. [DOI] [PubMed] [Google Scholar]
- 41. Lopez-Gines C, Cerda-Nicolas M, Gil-Benso R, et al. Association of chromosome 7, chromosome 10 and EGFR gene amplification in glioblastoma multiforme. Clin Neuropathol. 2005;24(5):209–218. [PubMed] [Google Scholar]
- 42. Malhotra A, Lindberg MR, Faust GG, et al. Breakpoint profiling of 64 cancer genomes reveals numerous complex rearrangements spawned by homology-independent mechanisms. Genome Res. 2013;235:762–776. [DOI] [PMC free article] [PubMed] [Google Scholar]