

Abstract
While clues have existed that endosomal trafficking is associated with Alzheimer’s disease (AD), whether it plays a central role in the disease and if so how has remained unknown. Here we rely on recent genetic and cellular findings to construct a model proposing that traffic jams in the early endosome can act as an upstream pathogenic hub in AD. We also rely on an independent series of findings to suggest how the traffic jams can act as a unified mediator of downstream pathophysiology. The model predicts, therefore, that interventions designed to unjam the endosome carry high therapeutic promise.
Four Classes of Genes Linked to Alzheimer’s Disease
The turn of the century represents a convenient timestamp around which the genetic investigation into Alzheimer’s disease (AD) can be organized. Available genetic tools at the end of the 20th century were best suited to isolate Mendelian-inherited mutations, and when applied to the rare autosomal-dominant forms of AD identified mutations in genes encoding the amyloid precursor protein (APP) and the presenilins, presenilin1 (PSEN1) & presenilin 2 (PSEN2) (Hardy and Selkoe, 2002). During the early part of the 21st century, new tools, large-scale samples, and computational prowess allowed the focus to shift to the more common late-onset ‘sporadic’ form of AD, which accounts for over 95% of all cases. These genetic studies have revealed approximately two dozen genes linked to late-onset AD (LOAD) (Guerreiro et al., 2013; Karch and Goate, 2015; Naj et al., 2017). Remarkably, the genes cohere into three general biological classes: ‘cholesterol metabolism’ genes, ‘immune response’ genes, and ‘endosomal trafficking’ genes. Thus, together with the class of ‘APP processing’ genes associated with autosomal-dominant disease, there are now 4 main gene classes that are linked to AD.
In this Opinion, we rely on recent insight into the function of these genes together with their intraneuronal effects to construct a testable pathogenic model of disease that is centered on ‘endosomal traffic jams’ (Fig. 1). The model proposes that endosomal traffic jams represents a pathogenic hub onto which nearly all AD genes can directly or indirectly converge, and suggests that this hub can act as a final common pathway through which many downstream pathophysiological effects can be mediated. If validated, this model predicts that immunotherapies directed against extracellular amyloid plaques might fail even if administered early on, and suggests alternative cell biological targets for therapeutic interventions.
Figure 1. A pathogenic hub of Alzheimer’s centered on endosomal traffic jams.
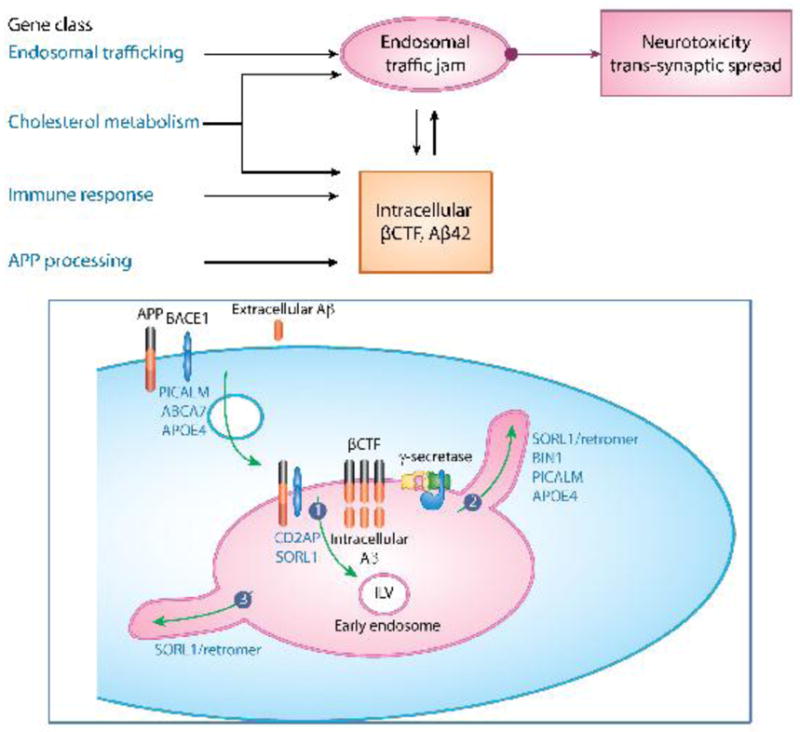
As outlined in the flow diagram (upper panel) and as illustrated in the figure (lower panel), the four gene classes associated with Alzheimer’s disease are directly or indirectly linked to endosomal traffic jams. These traffic jams can occur by altering the balance of membrane traffic into the early endosomes via the endocytosis pathway (indicated by the green arrow in the figure), or more commonly by altering one of the three traffic pathways out of the early endosome (indicated by the numbered green arrows): 1. The degradation pathway, which is initiated by sorting cargo to the intraluminal vesicle (ILV). 2. The ‘recycling’ pathway to the cell surface. 3. The ‘retrograde’ pathway back to the trans-Golgi network. The four gene classes are:
Endosomal Trafficking Genes. Typified by SORL1, BIN1, CD2AP and PICALM, this class directly affects the balance of membrane trafficking in and mainly out of the early endosome, upstream to amyloid accumulation. A downstream consequence is to increase APP and/or BACE1 at the endosomal membrane, leading to intracellular accumulation of βCTF and Aβ42, or to a decrease in the intraneuronal clearance of Aβ42. As illustrated, βCTF and/or Aβ42 can feed back and worsen endosomal traffic jams, creating a vicious cycle
Cholesterol Metabolism Genes. Typified by APOE4, this class can decrease the clearance of extracellular Aβ42, which can lead to increases in intracellular Aβ42. Additionally, this class can increase endocytosis or more likely reduce endosomal recycling, with a direct effect on endosomal traffic jams.
Immune response genes. Typified by TREM2 this class can decrease the clearance of extracellular Aβ42, leading to increases in intracellular Aβ42, which in turn can contribute to endosomal traffic jams.
APP processing genes. Including mutations in APP and the presenilins, this class leads to an intracellular accumulation of Aβ42 and/or βCTF and, which can act as drivers of endosomal traffic jams.
ENDOSOMAL TRAFFIC JAMS IN ALZHEIMER’S DISEASE
The first indication that endosomal traffic jams can occur in AD came from a careful microscopic analysis of postmortem brains showing that abnormal enlargement of the early endosome occurs with near diagnostic precision (Cataldo et al., 2000). The early endosome is a central hub in membrane trafficking, particularly in neurons (Kennedy and Ehlers, 2006), and cellular mechanisms carefully regulate the balance of transport in and out of this organelle (Chi et al., 2015; Pfeffer, 2013). Enlarged endosomes represent an imbalance in endosomal trafficking and this phenotype is therefore one, although not the only, manifestation of endosomal traffic jams. Together with AD’s histological hallmarks, amyloid plaques and neurofibrillary tangles, enlarged endosomes have now emerged as a cytopathological hallmark of the disease. For example, besides AD’s histological hallmarks, enlarged endosomes are characteristic observations in IPSC-derived neurons generated from late-onset sporadic AD patients (Israel et al., 2012) and in neurons or organoids derived from autosomal-dominant AD cases (Israel et al., 2012; Raja et al., 2016).
What causes endosomal traffic jams in AD? As elaborated in the next section, until recently the answer to this question could simply be that traffic jams reflect a secondary consequence of intracellular accumulation of APP fragments. To briefly review (Hardy and Selkoe, 2002), the amyloidogenic processing of APP into its fragments begins inside neurons by its cleavage by ‘β-amyloid cleaving enzyme 1′ (BACE1), whose products include the ‘c-terminal fragment’ (βCTF) (Fig. 1). This intermediate fragment is a substrate for γ-secretase, a multiprotein enzyme that contains the presenilins, whose products inside neurons include a range of ‘amyloid β’ (Aβ) peptides of varying amino acid lengths, but most commonly Aβ40 and Aβ42.
Intracellular accumulation of APP fragments occurs in all forms of AD. In autosomal-dominant forms, APP mutations commonly increase the production of its cleaved products in neurons, and since presenilin mutations cause a loss of enzymatic function, most -- though not all -- cause an increase in γ-secretase’s substrate, interneuronal βCTF (De Strooper, 2007; Li et al., 2016; Woodruff et al., 2013). The presenilin mutations’ effect on Aβ production is more complicated, but all seem to cause a relative increase in Aβ42 (De Strooper, 2007) inside neurons, and Aβ42 is thought to be the more toxic form of the peptide. Aβ42 also accumulates inside neurons in late-onset sporadic forms of the disease (Gouras et al., 2010). In some cases this accumulation occurs because of increased APP processing (Mattsson et al., 2016), which is associated with increased interneuronal βCTF and Aβ42, while in others it can occur as a secondary consequence of reduced extracellular clearance (Castellano et al., 2011) and increased neuronal uptake.
Evidence suggests that the intraneuronal accumulation of APP fragments, either βCTF and/or Aβ42, are toxic to the endosome and linked to endosomal trafficking (Fig. 1). There is near consensus that it is in the endosomal membrane of neurons where BACE1 is found and where APP is most likely to be cleaved by BACE1 (Small and Gandy, 2006). Although still debated, most studies suggest that βCTF is also commonly cleaved in the endosome (Kaether et al., 2006; Sannerud et al., 2016; Small and Gandy, 2006), liberating Aβ into the lumen of endosomes from where it can be secreted to the extracellular space. In scenarios where there is reduced clearance of extracellular Aβ42, that species can secondarily gain access to the endosomal lumen by neuronal endocytosis (e.g., (Kanekiyo et al., 2013).
What are the lines of evidence suggesting that the endosome is the site where APP fragments are likely to confer their intracellular toxicity? Indirect evidence comes from the fact that the endosome is the intraneuronal location where APP fragments typically accumulate, and it is in enlarged endosomes where Aβ is typically found (Cataldo et al., 2004). More direct evidence comes from (1) studies in cell culture and animal models showing that the intracellular accumulation of βCTF can cause endosomal enlargement (Jiang et al., 2010; Xu et al., 2016); and (2) a seminal study by the laboratory of the late Sue Lindquist that established that intracellular Aβ42 toxicity is differentially linked to endosomal trafficking (Treusch et al., 2011). Together, therefore, evidence exists linking intracellular APP fragments to endosomal traffic jams. Unclear, however, are the precise mechanisms for how APP fragments cause endosomal dysfunction. βCTF has been proposed to do so by disrupting membrane permeability (Jiang et al., 2010) and this might apply to intracellular Aβ42 as well. Nevertheless, and irrespective of the underlying mechanism, the Lindquist study suggests that endosomal trafficking genes, some linked to AD, are the dominant class that differentially regulates intracellular Aβ42 toxicity.
Taken together, as illustrated in the model (Fig. 1), endosomal traffic jams can, at least in part, be driven by the intracellular accumulation of APP fragments, either βCTF and/or Aβ42.
ENDOSOMAL TRAFFIC JAMS CAN OCCUR AS AN UPSTREAM EVENT
The identification of the ‘endosomal trafficking’ class of genes as being strongly linked to Alzheimer’s disease establishes that endosomal traffic jams can in principle occur as a primary event in AD, upstream to the accumulation of intracellular APP fragments (Fig. 1). The genes that best represent this class are SORL1, BIN1, PICALM, and CD2AP.
SORL1 in particular has been extensively investigated and validated. In contrast to other hits from gene-wide association studies, which typically confer a small increase in AD risk, SORL1 variants have been found that confer a five-fold risk (Vardarajan et al., 2015; Verheijen et al., 2016), on par with APOE4 carriers. More importantly, recent studies suggest that rare SORL1 mutations are actually causal mutations (Pottier et al., 2012; Vardarajan et al., 2015), akin to the autosomal-dominant mutations in APP or PSENs (Holstege et al., 2017).
An in-depth analysis of the studies that have investigated the function of the encoded proteins of this family of genes suggests that they affect the balance of membrane traffic into the early endosome, but even more so out of it. In general, there are three primary trafficking routes out of the early endosome (Fig. 1): the ‘recycling’ pathway, which delivers cargo directly to the cell surface, or via an intermediate endocytic organelle, the recycling endosome; the ‘retrograde’ pathway, which delivers cargo from the early endosome back to the trans-Golgi network; and the ‘degradation’ pathway, where cargo in the early endosome is delivered to intraluminal vesicles (ILVs), a first step by which that cargo is sorted for lysosomal degradation.
These trafficking outflow pathways have been linked genetically to AD. SORL1 (also called SORLA or LR11) is a member of a family Vps10-containing receptors that are trafficked by retromer (Rogaeva et al., 2007; Small and Gandy, 2006; Small et al., 2005), a multi-modular protein assembly that transports cargo out of the early endosome via the recycling and retrograde pathways (Small and Petsko, 2015). SORL1 has also been shown to traffic cargo via the third degradation pathway, in a retromer-independent manner (Dumanis et al., 2015). BIN1 functions in trafficking cargo out of the early endosome via the recycling pathway back to the cell surface (Pant et al., 2009), and CD2AP appears to mediate a primary sorting step in trafficking cargo out of the early endosome down the degradation pathway (Cormont et al., 2003). While most studies suggest that the dominant function of PICALM is to regulate traffic into the endosome (Xu et al., 2015), some studies have suggested that it also plays a role in endosomal recycling (Matsudaira et al., 2015; Petralia and Yao, 2007). We consider it pathogenically informative that these genes all converge on a single and specific intracellular organelle, the early endosome, and not the many other compartments of the endocytic system—i.e., the late endosome, the multivesicular body, or the lysosome.
The conclusion that disease-associated variants in this class of AD-linked genes directly cause endosomal traffic jams is supported by studies that have shown that primary alterations in retromer (Bhalla et al., 2012), SORL1 (Offe et al., 2006), BIN1 (Pant et al., 2009), and CD2AP (Cormont et al., 2003) all cause endosomal enlargements. The fact that some SORL1 variants are causal mutations (Holstege et al., 2017), on par with mutations in APP and PSENs, provides the strongest evidence that endosomal traffic jams can act as upstream drivers of AD pathogenesis.
At the same time, however, this class of genes has a secondary consequence of increasing intracellular APP fragments by regulating the levels of APP and/or BACE1 in the early endosome (Dumanis et al., 2015; Fjorback et al., 2012; Miyagawa et al., 2016; Ubelmann et al., 2016) and APP’s amyloidogenic cleavage (Andersen et al., 2005; Dumanis et al., 2015; Fjorback et al., 2012; Miyagawa et al., 2016; Offe et al., 2006; Rogaeva et al., 2007; Thomas et al., 2016; Ubelmann et al., 2016; Xiao et al., 2012) (Fig. 1). Additionally, SORL1 can also directly bind intra-endosomal Aβ (Dumanis et al., 2015), and thus further regulate intracellular Aβ42 levels by diverting this peptide toward the degradation pathway (Caglayan et al., 2014; Dumanis et al., 2015).
Taken together, as illustrated in the model (Fig. 1), endosomal trafficking genes support the proposed principle that endosomal traffic jams can occur as an upstream pathogenic event, which as a secondary consequence can lead to accumulation of intracellular APP fragments. Since late-onset AD is driven by a complex interplay of genes and environmental risk factors, we assume that besides genetics, other risk factors can also affect endosomal trafficking. For example, type II diabetes is one of the strongest risk factors for AD, and studies in animal models have shown how serological defects in this diabetes can cause retromer deficiencies in the hippocampus (Morabito et al., 2014).
Whether driven by genes or by the environment, the accumulation of intracellular APP fragments caused by defects in endosomal trafficking can in turn exacerbate endosomal traffic jams, as reviewed above. The model therefore proposes a feedback loop between traffic jams and intracellular amyloid, a vicious cycle that we believe is critical in the disease.
OTHER GENES AND THEIR LINKS TO ENDOSOMAL TRAFFICKING
The APOE4 allele best represents the class of cholesterol metabolism genes associated with AD, and TREM2 mutations best exemplify the class of immune response genes, both in terms of genetic effect size and the mechanistic insight about their function (Guerreiro et al., 2013; Karch and Goate, 2015; Naj et al., 2017). While there are still outstanding questions, the dominant view is that one main effect of both genes is to reduce the clearance of extracellular Aβ (Colonna and Wang, 2016; Kim et al., 1998).
Extracellular Aβ is known to be endocytosed into neurons (Dafnis et al., 2016; Kanekiyo et al., 2013), and an increase in the concentration of extracellular Aβ will likely cause a secondary increase in its intracellular levels. Indeed, studies have shown that compared to other APOE alleles, APOE4 increases the intracellular uptake of Aβ in neurons (Dafnis et al., 2016), and as reviewed above intracellular Aβ has been linked to endosomal trafficking (Treusch et al., 2011). Consistent with this formulation, postmortem studies have observed that the APOE4 genotype worsens the enlargement of endosomes in the neurons of AD brains, in regions relatively free of extracellular amyloid plaque deposition (Cataldo et al., 2000).
Interestingly, a similar association between APOE4 genotype and enlarged endosomes has been observed in ischemic brains (McColl et al., 2003), suggesting that APOE4 might be linked to endosomal traffic jams independent of amyloid. Mechanistic support for this interpretation comes from lipoprotein and cholesterol metabolism studies in non-neuronal cells that have investigated the differential effect of APOE vs. other apolipoproteins, or the effects of the APOE3 allele vs. APOE4. Compared to other apolipoproteins, APOE is more likely to be trafficked through the endosomal recycling pathway (Heeren et al., 2004), while compared to APOE3, APOE4 interferes with endosomal recycling (Heeren et al., 2006; Heeren et al., 2004). This differential effect of APOE4 vs. APOE3 has been extended into neurons, showing that APOE4 reduces endosomal recycling of cargo to the neuronal cell surface (Chen et al., 2010; DeKroon and Armati, 2001). Another study also suggested that APOE4 might accelerate endocytosis into the endosome (Ye et al., 2005). Since APOE4 exemplifies the class of cholesterol metabolism genes, it is noteworthy that altering the cholesterol levels of neurons (Marquer et al., 2014), or cholesterol transport within neurons (Jin et al., 2004), induces enlarged endosomes, reflective of putative endosomal traffic jams.
Taken together, as illustrated in the model, the class of cholesterol metabolism genes is proposed to link to endosomal traffic jams through two pathways. The first, which we consider the dominant link, is by affecting extracellular Aβ. The second is by directly affecting the trafficking into endosomes, and even more likely, out of them.
ENDOSOMAL TRAFFIC JAMS AND DOWNSTREAM PATHOPHYSIOLOGY
Synaptic dysfunction in AD is now understood to be an early manifestation of neurotoxicity (Selkoe, 2002). A range of studies in postmortem AD brains (Yasuda et al., 1995), animal models and cell culture (Guntupalli et al., 2016; Hsieh et al., 2006) suggest that a reduction in glutamate receptors is a key early feature of synaptic dysfunction in the disease. Notably, one of the most important functions of the neuronal early endosome is its role in glutamate receptor recycling. At the early endosome, endocytosed receptors are diverted away from the degradation pathway, and transported to the recycling endosome (Fig. 2). From there, they are trafficked back to the postsynaptic surface (Ehlers, 2000). Jamming outflow from the early endosome can lead to a reduction in glutamate receptor recycling, independent of amyloid, as shown in the case of retromer deficiency (Choy et al., 2014). Interestingly, APOE4 has also been shown to cause a reduction in glutamate receptor recycling (Chen et al., 2010), supporting the possibility, discussed above, that APOE4 might be part of the class of endosomal trafficking genes. Thus, endosomal traffic jams can act as final common pathway mediating synaptic toxicity by reducing glutamate receptors at the cell surface of neurons (Fig. 2).
Figure 2. Endosomal traffic jams can mediate Alzheimer’s pathophysiology.

As outlined in the flow diagram (upper panel) and illustrated in the figure (lower panel) two disease-relevant pathophysiological consequences occur in the setting of traffic jams:
A reduction of glutamate receptor recycling. Traffic jams in the early endosome reduces the transport of glutamate receptors to the postsynaptic surface of neurons, via recycling endosomes, impairing synaptic health.
An Increase in pathogenic exosomes. Traffic jams in the early endosome either increases the number of intraluminal vesicles (ILVs) in the multivesicular body thereby increasing the number of released exosomes; or, increases the content of exosomes with, for example, βCTF, Aβ or tau. Either increasing exosomal number or increasing exosomal content can accelerate anatomical spread of disease.
Another pathophysiology of AD that might be mediated by endosomal traffic jams is ‘trans-synaptic spread’. Exosomes have been proposed as a likely candidate to act as a carrier of amyloid (Rajendran et al., 2006) and tau as it spreads from a region to its neighbor (Polanco et al., 2016). Exosomes are intraluminar vesicles (ILVs) observed in early endosomes that progressively accumulate in the endosomal lumen as this compartment matures into a multivesicular endosome (MVB). Exosomes are formed by the invagination and scission of the limiting membrane of the endosome towards the endosomal lumen. These nanoparticles, of 50 to 150nm in diameter, can be released into the extracellular environment upon the fusion of the MVB with the plasma membrane. In neurodegenerative diseases they are capable of spreading and delivering pathogenic proteins to neighboring cells (Fig. 2).
Because of the site and manner of their production, exosomes contain proteins that accumulate on the endosomal membrane, while their lumen contains engulfed cytosolic molecules. Upon APP’s cleavage by BACE1, βCTF accumulates in the limiting membrane of the early endosome, before being sorted into ILVs (Perez-Gonzalez et al., 2012; Sharples et al., 2008). Endosomal jamming of APP and/or BACE1 leads to an accumulation of βCTF in endosomal membranes, and as shown for example in retromer deficiency (Sullivan et al., 2011), can then lead to an increase in exosomal βCTF (Sullivan et al., 2011). An interesting recent observation is that tau can also be found in exosomes, both in patients (Fiandaca et al., 2015; Saman et al., 2012) and animal models of AD (Polanco et al., 2016). This likely occurs because cytosolic tau, both full-length and its various processed subspecies, are incorporated into ILVs as they bud off from the limiting membrane of the endosome, ending up in the lumen of secreted exosomes.
Besides mediating synaptic toxicity and exosomal spread of disease, we briefly note that some studies have linked retromer trafficking (Small and Petsko, 2015), BIN1 (Calafate et al., 2016; Chapuis et al., 2013; Zhou et al., 2014), and CD2AP (Shulman et al., 2014) to tau pathology, potentially independent of amyloid, and retromer has also been linked to immune response genes, such as TREM2, that are phagocytic receptors trafficked in microglia (Lucin et al., 2013; Small and Petsko, 2015; Sole-Domenech et al., 2016; Yin et al., 2016). A detailed discussion of these additional pathophysiological links is considered outside the scope of this Opinion.
THERAPUTIC IMPLICATIONS
A number of years ago Gunnar Gouras’ laboratory made the seminal observation that Aβ peptides can accumulate intraneuronally (Gouras et al., 2010; Gouras et al., 2000), and more specifically within the early endosomes. While initially met with resistance, this observation is now supported by an overwhelming body of literature (as reviewed in (Gouras et al., 2010). Here too genetics has held sway, and the observation is supported by the fact that genetic defects in both LOAD and autosomal-dominant AD can cause a primary accumulation of endosomal βCTF and Aβ42.
A main component of our model is linking intracellular amyloid to what we consider a pathogenic hub and driver of disease, endosomal traffic jams, and in so doing places the primary site of amyloid toxicity inside the cell. This represents a shift from the ‘amyloid hypothesis’, which stipulates that it is amyloid deposition in the extracellular space that is the primary site of neuroxicity (Hardy and Selkoe, 2002). While this might seem like a subtle reformulation of the original hypothesis, it can resolve apparent challenges to the hypothesis and has profound therapeutic implications.
One of the greatest challenges to the amyloid hypothesis and its commitment to extracellular amyloid is the clear anatomical discordance between the distribution of plaque load and evidence for neurotoxicity (e.g, (Altmann et al., 2015). The clearest example of this discordance is in the medial temporal lobe. Recent imaging studies have extended prior observations first suggested by histological studies (Braak and Braak, 1991), documenting that this region typically accumulates near the lowest level of amyloid plaques throughout the cortex (e.g, (Altmann et al., 2015), yet by most indicators experiences the earliest and clearest evidence of neurotoxicity (Altmann et al., 2015; Khan et al., 2014). In fact some studies suggest that there might be an inverse relationship between levels of amyloid plaques and indicators of neurotoxicity (Altmann et al., 2015; Bischof et al., 2016). While the medial temporal lobe has low extracellular amyloid deposition, studies indicate that its neurons accumulate intracellular amyloid (Cataldo et al., 2004; Gouras et al., 2000) providing a better anatomical match to sites of dysfunction, and supporting our assumption that intracellular amyloid acts as a primary neurotoxin.
Why is there an anatomical mismatch between intracellular amyloid and extracellular amyloid plaques? The answer remains a mystery but one possibility is that amyloid plaques act as a sink for toxic but soluble Aβ, and if so it is possible that areas with highest plaque load might be anticipated to have the lowest intracellular amyloid. This idea is difficult to test, but some evidence in its support comes from a detailed analysis in animal models, which has documented an inverse relationship between intraneuronal Aβ and extracellular amyloid plaques (Oddo et al., 2006). But by whichever mechanism, this formulation predicts that amyloid immunotherapy, which likely has its greatest effect on extracellular amyloid plaques, would not necessarily affect intracellular amyloid - or, in the extreme case, by removing the chemical sink might even lead to increases in intracellular amyloid. In either case, this might account for why amyloid immunotherapies have thus far been unsuccessful. A shift in focus from extracellular to intracellular amyloid does suggest that reducing the productions of intracellular APP fragments might be more efficacious. Support for this prediction comes from a protective APP mutant that has been shown to reduce the affinity of that protein to BACE1 (Jonsson et al., 2012), thereby reducing the intracellular production of βCTF and Aβ and reducing the risk for developing LOAD.
The pathogenic model proposed here, however, suggests that interventions that are directly designed to increase flow through the early endosomes might be a better, or at least an alternative, therapeutic approach. Certainly they would be indicated in patients who have primary defects in endosomal trafficking, whether caused by genetics and/or the environment, where reducing intracellular amyloid would be therapeutically insufficient, but potentially for other causes as well. Endosomal enlargement appears to be commonly observed in AD (Cataldo et al., 2000; Israel et al., 2012; Raja et al., 2016), even before the clear evidence of extracellular amyloid accumulation. Therefore, there is reason to believe that endosomal traffic jams occur either as a primary event in many AD cases, or even if secondary occur very early in the disease process, mediating downstream pathophysiology. A recent failure of a clinical trial using a BACE1 inhibitor (Mullard, 2017), supports the formulation that drugs that simply reduce intracellular amyloid might miss the primary target or simply be given too late.
Proof-of-principle for targeting endosomal traffic jams by agents that increase endosomal flow exists in cell culture. Pharmacological chaperones that increase retromer levels have been shown to increase the flow of both SORL1 and APP out of the early endosome (Mecozzi et al., 2014), and to reduce the neuronal accumulation of βCTF and Aβ.
Concluding Remarks
An emerging goal in the AD field is to unify the four main gene classes identified in the context of late-onset sporadic AD into a single hypothesis (Selkoe and Hardy, 2016). It is interesting to speculate how the dominant hypothesis in AD might have differed if the order of genetic discoveries would have been reversed— i.e., first the discovery of the plethora of genes linked to late-onset sporadic AD, followed by genes underlying Mendelian forms of the disease. We posit that regardless of the historical ordering of these discoveries, a misprocessing of APP would certainly be included as a key biochemical event. However, if the late-onset genes had been discovered first, we believe that endosomal trafficking defects would be considered a central cellular event, around which APP misprocessing and its neurotoxic fragments would then be organized within its intracellular context.
We propose here a reformulation of AD’s pathophysiology, which assigns endosomal traffic jams a central organizing pathophysiological event, rather than extracellular amyloid. We argue that this reformulation reconciles inconsistencies of the amyloid hypothesis, and can help explain the poor record of therapeutic interventions that have emerged from the amyloid hypothesis. We emphasize that this reformulation does not reject the importance of ‘amyloid’. We believe that the genetics of early onset autosomal-dominant disease firmly establishes the importance of incorporating APP processing, and its cleaved fragments, into a unified pathogenic model of AD. As reviewed, however, it seems now clear that APP processing occurs within neurons, where its fragments first accumulate intracellularly and where they are shown to cause endosomal trafficking defects. These fragments are ultimately secreted into the extracellular space, where soluble Aβ can aggregate and accumulate into extracellular amyloid plaques. As we discussed, olgomeric Aβ, when not incorporated into extracellular plaques, is free to be endocytosed into neurons, where it can worsen endosomal traffic jams. According to this formulation, while extracellular amyloid plaques are not the prime drivers of disease, they nevertheless can still act as ‘reporters’ of intracellular pathology and can have high diagnostic and prognostic value.
While additional work is certainly required to test specific components of these competing central events, they are unlikely to be resolved by studies in model systems alone (see Outstanding Questions). Rather, these competing views are best tested in patients. The distinction between intracellular vs. extracellular amyloid might be tested in ongoing trials, in patients who receive BACE1 inhibitors on the one hand, or immunotherapies on the other. However, future studies that administer agents that directly target endosomal traffic jams are required to test the central pathogenic hub proposed here.
Outstanding Questions.
Biomarkers of endosomal traffic jams are needed, to ask: What is the precise prevalence and time course of endosomal traffic jams?
Will patients with genetic or biomarker evidence of endosomal traffic jams be more resistant to amyloid or tau therapies?
Neuroimaging biomarkers of intracellular amyloid are needed, to ask: In contrast to extracellular amyloid, do maps of intracellular amyloid better overlap with sites of brain dysfunction?
Would a manipulation that causes regional endosomal traffic jams accelerate trans-synaptic spread of the disease?
Trends Box.
While defects in intracellular trafficking have long been suspected in Alzheimer’s disease, recent genetic findings localize the defect, specifically, to trafficking in and out of endosomes.
Endosomal traffic jams can be caused directly, for example by endosomal genetic defects, or secondarily, by any defect that increased intracellular amyloid.
Endosomal traffic jams can mediate downstream toxicity, by reducing glutamate receptor recycling, leading to synaptic dysfunction, and potentially by increasing trans-synaptic spread by altering the content of exosomes.
Endosomal traffic jams can be considered a validated ‘cell biological’ target for Alzheimer’s drug discovery.
Because endosomal traffic jams occur very early in the disease, and might occur in an amyloid-independent manner, we argue that drugs that unjam the endosome carry high therapeutic promise.
Acknowledgments
This study was partly supported by NIH R01 grants AG034618 and AG035015.
Footnotes
Disclaimer statement
Scott Small and Gregory Petsko are on the advisory board of MeiraGTx. Scott Small is also on the Scientific Advisory Board of Denali Therapeutics and Janssen Pharmaceutical. Gregory Petsko has consulting arrangements with Amicus Therapeutics, Proclara Biosciences and QR Pharma, Inc.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Altmann A, Ng B, Landau SM, Jagust WJ, Greicius MD Alzheimer’s Disease Neuroimaging I. Regional brain hypometabolism is unrelated to regional amyloid plaque burden. Brain. 2015;138:3734–3746. doi: 10.1093/brain/awv278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen OM, Reiche J, Schmidt V, Gotthardt M, Spoelgen R, Behlke J, von Arnim CA, Breiderhoff T, Jansen P, Wu X, et al. Neuronal sorting protein-related receptor sorLA/LR11 regulates processing of the amyloid precursor protein. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:13461–13466. doi: 10.1073/pnas.0503689102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhalla A, Vetanovetz CP, Morel E, Chamoun Z, Di Paolo G, Small SA. The location and trafficking routes of the neuronal retromer and its role in amyloid precursor protein transport. Neurobiol Dis. 2012;47:126–134. doi: 10.1016/j.nbd.2012.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischof GN, Jessen F, Fliessbach K, Dronse J, Hammes J, Neumaier B, Onur O, Fink GR, Kukolja J, Drzezga A, et al. Impact of tau and amyloid burden on glucose metabolism in Alzheimer’s disease. Annals of clinical and translational neurology. 2016;3:934–939. doi: 10.1002/acn3.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol (Berl) 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- Caglayan S, Takagi-Niidome S, Liao F, Carlo AS, Schmidt V, Burgert T, Kitago Y, Fuchtbauer EM, Fuchtbauer A, Holtzman DM, et al. Lysosomal sorting of amyloid-beta by the SORLA receptor is impaired by a familial Alzheimer’s disease mutation. Science translational medicine. 2014;6:223ra220. doi: 10.1126/scitranslmed.3007747. [DOI] [PubMed] [Google Scholar]
- Calafate S, Flavin W, Verstreken P, Moechars D. Loss of Bin1 Promotes the Propagation of Tau Pathology. Cell reports. 2016;17:931–940. doi: 10.1016/j.celrep.2016.09.063. [DOI] [PubMed] [Google Scholar]
- Castellano JM, Kim J, Stewart FR, Jiang H, DeMattos RB, Patterson BW, Fagan AM, Morris JC, Mawuenyega KG, Cruchaga C, et al. Human apoE isoforms differentially regulate brain amyloid-beta peptide clearance. Science translational medicine. 2011;3:89ra57. doi: 10.1126/scitranslmed.3002156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldo AM, Petanceska S, Terio NB, Peterhoff CM, Durham R, Mercken M, Mehta PD, Buxbaum J, Haroutunian V, Nixon RA. Abeta localization in abnormal endosomes: association with earliest Abeta elevations in AD and Down syndrome. Neurobiol Aging. 2004;25:1263–1272. doi: 10.1016/j.neurobiolaging.2004.02.027. [DOI] [PubMed] [Google Scholar]
- Cataldo AM, Peterhoff CM, Troncoso JC, Gomez-Isla T, Hyman BT, Nixon RA. Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer’s disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. The American journal of pathology. 2000;157:277–286. doi: 10.1016/s0002-9440(10)64538-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapuis J, Hansmannel F, Gistelinck M, Mounier A, Van Cauwenberghe C, Kolen KV, Geller F, Sottejeau Y, Harold D, Dourlen P, et al. Increased expression of BIN1 mediates Alzheimer genetic risk by modulating tau pathology. Molecular psychiatry. 2013;18:1225–1234. doi: 10.1038/mp.2013.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Durakoglugil MS, Xian X, Herz J. ApoE4 reduces glutamate receptor function and synaptic plasticity by selectively impairing ApoE receptor recycling. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:12011–12016. doi: 10.1073/pnas.0914984107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi RJ, Harrison MS, Burd CG. Biogenesis of endosome-derived transport carriers. Cellular and molecular life sciences: CMLS. 2015;72:3441–3455. doi: 10.1007/s00018-015-1935-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choy RW, Park M, Temkin P, Herring BE, Marley A, Nicoll RA, von Zastrow M. Retromer mediates a discrete route of local membrane delivery to dendrites. Neuron. 2014;82:55–62. doi: 10.1016/j.neuron.2014.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colonna M, Wang Y. TREM2 variants: new keys to decipher Alzheimer disease pathogenesis. Nature reviews Neuroscience. 2016;17:201–207. doi: 10.1038/nrn.2016.7. [DOI] [PubMed] [Google Scholar]
- Cormont M, Meton I, Mari M, Monzo P, Keslair F, Gaskin C, McGraw TE, Le Marchand-Brustel Y. CD2AP/CMS regulates endosome morphology and traffic to the degradative pathway through its interaction with Rab4 and c-Cbl. Traffic. 2003;4:97–112. doi: 10.1034/j.1600-0854.2003.40205.x. [DOI] [PubMed] [Google Scholar]
- Dafnis I, Argyri L, Sagnou M, Tzinia A, Tsilibary EC, Stratikos E, Chroni A. The ability of apolipoprotein E fragments to promote intraneuronal accumulation of amyloid beta peptide 42 is both isoform and size-specific. Scientific reports. 2016;6:30654. doi: 10.1038/srep30654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Strooper B. Loss-of-function presenilin mutations in Alzheimer disease. Talking Point on the role of presenilin mutations in Alzheimer disease. EMBO reports. 2007;8:141–146. doi: 10.1038/sj.embor.7400897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeKroon RM, Armati PJ. The endosomal trafficking of apolipoprotein E3 and E4 in cultured human brain neurons and astrocytes. Neurobiol Dis. 2001;8:78–89. doi: 10.1006/nbdi.2000.0362. [DOI] [PubMed] [Google Scholar]
- Dumanis SB, Burgert T, Caglayan S, Fuchtbauer A, Fuchtbauer EM, Schmidt V, Willnow TE. Distinct Functions for Anterograde and Retrograde Sorting of SORLA in Amyloidogenic Processes in the Brain. J Neurosci. 2015;35:12703–12713. doi: 10.1523/JNEUROSCI.0427-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers MD. Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron. 2000;28:511–525. doi: 10.1016/s0896-6273(00)00129-x. [DOI] [PubMed] [Google Scholar]
- Fiandaca MS, Kapogiannis D, Mapstone M, Boxer A, Eitan E, Schwartz JB, Abner EL, Petersen RC, Federoff HJ, Miller BL, et al. Identification of preclinical Alzheimer’s disease by a profile of pathogenic proteins in neurally derived blood exosomes: A case-control study. Alzheimer’s & dementia: the journal of the Alzheimer’s Association. 2015;11:600–607. e601. doi: 10.1016/j.jalz.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fjorback AW, Seaman M, Gustafsen C, Mehmedbasic A, Gokool S, Wu C, Militz D, Schmidt V, Madsen P, Nyengaard JR, et al. Retromer binds the FANSHY sorting motif in SorLA to regulate amyloid precursor protein sorting and processing. J Neurosci. 2012;32:1467–1480. doi: 10.1523/JNEUROSCI.2272-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouras GK, Tampellini D, Takahashi RH, Capetillo-Zarate E. Intraneuronal beta-amyloid accumulation and synapse pathology in Alzheimer’s disease. Acta neuropathologica. 2010;119:523–541. doi: 10.1007/s00401-010-0679-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouras GK, Tsai J, Naslund J, Vincent B, Edgar M, Checler F, Greenfield JP, Haroutunian V, Buxbaum JD, Xu H, et al. Intraneuronal Abeta42 accumulation in human brain. The American journal of pathology. 2000;156:15–20. doi: 10.1016/s0002-9440(10)64700-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerreiro R, Bras J, Hardy J. SnapShot: genetics of Alzheimer’s disease. Cell. 2013;155:968–968. e961. doi: 10.1016/j.cell.2013.10.037. [DOI] [PubMed] [Google Scholar]
- Guntupalli S, Widagdo J, Anggono V. Amyloid-beta-Induced Dysregulation of AMPA Receptor Trafficking. Neural plasticity. 2016;2016:3204519. doi: 10.1155/2016/3204519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Heeren J, Beisiegel U, Grewal T. Apolipoprotein E recycling: implications for dyslipidemia and atherosclerosis. Arteriosclerosis, thrombosis, and vascular biology. 2006;26:442–448. doi: 10.1161/01.ATV.0000201282.64751.47. [DOI] [PubMed] [Google Scholar]
- Heeren J, Grewal T, Laatsch A, Becker N, Rinninger F, Rye KA, Beisiegel U. Impaired recycling of apolipoprotein E4 is associated with intracellular cholesterol accumulation. J Biol Chem. 2004;279:55483–55492. doi: 10.1074/jbc.M409324200. [DOI] [PubMed] [Google Scholar]
- Holstege H, van der Lee SJ, Hulsman M, Wong TH, van Rooij JG, Weiss M, Louwersheimer E, Wolters FJ, Amin N, Uitterlinden AG, et al. Characterization of pathogenic SORL1 genetic variants for association with Alzheimer’s disease: a clinical interpretation strategy. European journal of human genetics: EJHG. 2017 doi: 10.1038/ejhg.2017.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh H, Boehm J, Sato C, Iwatsubo T, Tomita T, Sisodia S, Malinow R. AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron. 2006;52:831–843. doi: 10.1016/j.neuron.2006.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israel MA, Yuan SH, Bardy C, Reyna SM, Mu Y, Herrera C, Hefferan MP, Van Gorp S, Nazor KL, Boscolo FS, et al. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature. 2012;482:216–220. doi: 10.1038/nature10821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Mullaney KA, Peterhoff CM, Che S, Schmidt SD, Boyer-Boiteau A, Ginsberg SD, Cataldo AM, Mathews PM, Nixon RA. Alzheimer’s-related endosome dysfunction in Down syndrome is Abeta-independent but requires APP and is reversed by BACE-1 inhibition. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:1630–1635. doi: 10.1073/pnas.0908953107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin LW, Shie FS, Maezawa I, Vincent I, Bird T. Intracellular accumulation of amyloidogenic fragments of amyloid-beta precursor protein in neurons with Niemann-Pick type C defects is associated with endosomal abnormalities. The American journal of pathology. 2004;164:975–985. doi: 10.1016/s0002-9440(10)63185-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson T, Atwal JK, Steinberg S, Snaedal J, Jonsson PV, Bjornsson S, Stefansson H, Sulem P, Gudbjartsson D, Maloney J, et al. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature. 2012;488:96–99. doi: 10.1038/nature11283. [DOI] [PubMed] [Google Scholar]
- Kaether C, Schmitt S, Willem M, Haass C. Amyloid precursor protein and Notch intracellular domains are generated after transport of their precursors to the cell surface. Traffic. 2006;7:408–415. doi: 10.1111/j.1600-0854.2006.00396.x. [DOI] [PubMed] [Google Scholar]
- Kanekiyo T, Cirrito JR, Liu CC, Shinohara M, Li J, Schuler DR, Shinohara M, Holtzman DM, Bu G. Neuronal clearance of amyloid-beta by endocytic receptor LRP1. J Neurosci. 2013;33:19276–19283. doi: 10.1523/JNEUROSCI.3487-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karch CM, Goate AM. Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biological psychiatry. 2015;77:43–51. doi: 10.1016/j.biopsych.2014.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy MJ, Ehlers MD. Organelles and trafficking machinery for postsynaptic plasticity. Annual review of neuroscience. 2006;29:325–362. doi: 10.1146/annurev.neuro.29.051605.112808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan UA, Liu L, Provenzano FA, Berman DE, Profaci CP, Sloan R, Mayeux R, Duff KE, Small SA. Molecular drivers and cortical spread of lateral entorhinal cortex dysfunction in preclinical Alzheimer’s disease. Nature neuroscience. 2014;17:304–311. doi: 10.1038/nn.3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DH, Inagaki Y, Suzuki T, Ioka RX, Yoshioka SZ, Magoori K, Kang MJ, Cho Y, Nakano AZ, Liu Q, et al. A new low density lipoprotein receptor related protein, LRP5, is expressed in hepatocytes and adrenal cortex, and recognizes apolipoprotein E. J Biochem. 1998;124:1072–1076. doi: 10.1093/oxfordjournals.jbchem.a022223. [DOI] [PubMed] [Google Scholar]
- Li N, Liu K, Qiu Y, Ren Z, Dai R, Deng Y, Qing H. Effect of Presenilin Mutations on APP Cleavage; Insights into the Pathogenesis of FAD. Frontiers in aging neuroscience. 2016;8:51. doi: 10.3389/fnagi.2016.00051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucin KM, O’Brien CE, Bieri G, Czirr E, Mosher KI, Abbey RJ, Mastroeni DF, Rogers J, Spencer B, Masliah E, et al. Microglial beclin 1 regulates retromer trafficking and phagocytosis and is impaired in Alzheimer’s disease. Neuron. 2013;79:873–886. doi: 10.1016/j.neuron.2013.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquer C, Laine J, Dauphinot L, Hanbouch L, Lemercier-Neuillet C, Pierrot N, Bossers K, Le M, Corlier F, Benstaali C, et al. Increasing membrane cholesterol of neurons in culture recapitulates Alzheimer’s disease early phenotypes. Mol Neurodegener. 2014;9:60. doi: 10.1186/1750-1326-9-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsudaira T, Niki T, Taguchi T, Arai H. Transport of the cholera toxin B-subunit from recycling endosomes to the Golgi requires clathrin and AP-1. Journal of cell science. 2015;128:3131–3142. doi: 10.1242/jcs.172171. [DOI] [PubMed] [Google Scholar]
- Mattsson N, Insel PS, Palmqvist S, Stomrud E, van Westen D, Minthon L, Zetterberg H, Blennow K, Hansson O. Increased amyloidogenic APP processing in APOE varepsilon4-negative individuals with cerebral beta-amyloidosis. Nature communications. 2016;7:10918. doi: 10.1038/ncomms10918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McColl BW, Graham DI, Weir CJ, White F, Horsburgh K. Endocytic pathway alterations in human hippocampus after global ischemia and the influence of APOE genotype. The American journal of pathology. 2003;162:273–281. doi: 10.1016/S0002-9440(10)63818-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mecozzi VJ, Berman DE, Simoes S, Vetanovetz C, Awal MR, Patel VM, Schneider RT, Petsko GA, Ringe D, Small SA. Pharmacological chaperones stabilize retromer to limit APP processing. Nat Chem Biol. 2014 doi: 10.1038/nchembio.1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyagawa T, Ebinuma I, Morohashi Y, Hori Y, Young Chang M, Hattori H, Maehara T, Yokoshima S, Fukuyama T, Tsuji S, et al. BIN1 regulates BACE1 intracellular trafficking and amyloid-beta production. Human molecular genetics. 2016;25:2948–2958. doi: 10.1093/hmg/ddw146. [DOI] [PubMed] [Google Scholar]
- Morabito MV, Berman DE, Schneider RT, Zhang Y, Leibel RL, Small SA. Hyperleucinemia causes hippocampal retromer deficiency linking diabetes to Alzheimer’s disease. Neurobiol Dis. 2014;65:188–192. doi: 10.1016/j.nbd.2013.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullard A. BACE inhibitor bust in Alzheimer trial. Nature reviews Drug discovery. 2017;16:155. doi: 10.1038/nrd.2017.43. [DOI] [PubMed] [Google Scholar]
- Naj AC, Schellenberg GD Alzheimer’s Disease Genetics C. Genomic variants, genes, and pathways of Alzheimer’s disease: An overview. American journal of medical genetics Part B, Neuropsychiatric genetics: the official publication of the International Society of Psychiatric Genetics. 2017;174:5–26. doi: 10.1002/ajmg.b.32499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Smith IF, Green KN, LaFerla FM. A dynamic relationship between intracellular and extracellular pools of Abeta. The American journal of pathology. 2006;168:184–194. doi: 10.2353/ajpath.2006.050593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Offe K, Dodson SE, Shoemaker JT, Fritz JJ, Gearing M, Levey AI, Lah JJ. The lipoprotein receptor LR11 regulates amyloid beta production and amyloid precursor protein traffic in endosomal compartments. J Neurosci. 2006;26:1596–1603. doi: 10.1523/JNEUROSCI.4946-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pant S, Sharma M, Patel K, Caplan S, Carr CM, Grant BD. AMPH-1/Amphiphysin/Bin1 functions with RME-1/Ehd1 in endocytic recycling. Nature cell biology. 2009;11:1399–1410. doi: 10.1038/ncb1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Gonzalez R, Gauthier SA, Kumar A, Levy E. The exosome secretory pathway transports amyloid precursor protein carboxyl-terminal fragments from the cell into the brain extracellular space. J Biol Chem. 2012;287:43108–43115. doi: 10.1074/jbc.M112.404467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petralia RS, Yao PJ. AP180 and CALM in the developing hippocampus: expression at the nascent synapse and localization to trafficking organelles. The Journal of comparative neurology. 2007;504:314–327. doi: 10.1002/cne.21454. [DOI] [PubMed] [Google Scholar]
- Pfeffer SR. A nexus for receptor recycling. Nature cell biology. 2013;15:446–448. doi: 10.1038/ncb2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polanco JC, Scicluna BJ, Hill AF, Gotz J. Extracellular Vesicles Isolated from the Brains of rTg4510 Mice Seed Tau Protein Aggregation in a Threshold-dependent Manner. J Biol Chem. 2016;291:12445–12466. doi: 10.1074/jbc.M115.709485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pottier C, Hannequin D, Coutant S, Rovelet-Lecrux A, Wallon D, Rousseau S, Legallic S, Paquet C, Bombois S, Pariente J, et al. High frequency of potentially pathogenic SORL1 mutations in autosomal dominant early-onset Alzheimer disease. Molecular psychiatry. 2012;17:875–879. doi: 10.1038/mp.2012.15. [DOI] [PubMed] [Google Scholar]
- Raja WK, Mungenast AE, Lin YT, Ko T, Abdurrob F, Seo J, Tsai LH. Self-Organizing 3D Human Neural Tissue Derived from Induced Pluripotent Stem Cells Recapitulate Alzheimer’s Disease Phenotypes. PLoS One. 2016;11:e0161969. doi: 10.1371/journal.pone.0161969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendran L, Honsho M, Zahn TR, Keller P, Geiger KD, Verkade P, Simons K. Alzheimer’s disease beta-amyloid peptides are released in association with exosomes. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:11172–11177. doi: 10.1073/pnas.0603838103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogaeva E, Meng Y, Lee JH, Gu Y, Kawarai T, Zou F, Katayama T, Baldwin CT, Cheng R, Hasegawa H, et al. The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat Genet. 2007 doi: 10.1038/ng1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saman S, Kim W, Raya M, Visnick Y, Miro S, Saman S, Jackson B, McKee AC, Alvarez VE, Lee NC, et al. Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J Biol Chem. 2012;287:3842–3849. doi: 10.1074/jbc.M111.277061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sannerud R, Esselens C, Ejsmont P, Mattera R, Rochin L, Tharkeshwar AK, De Baets G, De Wever V, Habets R, Baert V, et al. Restricted Location of PSEN2/gamma-Secretase Determines Substrate Specificity and Generates an Intracellular Abeta Pool. Cell. 2016;166:193–208. doi: 10.1016/j.cell.2016.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO molecular medicine. 2016;8:595–608. doi: 10.15252/emmm.201606210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharples RA, Vella LJ, Nisbet RM, Naylor R, Perez K, Barnham KJ, Masters CL, Hill AF. Inhibition of gamma-secretase causes increased secretion of amyloid precursor protein C-terminal fragments in association with exosomes. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2008;22:1469–1478. doi: 10.1096/fj.07-9357com. [DOI] [PubMed] [Google Scholar]
- Shulman JM, Imboywa S, Giagtzoglou N, Powers MP, Hu Y, Devenport D, Chipendo P, Chibnik LB, Diamond A, Perrimon N, et al. Functional screening in Drosophila identifies Alzheimer’s disease susceptibility genes and implicates Tau-mediated mechanisms. Human molecular genetics. 2014;23:870–877. doi: 10.1093/hmg/ddt478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small SA, Gandy S. Sorting through the cell biology of Alzheimer’s disease: intracellular pathways to pathogenesis. Neuron. 2006;52:15–31. doi: 10.1016/j.neuron.2006.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small SA, Kent K, Pierce A, Leung C, Kang MS, Okada H, Honig L, Vonsattel JP, Kim TW. Model-guided microarray implicates the retromer complex in Alzheimer’s disease. Annals of neurology. 2005;58:909–919. doi: 10.1002/ana.20667. [DOI] [PubMed] [Google Scholar]
- Small SA, Petsko GA. Retromer in Alzheimer disease, Parkinson disease and other neurological disorders. Nature reviews Neuroscience. 2015;16:126–132. doi: 10.1038/nrn3896. [DOI] [PubMed] [Google Scholar]
- Sole-Domenech S, Cruz DL, Capetillo-Zarate E, Maxfield FR. The endocytic pathway in microglia during health, aging and Alzheimer’s disease. Ageing research reviews. 2016;32:89–103. doi: 10.1016/j.arr.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan CP, Jay AG, Stack EC, Pakaluk M, Wadlinger E, Fine RE, Wells JM, Morin PJ. Retromer disruption promotes amyloidogenic APP processing. Neurobiol Dis. 2011;43:338–345. doi: 10.1016/j.nbd.2011.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas RS, Henson A, Gerrish A, Jones L, Williams J, Kidd EJ. Decreasing the expression of PICALM reduces endocytosis and the activity of beta-secretase: implications for Alzheimer’s disease. BMC neuroscience. 2016;17:50. doi: 10.1186/s12868-016-0288-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treusch S, Hamamichi S, Goodman JL, Matlack KE, Chung CY, Baru V, Shulman JM, Parrado A, Bevis BJ, Valastyan JS, et al. Functional links between Abeta toxicity, endocytic trafficking, and Alzheimer’s disease risk factors in yeast. Science. 2011;334:1241–1245. doi: 10.1126/science.1213210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubelmann F, Burrinha T, Salavessa L, Gomes R, Ferreira C, Moreno N, Guimas Almeida C. Bin1 and CD2AP polarise the endocytic generation of beta-amyloid. EMBO reports. 2016 doi: 10.15252/embr.201642738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vardarajan BN, Zhang Y, Lee JH, Cheng R, Bohm C, Ghani M, Reitz C, Reyes-Dumeyer D, Shen Y, Rogaeva E, et al. Coding mutations in SORL1 and Alzheimer disease. Annals of neurology. 2015;77:215–227. doi: 10.1002/ana.24305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verheijen J, Van den Bossche T, van der Zee J, Engelborghs S, Sanchez-Valle R, Llado A, Graff C, Thonberg H, Pastor P, Ortega-Cubero S, et al. A comprehensive study of the genetic impact of rare variants in SORL1 in European early-onset Alzheimer’s disease. Acta neuropathologica. 2016;132:213–224. doi: 10.1007/s00401-016-1566-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff G, Young JE, Martinez FJ, Buen F, Gore A, Kinaga J, Li Z, Yuan SH, Zhang K, Goldstein LS. The presenilin-1 DeltaE9 mutation results in reduced gamma-secretase activity, but not total loss of PS1 function, in isogenic human stem cells. Cell reports. 2013;5:974–985. doi: 10.1016/j.celrep.2013.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Q, Gil SC, Yan P, Wang Y, Han S, Gonzales E, Perez R, Cirrito JR, Lee JM. Role of phosphatidylinositol clathrin assembly lymphoid-myeloid leukemia (PICALM) in intracellular amyloid precursor protein (APP) processing and amyloid plaque pathogenesis. J Biol Chem. 2012;287:21279–21289. doi: 10.1074/jbc.M111.338376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Tan L, Yu JT. The Role of PICALM in Alzheimer’s Disease. Molecular neurobiology. 2015;52:399–413. doi: 10.1007/s12035-014-8878-3. [DOI] [PubMed] [Google Scholar]
- Xu W, Weissmiller AM, White JA, 2nd, Fang F, Wang X, Wu Y, Pearn ML, Zhao X, Sawa M, Chen S, et al. Amyloid precursor protein-mediated endocytic pathway disruption induces axonal dysfunction and neurodegeneration. The Journal of clinical investigation. 2016;126:1815–1833. doi: 10.1172/JCI82409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda RP, Ikonomovic MD, Sheffield R, Rubin RT, Wolfe BB, Armstrong DM. Reduction of AMPA-selective glutamate receptor subunits in the entorhinal cortex of patients with Alzheimer’s disease pathology: a biochemical study. Brain research. 1995;678:161–167. doi: 10.1016/0006-8993(95)00178-s. [DOI] [PubMed] [Google Scholar]
- Ye S, Huang Y, Mullendorff K, Dong L, Giedt G, Meng EC, Cohen FE, Kuntz ID, Weisgraber KH, Mahley RW. Apolipoprotein (apo) E4 enhances amyloid beta peptide production in cultured neuronal cells: apoE structure as a potential therapeutic target. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:18700–18705. doi: 10.1073/pnas.0508693102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin J, Liu X, He Q, Zhou L, Yuan Z, Zhao S. Vps35-dependent recycling of Trem2 regulates microglial function. Traffic. 2016 doi: 10.1111/tra.12451. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Hayashi I, Wong J, Tugusheva K, Renger JJ, Zerbinatti C. Intracellular clusterin interacts with brain isoforms of the bridging integrator 1 and with the microtubule-associated protein Tau in Alzheimer’s disease. PLoS One. 2014;9:e103187. doi: 10.1371/journal.pone.0103187. [DOI] [PMC free article] [PubMed] [Google Scholar]