

Abstract
Many different types of soft and solid tumors have now been sequenced, and meta-analyses suggest that genomic variation across tumors scales with the stiffness of the tumors’ tissues of origin. The opinion expressed here is based on a review of current genomics data, and it considers multiple ‘mechanogenomics’ mechanisms to potentially explain this scaling of mutation rate with tissue stiffness. Since stiff solid tissues have higher density of fibrous collagen matrix, which should decrease tissue porosity, cancer cell proliferation could be affected and so could invasion into stiff tissues as the nucleus is squeezed sufficiently to enhance DNA damage. Diversification of a cancer genome after constricted migration is now clear. Understanding genome changes that give rise to neo-antigens is important to selection as well as to the development of immunotherapies, and we discuss engineered monocytes/macrophages as particularly relevant to understanding infiltration into solid tumors.
Graphical Abstract
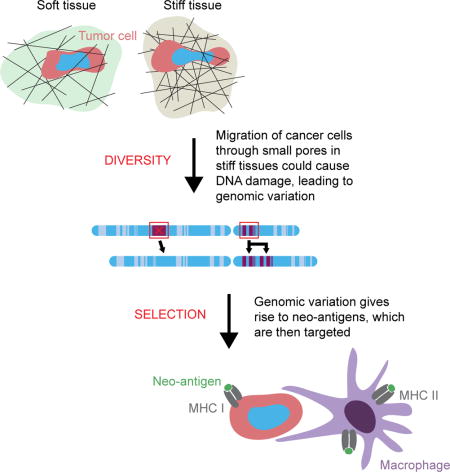
Introduction
Tumors are often palpably stiffer than nearby normal tissue [1], with stiffness of breast and liver, among other organs, correlating with cancer risk [2,3]. Tissue stiffness likely contributes in normal cells to motility [4] and differentiation [5], and in cancer cells to invasion [6] and various epigenetic mechanisms [7], including stiffness-dependent nuclear localization of oncogenic factors (e.g. YAP) [8]. It is unclear, however, if a physical attribute of the microenvironment such as stiffness could contribute—in a ‘mechanogenomics’ type of process—to any of the many genetic changes that typically occur in cancer.
Meta-analyses of recently published cancer mutation data are presented first in this current opinion article, and the trends begin to suggest that—beyond some initial driver mutation(s)—the large genomic variation across diverse cancers scales with tissue stiffness. Stiffness-dependent cell biological mechanisms for genome variation are needed to substantiate any such correlation, and some molecular mechanisms are now emerging. We focus on one possible mechanism based on the fact that stiffer tissues, including tumors, are enriched in collagen [9], and many studies of collagen gels show that denser collagen has smaller matrix pores [10]. Thus, as cancer cells invasively migrate into stiff, small-pore surroundings, the nucleus is damaged, which might ultimately contribute to genomic diversity.
Invasion is a defining task of a cancer cell; the equal but opposite challenge of an immune cell—therapeutic or otherwise—is to confront stiffness barriers and infiltrate a wound or disease site in order to attack ‘non-self’. In the cancer context, genome variation can produce novel protein sequences that might be perceived by the immune system as ‘neo-antigens’. Such sequences are by definition absent from normal cells, and so can be used to identify and eliminate cancerous cells if the neo-antigen signals are sufficiently potent, accessible, and foreign to overwhelm ‘self’ recognition [11]. A moonshot-scale effort now seeks to employ neo-antigens in various immunotherapy approaches. Some therapies use engineered T-cells to target neo-antigens on the cancer cell membrane [12], while other therapies exploit the major histocompatibility complex (MHC)—class I and class II—to target nuclear and cytoplasmic neo-antigens [13–15]. Monocytes and macrophages are the focus here and are particularly interesting for targeting to neo-antigens because these phagocytic cells exhibit a robust ability to infiltrate solid tissues, including tumors. The microenvironment-dependent plasticity of such cells, which is now being mapped by modern systems biology methods, could also be triggered, in part, by the stiffness or solidity of the tissue.
Genomic variation scales with tissue stiffness
Advances in genome sequencing have enabled cataloguing of the genomic variations that occur in cancers of many different types [11,16,17], and although oxidation artifacts can complicate such methods [18], somatic mutation rates are being collected in databases such as The Cancer Genome Atlas (TCGA) run by the National Cancer Institute (NCI). For the healthy tissues of origin of 36 types of cancer, tissue microelasticity data were culled from numerous recent papers [11,19–38] that used a variety of physical methods, including atomic force microscopy (AFM), micro-indentation probes, micropipette aspiration, and imaging-based elastography (Table 1). Whereas AFM pushes on cells and tissues at the ~100-nanometer to multi-micron length scales in order to provide a measure of a microenvironment’s stiffness, the larger length scale imaging-based elastography methods that perturb and monitor by magnetic resonance imaging, for example, typically probe on a millimeter length scale that encompasses many cells and the matrix between them; in principle, all of these types of measurements should be made on fresh tissue, since the former add up to the latter. However, measurements on cultured cells are likely to have little relevance to the tumor, because culture conditions such as gel stiffness influence cell mechanics [5]. Importantly, based on current tissue measurements, meta-analyses of genomics indicate that cancers arising in stiff tissues, such as lung and skin, exhibit 30-fold higher somatic mutation rates (as median per sequenced megabase) than cancers arising in soft tissues, such as marrow and brain (Fig. 1A). Importantly, the stiffness of a typical brain tumor or marrow tumor never increases to that of a typical bone tumor microenvironment even though tumors often stiffen — or, less frequently, soften — in tumorigenesis [1]. The hierarchy of normal tissue stiffness is therefore likely to prevail in cancer: that is, brain is softer than liver, which is softer then bone, etc. — regardless of cancer or not.
Table 1.
Cancer types and the microelasticities of the healthy tissues in which they arise
| Cancer type | Normal tissue stiffness (kPa) |
|
|---|---|---|
|
|
|
|
| Pilocytic astrocytoma | 0.4 [9] | |
| Acute myeloid leukemia (AML) | 0.3 [25] | |
| Acute lymphoblastic leukemia (ALL) | 0.3 [25] | |
| Chronic lymphocytic leukemia (CLL) | 0.3 [25] | |
| Medulloblastoma (MB) | 0.4 [9] | |
| Carcinoid | 0.4 [9] | |
| Neuroblastoma | 0.4 [9] | |
| Thyroid | 2.2 [25] | |
| Glioma low grade | 0.4 [9] | |
| Glioblastoma | 0.4 [9] | |
| Breast | 0.4 – 1.1 [27] | |
| Lymphoma B cell | 0.3 [25] | |
| Multiple myeloma | 0.3 [25] | |
| Kidney chromophobe | 2.6 [9] | |
| Prostate | 3.0 – 3.8 [28,29] | |
| Ovary | 2.5 [30] | |
| Kidney papillary cell | 2.6 [9] | |
| Kidney clear cell | 2.6 [9] | |
| Pancreas | 2.7 [31] | |
| Liver | 1.3 [9] | |
| Endometrium | 1.3 [28] | |
| Head and neck | ||
| Uterus | 1.3 [28] | |
| Cervix | 1.6 [32] | |
| Colorectum | 0.9 [33] | |
| Esophagus | 4.7 [34] | |
| Lung small cell | 5.9 [9] | |
| Stomach | 1.3 [35] | |
| Bladder | 3.2 [36] | |
| Lung adenocarcinoma | 5.9 [9] | |
| Lung squamous | 5.9 [9] | |
| Melanoma | 3.8 – 6.4 [9,37] | |
| Squamous cell carcinoma | 3.8 – 6.4 [9,37] | |
| Basal cell carcinoma | 3.8 – 6.4 [9,37] | |
|
|
|
|
| Childhood cancers |
ALL | 0.3 [25] |
| MB | 0.4 [9] | |
| Rhabdomyosarcoma | 11.9 - 25.7 [9,38] | |
| Osteosarcoma | 34.3 [9] | |
Fig. 1. Genomic variation increases versus tissue stiffness across cancers and with melanoma progression.
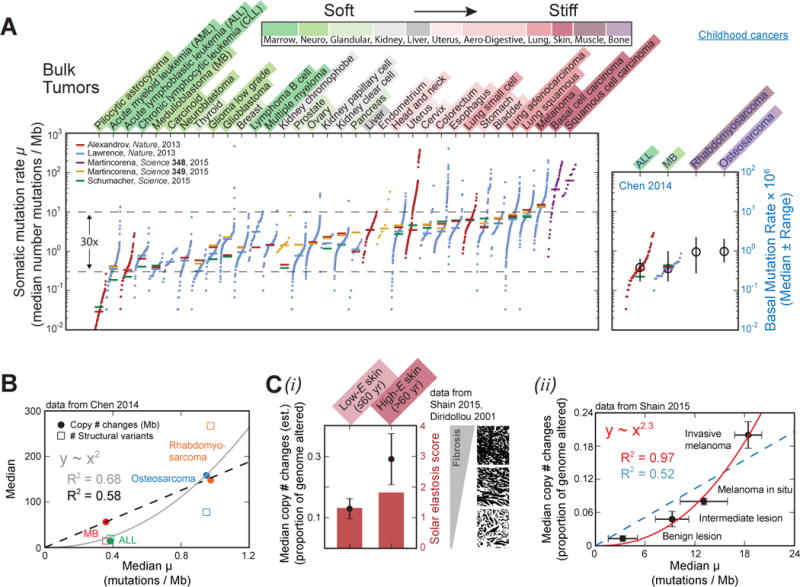
(A) Across different cancer types, somatic mutation rate—the median number of somatic substitutions and small insertions/deletions per megabase—increases with normal tissue stiffness. The same trend persists, albeit more weakly, among childhood cancers. The stiffness scale varies >10-fold from softer tissues (green), such as marrow and brain, to stiffer tissues (red), including lung, skin, muscle, and bone.
(B) Large-scale, chromosome-level mutations likewise increase with stiffness: childhood cancers in stiffer tissues have 10-to-20-fold more chromosome copy number changes and structural variants than do cancers in softer tissues, while somatic mutation rates differ much less.
(C) (i) Melanomas from patients of ≤60 years have fewer copy number changes than melanomas from patients over 60. The younger patients also have softer, less fibrotic skin, as inferred from their lower average solar elastosis score; solar elastosis is the thickening of skin due to prolonged sun exposure. (ii) In skin cancer genomes, chromosome copy number changes increase strongly with somatic mutation rate and lesion stiffness, with all highest in “invasive melanoma.”
Childhood muscle and bone cancers have only slightly elevated somatic mutation rates as compared to childhood marrow and brain cancers, but they have >10-fold more chromosome copy number changes and structural variants [23] (Fig. 1B). This disparity suggests that large-scale, chromosome-level amplifications and deletions — more so than somatic mutations — are signatures of some mutational processes that associate with tissue stiffness. In adult melanoma, fibrotic skin tends to be stiffer and exhibit more chromosome copy number changes than softer, less fibrotic skin [24,39] (Fig. 1C-i). Moreover, these copy number changes increase even faster with stiffness than do somatic mutation rates, and all mutations are most abundant in invasive melanoma [24] (Fig. 1C-ii). The relationship between chromosome-level mutations and stiffness thus holds even within a given tissue type, suggesting—in our opinion—a correlation between mutations and stiffness that cannot be entirely explained away by exposure to carcinogens.
Mechanical causes of mutation in the correlation of genomic variation with tissue stiffness
Scaling of genomic variation with tissue stiffness could result from at least three possible mechanical sources of mutations. First, stiff matrix enhances cell proliferation, as has been shown by an increase in BrdU incorporation with substrate stiffness in 2D cultures of normal human smooth muscle and breast epithelial cells as well as mouse embryonic fibroblasts [40]. DNA replication in each cell division cycle carries with it some risk of spontaneous mutation [20,41], accounting for about 67% of mutations in human cancers [42]. Since these mutations accumulate over successive generations, more proliferation should mean more changes to the genome. Nowak and Waclaw [43] recently pointed out for the case of one-hit, oncogenic initiation that cancer risk scales (in log-log plots) with the division rate of the resident tissue stem cells with a power law of 0.53, which is lower than linear scaling as expected for a simple stem cell contribution [42]. Tissue geometry was speculated to suppress the ‘effective number of stem cell divisions’ [43]. More study is needed, especially in 3D, because a 3D stiff surrounding could, for example, physically impact the fidelity of replication and chromosome segregation during mitosis.
A second conceivable explanation for the scaling relationship is that stiffness increases the frequency of nuclear envelope rupture [44]. Such rupture causes transient leakage into the nucleus of cytoplasmic factors, including perhaps nucleases, that might damage DNA and contribute to genome instability [45]. However, the increase of rupture frequency with substrate stiffness has been observed only in cells with defects in lamin-A, which is one of the three intermediate filament proteins that confer strength and stability to the nucleus. Yet, cancer types vary widely in their lamin-A expression levels: it is downregulated in leukemia as well as in breast and lung cancers, whereas it is upregulated in colorectal and skin cancers (for review: [46]). Lamin-A is highly mutated in multiple laminopathies, but cancer risk is not reported to be elevated. The inconsistency in lamin-A levels across cancer types argues against a simple stiffness-induced nuclear rupture hypothesis.
A third explanation that we find more promising is based on invasion of cells through stiff tissues, given that invasion is a ‘hallmark’ of cancer. Tissue stiffness increases with fibrous protein (e.g. collagen) concentration [9], which, in turn, anti-correlates with extracellular matrix pore size [10] (Fig. 2A). Hence, cancer cells invading normal tissue, as during tumor growth [47], encounter higher collagen matrix levels and smaller pores in stiffer tissues than in softer ones [48]. Squeezing through small pores—but not larger ones—greatly deforms the nuclei of invading cancer cells [49] and has a number of consequences. For one, constricted migration segregates mobile nuclear factors away from DNA [50]. Among cells in static culture, hetero/euchromatin occupies roughly 50–70% of the nuclear volume per previous estimates from molecular mobility [51], and we have shown for various cancer cell lines that the chromatin volume fraction can increase locally to 100% as the nucleus enters a small constriction. Conversely, all mobile proteins in the nucleus, including those that function as key DNA repair proteins, are always seen to deplete within the constriction [50]. Such unavoidable ‘squeeze-out’ of mobile nuclear factors away from the constriction, where DNA concentration is highest, has important implications for the repair of DNA damage that might occur during replication, for example. Inactivating mutations in repair factors such as BRCA1 and BRCA2 are well-established risk factors for cancer and are sufficient cause for prophylactic mastectomy (i.e. preventative surgery), and so transient partial depletion of such factors could increase mutational probabilities.
Fig. 2. Tissue stiffness increases with matrix density, which anti-correlates with interstitial pore size.
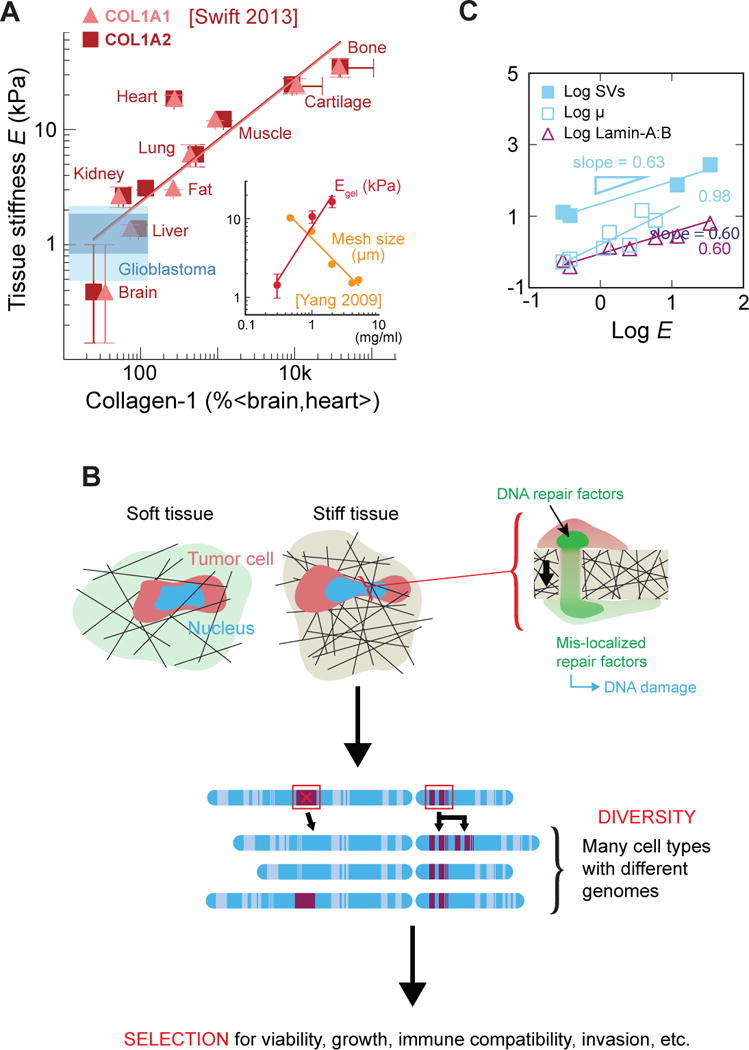
(A) Normal tissue microelasticity scales with collagen concentration. Blue shading indicates the range of stiffness values for 22 glioblastoma tumors, measured using multifrequency magnetic resonance elastography [98]. Evidently, brain tumors remain soft in vivo despite intra-tumor heterogeneity. (inset plot) At the collagen concentration at which pure collagen-1 gels exhibit a high tissue-like stiffness, they have a mesh size of a few microns or less. (B) Cancer cells sustain severe nuclear stress during tumorigenic invasion into small holes in stiff, fibrous tissues. This stress causes global loss of DNA repair factors via both ‘squeeze-out’ and nuclear envelope rupture. Perhaps due to repair loss, migrated cells exhibit elevated DNA damage, which ultimately leads to genome instability. (C) Somatic mutation rate (μ), structural variant (SV) number, and lamin-A expression all correlate with tissue stiffness.
In addition to inevitable squeeze-out of mobile repair factors, constriction can also cause rupture of the nuclear lamina [49]. Further studies with various cancer cell lines, immortalized epithelial cells, and primary dendritic cells, showed that migration through narrow channels can rupture the nuclear envelope and thereby permit cytoplasmic accumulation of GFP-tagged nuclear localization signal (NLS) constructs [52,53]. Rupture—and the ensuing nucleocytoplasmic exchange—occurs more frequently after knockdown of lamin-A and is followed by focal enrichment of an overexpressed DNA damage factor, which the authors interpreted as evidence of DNA damage and speculated on the entry of cytoplasmic nucleases [52,53]. If nuclease entry were responsible for constriction-induced DNA damage, then we would expect to see enrichment of damage foci near the site of nuclear envelope rupture. However, recent pore migration studies of an osteosarcoma line (U2OS), a lung carcinoma line (A549), and primary human mesenchymal stem cells have all shown by multiple measures of DNA damage (especially foci of γH2AX and phospho-ATM) that constricted migration produces a pan-nucleoplasmic distribution of DNA damage foci. This distribution suggests a global—rather than rupture site-specific—DNA damage mechanism; the distribution is consistent with transient knockdown of DNA repair proteins [50,54]. Similar depletion on the hours-long timescale of migration could likewise delay repair of replication errors, leading to the observed accumulation of DNA damage in migrated cells.
Importantly, the recent studies of DNA damage incurred during constricted migration also provided the first evidence of propagatable mutations. The genomes of serially migrated clones were analyzed by comparative genome hybridization arrays (aCGH), single-nucleotide polymorphism arrays (SNPa), and whole-exome sequencing (as well as RNA sequencing). Compared to unmigrated control clones, the migrated cells exhibited elevated chromosome copy number changes [54], suggesting that such chromosome-level abnormalities are characteristic of constricted migration. Recall that our meta-analysis showed that copy number changes and structural variants scale with normal tissue stiffness, perhaps more so than somatic mutations (Fig. 1B, C). Hence, constricted migration and stiffness seem to share a mutational signature, namely large-scale genome instability. This signature also resembles that of osteosarcomas and breast and ovarian cancers with BRCA deficiencies [55], although more such analysis is needed. Taken altogether, these genomic analyses hint at a connection between stiffness, constricted migration, and repair factor depletion (Fig. 2C). Thus, these studies tentatively support the hypothesis that loss of DNA repair during migration of cancer cells through small pores in fibrous matrix could underlie the scaling relation between mutation rate and tissue stiffness (Fig. 1).
Genomic variation gives rise to targetable neo-antigens
Genome changes, including those induced by a stiffness-related mechanism, can affect gene expression and lead to protein changes, which can contribute to a cancerous phenotype or merely be recognizable passenger mutations [11,16,17]. For example, in our studies of genome variation caused by constricted migration, one clone acquired after migration a spindle shape and migrated through pores much faster than other clones. Further experiments attributed this distinct phenotype to upregulation of the transcription factor GATA4 [54], which influences a program for microtubule organization. Microtubules are well known to be the most rigid polymers in cells and help direct cell migration.
Stiffness can also directly affect the expression of genes, so in a cancer like melanoma where tissue stiffness increases with invasiveness (Fig. 1C), some genes are expected to be upregulated. If these genes are also mutated — by a ‘mechanogenomics’ process or otherwise, then they could present neo-antigens to the immune system. Neo-antigens, or altered proteins, are ‘foreign’ in being distinct from anything in healthy cells and can thus be used therapeutically to target diseased cells. As an example, tissue stiffness causes systematic upregulation of lamin-A over a 20-to-30-fold range [9] (Fig. 2B). Mutations in lamin-A have been reported in The Cancer Genome Atlas (TCGA): one case study showed about 4% of 287 melanoma patients exhibited either amplifications (2%) or mis-sense passenger mutations (2%) in lamin-A’s coding sequence, with no statistically significant impact on patient survival (http://bit.ly/2oUMGyL). Lamin-A is nonetheless one conceivable source of neo-antigen that – when mutated – associates with tissue stiffness. Future studies of such upregulated, mutated genes should yield other candidates.
Efforts to therapeutically target neo-antigens, including those that arise in a stiffness-dependent way, are complicated by intratumor heterogeneity: different cells from a single tumor have been found to vary widely in their somatic mutations [20,56]. This heterogeneity reduces the probability of finding a ubiquitous, targetable mutation present in all of a patient’s cancer cells. Promising candidates—mutations that are relatively common among tumor cells—are those that occur early in cancer development [57], as tumor heterogeneity increases with cancer progression [56]. Conceivably, heterogeneity rises because cancer cells sustain additional mutations as they invade small pores in surrounding tissue during tumor growth. Of course, therapies designed against even the most widely expressed neo-antigen will likely produce negative selection, leading to the survival of cancer cells that are largely or wholly deficient in that neo-antigen. The ideal therapy must therefore target several different neo-antigens, which requires cancer cells to have a high mutation burden since not all neo-antigens are targetable [58]. Indeed, in non-small cell lung cancer, high mutation load is associated with improved clinical response to immunotherapy [59].
Though next-generation sequencing offers a means to identify neo-antigens, more development is needed before this technology can be implemented clinically, as neo-antigens vary within individual tumors as well as between patients [60–62]. Cancers of a given type often share mutated driver genes [63], which yield similar abnormal protein phenotypes, but changes can vary between patients. Hence, therapies for different patients must target different peptide sequences [11,64,65]. This variability makes it necessary to isolate and sequence every individual tumor to identify its unique neo-antigen profile, which remains a resource-intensive challenge for current sequencing [66]. If the technology continues to advance, it is exciting to consider that personalized neo-antigen-based therapy could enter clinical practice [67,68].
Diverse neo-antigen-based immunotherapies are currently under development
Neo-antigen-based therapies can take various approaches. Some therapies target proteins expressed on the plasma membrane, relying on surface protein expression level to distinguish cancerous cells from healthy ones. In one of the most-used such therapies, chimeric antigen receptor (CAR) T cells target the B-lymphocyte antigen CD19 [12]. While CD19 is expressed on over 95% of B-cell malignancies, it is also expressed on healthy B cells, making the latter susceptible to off-target effects [69]. However, the ongoing discovery of surface neo-antigens, such as mucin-1 (MUC1, which is also in the extracellular matrix and leaks into serum), that have irregular glycosylation patterns in cancer cells makes it possible to engineer CARs to target these unique glycosylations, thus minimizing deleterious side effects [70,71]. Most neo-antigens are not expressed on the plasma membrane, but rather in the nucleus or cytoplasm of the cell [72], so chemotherapeutics, which readily penetrate the cell membrane, can be effective against them. However, chemotherapeutic agents are difficult to modify against different peptide sequences since any change in their structural chemistry can radically alter pharmacokinetics. Although the chemotherapy drug Vemurafenib effectively targets the BRAF V600 mutation, it is ineffective against other BRAF mutations [73,74], which reinforces the need to identify each patient’s unique mutation profile.
Other approaches use nuclear and cytoplasmic neo-antigens to develop vaccines and checkpoint inhibitors [13]. These approaches take advantage of the major histocompatibility complex class I (MHC I), which presents peptide fragments—8–10 amino acids long—that are continuously screened by the immune system for foreign peptide sequences [75]. Unfortunately, mutated peptide sequences in cancerous cells often go undetected because they either have poor MHC I affinity, differ little from their wild-type counterparts, or are abetted by high levels of the cancer cell-derived immune inhibitory ligand PD-L1 [17,76–80]. PD-L1 inhibitors can counteract this tumor-induced immune suppression [81]. Sequencing is now being used to identify cancerous mutations in oncogenic drivers like KRAS and p53 and that might also have high MHC I affinity as needed for vaccination therapy [82,83].
An alternative vaccination approach exploits MHC class II molecules expressed by macrophages [14,15]. Like MHC I, MHC II presents peptide fragments at the cell surface. But whereas detection of a foreign sequence triggers cell destruction by the immune system in the MHC I case, it triggers activation of the adaptive immune system—against other cells weakly presenting that same foreign sequence—in the MHC II case. Macrophages use MHC II to present peptide sequences from foreign organisms or viruses that they have phagocytosed. Unfortunately, macrophages do not eat cancerous cells in part because the latter overexpress CD47 [84], which is a ‘marker of self’ that inhibits phagocytosis [85]. The immune system is therefore not activated by MHC II presentation of neo-antigens [86]. However, macrophages have long been engineered ex vivo for anti-cancer purposes and can be made to express patient-specific cancer neo-antigen peptide sequences loaded into MHC II [15]. Upon injection back into the patient, these engineered macrophages activate the immune system against cancer cells that display the special peptide sequence. Numerous clinical trials conducted over the past two decades demonstrate that adoptive transfer of macrophages into humans shows little to no toxicity at doses of 1.5 billion cells [87–89]. Strategic engineering of ‘self’ markers like CD47 should further reduce autoimmune toxicity by eliminating the need for systemic inhibition of CD47, which leads to rapid loss of red blood cells (RBCs) and in some mice to autoimmune responses, including production of anti-RBC antibodies [84,90].
One new macrophage-based therapy aims to exploit neo-antigens without the need for extensive sequencing or artificial targeting vectors (e.g. CARs). Tumor associated macrophages (TAMs) can, through inhibition of CD47, phagocytose cancer cells [91], and then present neo-antigens from those cells to activate the adaptive immune system [86,92]. However, TAMs promote tumor growth, are weakly phagocytic even with CD47 disruption, and have low MHC II expression, which hinders their ability to activate an adaptive immune response [15,93,94]. A cell therapy approach using marrow-derived monocytes and macrophages could be an alternative, as these cells have low SIRPα (i.e. the inhibitory receptor that interacts with CD47) and high MHC II expression [95]. In addition, marrow-derived monocytes and macrophages are highly migratory and can traffic into solid tumors [96] (Fig. 3A). However, like cancer cells invading nearby tissue, these ‘invading’ immune cells might also suffer DNA damage, with consequences yet unknown. The absence of oncogenic driver mutations probably limits the cancerous potential of infiltrating monocytes and macrophages; any DNA damage in these cells might instead contribute to senescence [97].
Fig. 3. Monocyte/macrophage-based immunotherapies target neo-antigens while exploiting the ability of phagocytic cells to infiltrate solid tumor tissues.
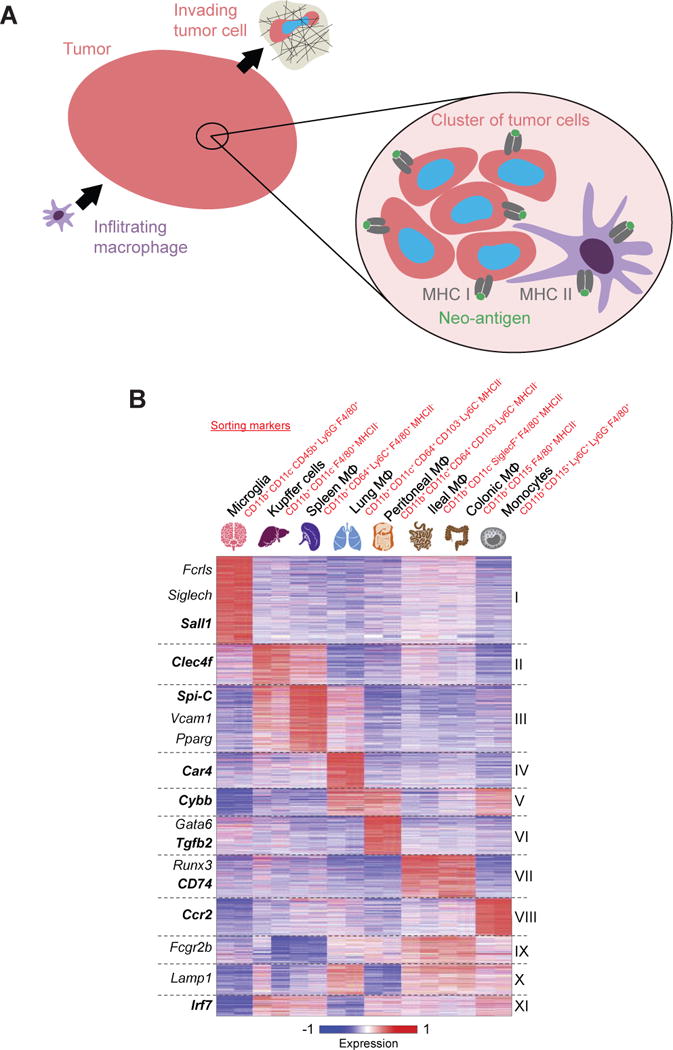
(A) Immune cells must first migrate into tumors in order to attack ‘non-self’ cancer cells. Then, macrophages use MHC II to present neo-antigens from cancer cells that they have phagocytosed. (B) Systems biology approaches are beginning to illuminate the microenvironment-dependent plasticity of monocytes and macrophages. Adapted from [95].
Lastly, while novel cell therapy approaches with infiltrating immune cells seems an encouraging but challenging future direction for the field of neo-antigen-based immunotherapy, the microenvironment-dependent plasticity of such cells is also emerging from modern systems biology methods (Fig. 3B) and must be factored into cell function. Interestingly, for monocytes and macrophages, at least some genes in published profiles (e.g. lamin-A) increase with tissue stiffness, consistent with mechanically regulated epi-genetic processes in normal cells [9] and likely also in cancer cells [7,8]. Further RNA profiling of tissue macrophages, including TAMs, needs to be done using identical markers across different tissues, as macrophage transcriptomics would be expected to change when sorted on different markers (Fig. 3B).
Conclusion
The meta-analysis here of recently published sequencing data reveals that somatic mutation rate increases with normal tissue stiffness across cancer types, while the rate of larger-scale, chromosome-level mutations increases even faster. Among various hypotheses that seek to explain this scaling relationship, the one that we consider most promising holds that stiffer tissues have smaller extracellular matrix pores, which can increase DNA damage in invading cancer cells, leading perhaps to genomic variation. In the case of immune cells that might infiltrate solid tumors—and go on to recognize neo-antigens—similar damage has not yet been observed. Since such healthy cells lack driver mutations, they are unlikely to become oncogenic, but they can be expected to differentiate, perhaps even in relation to tissue stiffness. (3300 words, not including figure legends)
Highlights.
The number of mutations in a tumor increases with the stiffness of a tissue.
Tissue stiffness might increase DNA damage in division and migration.
Copy number changes after constricted migration have been documented.
Macrophages are highly infiltrative and might recognize some neo-antigens.
Acknowledgments
The authors were supported by the National Cancer Institute of the National Institutes of Health under PSOC Award Number U54 CA193417 and by the National Heart Lung and Blood Institute of the National Institutes of Health under Award Number R01 HL124106. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, Fong SFT, Csiszar K, Giaccia A, Weninger W, et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2010;139:891–906. doi: 10.1016/j.cell.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boyd NF, Guo H, Martin LJ, Sun L, Stone J, Fishell E, Jong RA, Hislop G, Chiarelli A, Minkin S, et al. Mammographic density and the risk and detection of breast cancer. N Engl J Med. 2007;356:227–236. doi: 10.1056/NEJMoa062790. [DOI] [PubMed] [Google Scholar]
- 3.Singh S, Fujii LL, Murad MH, Wang Z, Asrani SK, Ehman RL, Kamath PS, Talwalkar JA. Liver stiffness is associated with risk of decompensation, liver cancer, and death in patients with chronic liver diseases: A systematic review and meta-analysis. Clin Gastroenterol Hepatol. 2014;11:1573–1584. doi: 10.1016/j.cgh.2013.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pelham RJ, Wang Y. Cell locomotion and focal adhesions are regulated by substrate flexibility. PNAS. 1997;94:13661–13665. doi: 10.1073/pnas.94.25.13661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell. 2006;126:677–689. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 6.Przybyla L, Muncie JM, Weaver VM. Mechanical control of epithelial-to-mesenchymal transitions in development and cancer. Annu Rev Cell Dev Biol. 2016;32:527–554. doi: 10.1146/annurev-cellbio-111315-125150. [DOI] [PubMed] [Google Scholar]
- 7.Spencer VA, Xu R, Bissell MJ. Extracellular matrix, nuclear and chromatin structure, and gene expression in normal tissues and malignant tumors: A work in progress. Adv Cancer Res. 2007;97:275–294. doi: 10.1016/S0065-230X(06)97012-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, et al. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474:179–183. doi: 10.1038/nature10137. [DOI] [PubMed] [Google Scholar]
- 9.Swift J, Ivanovska IL, Buxboim A, Harada T, Dingal PCDP, Pinter J, Pajerowski JD, Spinler KR, Shin J, Tewari M, et al. Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science. 2013;341:1240104. doi: 10.1126/science.1240104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang Y, Leone LM, Kaufman LJ. Elastic moduli of collagen gels can be predicted from two-dimensional confocal microscopy. Biophys J. 2009;97:2051–2060. doi: 10.1016/j.bpj.2009.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11••.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348:69–74. doi: 10.1126/science.aaa4971. This review both provides somatic mutation prevalence for 30 cancer types and relates those mutational loads to the formation of neo-antigen repertoires. It further discusses the exploitation of neo-antigens in cancer immunotherapy. [DOI] [PubMed] [Google Scholar]
- 12.Ramos CA, Savoldo B, Dotti G. CD19-CAR trials. 2015;20:112–118. doi: 10.1097/PPO.0000000000000031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ly LV, Sluijter M, van der Burg SH, Jager MJ, van Hall T. Effective cooperation of monoclonal antibody and peptide vaccine for the treatment of mouse melanoma. J Immunol. 2013;190:489–496. doi: 10.4049/jimmunol.1200135. [DOI] [PubMed] [Google Scholar]
- 15.Liu Q, Wen W, Tang L, Qin C-J, Lin Y, Zhang H-L, Ashton C, Wu H-P, Ding J, Dong W, et al. Inhibition of SIRPα in dendritic cells potentiates potent antitumor immunity. Oncoimmunology. 2016;5 doi: 10.1080/2162402X.2016.1183850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martin SD, Coukos G, Holt RA, Nelson BH. Targeting the undruggable: Immunotherapy meets personalized oncology in the genomic era. Ann Oncol. 2015;26:2367–2374. doi: 10.1093/annonc/mdv382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matsushita H, Sato Y, Karasaki T, Nakagawa T, Kume H, Ogawa S, Homma Y, Kakimi K. Neoantigen load, antigen presentation machinery, and immune signatures determine prognosis in clear cell renal cell carcinoma. Cancer Immunol Res. 2016;4:463–471. doi: 10.1158/2326-6066.CIR-15-0225. [DOI] [PubMed] [Google Scholar]
- 18.Costello M, Pugh TJ, Fennell TJ, Stewart C, Lichtenstein L, Meldrim JC, Fostel JL, Friedrich DC, Perrin D, Dionne D, et al. Discovery and characterization of artifactual mutations in deep coverage targeted capture sequencing data due to oxidative DNA damage during sample preparation. Nucleic Acids Res. 2013;41 doi: 10.1093/nar/gks1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale A-L, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–218. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martincorena I, Roshan A, Gerstung M, Ellis P, Loo P Van, Mclaren S, Wedge DC, Fullam A, Alexandrov LB, Tubio JM, et al. High burden and pervasive positive selection of somatic mutations in normal human skin. Science. 2015;348:880–886. doi: 10.1126/science.aaa6806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martincorena I, Campbell PJ. Somatic mutation in cancer and normal cells. Science. 2015;349:961–968. doi: 10.1126/science.aab4082. [DOI] [PubMed] [Google Scholar]
- 23••.Chen X, Bahrami A, Pappo A, Easton J, Dalton J, Hedlund E, Ellison D, Shurtleff S, Wu G, Wei L, et al. Recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma. Cell Rep. 2014;7:104–112. doi: 10.1016/j.celrep.2014.03.003. To characterize the genomic landscape of pediatric osteosarcoma, the authors perform whole-genome sequencing of 20 osteosarcoma tumors and matched normal tissues. Their findings reveal that osteosarcoma has an elevated rate of structural variants and copy number variations compared to other childhood cancers. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24••.Shain AH, Yeh I, Kovalyshyn I, Sriharan A, Talevich E, Gagnon A, Dummer R, North J, Pincus L, Ruben B, et al. The genetic evolution of melanoma from precursor lesions. N Engl J Med. 2015;373:1926–1936. doi: 10.1056/NEJMoa1502583. The authors sequence ~300 cancer-relevant genes in 37 melanocytic neoplasms, which fall into histologic categories ranging from “benign” to “invasive melanoma.” They find that the mutational patterns of these neoplasms correlate with their histopathological features. [DOI] [PubMed] [Google Scholar]
- 25.Shin J, Buxboim A, Spinler KR, Swift J, Christian DA, Hunter CA, Léon C, Gachet C, Dingal PCDP, Ivanovska IL, et al. Contractile forces sustain and polarize hematopoiesis from stem and progenitor cells. Cell Stem Cell. 2014;14:81–93. doi: 10.1016/j.stem.2013.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Prabhune M, Belge G, Dotzauer A, Bullerdiek J, Radmacher M. Comparison of mechanical properties of normal and malignant thyroid cells. Micron. 2012;43:1267–1272. doi: 10.1016/j.micron.2012.03.023. [DOI] [PubMed] [Google Scholar]
- 27.Lopez JI, Kang I, You W-K, McDonald DM, Weaver VM. In situ force mapping of mammary gland transformation. Integr Biol. 2011;3:910–921. doi: 10.1039/c1ib00043h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lekka M, Gil D, Pogoda K, Dulinska-Litewka J, Jach R, Gostek J, Klymenko O, Prauzner-Bechcicki S, Stachura Z, Wiltowska-Zuber J, et al. Cancer cell detection in tissue sections using AFM. Arch Biochem Biophys. 2012;518:151–156. doi: 10.1016/j.abb.2011.12.013. [DOI] [PubMed] [Google Scholar]
- 29.Hoyt K, Castaneda B, Zhang M, Nigwekar P, di Sant’Agnese PA, Joseph JV, Strang J, Rubens DJ, Parker KJ. Tissue elasticity properties as biomarkers for prostate cancer. Cancer Biomarkers. 2008;4:213–225. doi: 10.3233/cbm-2008-44-505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu W, Mezencev R, Kim B, Wang L, Mcdonald J, Sulchek T. Cell stiffness is a biomarker of the metastatic potential of ovarian cancer cells. PLoS One. 2012;7:e46609. doi: 10.1371/journal.pone.0046609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cross SE, Jin Y-S, Lu Q-Y, Rao J, Gimzewski JK. Green tea extract selectively targets nanomechanics of live metastatic cancer cells. Nanotechnology. 2011;22:215101. doi: 10.1088/0957-4484/22/21/215101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guz N, Dokukin M, Kalaparthi V, Sokolov I. If cell mechanics can be described by elastic modulus: Study of different models and probes used in indentation experiments. Biophysical. 2014;107:564–575. doi: 10.1016/j.bpj.2014.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kawano S, Kojima M, Higuchi Y, Sugimoto M, Ikeda K, Sakuyama N, Takahashi S, Hayashi R, Ochiai A, Saito N. Assessment of elasticity of colorectal cancer tissue, clinical utility, pathological and phenotypical relevance. Cancer Sci. 2015;106:1232–1239. doi: 10.1111/cas.12720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fuhrmann A, Staunton JR, Nandakumar V, Banyai N, Davies PCW, Ros R. AFM stiffness nanotomography of normal, metaplastic and dysplastic human esophageal cells. Phys Biol. 2011;8:15007. doi: 10.1088/1478-3975/8/1/015007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lim Y-J, Deo D, Singh TP, Jones DB, De S. In situ measurement and modeling of biomechanical response of human cadaveric soft tissues for physics-based surgical simulation. Surg Endosc. 2009;23:1298–1307. doi: 10.1007/s00464-008-0154-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lekka M, Pogoda K, Gostek J, Klymenko O, Prauzner-Bechcicki S, Wiltowska-Zuber J, Jaczewska J, Lekki J, Stachura Z. Cancer cell recognition – mechanical phenotype. Micron. 2012;43:1259–1266. doi: 10.1016/j.micron.2012.01.019. [DOI] [PubMed] [Google Scholar]
- 37.Petrie RJ, Gavara N, Chadwick RS, Yamada KM. Nonpolarized signaling reveals two distinct modes of 3D cell migration. J Cell Biol. 2012;197:439–455. doi: 10.1083/jcb.201201124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mathur AB, Collinsworth AM, Reichert WM, Kraus WE, Truskey GA. Endothelial, cardiac muscle and skeletal muscle exhibit different viscous and elastic properties as determined by atomic force microscopy. J Biomech. 2001;34:1545–1553. doi: 10.1016/s0021-9290(01)00149-x. [DOI] [PubMed] [Google Scholar]
- 39.Diridollou S, Vabre V, Berson M, Black D, Lagarde JM, Gregoire JM, Gall Y. Skin ageing: Changes of physical properties of human skin in vivo. Int J Cosmet Sci. 2001;23:353–362. doi: 10.1046/j.0412-5463.2001.00105.x. [DOI] [PubMed] [Google Scholar]
- 40.Klein EA, Castagnino P, Kothapalli D, Yin L, Byfield FJ, Xu T, Levental I, Hawthorne E, Janmey PA, Assoian RK. Cell cycle control by physiological matrix elasticity and in vivo tissue stiffening. Curr Biol. 2009;19:1511–1518. doi: 10.1016/j.cub.2009.07.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seshadri R, Kutlaca RJ, Trainor K, Matthews C, Morley AA. Mutation rate of normal and malignant human lymphocytes. Cancer Res. 1987;47:407–409. [PubMed] [Google Scholar]
- 42.Tomasetti C, Li L, Vogelstein B. Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention. Science. 2017;355:1330–1334. doi: 10.1126/science.aaf9011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nowak MA, Waclaw B. Genes, environment, and “bad luck”. Science. 2017;355:1266–1267. doi: 10.1126/science.aam9746. [DOI] [PubMed] [Google Scholar]
- 44.Tamiello C, Kamps MAF, van den Wijngaard A, Verstraeten VLRM, Baaijens FPT, Broers JLV, Bouten CCV. Soft substrates normalize nuclear morphology and prevent nuclear rupture in fibroblasts from a laminopathy patient with compound heterozygous LMNA mutations. Nucleus. 2013;4:61–73. doi: 10.4161/nucl.23388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Maciejowski J, Li Y, Bosco N, Campbell PJ, Maciejowski J, Li Y, Bosco N, Campbell PJ, Lange TDe. Chromothripsis and kataegis induced by telomere crisis. Cell. 2015;163:1641–1654. doi: 10.1016/j.cell.2015.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Irianto J, Pfeifer CR, Ivanovska IL, Swift J, Discher DE. Nuclear lamins in cancer. Cell Mol Bioeng. 2016;9:258–267. doi: 10.1007/s12195-016-0437-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liotta LA, Steeg PS, Stetler-Stevenson WG. Cancer metastasis and angiogenesis: An imbalance of positive and negative regulation. Cell. 1991;64:327–336. doi: 10.1016/0092-8674(91)90642-c. [DOI] [PubMed] [Google Scholar]
- 48.Irianto J, Pfeifer CR, Xia Y, Discher DE. SnapShot: Mechanosensing matrix. Cell. 2016;165:1820–1820.e1. doi: 10.1016/j.cell.2016.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49•.Harada T, Swift J, Irianto J, Shin JW, Spinler KR, Athirasala A, Diegmiller R, Dingal PCDP, Ivanovska IL, Discher DE. Nuclear lamin stiffness is a barrier to 3D migration, but softness can limit survival. J Cell Biol. 2014;204:669–682. doi: 10.1083/jcb.201308029. The authors demonstrate constricted migration causes nuclear lamina damage and cell death that increases with lamin-A knockdown. The explanation provided the first evidence of a role for DNA damage and repair. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Irianto J, Pfeifer CR, Bennett RR, Xia Y, Ivanovska IL, Liu AJ, Greenberg RA, Discher DE. Nuclear constriction segregates mobile nuclear proteins away from chromatin. Mol Biol Cell. 2016;27:4011–4020. doi: 10.1091/mbc.E16-06-0428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bancaud A, Huet S, Daigle N, Mozziconacci J, Beaudouin J, Ellenberg J. Molecular crowding affects diffusion and binding of nuclear proteins in heterochromatin and reveals the fractal organization of chromatin. EMBO J. 2009;28:3785–3798. doi: 10.1038/emboj.2009.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52•.Denais CM, Gilbert RM, Isermann P, Mcgregor AL, te Lindert M, Weigelin B, Davidson PM, Friedl P, Wolf K, Lammerding J. Nuclear envelope rupture and repair during cancer cell migration. Science. 2016;352:353–358. doi: 10.1126/science.aad7297. The authors confirm that migration of cancer cell lines and fibroblasts through narrow channels causes nuclear lamina rupture. Transient rupture of the envelope leads to leakage of nuclear and cytoplasmic constructs as well as local enrichment of the overexpressed DNA damage repair protein RFP-53BP1. Rupture repair is mediated by the ESCRT III complex. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53•.Raab M, Gentili M, de Belly H, Thiam HR, Vargas P, Jimenez AJ, Lautenschlaeger F, Voituriez R, Lennon-Dumenil AM, Manel N, et al. ESCRT III repairs nuclear envelope ruptures during cell migration to limit DNA damage and cell death. Science. 2016;352:359–362. doi: 10.1126/science.aad7611. The authors confirm that constricted migration of cancer and dendritic cells leads to nuclear lamina rupture. Transient rupture of the envelope leads to leakage of nuclear and cytoplasmic constructs as well as local enrichment of the overexpressed DNA damage repair protein 53BP1-GFP. Rupture repair is mediated by the ESCRT III complex. The authors speculate that rupture-induced leakage of cytoplasmic factors into the nucleus causes double-strand breaks. [DOI] [PubMed] [Google Scholar]
- 54•.Irianto J, Xia Y, Pfeifer CR, Athirasala A, Ji J, Alvey C, Tewari M, Bennett RR, Harding SM, Liu AJ, et al. DNA damage follows repair factor depletion and portends genome variation in cancer cells after pore migration. Curr Biol. 2017;27:210–223. doi: 10.1016/j.cub.2016.11.049. The authors demonstrate constricted migration causes DNA damage in a mechanism consistent with repair factor loss. This is the first paper to document genomic changes in cell migration. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kovac M, Blattmann C, Ribi S, Smida J, Mueller NS, Engert F, Castro-Giner F, Weischenfeldt J, Kovacova M, Krieg A, et al. Exome sequencing of osteosarcoma reveals mutation signatures reminiscent of BRCA deficiency. Nat Commun. 2015 doi: 10.1038/ncomms9940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56••.Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–892. doi: 10.1056/NEJMoa1113205. A critical study that quantifies genomic heterogeneity from a single tumor that arises from somatic mutations. The authors conclude that 63 to 69% of all somatic mutations are not detectable across regions of a tumor and that phylogenetic reconstruction shows branched evolutionary tumor growth. This highlights the difficulty of developing therapies that target neo-antigens which are ubiquitiously expressed in cancer cells within a single tumor. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.de Bruin EC, Mcgranahan N, Mitter R, Salm M, Wedge DC, Yates L, Jamal-Hanjani M, Shafi S, Murugaesu N, Rowan AJ, et al. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science. 2013;346:251–256. doi: 10.1126/science.1253462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Martin SD, Brown SD, Wick DA, Nielsen JS, Kroeger DR, Twumasi-Boateng K, Holt RA, Nelson BH. Low mutation burden in ovarian cancer may limit the utility of neoantigen-targeted vaccines. PLoS One. 2016;11 doi: 10.1371/journal.pone.0155189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59••.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–128. doi: 10.1126/science.aaa1348. This study solidifies the link between checkpoint inhibitor efficacy and mutation burden in lung cancer patients. Human cancer cells present neo-antigens but also overexpress T cell inhibitors, like PD-L1, which prevent an immune response. PD-L1-treated patients with higher mutation burden respond significantly better than those with low mutation burden. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Heemskerk B, Kvistborg P, Schumacher TN. The cancer antigenome. EMBO J. 2013;32:194–203. doi: 10.1038/emboj.2012.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Robbins PF, Lu Y-C, El-Gamil M, Li YF, Gross C, Gartner J, Lin JC, Teer JK, Cliften P, Tycksen E, et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med. 2013;19:747–752. doi: 10.1038/nm.3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rajasagi M, Shukla SA, Fritsch EF, Keskin DB, DeLuca D, Carmona E, Zhang W, Sougnez C, Cibulskis K, Sidney J, et al. Systematic identification of personal tumor-specific neoantigens in chronic lymphocytic leukemia. Blood. 2014;124:453–463. doi: 10.1182/blood-2014-04-567933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mcgranahan N, Favero F, de Bruin EC, Birkbak NJ, Szallasi Z, Swanton C. Clonal status of actionable driver events and the timing of mutational processes in cancer evolution. Sci Transl Med. 2015;7 doi: 10.1126/scitranslmed.aaa1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Segal NH, Parsons DW, Peggs KS, Velculescu V, Kinzler KW, Vogelstein B, Allison JP. Epitope landscape in breast and colorectal cancer. Cancer Res. 2008;68:889–892. doi: 10.1158/0008-5472.CAN-07-3095. [DOI] [PubMed] [Google Scholar]
- 65.Jo P, König A, Schirmer M, Kitz J, Conradi L-C, Azizian A, Bernhardt M, Wolff HA, Grade M, Ghadimi M, et al. Heterogeneity of KRAS mutation status in rectal cancer. PLoS One. 2016;20 doi: 10.1371/journal.pone.0153278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li Y, Bare LA, Bender RA, Sninsky JJ, Wilson LS, Devlin JJ, Waldman FM. Cost effectiveness of sequencing 34 cancer-associated genes as an aid for treatment selection in patients with metastatic melanoma. Mol Diagn Ther. 2015;19:169–177. doi: 10.1007/s40291-015-0140-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.LeBlanc VG, Marra MA. Next-generation sequencing approaches in cancer: Where have they brought us and where will they take us? Cancers (Basel) 2015;7:1925–1958. doi: 10.3390/cancers7030869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schwaederle M, Parker BA, Schwab RB, Fanta PT, Boles SG, Daniels GA, Bazhenova LA, Subramanian R, Coutinho AC, Ojeda-Fournier H, et al. Molecular tumor board: The University of California San Diego Moores Cancer Center experience. Oncologist. 2014;19:631–636. doi: 10.1634/theoncologist.2013-0405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Turtle CJ, Hanafi L-A, Berger C, Gooley TA, Cherian S, Hudecek M, Sommermeyer D, Melville K, Pender B, Budiarto TM, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest. 2016;126:2123–2138. doi: 10.1172/JCI85309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Posey AD, Schwab RD, Boesteanu AC, Steentoft C, Mandel U, Engels B, Stone JD, Madsen TD, Schreiber K, Haines KM, et al. Engineered CAR T cells targeting the cancer-associated Tn-glycoform of the membrane mucin MUC1 control adenocarcinoma. Immunity. 2016;44:1444–1454. doi: 10.1016/j.immuni.2016.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sabbatini PJ, Ragupathi G, Hood C, Aghajanian CA, Juretzka M, Iasonos A, Hensley ML, Spassova MK, Ouerfelli O, Spriggs DR, et al. Pilot study of a heptavalent vaccine-keyhole limpet hemocyanin conjugate plus QS21 in patients with epithelial ovarian, fallopian tube, or peritoneal cancer. Clin Cancer Res. 2007;13:4170–4177. doi: 10.1158/1078-0432.CCR-06-2949. [DOI] [PubMed] [Google Scholar]
- 72.Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–339. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McArthur GA, Chapman PB, Robert C, Larkin J, Haanen JB, Dummer R, Ribas A, Hogg D, Hamid O, Ascierto PA, et al. Safety and efficacy of vemurafenib in BRAFV600E and BRAFV600K mutation-positive melanoma (BRIM-3): Extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol. 2014;15:323–332. doi: 10.1016/S1470-2045(14)70012-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Klein O, Clements A, Menzies AM, O’Toole S, Kefford RF, Long GV. BRAF inhibitor activity in V600R metastatic melanoma – Response. Eur J Cancer. 2013;49:1797–1798. doi: 10.1016/j.ejca.2013.02.010. [DOI] [PubMed] [Google Scholar]
- 75.Schumacher T, Bunse L, Pusch S, Sahm F, Wiestler B, Quandt J, Menn O, Osswald M, Oezen I, Ott M, et al. A vaccine targeting mutant IDH1 induces antitumour immunity. Nature. 2014;512:324–327. doi: 10.1038/nature13387. [DOI] [PubMed] [Google Scholar]
- 76.Wick DA, Webb JR, Nielsen JS, Martin SD, Kroeger DR, Milne K, Castellarin M, Twumasi-Boateng K, Watson PH, Holt RA, et al. Surveillance of the tumor mutanome by T cells during progression from primary to recurrent ovarian cancer. Clin Cancer Res. 2014;20:1125–1134. doi: 10.1158/1078-0432.CCR-13-2147. [DOI] [PubMed] [Google Scholar]
- 77.Paul S, Weiskopf D, Angelo MA, Sidney J, Peters B, Sette A. NIH Public Access. J Immunol. 2013;191:5831–5839. doi: 10.4049/jimmunol.1302101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Assarsson E, Sidney J, Oseroff C, Pasquetto V, Bui H, Frahm N, Brander C, Peters B, Grey H, Sette A. A quantitative analysis of the variables affecting the repertoire of T cell specificities recognized after vaccinia virus infection. J Immunol. 2007;178:7890–7901. doi: 10.4049/jimmunol.178.12.7890. [DOI] [PubMed] [Google Scholar]
- 79.Verdegaal EME, de Miranda NFCC, Visser M, Harryvan T, van Buuren MM, Andersen RS, Hadrup SR, van der Minne CE, Schotte R, Spits H, et al. Neoantigen landscape dynamics during human melanoma–T cell interactions. Nature. 2016;536:91–95. doi: 10.1038/nature18945. [DOI] [PubMed] [Google Scholar]
- 80.Strickland KC, Howitt BE, Shukla SA, Rodig S, Ritterhouse LL, Liu JF, Garber JE, Chowdhury D, Wu CJ, D’Andrea AD, et al. Association and prognostic significance of BRCA1/2-mutation status with neoantigen load, number of tumor-infiltrating lymphocytes and expression of PD-1/PD-L1 in high grade serous ovarian cancer. Oncotarget. 2016;7:13587–13598. doi: 10.18632/oncotarget.7277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2016;12:252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rahma OE, Hamilton JM, Wojtowicz M, Dakheel O, Bernstein S, Liewehr DJ, Steinberg SM, Khleif SN. The immunological and clinical effects of mutated ras peptide vaccine in combination with IL-2, GM-CSF, or both in patients with solid tumors. J Transl Med. 2014;12:55–67. doi: 10.1186/1479-5876-12-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Carbone DP, Ciernik IF, Kelley MJ, Smith MC, Nadaf S, Kavanaugh D, Maher VE, Stipanov M, Contois D, Johnson BE, et al. Immunization with mutant p53- and K-ras-derived peptides in cancer patients: Immune response and clinical outcome. J Clin Oncol. 2005;23:5099–5107. doi: 10.1200/JCO.2005.03.158. [DOI] [PubMed] [Google Scholar]
- 84.Willingham SB, Volkmer J-P, Gentles AJ, Sahoo D, Dalerba P, Mitra SS, Wang J, Contreras-Trujillo H, Martin R, Cohen JD, et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. PNAS. 2012;109:6662–6667. doi: 10.1073/pnas.1121623109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tsai RK, Discher DE. Inhibition of “self” engulfment through deactivation of myosin-II at the phagocytic synapse between human cells. J Cell Biol. 2008;180:989–1003. doi: 10.1083/jcb.200708043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86•.Liu X, Pu Y, Cron K, Deng L, Kline J, Frazier WA, Xu H, Peng H, Fu Y-X, Xu MM. CD47 blockade triggers T cell-mediated destruction of immunogenic tumors. Nat Med. 2015;21:1209–1215. doi: 10.1038/nm.3931. The authors demonstrate that shrinkage of tumors via CD47 inhibition is not completely dependent on macrophage destruction of cancer cells, but rather on their ability to activate T cells. Therefore, macrophages may be able to naturally screen neo-antigens and present them to T cells and perhaps B cells, leading to the activation of these cells against malignancies. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Andreesen R, Hennemann B, Krause SW. Adoptive immunotherapy of cancer using monocyte-derived macrophages: rationale, current status, and perspectives. J Leukoc Biol. 1998;64:419–426. doi: 10.1002/jlb.64.4.419. [DOI] [PubMed] [Google Scholar]
- 88.Hennemann B, Beckmann G, Eichelmann A, Rehm A, Andreesen R. Phase I trial of adoptive immunotherapy of cancer patients using monocyte-derived macrophages activated with interferon g and lipopolysaccharide. Cancer Immunol Immunother. 1998;45:250–256. doi: 10.1007/PL00006671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Faradji A, Bohbot A, Frost H, Schmitt-Goguel M, Siffert JC, Dufour P, Eber M, Lallot C, Wiesel ML, Bergerat JP, et al. Phase I study of liposomal MTP-PE-activated autologous monocytes administered intraperitoneally to patients with peritoneal carcinomatosis. J Clin Oncol. 1991;9:1251–1260. doi: 10.1200/JCO.1991.9.7.1251. [DOI] [PubMed] [Google Scholar]
- 90.Oldenborg P, Zheleznyak A, Fang Y, Lagenaur CF, Gresham HD, Lindberg FP. Role of CD47 as a marker of self on red blood cells. Science. 2000;288:2051–2054. doi: 10.1126/science.288.5473.2051. [DOI] [PubMed] [Google Scholar]
- 91.Weiskopf K, Ring AM, Ho CCM, Volkmer J-P, Levin AM, Volkmer AK, Özkan E, Fernhoff NB, van de Rijn M, Weissman IL, et al. Engineered SIRPa variants as immunotherapeutic adjuvants to anticancer antibodies. Science. 2013;341:88–91. doi: 10.1126/science.1238856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tseng D, Volkmer J-P, Willingham SB, Contreras-Trujillo H, Fathman JW, Fernhoff NB, Seita J, Inlay MA, Weiskopf K, Miyanishi M, et al. Anti-CD47 antibody–mediated phagocytosis of cancer by macrophages primes an effective antitumor T-cell response. PNAS. 2013;110:11103–11108. doi: 10.1073/pnas.1305569110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lu-Emerson C, Snuderl M, Kirkpatrick ND, Goveia J, Davidson C, Huang Y, Riedemann L, Taylor J, Ivy P, Duda DG, et al. Increase in tumor-associated macrophages after antiangiogenic therapy is associated with poor survival among patients with recurrent glioblastoma. Neuro Oncol. 2013;15:1079–1087. doi: 10.1093/neuonc/not082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rodríguez D, Silvera R, Carrio R, Nadji M, Caso R, Rodríguez G, Iragavarapu-Charyulu V, Torroella-Kouri M. Tumor microenvironment profoundly modifies functional status of macrophages: Peritoneal and tumor-associated macrophages are two very different subpopulations. Cell Immunol. 2013;283:51–60. doi: 10.1016/j.cellimm.2013.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, Jung S, Amit I. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell. 2014;159:1312–1326. doi: 10.1016/j.cell.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Pan Y, Tan Y, Wang M, Zhang J, Zhang B, Yang C, Ding Z, Dong L, Wang H. Signal regulatory protein a is associated with tumor-polarized macrophages phenotype switch and plays a pivotal role in tumor progression. Hepatology. 2013;58:680–691. doi: 10.1002/hep.26391. [DOI] [PubMed] [Google Scholar]
- 97.von Zglinicki T, Saretzki G, Ladhoff J, d’Adda di Fagagna F, Jackson SP. Human cell senescence as a DNA damage response. Mech Ageing Dev. 2005;126:111–117. doi: 10.1016/j.mad.2004.09.034. [DOI] [PubMed] [Google Scholar]
- 98.Streitberger K-J, Reiss-Zimmermann M, Freimann FB, Bayerl S, Guo J, Arlt F, Wuerfel J, Hoffmann K-T, Sack I. High-resolution mechanical imaging of glioblastoma by multifrequency magnetic resonance elastography. PLoS One. 2014;9 doi: 10.1371/journal.pone.0110588. [DOI] [PMC free article] [PubMed] [Google Scholar]