

Abstract
Background
Serum amyloid A (SAA) induces inflammation and apoptosis in kidney cells and associates with pathologic changes of diabetic kidney disease (DKD). Higher serum SAA concentrations were previously associated with increased risk of end-stage renal disease (ESRD) and death in persons with type 2 diabetes and advanced DKD. We explored the prognostic value of SAA in American Indians with type 2 diabetes without DKD or with early DKD.
Methods
SAA concentration was measured in serum samples obtained at the start of follow-up. Multivariate proportional hazards models were employed to examine the magnitude of the risk of ESRD or death across tertiles of SAA concentration after adjustment for traditional risk factors. The C statistic was used to assess the additional predictive value of SAA relative to traditional risk factors.
Results
Of 256 participants (mean ± SD glomerular filtration rate [iothalamate]=148±45 ml/min, and median [IQR] urine albumin/creatinine=39 [14–221] mg/g), 76 developed ESRD and 125 died during median follow-up of 15.2 and 15.7 years, respectively. After multivariable proportional hazards regression, participants in the two highest SAA tertiles combined exhibited a 53% lower risk of ESRD (Hazard Ratio [HR]=0.47, 95%CI 0.29–0.78), and a 30% lower risk of death (HR=0.70, 95%CI 0.48–1.02), compared with participants in the lowest SAA tertile, although the lower risk of death was not statistically significant. Addition of SAA to the ESRD model increased the C statistic from 0.814 to 0.815 (P=0.005).
Conclusions
Higher circulating SAA concentration is associated with a reduced risk of ESRD in American Indians with type 2 diabetes.
Keywords: Biomarkers, Diabetes, Mortality, Nephropathy
Introduction
Serum amyloid A (SAA) is a potent pro-inflammatory protein produced in the liver, fat, and kidneys. Local production in the kidneys occurs predominantly in podocytes, mesangial cells [1], and proximal tubular cells [2, 3]. SAA promotes expression of inflammatory cytokines by fibroblasts [4], macrophages [5], and podocytes [6]. Extensive SAA protein deposition in glomeruli and the tubulointerstitium, and higher SAA concentrations in blood, are found in two different mouse models of diabetic kidney disease (DKD) compared to controls [6], as well as in people with type 2 diabetes and advanced DKD, characterized by clinical proteinuria or decreased glomerular filtration rate (GFR), versus those without kidney disease [7]. On the other hand, SAA enhances tubular cell proliferation in a rodent model of acute kidney injury (AKI) [8], suggesting that it is also capable of inducing kidney repair under some circumstances.
We examined the association of serum levels of SAA with ESRD and all-cause mortality in American Indians with type 2 diabetes who did not have advanced DKD. This group was comprised of individuals with a spectrum of kidney function from near-normal GFR to glomerular hyperfiltration, or early DKD primarily characterized by elevated albuminuria.
Materials and Methods
Study Subjects and Design
From 1965–2007, Pima Indians from the Gila River Indian Community participated in a longitudinal study of diabetes and diabetic complications. Each member of this community who was at least 5 years of age was invited to undergo a research examination approximately every 2 years. Diabetes was diagnosed by a 2-hour post-load plasma glucose concentration ≥200 mg/dl at these biennial examinations, or when the diagnosis was documented in the medical record. For the present study, we selected participants from this longitudinal population-based study who were ≥18 years old, had type 2 diabetes, participated in one of two longitudinal studies of kidney function that included measurements of GFR by the urinary clearance of iothalamate [9, 10], and had a GFR >30 ml/min at study entry. The date of diabetes diagnosis for each participant was ascertained from the longitudinal study, and all other variables used in the analyses were measured at the first kidney function study at which stored serum was available for measurement of SAA. This kidney function study was considered as the baseline. Baseline examinations were conducted between May 1990 and May 2005.
Participants were followed until ESRD, death, or December 31, 2015. Vital status and development of ESRD were ascertained independently of the research examinations. ESRD was defined by the initiation of chronic kidney replacement therapy or death from DKD. Underlying causes of death were determined from death certificates. This study was approved by the Institutional Review Boards of the National Institute of Diabetes and Digestive and Kidney Diseases and Providence Health Care. Each participant signed an informed consent document.
Clinical and Anthropometric Measures
Blood pressure was measured while the participant was resting in the seated position. Mean arterial pressure (MAP) was calculated as (2×diastolic blood pressure + systolic blood pressure)/3. Total serum cholesterol and serum triglyceride concentrations were measured by an enzymatic method. Glycated hemoglobin (HbA1c) was measured by high performance liquid chromatography. This method was also used to measure the concentration of non-radioactive iothalamate for GFR determination. Urine albumin concentration was measured by nephelometric immunoassay and urine creatinine concentration by a modified Jaffé reaction (Siemens, Erlangen, Germany). Urine albumin concentrations below the detection limit of the assay (≤6.8 mg/L) were set to 6.8 mg/L in the analyses. Albuminuria was estimated by computing the urine albumin-to-creatinine ratio (ACR). Macroalbuminuria was defined as ACR ≥300 mg/g, microalbuminuria as ACR 30 to <300 mg/g, and normoalbuminuria as ACR <30 mg/g.
SAA Assay
Samples were collected under standardized conditions and stored at −80°C, undergoing only one freeze-thaw cycle prior to assay in the year 2015. Storage time of baseline samples prior to performance of biomarker assays was bimodal because specimens were derived from two different study cohorts that underwent identical kidney function testing [9, 10]. Of the 256 participants included in this study, 144 were from the first study cohort, and 112 were from the second study cohort. Median storage time for the first cohort was 24.2 years (interquartile range [IQR] 23.7–24.6 years) and for the second cohort was 15.0 years (IQR 14.7–15.3 years). Insoluble materials were removed from the serum by centrifugation for 10 minutes (1000 × g) prior to performing the assay. SAA was measured by an enzyme-linked immunosorbent assay for the predominant human isoform in the blood and kidneys, SAA isoform 1 (SAA1). Samples were analyzed in duplicate on a 96-well plate, and the colorimetric product was quantified by absorbance at 460 nm on an absorbance plate reader (Tecan Group, Männedorf, Switzerland). Concentrations were calculated by comparison with a standard curve generated on each plate. The lower limit of detection (LOD) was 0.0016 mg/L. The coefficient of variation was independently verified for the present analyses, with intra-assay coefficients of variation of 4.2% in samples from study participants. Reproducibility of the SAA assays was assessed by intra- class correlation of measurements from 33 duplicate samples blinded to the performance laboratory. Intra-class correlation coefficient for SAA was 0.599 reflecting acceptable agreement.
Statistical Analysis
Participant characteristics were expressed as mean ± SD, median (IQR), or n (%). Spearman’s correlations were used to assess the relationship of serum SAA concentrations with clinical variables. For regression analyses, ACR was log2-transformed. SAA concentrations by sex, albuminuria categories, and GFR tertiles were compared by the Kruskall-Wallis test.
Kaplan-Meier survival curves for the outcomes of ESRD and death were plotted by tertiles of SAA concentration (tertile 1<0.937 mg/L; tertile 2, 0.937≤SAA<1.62 mg/L, and tertile 3, SAA≥1.62 mg/L). Because the cumulative incidence function for each outcome was comparable in the two higher tertiles of SAA concentration (P=0.261 for ESRD, P=0.848 for death) these two tertiles were combined and compared with the lowest tertile in subsequent analyses. Log-rank statistics were calculated to examine differences in the probability of reaching the specified outcomes in these categories. Pointwise confidence intervals were computed for the cumulative incidence at 15 years of follow-up, using a log-log transformation of the survivor function [11]. Two proportional hazards regression models were then considered: (A) univariate; (B) adjusted for age, sex, diabetes duration, BMI, HbA1c, MAP, renin-angiotensin-system (RAS) inhibitor use, study cohort, GFR and the logarithm of ACR. We used absolute measurements of GFR in these analyses, because the study included overweight and obese participants and indexing for body surface area may significantly underestimate their actual GFR [12]. Because the effect of SAA on the health outcomes was not linear, we reported the categorical results, again comparing the combined upper tertiles with the lowest tertile. Proportionality assumptions were met by each covariate when examined using Schoenfeld residuals. The extent to which SAA enhanced prediction of ESRD was assessed by generalized C statistics after accounting for variable follow-up times [13]. Comparisons between nested models that included or excluded SAA were assessed by likelihood ratio tests [13, 14]. In addition, the relative integrated discrimination improvement (rIDI) index was calculated to assess the improvement in 15-year ESRD risk prediction of SAA relative to traditional risk factors [15]. The 15-year risk was selected as it approximates the median follow-up time for the cohort. The 95% CIs for the rIDI was computed based on 10,000 bootstrap samples.
We conducted a sensitivity analysis by using the competing risk model of Fine and Gray to estimate the subdistribution hazard ratios for ESRD, while accounting for the competing risk of pre-ESRD deaths [16].
Statistical analyses were performed with SAS version 9.3 (SAS Institute, Cary, NC). P values <0.05 were considered statistically significant, and 95% confidence intervals were calculated for our regression estimates.
Results
Baseline Characteristics
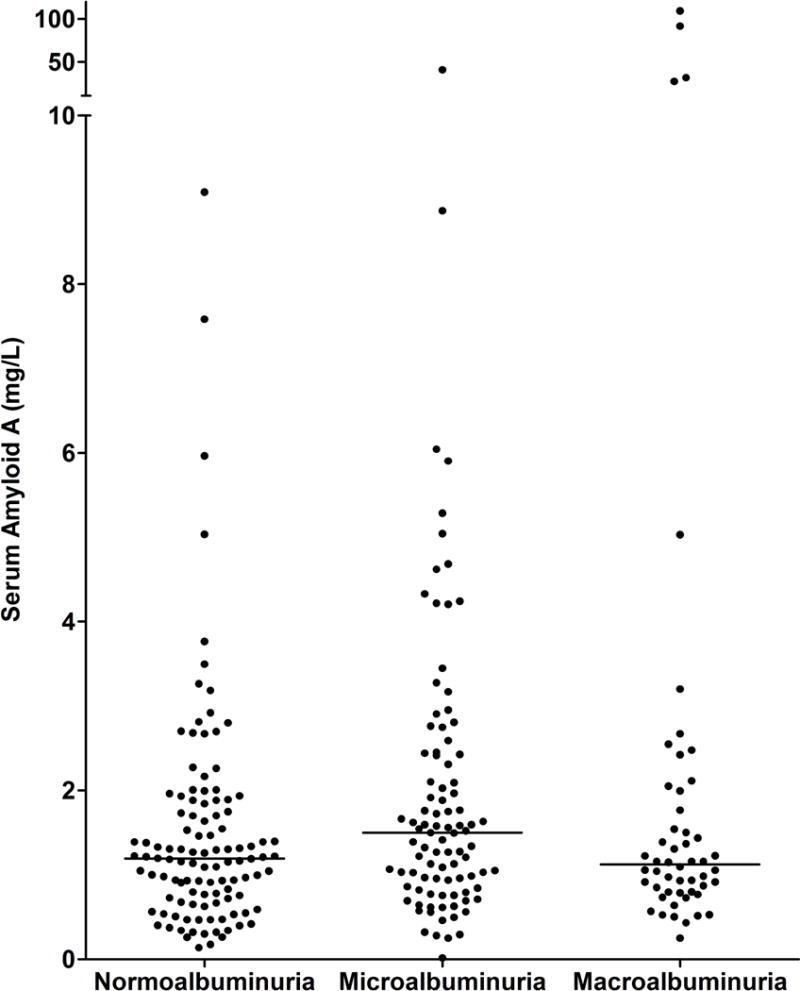
Clinical and biological characteristics of the 256 participants at baseline are summarized in Table 1 according to tertiles of SAA concentrations. Mean age of the participants was 42.5 ± 10.4 years, mean diabetes duration was 11.4 ± 6.7 years, mean HbA1c was 9.5 ± 2.3%, mean GFR was 148 ± 45 ml/min, and median urine ACR was 39 mg/g (IQR=14–221 mg/g). One-hundred-eighteen participants (46%) had hyperfiltration, defined by a GFR ≥154 ml/min, a value two standard deviations above the mean GFR in Pima Indians with normal glucose tolerance. The proportion of participants with hyperfiltration was lowest in the lowest tertile of SAA concentration. Nevertheless, serum SAA concentrations did not differ significantly by tertiles of GFR (Figure 1, P=0.099). One-hundred-ten participants (43%) had normoalbuminuria, 94 (37%) had microalbuminuria, and 52 (20 %) had macroalbuminuria. Serum SAA concentrations were similar in men and women (1.30 [0.84–2.00] mg/L vs. 1.13 [0.72–1.73] mg/L, P = 0.078), and did not differ significantly by albuminuria category (Figure 2, P=0.073). Six of the participants had GFR <60 ml/min (range=26–59 ml/min), and all but one of these had macroalbuminuria. Five participants had extremely high SAA concentrations compared with the rest of the cohort, and one had a concentration that was 1/10 of the next lowest concentration in the cohort.
Table 1.
Baseline clinical and biological characteristics of the study cohort according to tertiles of SAA concentration
| Variable | All (n=256) |
Tertile 1 (n=85) |
Tertile 2 (n=86) |
Tertile 3 (n=85) |
|---|---|---|---|---|
| SAA (mg/L)a | 1.22 (0.01–109.83) | 0.63 (0.01–0.937) | 1.22 (0.937–1.62) | 2.59 (1.62–109.83) |
| Age (years) | 42.5 ± 10.4 | 44.3 ± 9.7 | 42.8 ± 10.7 | 40.4 ± 10.6 |
| Women;men (men %) | 173;83 (32) | 49;36 (42) | 60;26 (30) | 64;21 (24) |
| Body mass index (kg/m2) | 35 ± 8 | 33 ± 7 | 35 ± 8 | 37 ± 9 |
| Diabetes duration (years) | 11.4 ± 6.7 | 12.8 ± 6.6 | 11.9 ± 7 | 9.6 ± 6.1 |
| Systolic blood pressure (mmHg) | 121 ± 15 | 122 ± 15 | 121 ± 15 | 120 ± 14 |
| Diastolic blood pressure (mmHg) | 76 ± 9 | 76 ± 10 | 75 ± 9 | 75 ± 9 |
| RAS inhibitor use | 57 (22) | 22 (26) | 17 (20) | 18 (21) |
| Lipid lowering drug use | 4 (1.6) | 1(0.4) | 1(0.4) | 2(0.8) |
| HbAlc (%) | 9.5 ± 2.3 | 9.3 ± 2.5 | 9.7 ± 2.2 | 9.4 ± 2.2 |
| Glomerular filtration rate (ml/min) | 148 ± 45 | 154 ± 50 | 149 ± 45 | 142 ± 40 |
| Hyperfiltration (>154 ml/min)b | 118 (46) | 43 (51) | 42 (49) | 33 (39) |
| Glomerular filtration rate (ml/min/1.73m2) | 128 ± 37 | 132 ± 41 | 130 ± 38 | 122 ± 32 |
| Urine albumin/creatinine (mg/g)c | 39 (14–221) | 33 (16–222) | 39 (14–191) | 50 (12–219) |
| Total cholesterol (mg/dL)c | 164 (142–193) | 160 (141–188.5) | 172 (148–200) | 159 (138–187) |
| HDL-Cholesterol (mg/dL)d | 40 (35–45) | 45 (37–46) | 39 (32–46) | 39 (36–43) |
| LDL-Cholesterol (mg/dL)e | 95 (79–119) | 90 (75–117) | 102 (86–124) | 95 (79–115) |
| Triglycerides (mg/dL)c | 132 (101–214) | 128 (94–217) | 139 (103–214) | 125 (101–189) |
Data are means±SD, medians (25th –75th percentiles) or n (%).
Data are medians (range).
Hyperfiltration is defined by a measured GFR ≥154 ml/min, a value 2 standard deviations above the mean GFR in Pima Indians with normal glucose tolerance.
4 missing values.
123 missing values.
142 missing values.
Fig. 1.
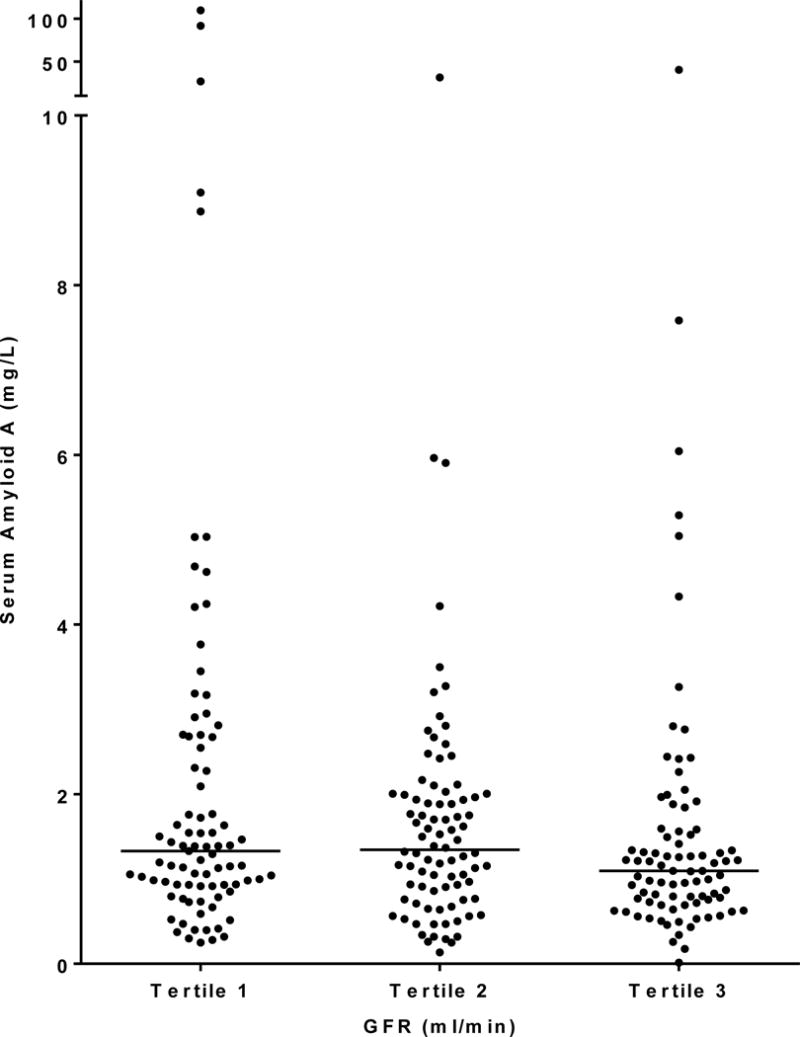
Distributions of SAA concentration by tertiles of GFR at baseline. GFR range by tertile is 26–129 ml/min for tertile 1, 129–167 ml/min for tertile 2, and 168–265 ml/min for tertile 3. Medians are represented by horizontal lines.
Fig. 2.
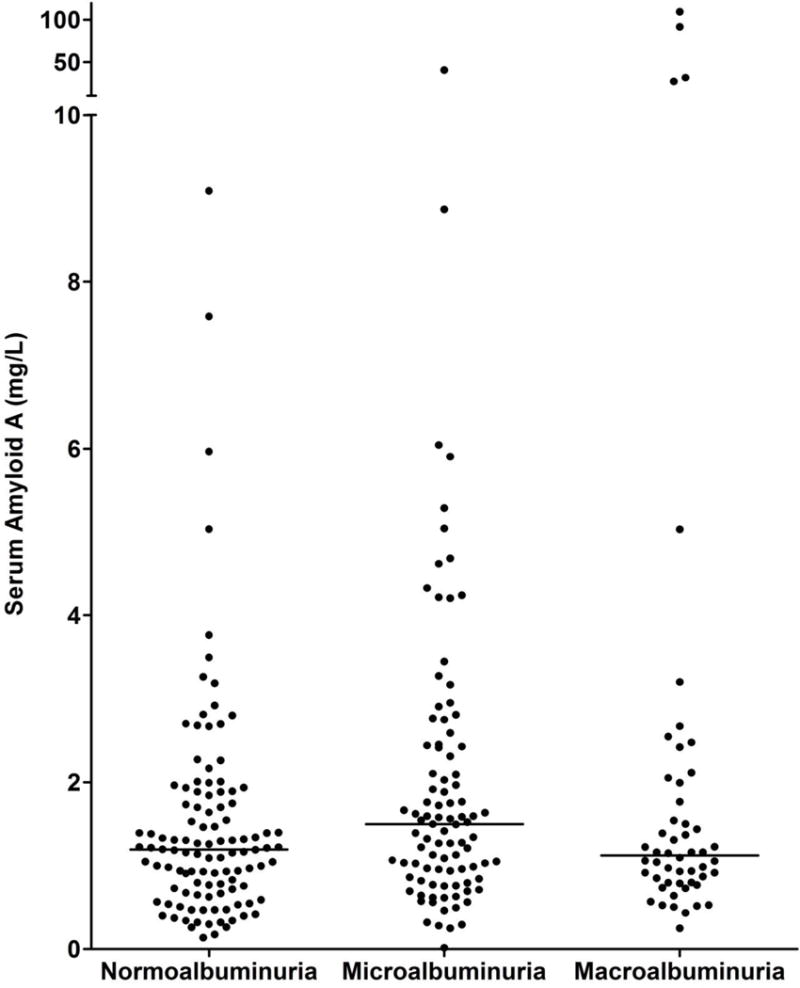
Distributions of SAA concentration by level of albuminuria at baseline. Medians are represented by horizontal lines.
SAA concentration correlated negatively with age (r=−0.15, P=0.014), diabetes duration (r−.19, P=0.002), and GFR (r=−0.13, P=0.044) and positively with BMI (r=0.24, P<0.001); it was not correlated with ACR (r=0.07, P=0.292), MAP (r=−0.05, P=0.473), HbA1c (r=0.02, P=0.806), total cholesterol (r=−0.03, P=0.604), or serum triglyceride concentrations (r=−0.003, P=0.973). In the subset of patients with available cholesterol fractions, SAA concentration was not correlated with either HDL cholesterol (n=123, r=−0.09, P=0.288) or LDL cholesterol (n=114, r=−0.04, P=0.647).
SAA and Risk of ESRD or Death
Participants were followed for a median of 15.2 years (IQR=9.5–17.0 years) for ESRD and 15.7 years (13.6–21.8 years) for death. During follow-up, 76 participants developed ESRD and 125 died (15 from malignancy, 31 from cardiovascular disease (CVD), 18 from alcoholic liver disease, 16 from diabetic nephropathy, 13 from infection, 23 from other natural causes, and 9 from external causes). Fifty-two participants died after developing ESRD. Both ESRD (log rank test P=0.050) and death (log rank test P=0.006) occurred more frequently in the lowest tertile of SAA concentration (SAA<0.937 mg/L) compared to the higher SAA concentration tertiles (SAA≥0.937 mg/L). The unadjusted cumulative incidence of ESRD at 15 years of follow-up was 35.8% (95% CI 26.1–47.8) and 23.2% (17.4–30.6), and the 15-year cumulative mortality was 40.5% (95% CI 30.5–51.8) and 23.6% (95% CI 17.9–30.8) in the lowest and the two highest tertiles of SAA combined (Figure 3). The incidence of ESRD and death according to SAA tertiles are presented in the Supplementary Table.
Fig. 3.
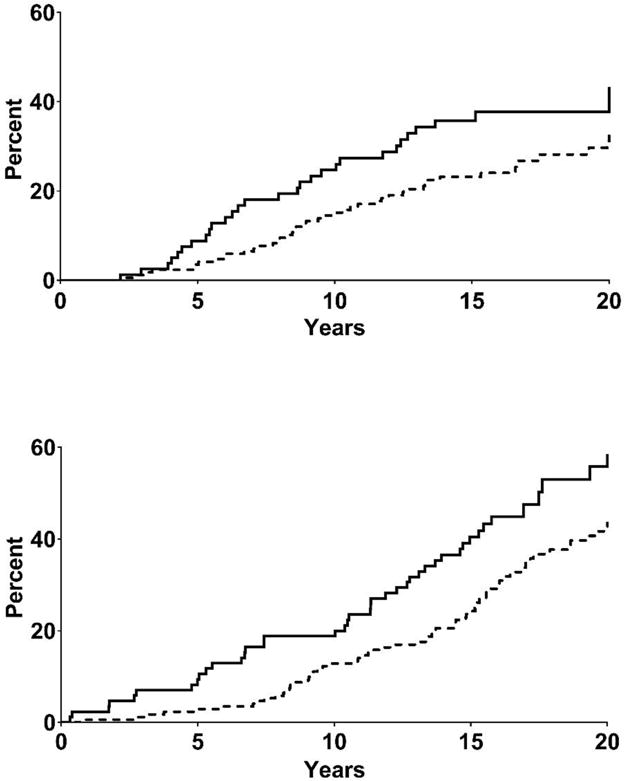
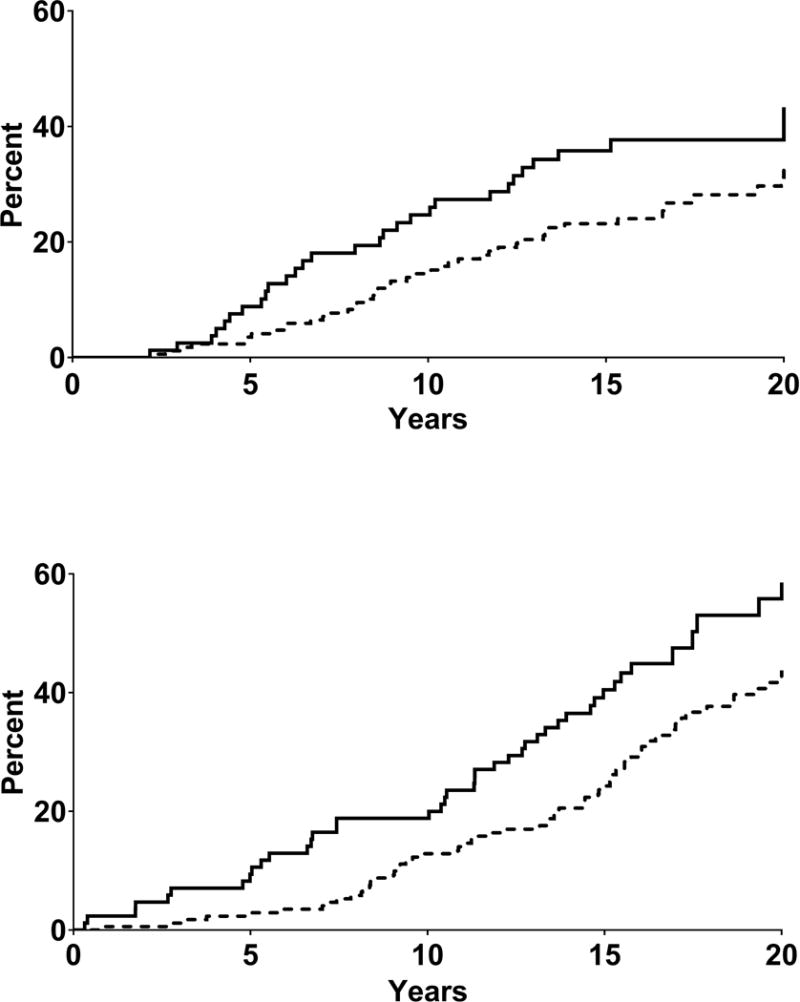
Cumulative incidence of end stage renal disease (upper panel) and all-cause death (lower panel). Kaplan-Meier survival curves are displayed according to SAA concentration at baseline. Solid line indicates lowest SAA concentration tertile (SAA<0.937 mg/L), and dotted line indicates the two highest SAA concentration tertiles combined (second tertile SAA≥ 0.937<1.62 mg/L and third tertile SAA ≥ 1.62 mg/L). (P-value from log-rank test, ESRD=0.050; all-cause death=0.006). The unadjusted proportional hazards regression model was not statistically significant for ESRD (HR=0.63, 95% CI 0.40–1.004), and was for death (HR=0.60, 95% CI 0.42–0.87). After adjustment for traditional risk factors, the proportional hazards regression model was statistically significant for ESRD (HR=0.47, 95% CI 0.29-0.78), and was not for death (HR=0.70, 95% CI 0.48-1.02).
In the unadjusted proportional hazards regression model, the hazard ratio (HR) for ESRD was 0.63 (95% CI 0.40–1.004) in the highest two SAA tertiles compared with the lowest SAA tertile (Table 2). After adjustment for traditional risk factors, the relationship between SAA concentration and ESRD strengthened and remained statistically significant (HR=0.47, 95% CI 0.29–0.78), with the top two tertiles of SAA concentration combined having a 53% lower risk of ESRD than the lowest tertile. In this model, higher baseline GFR predicted a lower risk of ESRD (HR per 10 ml/min GFR increment=0.90, 95% CI 0.84–0.96). The exclusion of the 6 patients with extreme SAA values did not modify this finding (HR=0.44, 95% CI 0.26–0.73).
Table 2.
Hazard ratios (HRs) and 95% confidence intervals for ESRD and all-cause death. HRs are expressed for the two highest tertiles in comparison with the lowest tertile of SAA
| Events/Person-Years | Unadjusted
|
Adjusteda
|
|||
|---|---|---|---|---|---|
| HR (95% CI) | P value | HR (95% CI) | P value | ||
| Primary Cox analysis (n=256) | |||||
| ESRD | 76/3715 | 0.63 (0.40–1.004) | 0.052 | 0.47 (0.29–0.78) | 0.004 |
| Death | 125/4155 | 0.60 (0.42–0.87) | 0.006 | 0.70 (0.48–1.02) | 0.060 |
| Fine and Gray competing risk analysis (n=256) | |||||
| ESRD | 76/3715 | 0.71 (0.45–1.13) | 0.148 | 0.51 (0.31–0.85) | 0.010 |
| Cox analysis after exclusion of 6 participants with baseline GFR <60 ml/min (n=250) | |||||
| ESRD | 72/3674 | 0.65 (0.40–1.05) | 0.076 | 0.49 (0.29–0.82) | 0.006 |
| Death | 12/4105 | 0.60 (0.41–0.86) | 0.006 | 0.69 (0.46–1.004) | 0.052 |
Adjustment for age, sex, RAS inhibitor use, study cohort, diabetes duration, MAP, HbA1c, BMI, GFR and ACR.
In the unadjusted proportional hazards regression model, the HR for death was 0.60 (95% CI 0.42–0.87) in the highest two SAA tertiles compared with the lowest SAA tertile (Table 2). After adjusting for traditional risk factors, the HR was 0.70 (95% CI 0.48–1.02) in the two highest SAA tertiles compared with the lowest SAA tertile, reflecting a 30% reduction in the risk of death, although the results for death were not statistically significant. In this model, a higher baseline GFR predicted a lower risk of death (HR=0.95, 95% CI 0.90–1.00). The exclusion of the 6 patients with extreme SAA values did not substantially modify the magnitude of the relationship (HR=0.66, 95% CI 0.44–0.97).
The inclusion of SAA in the fully adjusted proportional hazards regression model increased the C statistic for predicting ESRD from 0.814 to 0.815 (P=0.005) and for predicting death from 0.701 to 0.712 (P=0.064) compared with the model that did not include SAA. The inclusion of SAA, however, did not significantly improve the rIDI for predicting ESRD (3.4% [95% CI: −0.7–10.8]; P=0.198) or death (1.3% [95% CI: −0.9–14.8]; P=0.660) after 15 years of follow-up.
Sensitivity Analyses
When examining the competing risk of mortality in a Fine and Gray analysis, SAA remained independently associated with the risk of ESRD (subhazard HR=0.51, 95% CI 0.31–0.85). Conclusions of the study were unchanged when the six participants with GFR<60 ml/min at baseline were excluded from the analysis (Table 2).
Discussion
A higher serum concentration of SAA in American Indians with type 2 diabetes predicted a reduced risk of ESRD, but not mortality, over approximately 15 years of follow-up. We found a modest inverse univariate correlation between SAA concentration and GFR in this study, suggesting that greater clearance of SAA from the circulation was occurring in those with higher GFR. On the other hand, higher GFR was associated with a lower risk of ESRD or death even after adjustment for SAA in the multivariable model. The extent to which hyperfiltration in persons with diabetes predicts adverse health outcomes is uncertain [17], but our findings suggest that the relationship between SAA and adverse health outcomes may be influenced by the level of GFR in individuals with type 2 diabetes and early or no DKD. The exact nature of this influence, however, is unknown. Nevertheless, after accounting for traditional DKD risk factors, including GFR and urine ACR, the magnitude of improvement in risk assessment by the addition of SAA to the multivariable model was small and did not improve discrimination for ESRD or death in a clinically meaningful manner in these individuals without DKD or with early DKD. These findings suggest that in the absence of DKD or in early DKD, higher SAA concentrations may have renoprotective effects that warrant further study.
The present findings differ from those from the Goldenstate study [7], a study of coronary artery calcification in participants with type 2 diabetes and advanced DKD, in whom higher serum concentrations of SAA were associated with a higher risk of the combined endpoint of ESRD or death and with all-cause death alone but not with ESRD alone. Although both studies specifically measured SAA isoform 1 concentration, some key differences between these two studies may account for the disparate findings. Participants in the Goldenstate study were generally older, predominantly Mexican-American or Black, had higher blood pressure and more frequent use of RAS inhibitors. Compared with the American Indian cohort, they also had more advanced DKD with much lower estimated GFR (mean eGFR±SD in Goldenstate = 56±22 ml/min/1.73m2 versus mean eGFR±SD in American Indian study = 128±37 ml/min/1.73m2), markedly higher urine ACR (median = 1861 mg/g in Goldenstate study versus 39 mg/g in American Indian cohort), and higher rates of ESRD and all-cause mortality. Moreover, the 67th percentile of SAA concentration in the Goldenstate study (1.0 mg/L) was similar to the 33rd percentile (0.937 mg/L) in the American Indian cohort, indicating a very different distribution of SAA in the two studies. Lower concentrations of SAA isoform 1 were previously reported in healthy volunteers than in those with type 2 diabetes or with type 2 diabetes and DKD, defined by the presence of proteinuria [6]. Another study in which all isoforms were measured also demonstrated lower concentrations in healthy nondiabetic participants than in those with type 2 diabetes or with type 2 diabetes and DKD, defined either by albuminuria or by the presence of structural lesions [18]. Together, these studies suggest an association between higher SAA concentrations and progressive DKD [6, 18]. Given these findings, the observation in the present study that a higher concentration of SAA isoform 1 predicts a lower risk of ESRD in persons who had no or early DKD at baseline requires further investigation.
SAA was previously identified as a marker for CVD-specific mortality, in part because it transforms HDL cholesterol from a vasoprotective to a pro-atherosclerotic lipoprotein [19, 20]. In the Ludwigshafen Risk and Cardiovascular Health (LURIC) study, which included 3310 patients (3.5% with diabetes), higher SAA concentrations were strongly associated with all-cause and cardiovascular mortality, and higher concentrations were typically observed in older individuals, those with diabetes, and those with lower GFR [19]. We also found an inverse correlation between SAA and GFR, but only a weak inverse correlation with age in our younger American Indian cohort, which had only six participants with GFR <60 ml/min at baseline. Although cause of death was not available in the Goldenstate cohort, we would expect a higher level of CVD mortality in this cohort than in the American Indians, because of their older age and advanced DKD, and the lower frequency of CVD generally in this Southwestern American Indian population [21]. These factors may help explain our finding of no association between SAA concentration and mortality, despite the positive association in the Goldenstate study and in the previous studies of CVD [19, 20]. The findings from the present study, however, do accord with an animal study, in which intravenous cytotherapy with SAA-expressing cells improved kidney function and enhanced tubular cell proliferation in a rodent AKI model [8]. These data suggest that acute inflammation, marked by elevation of SAA, may have a protective role in settings such as AKI without background kidney disease. Thus, it may be that in diabetes without DKD or in early DKD, production of SAA could protect against kidney-disease-accelerating experiences such as episodes of AKI. Conversely, once DKD is sufficiently advanced, SAA may contribute to pro-inflammatory mechanisms of DKD progression. Elucidation of conditions under which such putative disease transitions may occur are important to advance understanding of relationships between underlying inflammation and DKD.
SAA can be considered an adipokine, since it is produced in fat [22] even though it is also produced by liver and kidneys. It abrogates the anti-inflammatory properties of HDL-cholesterol [23, 24], and promotes CVD complications of obesity [25], and insulin-resistance [26]. We found that serum SAA concentration correlated positively with BMI, as already shown by others [26], but not with total serum cholesterol or triglycerides. Acute inflammatory stress can lead to as much as a 1,000-fold increase of blood levels of SAA, largely produced by the liver, within 5–6 hours [27, 28]. SAA is immunologically active, promoting expression of cytokines such as interleukin-1 beta, interleukin-6, interleukin-8, and tumor necrosis factor (TNF) alpha by fibroblasts [4] and macrophages [5]. SAA knockout mice fed a high-fat, high-sucrose diet displayed decreased expression of TNF and monocyte chemoattractant protein-1 and attenuated macrophage accumulation in visceral fat [29]. SAA is a ligand for many receptors involved in immune functions, including toll-like receptors and the receptor for advanced glycation end products [30, 31], and it can trigger pro-inflammatory cascades through activation of transcription factors such as nuclear factor kappa B, activator protein 1, and interferon regulatory factor 3 [6, 32]. Advanced glycation end products induce up-regulation of SAA in podocytes [6] and SAA itself can bind the receptor for advanced glycation end products [33]. Notably, SAA can also amplify its own expression in an autocrine manner, along with a host of the aforementioned pro-inflammatory factors [6, 34]. Conversely, in rodents without background kidney disease, SAA induces proximal tubule formation [35]. Therefore, under physiological conditions, the acute effects of SAA may predominate as a paracrine function to enhance cellular proliferation and repair in situations like AKI. With persistent inflammation and chronic disease, such as advanced DKD, local repair mechanisms may become insufficient with SAA instead promoting inflammatory injury, ultimately leading to progressive kidney damage.
In humans, SAA exists predominantly in two isoforms, SAA1 and SAA2 [36, 37]. Their distribution and function vary by disease state and tissue. Research is needed to determine the isoform(s) expressed in diabetes, in the kidney, as well as other tissues, and changes over the course of DKD. For example, the pro-inflammatory janus-kinase 2 (JAK-2) signaling pathway is a major activator of SAA gene expression [34]. Based on human kidney biopsy studies, the location and degree of JAK-2 activity changes predominantly from the glomerular compartment in Pima Indians of the present cohort with type 2 diabetes and no DKD or only early DKD to the tubulointerstitial compartment in white Europeans with type 2 diabetes and advanced DKD [38], consistent with a pattern of shift in inflammatory responses in DKD. Notably, in a clinical trial among people with DKD marked by macroalbuminuria and low estimated GFR, baricitinib, a novel JAK-2 inhibitor, reduced albuminuria, SAA, soluble TNF receptors 1 and 2, and other inflammatory biomarkers in concert, suggesting a potential therapeutic response [39]. Thus, biological effects of SAA and isoform expression appear to vary over the course of DKD.
The strengths of this study include the detailed characterization of the study cohort, the median follow-up of 15.2 years for ESRD and 15.7 years for death, and the considerable number of cases of ESRD and death. Samples were collected under standardized conditions and stored at −80°C, undergoing only one prior freeze-thaw cycle. A previous study reports no meaningful impact on data quality of the SAA assay after 5 freeze-thaw cycles [40]. The conclusions of the study were robust when examined by several analytical strategies. Limitations of the study include measurement of a single biomarker. C-reactive protein was previously measured in less than a third of the participants and was therefore not reported. In addition, cholesterol fractions, including HDL cholesterol and LDL cholesterol, were only available for half the participants. We did not assess biomarker stability over time, since SAA was only measured once in baseline samples, and we have no information on the impact of long-term storage on assay results. To better assess the potential role of SAA as a biomarker for DKD, we are currently undertaking a series of longitudinal measures of SAA both to examine variation over time and to search for changes in relationships with changes in kidney function. We do not have an explanation for the 6 outliers whose removal further strengthened the results. Survivor bias may have also influenced relationships of serum SAA concentrations with ESRD and death. The potentially large and rapid effect of acute inflammation on serum concentrations of SAA may affect its reliability as a biomarker of progressive DKD. However, levels of SAA in the blood due to acute inflammation are typically much higher than those observed in the present study.
In conclusion, higher serum SAA concentration was associated with lower risk of ESRD in American Indians with type 2 diabetes without DKD or with early DKD. By contrast, evidence from other studies suggests that SAA is not protective in those with more advanced DKD. Instead, higher SAA concentration is associated with all-cause mortality and ESRD, and serum levels of SAA respond to anti-inflammatory therapy in humans with DKD [38, 39]. If these observations are confirmed, identifying mechanisms that suppress early beneficial effects or promote harmful effects of SAA may enhance prediction of progressive DKD and provide new therapeutic targets.
Supplementary Material
Acknowledgments
This research was supported by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases, by Providence Sacred Heart Medical Center and Children’s Hospital, the Providence Foundation, and the Chronic Kidney Disease Biomarker Consortium-NIDDK Grant 5U01DK103225-02.
The authors thank the participants and the doctors, nurses, and support staff for their role in collecting and processing the data.
Footnotes
Disclosure Statement: K.R.T. has received consulting fees regarding therapies for DKD from Eli Lilly and Company, Boehringer Ingelheim, and Gilead. All other authors have no disclosures to report.
References
- 1.Meek RL, LeBoeuf RC, Saha SA, Alpers CE, Hudkins KL, Cooney SK, Anderberg RJ, Tuttle KR. Glomerular cell death and inflammation with high-protein diet and diabetes. Nephrol Dial Transplant. 2013;28:1711–1720. doi: 10.1093/ndt/gfs579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Meek RL, Eriksen N, Benditt EP. Serum amyloid A in the mouse. Sites of uptake and mRNA expression. Am J Pathol. 1989;135:411–419. [PMC free article] [PubMed] [Google Scholar]
- 3.Urieli-Shoval S, Cohen P, Eisenberg S, Matzner Y. Widespread expression of serum amyloid A in histologically normal human tissues: predominant localization to the epithelium. J Histochem Cytochem. 1998;46:1377–1384. doi: 10.1177/002215549804601206. [DOI] [PubMed] [Google Scholar]
- 4.Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, Fernandes-Alnemri T, Wu J, Monks BG, Fitzgerald KA, Hornung V, Latz E. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol. 2009;183:787–791. doi: 10.4049/jimmunol.0901363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Niemi K, Teirila L, Lappalainen J, Rajamaki K, Baumann MH, Oorni K, Wolff H, Kovanen PT, Matikainen S, Eklund KK. Serum amyloid A activates the NLRP3 inflammasome via P2X7 receptor and a cathepsin B-sensitive pathway. J Immunol. 2011;186:6119–6128. doi: 10.4049/jimmunol.1002843. [DOI] [PubMed] [Google Scholar]
- 6.Anderberg RJ, Meek RL, Hudkins KL, Cooney SK, Alpers CE, Leboeuf RC, Tuttle KR. Serum amyloid A and inflammation in diabetic kidney disease and podocytes. Lab Invest. 2015;95:697. doi: 10.1038/labinvest.2015.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dieter BP, McPherson SM, Afkarian M, de Boer IH, Mehrotra R, Short R, Barbosa-Leiker C, Alicic RZ, Meek RL, Tuttle KR. Serum amyloid a and risk of death and end-stage renal disease in diabetic kidney disease. J Diabetes Complications. 2016 doi: 10.1016/j.jdiacomp.2016.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kelly KJ, Kluve-Beckerman B, Zhang J, Dominguez JH. Intravenous cell therapy for acute renal failure with serum amyloid A protein-reprogrammed cells. Am J Physiol Renal Physiol. 2010;299:F453–464. doi: 10.1152/ajprenal.00050.2010. [DOI] [PubMed] [Google Scholar]
- 9.Nelson RG, Bennett PH, Beck GJ, Tan M, Knowler WC, Mitch WE, Hirschman GH, Myers BD. Development and progression of renal disease in Pima Indians with non-insulin-dependent diabetes mellitus. Diabetic Renal Disease Study Group. N Engl J Med. 1996;335:1636–1642. doi: 10.1056/NEJM199611283352203. [DOI] [PubMed] [Google Scholar]
- 10.Weil EJ, Fufaa G, Jones LI, Lovato T, Lemley KV, Hanson RL, Knowler WC, Bennett PH, Yee B, Myers BD, Nelson RG. Effect of losartan on prevention and progression of early diabetic nephropathy in American Indians with type 2 diabetes. Diabetes. 2013;62:3224–3231. doi: 10.2337/db12-1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kalbfleisch JD, Prentice RL. The statistical analysis of failure time data. New York, NY: John Wiley & Sons; 1980. [Google Scholar]
- 12.Delanaye P, Radermecker RP, Rorive M, Depas G, Krzesinski JM. Indexing glomerular filtration rate for body surface area in obese patients is misleading: concept and example. Nephrol Dial Transplant. 2005;20:2024–2028. doi: 10.1093/ndt/gfh983. [DOI] [PubMed] [Google Scholar]
- 13.Pencina MJ, D’Agostino RB. Overall C as a measure of discrimination in survival analysis: model specific population value and confidence interval estimation. Statistics in Medicine. 2004;23:2109–2123. doi: 10.1002/sim.1802. [DOI] [PubMed] [Google Scholar]
- 14.Pencina MJ, D’Agostino RB, Sr, D’Agostino RB, Jr, Vasan RS. Evaluating the added predictive ability of a new marker: from area under the ROC curve to reclassification and beyond. Stat Med. 2008;27:157–172. doi: 10.1002/sim.2929. discussion 207-112. [DOI] [PubMed] [Google Scholar]
- 15.Ninomiya T, Perkovic V, de Galan BE, Zoungas S, Pillai A, Jardine M, Patel A, Cass A, Neal B, Poulter N, Mogensen CE, Cooper M, Marre M, Williams B, Hamet P, Mancia G, Woodward M, Macmahon S, Chalmers J, Group AC Albuminuria and kidney function independently predict cardiovascular and renal outcomes in diabetes. J Am Soc Nephrol. 2009;20:1813–1821. doi: 10.1681/ASN.2008121270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fine JP, Gray RJ. A proportional hazards model for the subdistribution of a competing risk. J Am Stat Assoc. 1999;94:496–509. [Google Scholar]
- 17.Tonneijck L, Muskiet MHA, Smits MM, van Bommel EJ, Heerspink HJL, van Raalte DH, Joles JA. Glomerular hyperfiltration in diabetes: mechanisms, clinical significance, and treatment. J Am Soc Nephrol. 2017;28:1023–1039. doi: 10.1681/ASN.2016060666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dalla Vestra M, Mussap M, Gallina P, Bruseqhin M, Cerniqoi AM, Saller A, Plebani M, Fioretto P. Acute-phase markers of inflammation and glomerular structure in patients with type 2 diabetes. J Am Soc Nephrol. 2005;16(Suppl 1):S78–S82. doi: 10.1681/asn.2004110961. [DOI] [PubMed] [Google Scholar]
- 19.Zewinger S, Drechsler C, Kleber ME, Dressel A, Riffel J, Triem S, Lehmann M, Kopecky C, Saemann MD, Lepper PM, Silbernagel G, Scharnagl H, Ritsch A, Thorand B, de las Heras Gala T, Wagenpfeil S, Koenig W, Peters A, Laufs U, Wanner C, Fliser D, Speer T, Marz W. Serum amyloid A: high-density lipoproteins interaction and cardiovascular risk. Eur Heart J. 2015;36:3007–3016. doi: 10.1093/eurheartj/ehv352. [DOI] [PubMed] [Google Scholar]
- 20.O Hartaigh B, Bosch JA, Carroll D, Hemming K, Pilz S, Loerbroks A, Kleber ME, Grammer TB, Fischer JE, Boehm BO, Marz W, Thomas GN. Evidence of a synergistic association between heart rate, inflammation, and cardiovascular mortality in patients undergoing coronary angiography. Eur Heart J. 2013;34:932–941. doi: 10.1093/eurheartj/ehs396. [DOI] [PubMed] [Google Scholar]
- 21.Pavkov ME, Bennett PH, Sievers ML, Krakoff J, Williams DE, Knowler WC, Nelson RG. Predominant effect of kidney disease on mortality in Pima Indians with or without type 2 diabetes. Kidney Int. 2005;68:1267–1274. doi: 10.1111/j.1523-1755.2005.00523.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jernas M, Palming J, Sjoholm K, Jennische E, Svensson PA, Gabrielsson BG, Levin M, Sjogren A, Rudemo M, Lystig TC, Carlsson B, Carlsson LM, Lonn M. Separation of human adipocytes by size: hypertrophic fat cells display distinct gene expression. FASEB J. 2006;20:1540–1542. doi: 10.1096/fj.05-5678fje. [DOI] [PubMed] [Google Scholar]
- 23.Han CY, Tang C, Guevara ME, Wei H, Wietecha T, Shao B, Subramanian S, Omer M, Wang S, O’Brien KD, Marcovina SM, Wight TN, Vaisar T, de Beer MC, de Beer FC, Osborne WR, Elkon KB, Chait A. Serum amyloid A impairs the antiinflammatory properties of HDL. The Journal of clinical investigation. 2016;126:266–281. doi: 10.1172/JCI83475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weichhart T, Kopecky C, Kubicek M, Haidinger M, Doller D, Katholnig K, Suarna C, Eller P, Tolle M, Gerner C, Zlabinger GJ, van der Giet M, Horl WH, Stocker R, Saemann MD. Serum amyloid A in uremic HDL promotes inflammation. J Am Soc Nephrol. 2012;23:934–947. doi: 10.1681/ASN.2011070668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Z, Nakayama T. Inflammation, a link between obesity and cardiovascular disease. Mediators Inflamm. 2010;2010:535918. doi: 10.1155/2010/535918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang RZ, Lee MJ, Hu H, Pollin TI, Ryan AS, Nicklas BJ, Snitker S, Horenstein RB, Hull K, Goldberg NH, Goldberg AP, Shuldiner AR, Fried SK, Gong DW. Acute-phase serum amyloid A: an inflammatory adipokine and potential link between obesity and its metabolic complications. PLoS Med. 2006;3:e287. doi: 10.1371/journal.pmed.0030287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Whitehead AS, de Beer MC, Steel DM, Rits M, Lelias JM, Lane WS, de Beer FC. Identification of novel members of the serum amyloid A protein superfamily as constitutive apolipoproteins of high density lipoprotein. J Biol Chem. 1992;267:3862–3867. [PubMed] [Google Scholar]
- 28.De Buck M, Gouwy M, Wang JM, Van Snick J, Proost P, Struyf S, Van Damme J. The cytokine-serum amyloid A-chemokine network. Cytokine Growth Factor Rev. 2016;30:55–69. doi: 10.1016/j.cytogfr.2015.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.den Hartigh LJ, Wang S, Goodspeed L, Ding Y, Averill M, Subramanian S, Wietecha T, O’Brien KD, Chait A. Deletion of serum amyloid A3 improves high fat high sucrose diet-induced adipose tissue inflammation and hyperlipidemia in female mice. PLoS One. 2014;9:e108564. doi: 10.1371/journal.pone.0108564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cai H, Song C, Endoh I, Goyette J, Jessup W, Freedman SB, McNeil HP, Geczy CL. Serum amyloid A induces monocyte tissue factor. J Immunol. 2007;178:1852–1860. doi: 10.4049/jimmunol.178.3.1852. [DOI] [PubMed] [Google Scholar]
- 31.Belmokhtar K, Robert T, Ortillon J, Braconnier A, Vuiblet V, Boulagnon-Rombi C, Diebold MD, Pietrement C, Schmidt AM, Rieu P, Toure F. Signaling of Serum Amyloid A Through Receptor for Advanced Glycation End Products as a Possible Mechanism for Uremia-Related Atherosclerosis. Arterioscler Thromb Vasc Biol. 2016;36:800–809. doi: 10.1161/ATVBAHA.115.306349. [DOI] [PubMed] [Google Scholar]
- 32.Eklund KK, Niemi K, Kovanen PT. Immune functions of serum amyloid A. Crit Rev Immunol. 2012;32:335–348. doi: 10.1615/critrevimmunol.v32.i4.40. [DOI] [PubMed] [Google Scholar]
- 33.Sun L, Ye RD. Serum amyloid A1: Structure, function and gene polymorphism. Gene. 2016;583:48–57. doi: 10.1016/j.gene.2016.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pichler R, Afkarian M, Dieter BP, Tuttle KR. Immunity and Inflammation in Diabetic Kidney Disease: Translating Mechanisms to Biomarkers and Treatment Targets. Am J Physiol Renal Physiol. 2016 doi: 10.1152/ajprenal.00314.2016. ajprenal 00314 02016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kelly KJ, Kluve-Beckerman B, Dominguez JH. Acute-phase response protein serum amyloid A stimulates renal tubule formation: studies in vitro and in vivo. Am J Physiol Renal Physiol. 2009;296:F1355–1363. doi: 10.1152/ajprenal.90622.2008. [DOI] [PubMed] [Google Scholar]
- 36.Meek RL, Urieli-Shoval S, Benditt EP. Expression of apolipoprotein serum amyloid A mRNA in human atherosclerotic lesions and cultured vascular cells: implications for serum amyloid A function. Proc Natl Acad Sci U S A. 1994;91:3186–3190. doi: 10.1073/pnas.91.8.3186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Upragarin N, Landman WJ, Gaastra W, Gruys E. Extrahepatic production of acute phase serum amyloid A. Histol Histopathol. 2005;20:1295–1307. doi: 10.14670/HH-20.1295. [DOI] [PubMed] [Google Scholar]
- 38.Berthier CC, Zhang H, Schin M, Henger A, Nelson RG, Yee B, Boucherot A, Neusser MA, Cohen CD, Carter-Su C, Argetsinger LS, Rastaldi MP, Brosius FC, Kretzler M. Enhanced expression of Janus kinase-signal transducer and activator of transcription pathway members in human diabetic nephropathy. Diabetes. 2009;58:469–477. doi: 10.2337/db08-1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tuttle KR, Brosius FC. Title. Kidney International (in review) 2017 [Google Scholar]
- 40.Hazenberg BPC, Limburg PC, Bijzet J, van Rijswijk MH. Amyloid and amyloidosis. Kluwer Academic; Dordrecht: 1991. Monoclonal antibody based ELISA for human SAA; pp. 898–901. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.