

Abstract
Quantitative microscopy is needed to understand reactions or phenomena carried out by biological molecules such as enzymes, receptors, and membrane-localized proteins. Counting the biomolecules of interest in single organelles or cellular compartments is critical in these approaches. In this brief perspective, we focus on the development of quantitative fluorescence microscopies that measure the precise copy numbers of proteins in cellular organelles or purified samples. We introduce recent improvements in quantitative microscopies to overcome undercounting or overcounting errors in certain conditions. We conclude by discussing biological applications.
Introduction
Quantitative biological measurements have become increasing important. To address this need, new biochemical and biophysical methods have been introduced, while conventional approaches have been significantly advanced. For example, mass spectrometry has become ever more sensitive, and traditional workhorses of the biochemistry lab, such as Western Blotting, has been greatly improved for ascertaining the relative amounts of proteins present. Despite these improvements, bulk methods suffer from two inherent limitations: 1) Bulk measurements can only provide information about the average value, but not information regarding the distribution about the average (i.e. heterogeneity); and 2) Bulk approaches usually require calibration and the use of a standard or internal reference, and thus are relative measurements and do not offer absolute quantification. These two limitations can be overcome using single-molecule counting, which often is based on fluorescence microscopy. As a result, this perspective article focuses on fluorescence imaging based approaches.
Fluorescence microscopy requires the use of an imaging agent, such as dye-tagged antibodies or the use of fluorescent proteins (e.g. green fluorescent protein, known as GFP) that co-express in cells with the proteins of interest. Conventional fluorescence microscopy, however, has a critical downside in accurately measuring the number of proteins of interest in small organelles (< 100 nm) because of the limit in optical resolution (> ~ 200 nm for lateral resolution and > ~ 500 nm for the z-direction). This resolution limit prevents conventional fluorescence microscopy from resolving individual molecules of interest for counting in cellular compartments. Although methods to calibrate the measured intensity in fluorescence microscopy are available (vide infra), these are difficult to be applied to quantify low-copy-number proteins due to the stochastic nature of fluorescence emission from a few molecules. Non-fluorescence based microscopy has also been attempted for counting molecules, such as the use of immuno-gold combined with electron microscopy. This method, in which antibodies labeled with gold are detected by electron microscopy, can measure nanometer-scale localization of proteins in cellular compartments; however, precise counting of proteins is difficult with this technique because the antibodies do not label all target proteins which cause inaccurate and large standard deviations in measurements.
Therefore, researchers have been motivated to develop a new technique to count protein copies and to overcome most of the limitations of conventional optical and biochemical methods. After single-molecule detection was realized [2, 3], it was soon recognized that this breakthrough would allow researchers to gain new perspectives on biomolecular mechanisms [4-11]. We focus here on quantitative microscopy based on single-molecule fluorescence that counts cellular materials.
Several quantitative microscopy methods have been developed to estimate protein copies in cellular compartments. The first approach used beads of known brightness that corresponded to a known copy number of fluorescent proteins after calibration [12]. It carefully estimated the number of several representative proteins in postsynaptic density. However, it only works well for large copy numbers of proteins, usually of more than a hundred, because of large standard deviations in measurement when low copy numbers are involved.
After discovery of localization by photobleaching [13], sequential photobleaching steps of single-molecule fluorophores were used to determine functional stoichiometry of proteins [8, 14] and to count the number of proteins [15]. But it is also limited by the difficulty of correlating the photobleaching step size with copy number, especially as the copy number increases, due to coincident photobleaching of single fluorophores, non-homogeneous brightness, and poor signal-to-noise [16*].
To address the above challenges in quantitative microscopy, we developed statistical deconvolution quantitative microscopy based on single-molecule fluorescence. This approach covered the range from a single to a few tens of copies of proteins [17*]. Soon after, researchers recognized that photoswitchable organic dyes [18] or photoactivable fluorescent proteins [19] were useful for localization microscopy with nanometer-scale precision to quantify protein numbers. However, a critical issue has been the blinking of fluorophores.
In this perspective, we discuss ways to quantify the absolute number of proteins in single organelles or cellular compartments and introduce recent developments and improvements of single-molecule-based, high-resolution microscopies and their potential impact in biological studies.
Single-Molecule-based Statistical Deconvolution Quantitative Microscopy
Previously, researchers introduced counting methods for proteins of interest tagged with GFP by using single-molecule photobleaching microscopy (SMPM). Simple sequential photobleaching steps were used for determining the functional stoichiometry of membrane-localized proteins [8, 9, 14] and for counting protein copies [15]. However, copy numbers of more than 10 proteins in a punctum can be difficult to count by the photobleaching steps because multiple photobleaching steps, depending on the laser power, can occur simultaneously, although analytical tools have been proposed to address this issue [16*, 20, 21].
One potential limitation with using GFP is that co-expression of GFP with the proteins of interest may perturb the trafficking and expression level of the native protein, depending on the concentration of the native protein [12]. For crowded environments, such as the synaptic vesicle, we found the number of the protein VAMP 2 was lowered when tagged with GFP [22, 23]. To address this issue, fluorescent antibodies may be used to target and label the proteins of interest. In this case, however, care must be taken to remove any artifacts caused by non-specific binding of antibodies to off-target molecules in the specimen, which would introduce ‘overcounting’ errors; incomplete labeling of target proteins by antibodies would introduce ‘undercounting’ errors. Furthermore, it was difficult to apply single-molecule photobleaching to dye-tagged antibodies because each antibody had a different copy number of dyes [24]. There was no direct way to correlate the number of bleached dyes with the number of antibodies present unless a single dye was labeled to a single antibody [25], which was a difficult feat to accomplish.
In order to overcome these issues, as shown in Figure 1, we developed single-molecule-based statistical deconvolution quantitative microscopy combined with a microfluidic chamber to count copies of native proteins in individually purified ~ 40-nm-diameter synaptic vesicles (Figure 1a/b). The detailed analytical procedures are shown in Figure 1c-g [26**]. Our approach had several advantages: 1) Statistical deconvolution of fully-labeled samples compared with single-molecule-intensity distribution allowed us to estimate precisely the copy number of a protein of interest [17*]. 2) Our method did not need to consider the number of fluorophores on a single antibody [24] because it did not rely on photobleaching of fluorophores [17*]. 3) The method minimized the effect of incomplete labeling of antibodies to the target proteins because epitopes (antibody binding sites) were well exposed to the solution. 4) The microfluidic device allowed us to measure more than 10,000 small organelles per experiment. 5) We could minimize potential artifacts introduced by nonspecific binding of fluorescent-dye-labeled antibodies to the glass because the antibody labeling step was before the injection of sample into the chamber and because we required co-localization of different color antibodies targeted to the same synaptic vesicle for analysis [22, 26**].
Figure 1.
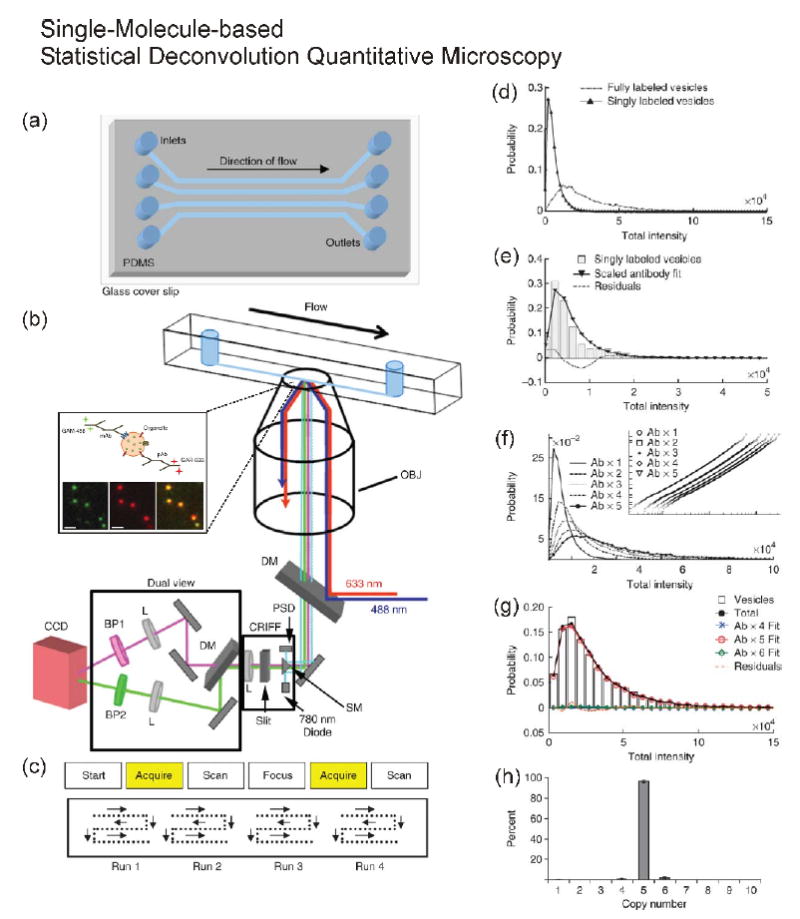
Schematic diagram of single-molecule-based statistical deconvolution quantitative microscopy combined with a microfluidic chip and an example of statistical deconvolution. (a) Microfluidic chip, (b) Total-internal-reflection-fluorescence (TIRF) microscopy, and (c) automatic scanning patterns for collecting images. The expansion of inside of the microfluidic chip in (b) shows synaptic vesicles labeled with primary and dye-tagged secondary antibodies in the imaging plane using TIRF microscopy. (d-h) The detailed procedures of counting of a fully labeled sample using single-antibody distribution. (d) Comparison of synaptic vesicles singly or fully labeled with primary and secondary antibodies. (e) Fitting of single-labeled synaptic vesicles. (f) Plotting of multiplication of the scaled single-antibody distribution to generate calibration curves for fitting. (g) A plot showing the fit for the protein SV2 on synaptic vesicles. (h) Results of fitting six different SV2 data sets. Reprinted with permission from Nature Protocol [26].
These merits allowed us to count a broad range in the number of proteins, from single protein to a few tens of copies of a protein, in the single organelles or cellular compartments. In the case of synaptic vesicles, for example, we found that synaptophysin and VAMP2 to be among the more abundant proteins, with an average number of around 13 and 10.5 copies per vesicle, respectively. The more interesting finding was that some proteins are highly monodisperse in copy number while others are not. The observation brings up an interesting question of whether neurodegenerative pathophysiological conditions, like Alzheimer’s disease, may change the degree of polydispersity of some of the synaptic vesicle proteins and thus affect neuronal communication in the brain. In addition to using dye-tagged antibodies, we also tested synaptic vesicles that expressed GFP [23]. After photobleaching of ~ 10 copies of GFP down to a single molecule of GFP to generate the single-molecule calibration, we counted the GFP number using the statistical deconvolution method as described above. While we applied our statistical deconvolution quantitative microscopy so far only to count proteins on isolated organelles, with the development of a photobleached internal standard [23], this method may be extended for use to count the number of proteins in individual puncta often observed in fluorescence microscopy of intact cells.
Single-Molecule Localization Microscopies (SMLM) for counting proteins
Localization microscopies are very powerful tools for visualizing and pinpointing the proteins of interest with nanometer-scale precision. They are especially useful for studying biological scaffold complexes or small organelles, whose sizes fall in the diffraction limit [18, 19, 27, 28, 29**]. This group of methods is based on the notion of photoswitchable single-molecule fluorescence and point-spread function localization: A single fluorophore will appear in an image as a blurred diffraction-limited spot, but the position of the fluorophore can be determined to within nanometers by performing centroid analysis, provided there aren’t any neighboring fluorophores within the diffraction-limited spot. By sequentially turning “on” and “off” different subsets of fluorophores and by noting the position of each fluorophore, a super-resolution image can be generated. The resolution is not dictated by the diffraction limit, but by the number of photons collected for centroid analysis. Resolutions down to a few nanometers can be achieved, albeit 10-20 nm is more common. To estimate accurate protein compositions using stochastic optical reconstruction microscopy (STORM) or direct STORM, however, issues of undercounting, caused by incomplete labeling by antibodies, and overcounting, caused by blinking (‘reversible transition from the fluorescent state into the non-fluorescent dark state’) of dyes on antibodies [18, 30], need to be accounted for and corrected. Furthermore, multiple fluorescent dyes on individual antibodies generated low precision for counting of proteins of interest because of an increase in the standard deviation. Similarly, for photoactivated localization microscopy (PALM), it is necessary to consider overcounting issues caused by blinking of photoactivatable proteins in sparsely distributed molecules in the imaging plane [31**] and undercounting issues by a population of nonfluorescent proteins due to misfolding of fluorescent proteins [12] and fluorescence mixing effect due to high molecular density [32**].
The simplest approach to correct the blinking events was to assume that blinking contributes linearly to the number of proteins of interest, e.g. double or triple counting by two or three blinks of the same molecules before the final photobleaching step. Later, researchers recognized that the counting error could be exponentially reduced by tolerance time (or ‘dark time’) defined by a time to distinguish individual molecules. The tolerance time is an empirical parameter for analysis that depends on the off-time of blinking of individual fluorophores, the species of fluorophores, the excitation light power, and acquisition frame rate [31**]. For instance, if the tolerance time is shorter than the off-time (‘dwell time of dark state”) of the real blinking events, it can generate overcounting errors because the algorithm does not have enough time to distinguish between two different molecules. In other words, not only tolerance time but also the activated molecular density would be critical factors for minimizing counting errors of the SMLMs. How can we deal with those issues?
To address this question, Lee and coworkers found an elegant way to optimize tolerance time and photoactivation time to control the number of active molecules using a photokinetic model [32**] although a model-independent approach was recently proposed [33]. After simulation of blinking events with a four-state model — nonactive (‘nonfluorescent’), active (‘fluorescent’), dark, and photobleached — Lee and coworkers applied rate constants obtained from the blinking model to correct for the contributions of individual blinking events to minimize the standard deviation (Figure 2a). The most important kinetic parameter was the transition rate from the dark state to the active state, which provided the right tolerance time to correct for overcounting error (Figure 2b). To reduce fluorescence mixing effect by too many activated molecules at the same time, which could lead to a molecular undercounting error, they found an optimal activation time (‘Fermi activation’, Figure 2c). Furthermore, if the tolerance time was rapidly increased to reduce overcounting errors, the undercounting error could be increased by missing real events. To optimize tolerance time without any biased analysis (Figure 2d) to reduce uncertainty (Figure 2e), an iterative approach was chosen to balance the overcounting and the undercounting errors caused by blinking and the fluorescence mixing effect, respectively. Finally they counted 33 molecules of FliM protein per bacterial flagellar motor with a very small standard deviation.
Figure 2.
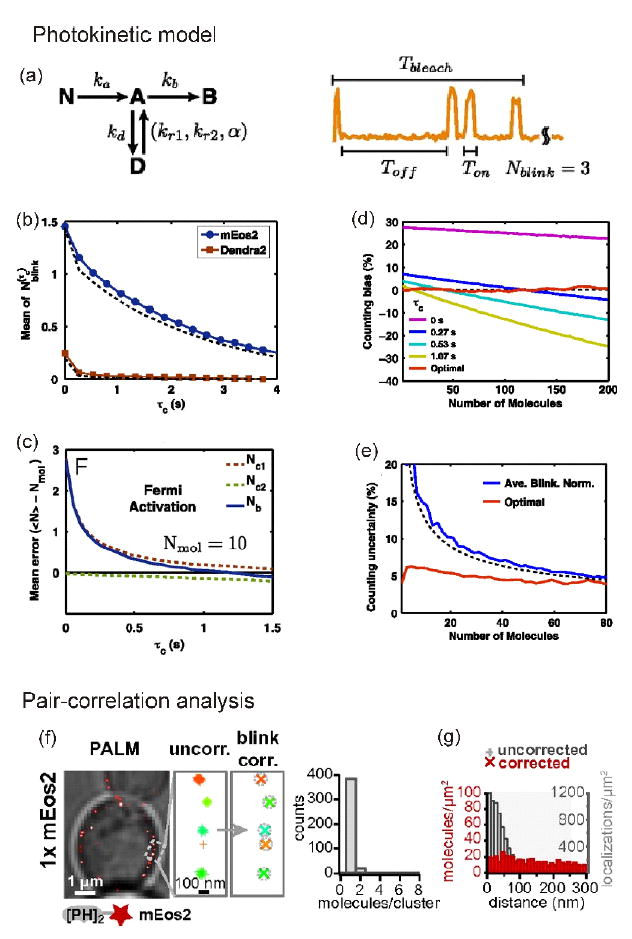
Counting of proteins by a PALM-based photokinetic model and pair-correlation function. (a) Photokinetic model of blinking of photoactivable fluorescent protein (PAFP). Nonactive form, N, of PAFP is converted to the active form, A, with rate ka, upon 405-nm laser illumination. In the presence of a 561-nm laser, the molecule emits light and can either go to dark, D, or to bleach, B, states with rates kd and kb, respectively. The molecule recovers from D to A state with two rates kr1 (fast) and kr2 (slow); α is the ratio between the contributions of the fast and slow rates. The recovery rates are dependent on the Toff. (b) Correlation between the number of blinking of PAFP (N) and tolerance time (τc). (c) Fermi activation for optimization of the number of active molecules. (d) Counting bias by tolerance time. (e) Counting uncertainty in the case of iterative way versus a simple average blinking normalization method. Reprinted from [32]. (f) Pair-correlation analysis to remove artificial clustering effect caused by blinking in a small region. Single mEos2-fused PI3P-sensitive PH probe (PH2-mEOs2). (g) After correction, the blinking molecules do not have colocalization artifacts (red bars) compared with before correction (gray bars). Reprinted from [34].
Compared with the previous photokinetic-modeling-dependent methods, a different approach was proposed to calibrate the contribution of mEos2 (monomeric photoactivable fluorescent protein of Eos2) blinking to quantify lipid binding sites on single vesicles in yeast [34*]. Extracting photokinetic parameters sometimes can be tricky because of the complexity of blinking behaviors and the variation in individual fluorophores. Puchner and coworkers prepared a single mEos2-fused, membrane localizing phosphatidylinositol 3-phosphate (PI3P)-sensitive PH domain of phospholipase C delta (PLCδ), which was randomly distributed in vesicle membrane of yeast. As expected, they observed a pronounced peak of distance distribution with a single mEos2 tagging the PH probe. This meant that blinking events potentially contributed to the distance distribution of the probes as an artificial clustering of proteins derived by blinking of fluorophores [35*]. To remove the artifact by blinking, they analyzed the same data with a pair-correlation function so they could generate a random distance distribution of the PH probes (Figure 2f/g). Finally, Puchner and coworkers reported that endocytosed vesicles in cytoplasm increase ~ 100-fold of PI3P binding sites in relation to their fusion [34*].
Although it showed the way to reduce miscounting of the number of proteins, the SMLMs combined with stochastic switching were still not suitable to count with excellent precision and accuracy the number of proteins in densely clustered regions that have more than 100 protein copies. The variation of environment-dependent photophysics of fluorophores, e.g. blinking rate and number of photons emitted from individual fluorophores [30, 36], made the counting difficult. To overcome this limitation, a simple and robust quantitative super-resolution method called quantitative points accumulation in nanoscale topography (qPAINT) was developed [37**]. Instead of the stochastic switching, qPAINT achieved transient binding of free floating dye-labeled short DNA sequences (‘imager strand’) to complementary target DNA sequences (‘docking strand’) tagged onto secondary antibody which can be complexed with primary antibodies targeted to proteins of interest (Figure 3a). The binding rate of imager strands to a docking strand was proportionally increased as a function of the concentration of imager stand because of the second-order binding reaction (Figure 3b). So, Jungmann and coworkers could quantify the number of target strands on the proteins of interest after conversion of the binding rate of imager strands. A potential use of the method was to detect many, discrete single molecules of the nuclear pore complex protein (Figure 3c-d) and an even more densely packed molecular complex with more than 100 protein copies in synaptic active zone (Figure 3e-f). Because qPAINT was independent of the blinking kinetics of fluorophores and photobleaching, it avoided typical undercounting errors caused by already photobleached dyes or incomplete labeling of antibodies as well as overcounting errors caused by blinking artifacts. Major barriers for the qPAINT method are still dependent on stoichiometric labeling of protein targets and efficiency of target labeling as well as the accessibility of probe strand to the target DNA because of the secondary structure in the single strand target DNA under certain conditions.
Figure 3.
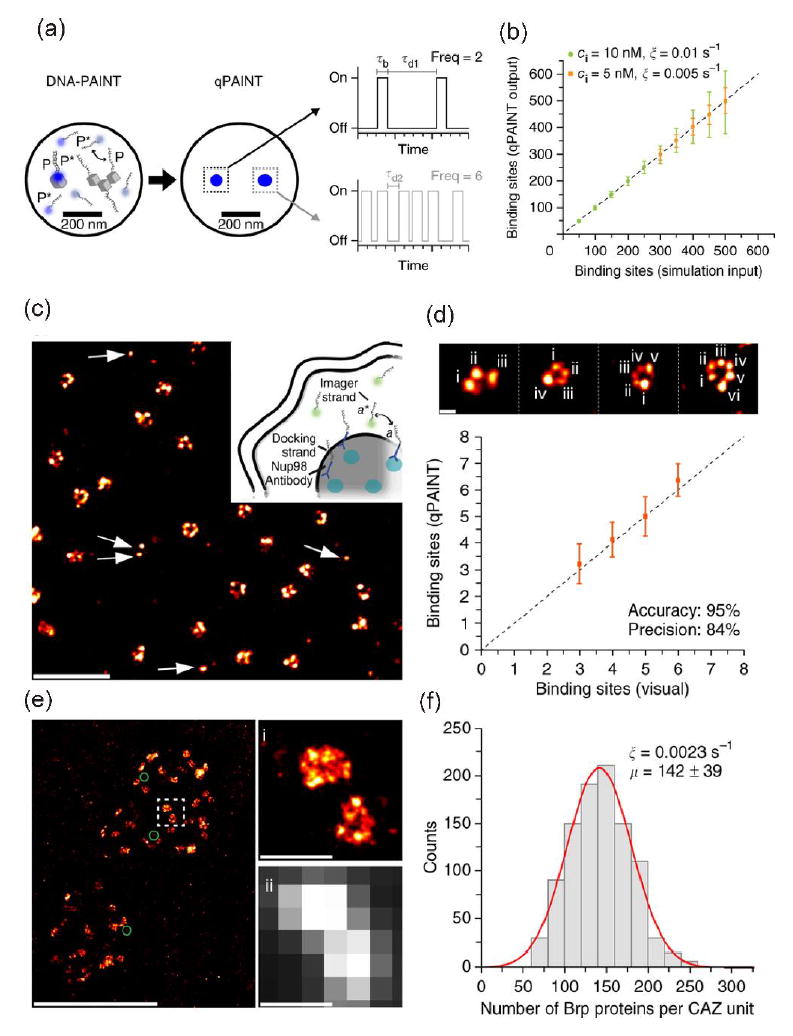
qPAINT. (a-b) Principle of qPAINT and simulated results. Imager and docking strands are P* and P, respectively. Standardization of the rate of binding (‘influx rate’ or ξ) of imager strand to a single docking strand attached to target. ξ = (kon×ci) = τd-1where kon, ci and τd are rate constant, known concentration of dye-labeled imager strand (P*), and dark time before binding, respectively. Then the rate (ξ) can be converted to a real number of binding sites, e.g. binding sites = τd/τd* = (ξ×τd*)-1 where τd* is the actual average dark time of the imager strand to real sample. For example, if ξ* and τd* are 0.005 s-1 and τd* = 1 s, the actual binding sites would be 200. (c-f) Examples with qPAINT method. (c and d) qPAINT quantification with nucleoporin Nup98 (anchored to the inner ring of the nuclear pore complex). (e and f) qPAINT quantification with Bruchpilot proteins (Brp) molecules which are components of the cytomatrix at the synaptic active zone (CAZ). The average Brp number is 142 ± 39 (mean ± s.d.). Reprinted from Nature Methods under their permission [37].
SMLM is suitable not only for protein counting but also for RNA or DNA counting
The counting method of RNA or DNA called Fluorescence In Situ Hybridization, or FISH, is more straightforward than counting methods for proteins because of complementary interaction between synthetic oligonucleotides and target mRNA sequences [10]. In the original demonstration of FISH, conventional epi-fluorescence microscopy was used to detect single mRNAs using multiple dye-labeled synthetic oligonucleotides to compensate for the detection sensitivity of a normal CCD camera. The signal was then optically deconvolved to pick out a single mRNA. The downside to this approach was the labeling of a single oligonucleotide with the multiple dyes (> 5) and the non-specific binding of the synthetic sequence to off-target mRNA molecules. After the development of single-dye labeling at 3’ termini of short oligonucleotides, an improved approach was proposed [38]. Forty-eight different single-dye-labeled oligonucleotides, to reduce off-target or non-binding effects, were used to detect a coding region of GFP; the 3’ untranslated region also was targeted with 4 oligonucleotides labeled with different colored dyes. By combining the two independent signals, the investigators counted the mRNAs which had both colors located in a same region. Using the advanced FISH, researchers have addressed the old question of the correlation between cell volume and RNA or DNA copy number [39]: The RNA copy number was dependent on the cell volume but not on the cell-cycle stage; as expected, the DNA copy number was volume-independent.
Influence of precise protein counting on biology and future directions
Many scientific findings are still qualitative even though quantitative approaches are strongly recommended to understand biochemical reactions. Quantitative microscopy allows us to show the heterogeneous distribution of biological materials inside individual cells or organelles which is important for understanding biological phenomena. In this perspective, we discussed recent improvements in calibration methods for SMLMs and new proposals for the precise and accurate counting methods. Despite these advances, several points should be addressed in near future.
First, it is necessary to improve the labeling of a specific target with a known stoichiometry of dyes to the protein of interest. Currently, the tagging strategies available include the six-peptide sequence for labeling with a FlAsH dye (‘FlAsH tag’) inserted into a specific domain of proteins or a small SNAP/CLIP tag on C- or N- termini of proteins of interest combined with cell-permeable dyes for live-cell imaging [29]. However, the photophysical properties of FlAsH and SNAP/CLIP dyes were not rigorously tested for quantitative microscopy.
The second approach is to insert a photoactivable fluorescent protein into the gene using genome editing technologies, such as the CRISPR-Cas9 system [40, 41, 42] and the zinc-finger nuclease [43, 44]. This approach can greatly benefit the counting of proteins with low copy numbers. For instance, it has been estimated that ~ 26 copies of dynamin can be recruited to clathrin-mediated endocytic vesicles in live cells [44]. In addition, Leonetti and colleagues have successfully tagged GFP onto endogenous human proteins using the CRISPR-Cas9 system [45].
Most of the current single-molecule counting microscopies have been concentrated on static information, that is, counting molecules on fixed samples. One obvious and important direction to improve these counting techniques is to expand their reach to measure dynamic events. However, dynamic molecular interactions in live cells can occur even within milliseconds [29**]. Counting single molecules at such fast rates will put severe demands on both the brightness of the probe and the sensitivity of the microscope. To this end, a small-sized semiconducting polymer dot [46], which has a high brightness and low bleaching rate, together with improved sCMOS technology that offer high quantum efficiency, low noise, and fast frame rates [47*], may help to improve the image acquisition speeds of current single-molecule counting microscopies. The ability to observe time-dependent redistribution of protein copy numbers with good time resolution down to milliseconds would be an important advance. Integration of the obtained quantitative measurements with the appropriate cellular and mathematical modeling, will surely provide deep insight into biological processes in many different types of biological samples [48*].
Concluding remarks
Over the past two decades, researchers have had many breakthroughs in overcoming the diffraction limit and developing quantitative microscopies with nanometer-scale spatial precision. Beyond the breaking of optical resolution, we and others have tackled the development of new quantitative microscopy methods that correct undercounting or overcounting errors so that the precise counting of proteins and nucleotides can be achieved in cellular compartments. Our statistical deconvolution algorithm and other approaches using single-molecule localization microscopy help to measure the precise protein copies in many complex samples and conditions. This perspective article also discussed how to resolve blinking issues of fluorophores using photokinetic modeling, pair-correlation analysis, qPAINT, and advanced FISH. Finally, we discussed future directions such as the counting and mapping of biomolecules in a time-dependent manner to understand dynamic events in cells.
Highlights.
Single-molecule based statistical deconvolution quantitative microscopy
Single-molecule localization microscopy: Overcoming under- or over-counting errors
Principle of qPAINT method to count the number of proteins of interest
Future directions for quantitative microscopy based on single-molecule fluorescence
Acknowledgments
We are grateful to the NIH (MH113333) and the University of Washington for support of this work.
Footnotes
Conflict of Interests
The authors declare no conflict of interests for this paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and recommended reading
Papers of particular interest have been highlighted as:
* of special interest
** of outstanding interest
- 1.Rissin DM, Kan CW, Campbell TG, Howes SC, Fournier DR, Song L, Piech T, Patel PP, Chang L, Rivnak AJ, Ferrell EP, Randall JD, Provuncher GK, Walt DR, Duffy DC. Single-molecule enzyme-linked immunosorbent assay detects serum proteins at subfemtomolar concentrations. Nat Biotechnol. 2010;28:595–599. doi: 10.1038/nbt.1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moerner WE, Kador L. Optical detection and spectroscopy of single molecules in a solid. Phys Rev Lett. 1989;62:2535–2538. doi: 10.1103/PhysRevLett.62.2535. [DOI] [PubMed] [Google Scholar]
- 3.Betzig E, Chichester RJ. Single molecule observed by near field scanning optical microscopy. Science. 1993;262:1422–1425. doi: 10.1126/science.262.5138.1422. [DOI] [PubMed] [Google Scholar]
- 4.Funatsu T, Harada Y, Tokunaga M, Saito K, Yanagida T. Imaging of single fluorescent molecules and individual ATP turnovers by single myosin molecules in aqueous solution. Nature. 1995;374:555–559. doi: 10.1038/374555a0. [DOI] [PubMed] [Google Scholar]
- 5.Ha T, Enderle T, Ogletree DF, Chemla DS, Selvin PR, Weiss S. Probing the interaction between two single molecules: fluorescence resonance energy transfer between a single donor and a single acceptor. Proc Natl Acad Sci USA. 1996;93:6264–6268. doi: 10.1073/pnas.93.13.6264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lu HP, Xun L, Xie XS. Single-molecule enzymatic dynamics. Science. 1998;282:1877–1882. doi: 10.1126/science.282.5395.1877. [DOI] [PubMed] [Google Scholar]
- 7.Huang B, Wu H, Bhaya D, Grossman A, Granier S, Kobilka BK, Zare RN. Counting low-copy number proteins in a single cell. Science. 2007;315:81–84. doi: 10.1126/science.1133992. [DOI] [PubMed] [Google Scholar]
- 8.Penna A, Demuro A, Yeromin AV, Zhang SL, Safrina O, Parker I, Cahalan MD. The CRAC channel consists of a tetramer formed by Stim-induced dimerization of Orai dimers. Nature. 2008;456:116–120. doi: 10.1038/nature07338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ji W, Xu P, Li Z, Lu J, Liu L, Zhan Y, Chen Y, Hille B, Xu T, Chen L. Functional stoichiometry of the unitary calcium-release-activated calcium channel. Proc Natl Acad Sci USA. 2008;105:13668–13673. doi: 10.1073/pnas.0806499105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Femino AM, Fay FS, Fogarty K, Singer RH. Visualization of single RNA transcript in situ. Science. 1998;280:585–590. doi: 10.1126/science.280.5363.585. [DOI] [PubMed] [Google Scholar]
- 11.Neely LA, Patel S, Garver J, Gallo M, Hackett M, McLaughlin S, Nadel M, Harris J, Gullans S, Rooke J. A single-molecule method for the quantitation of microRNA gene expression. Nat Methods. 2006;3:41–46. doi: 10.1038/nmeth825. [DOI] [PubMed] [Google Scholar]
- 12.Sugiyama Y, Kawabata I, Sobue K, Okabe S. Determination of absolute protein numbers in single synapses by a GFP-based calibration technique. Nat Methods. 2005;2:677–684. doi: 10.1038/nmeth783. [DOI] [PubMed] [Google Scholar]
- 13.Gordon MP, Ha T, Selvin PR. Single-molecule high-resolution imaging with photobleaching. Proc Natl Acad SCi USA. 2004;101:6462–6465. doi: 10.1073/pnas.0401638101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ulbrich MH, Isacoff EY. Subunit counting in membrane-bound proteins. Nat Methods. 2007;4:319–321. doi: 10.1038/NMETH1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leake MC, Chandler JH, Wadhams GH, Bai F, Berry RM, Armitage JP. Stoichiometry and turn over in single, functioning membrane protein complexes. Nature. 2006;443:355–358. doi: 10.1038/nature05135. [DOI] [PubMed] [Google Scholar]
- 16.Chen Y, Deffenbaugh NC, Anderson CT, Hancock WO. Molecular counting by photobleaching in protein complexes with many subunits: best practices and application to the cellulose synthesis complex. Mol Biol Cell. 2014;25:3630–3642. doi: 10.1091/mbc.E14-06-1146. * Simultaneous photobleaching of multiple dyes is hard to distinguish with common algorithm. Chen et al. proposed new algorithm to analyze photobleaching steps for counting protein copies. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mutch SA, Fujimoto BS, Kuyper CL, Kuo JS, Bajjalieh SM, Chiu DT. Deconvolving single-molecule intensity distributions for quantitative microscopy measurements. Biophys J. 2007;92:2926–2943. doi: 10.1529/biophysj.106.101428. * Describes how to overcome limitation of single-molecule photobleaching or localization microscopy, which uses antibody labeling to detect native proteins. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bates M, Huang B, Dempsey GT, Zhuang X. Multicolor super-resolution imaging with photo-switchable fluorescent probes. Science. 2007;317:1749–1753. doi: 10.1126/science.1146598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, Bonifacino JS, Davidson MW, Lippincott-Schwartz J, Hess HF. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006;313:1642–1645. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- 20.Liesche C, Grussmayer KS, Ludwig M, Worz S, Rohr K, Herten DP, Beaudouin J, Eils R. Automated analysis of single-molecule photobleaching data by statistical modeling of spot populations. Biophys J. 2015;109:2352–2362. doi: 10.1016/j.bpj.2015.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tsekouras K, Custer TC, Jashnsaz H, Walter NG, Pressé S. A novel method to accurately locate and count large numbers of steps by photobleaching. Mol Biol Cell. 2016;27:3601–3615. doi: 10.1091/mbc.E16-06-0404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mutch SA, Kensel-Hammes P, Gadd JC, Fujimoto BS, Allen RW, Schiro PG, Lorenz RM, Kuyper CL, Kuo JS, Bajjalieh SM, Chiu DT. Protein quantification at the single vesicle level reveals that a subset of synaptic vesicle proteins are trafficked with high precision. J Neurosci. 2011;31:1461–1470. doi: 10.1523/JNEUROSCI.3805-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gadd JC, Fujimoto BS, Bajjalieh SM, Chiu DT. Single-molecule fluorescence quantification with a photobleached internal standard. Anal Chem. 2012;84:10522–10525. doi: 10.1021/ac303032m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Panchuk-Voloshina N, Haugland RP, Bishop-Stewart J, Bhalgat MK, Millard PJ, Mao F, Leung WY, Haugland RP. Alexa dyes, a series of new fluorescent dyes that yield exceptionally bright, photostable conjugates. J Histochem Cytochem. 1999;47:1179–1188. doi: 10.1177/002215549904700910. [DOI] [PubMed] [Google Scholar]
- 25.Ehmann N, van de Linde S, Alon A, Ljaschenko D, Keung XZ, Holm T, Rings A, DiAntonio A, Hallermann S, Ashery U, Heckmann M, Sauer M, Kittel RJ. Quantitative super-resolution imaging of Bruchpilot distinguishes active zone states. Nat Commun. 2014;5:4650. doi: 10.1038/ncomms5650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mutch SA, Gadd JC, Fujimoto BS, Kensel-Hammes P, Schiro PG, Bajjalieh SM, Chiu DT. Determining the number of specific proteins in cellular compartments by quantitative microscopy. Nat Protoc. 2011;6:1953–1968. doi: 10.1038/nprot.2011.414. ** A protocol paper about statistical deconvolution quantitative microscopy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heilemann M, van de Linde S, Schuttpelz M, Kasper R, Seefeldt B, Mukherjee A, Tinnefeld P, Sauer M. Subdiffraction-resolution fluorescence imaging with conventional fluorescence probes. Angew Chem Int Ed Engl. 2008;47:6172–6176. doi: 10.1002/anie.200802376. [DOI] [PubMed] [Google Scholar]
- 28.Fürstenberg A, Heilemann M. Single-molecule localization microscopy-near-molecular spatial resolution in light microscopy with photoswitchable fluorophores. Phys Chem Chem Phys. 2013;15:14919–14930. doi: 10.1039/c3cp52289j. [DOI] [PubMed] [Google Scholar]
- 29.Turkowyd B, Virant D, Endesfelder U. From single molecules to life: microscopy at the nanoscale. Anal Bioanal Chem. 2016;408:6885–6911. doi: 10.1007/s00216-016-9781-8. ** A review about single-molecule localization microscopy and other super-resolution microscopies. This article explains principles of these microscopy methods as well as recent applications of and developments in these new techniques. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dempsey GT, Vaughan JC, Chen KH, Bates M, Zhuang X. Evaluation of fluorophores for optimal performance in localization-based super-resolution imaging. Nat Methods. 2011;8:1027–1036. doi: 10.1038/nmeth.1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Annibale P, Vanni S, Scarselli M, Rothlisberger U, Radenovic A. Quantitative photo activated localization microscopy: unraveling the effects of photoblinking. PLoS One. 2011;6:e22678. doi: 10.1371/journal.pone.0022678. ** Clear explanation about the dark time (or tolerance time) to correct overcounting error by photoblinking. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee SH, Shin JY, Lee A, Bustamante C. Counting single photoactivatable fluorescent molecules by photoactivated localization microscopy (PALM) Proc Natl Acad Sci USA. 2012;109:17436–17441. doi: 10.1073/pnas.1215175109. ** Photokinetic modeling to improve over- or under-counting errors of proteins of interest produced by blinking of fluorophores. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hummer G, Fricke F, Heilemann M. Model-independent counting of molecules in single-molecule localization microscopy. Mol Biol Cell. 2016;27:3637–3644. doi: 10.1091/mbc.E16-07-0525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Puchner EM, Walter JM, Kasper R, Huang B, Lim WA. Counting molecules in single organelles with superresolution microscopy allows tracking of the endosome maturation trajectory. Proc Natl Acad Sci USA. 2013;110:16015–16020. doi: 10.1073/pnas.1309676110. * Demonstrated pair-correlation analysis to correct overcounting errors by blinking of photoactivable fluorescent proteins. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Annibale P, Vanni S, Scaselli M, Rothlisberger U, Radenovic A. Identification of clustering artifacts in photoactivated localization microscopy. Nat Methods. 2011;8:527–528. doi: 10.1038/nmeth.1627. * Demonstration of clustering artifact by blinking of photoactivable fluorescent protein. [DOI] [PubMed] [Google Scholar]
- 36.Nieuwenhuizen RP, Bates M, Szymborska A, Lidke KA, Rieger B, Stallinga S. Quantitative localization microscopy: effects of photophysics and labeling stoichiometry. PLoS One. 2015;10:e0127989. doi: 10.1371/journal.pone.0127989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jungmann R, Avendano MS, Dai M, Woehrstein JB, Aqasti SS, Feiger Z, Rodal A, Yin P. Quantitative super-resolution imaging with qPAINT. Nat Mathods. 2016;13:439–442. doi: 10.1038/nmeth.3804. ** Overcomes limitations of current single-molecule localization microscopy based on blinking of fluorophores for counting of protein of interest. It uses concentration dependent annealing of single-dye labeled short DNA sequences to target DNA tagged on primary antibodies. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Raj A, van den Bogaard P, Rifkin SA, van Oudenaarden A, Tyagi S. Imaging individual mRNA molecules using multiple singly labeled probes. Nat Methods. 2008;5:877–879. doi: 10.1038/nmeth.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Padovan-Merhar O, Nair GP, Biaesch AG, Mayer A, Scarfone S, Foley SW, Wu AR, Churchman LS, Singh A, Raj A. Single mammalian cells compensate for differences in cellular volume and DNA copy number through independent global transcriptional mechanisms. Mol Cell. 2015;58:339–352. doi: 10.1016/j.molcel.2015.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lin S, Staahl BT, Alla RK, Doudna JA. Enhanced homology-directed human genome engineering by controlled timing of CRISPR-Cas9 delivery. eLife. 2014;3:e04766. doi: 10.7554/eLife.04766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim S, Kim D, Cho SW, Kim J, Kim JS. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014;24:1012–1019. doi: 10.1101/gr.171322.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Doyon JB, Zeitler B, Cheng J, Cherone JM, Santiago Y, Lee AH, Vo TD, Doyon Y, Miller JC, Paschon DE, Zhang L, Rebar EJ, Gregory PD, Urnov FD, Drubin DG. Rapid and efficient clathrin-meditated endocytosis revealed in genome-edited mammalian cells. Nat Cell Biol. 2011;13:331–337. doi: 10.1038/ncb2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grassart A, Cheng AT, Hong SH, Zhang F, Zenzer N, Feng Y, Briner DM, Davis GD, Malkov D, Drubin DG. Actin and dynamin2 dynamics and interplay during clathrin-medicated endocytosis. J Cell Biol. 2014;205:721–735. doi: 10.1083/jcb.201403041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leonetti MD, Sekine S, Kamiyama D, Weissman JS, Huang B. A scalable strategy for high-throughput GFP tagging of endogenous human proteins. Proc Natl Acad Sci USA. 2016;113:E3501–E3508. doi: 10.1073/pnas.1606731113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang T, Yu J, Gallina ME, Sun W, Rong Y, Chiu DT. Highly luminescent, fluorinated semiconducting polymer dots for cellular imaging and analysis. Chem Commun (Camb) 2013;49:8256–8258. doi: 10.1039/c3cc44048f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Juette MF, Terry DS, Wasserman MR, Altman RB, Zhou Z, Zhao H, Blanchard SC. Single-molecule imaging of non-equilibrium molecular ensembles on the millisecond timescale. Nat Methods. 2016;13:341–344. doi: 10.1038/nmeth.3769. * Demonstration of sCMOS detector for single molecule fluorescence imaging compared with current EMCCD camera. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jung SR, Seo JB, Deng Y, Asbury CL, Hille B, Koh DS. Contribution of protein kinases and β-arrestin to termination of protease-activated receptor 2 signaling. J Gen Physiol. 2016;147:255–271. doi: 10.1085/jgp.201511477. * An example of mathematical modeling of termination of G protein-coupled receptor (GPCR) signaling based on the quantitative estimation of kinetic parameters and counting of the number of plasma membrane localized GPCR. [DOI] [PMC free article] [PubMed] [Google Scholar]