

Summary
During interphase, the nuclear envelope (NE) serves as a selective barrier between cytosol and nucleoplasm. When vertebrate cells enter mitosis, the NE is dismantled in the process of nuclear envelope breakdown (NEBD). Disassembly of nuclear pore complexes (NPCs) is a key aspect of NEBD, required for NE permeabilization and formation of a cytoplasmic mitotic spindle. Here, we show that both CDK1 and polo-like kinase 1 (PLK1) support mitotic NPC disintegration by hyperphosphorylation of Nup98, the gatekeeper nucleoporin, and Nup53, a central nucleoporin linking the inner NPC scaffold to the pore membrane. Multisite phosphorylation of Nup53 critically contributes to its liberation from its partner nucleoporins, including the pore membrane protein NDC1. Initial steps of NPC disassembly in semi-permeabilized cells can be reconstituted by a cocktail of mitotic kinases including cyclinB-CDK1, NIMA, and PLK1, suggesting that the unzipping of nucleoporin interactions by protein phosphorylation is an important principle underlying mitotic NE permeabilization.
Keywords: NPC, mitosis, PLK1, phosphorylation, nucleoporin, CDK1, NPC disassembly, Nup98, Nup53, NDC1
Graphical Abstract
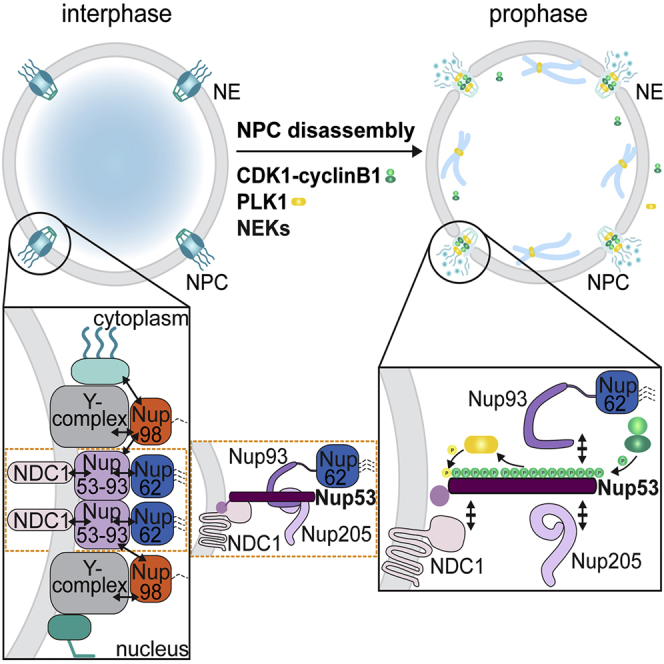
Highlights
-
•
PLK1 localizes to NPCs during prophase and promotes mitotic NPC disassembly
-
•
CDK1 and PLK1 mediate hyperphosphorylation of the central linker nucleoporin Nup53
-
•
Multisite phosphorylation of Nup53 promotes the disintegration of the central NPC
-
•
Mitotic kinases induce NPC disassembly in a reconstituted system
The disassembly of nuclear pore complexes (NPCs) is a key process for nuclear envelope breakdown at the onset of open mitosis in metazoan cells. Linder et al. show that multisite phosphorylation of the key interconnecting nucleoporin Nup53 by CDK1 and PLK1 is required for the timely disassembly of the central NPC framework.
Introduction
During entry into mitosis in mammalian cells, the complex structure of the nuclear envelope (NE) is disassembled to allow for the formation of the cytoplasmic mitotic spindle. Nuclear envelope breakdown (NEBD) is tightly coordinated with the cell cycle and involves the disassembly of nuclear pore complexes (NPCs), the depolymerization of the nuclear lamina, and the release of the NE membrane from chromatin (Champion et al., 2016). A decisive aspect of NEBD is the disintegration of NPCs, leading to permeabilization of the NE, and the mixing of nuclear and cytoplasmic components.
NPCs are huge macromolecular assemblies that are built by multiple copies of ∼30 different subunits, termed nucleoporins (Nups). Mammalian NPCs may contain up to 1,000 Nups and have a molecular weight of about ∼100 MDa. Two large nucleoporin subcomplexes, the inner ring complex (IRC) and the Y-complex, serve as modular, repetitive building blocks of the central NPC framework (Figure S1). These NPC scaffold complexes form three large ring-shaped structures that are attached to the pore membrane by interaction with transmembrane nucleoporins and the lipid bilayer. Attached to these rings are nucleoporins containing non-globular phenylalanine-glycine (FG) repeat domains, which functionalize the NPC as a transport channel and diffusion barrier. FG domains serve as binding sites for nuclear transport receptors and restrict the passive diffusion of inert macromolecules >30 kDa (Hoelz et al., 2016, Knockenhauer and Schwartz, 2016, Schmidt and Görlich, 2016). Nup98 is a crucial constituent of the NPC permeability barrier, which is formed by multivalent interactions of Nup98's self-cohesive FG repeat domain (Hülsmann et al., 2012).
While recent structural analyses have provided key insights into the organizational principles of the NPC scaffold (Hoelz et al., 2016), it not only remains enigmatic how nucleoporins are brought together for NPC formation, but also as to how these gigantic complexes can be rapidly disassembled at the onset of open mitosis. Deterioration of NE barrier function and release of Nup98 represent early signs of mitotic NPC disassembly in prophase (Dultz et al., 2008, Laurell and Kutay, 2011). Loss of the NE diffusion barrier and dissociation of Nup98 from NPCs are driven by the stepwise hyperphosphorylation of Nup98 by several mitotic kinases, including cyclin-dependent kinase 1 (CDK1)-cyclinB and members of the NIMA-related kinase (NEK) family (Laurell et al., 2011). However, it is unclear whether these kinases are sufficient to initialize NPC disassembly or whether they cooperate with additional factors. Furthermore, a mechanistic understanding of subsequent steps of NPC disassembly is lacking, especially with respect to the fate of the large NPC scaffold complexes.
Cellular reorganization during mitotic entry is orchestrated and directly driven by mitotic kinases. Besides cyclinB-CDK1 and NEK kinases, polo-like kinase 1 (PLK1) has emerged as a candidate promoting NEBD and potentially mitotic NPC disintegration. PLK1 activity is required for timely entry into prometaphase in human somatic cells (Lenart et al., 2007), and it promotes NEBD in mouse oocytes and Caenorhabditis elegans (Rahman et al., 2015, Solc et al., 2015). However, as PLK1 is involved in activation of cyclinB-CDK1 (Gavet and Pines, 2010, Lindqvist et al., 2009), it is difficult to differentiate between direct and indirect roles of PLK1 in promoting NEBD. Large-scale proteomic studies have revealed that several nucleoporins are phosphorylated on PLK1 consensus sites during mitosis (Kettenbach et al., 2011, Olsen et al., 2010, Santamaria et al., 2011), hinting at a direct role of PLK1 in NPC disassembly.
We set out to explore the function of PLK1 in mitotic NPC disassembly. Using an in vitro system that allows disentangling the role of mitotic kinases in NEBD, we demonstrate that PLK1 cooperates with CDK1 in mitotic NPC disassembly. We identify the scaffold nucleoporin Nup53 and the NPC gatekeeper Nup98 as two targets for mitotic multisite phosphorylation by CDK1 and PLK1, which promotes the dissociation of these interconnecting Nups from the NE. Reconstitution experiments with purified cyclinB-CDK, PLK1, and NIMA reveal that Nup phosphorylation is a major principle underlying NE permeabilization during NEBD.
Results
PLK1 Is Required for Efficient NPC Disassembly
To test whether PLK1 supports NPC disassembly, we applied a previously developed in vitro system that recapitulates mitotic NPC disintegration on nuclei of semi-permeabilized HeLa cells upon addition of mitotic HeLa cell extracts (Laurell et al., 2011, Marino et al., 2014). This quantitative visual assay allows studying both the kinetics of NE permeabilization based on nuclear influx of a fluorescently labeled dextran and the release of GFP-labeled nucleoporins from NPCs by time-lapse confocal microscopy (Figure 1A).
Figure 1.

Immunodepletion of PLK1 from Mitotic Extracts Delays NEBD In Vitro
(A) Schematic representation of the in vitro NPC disassembly assay.
(B) Mitotic cell extract (ME) was either mock-treated (control depletion with protein A/G sepharose) or depleted with anti-PLK1 antibodies. Extracts were supplemented with a 155 kDa fluorescent dextran and added to semi-permeabilized HeLa cells expressing 2GFP-Nup58. NPC disassembly was monitored by confocal time-lapse microscopy. Scale bar, 10 m.
(C) Quantification of dextran-positive nuclei over time. N = 3, n > 100 cells. Error bars, SEM.
(D) Quantification of 2GFP-Nup58 intensity at the NE. Error bars, SEM.
(E) Quantification of the average time point at which 50% of nuclei were dextran-positive (t50). Error bars, SD; ∗p < 0.05, unpaired t test, two-tailed.
(F) Immunoblot analysis of PLK1 immunodepletion.
(G) In vitro kinase assays with mock and PLK1-depleted extracts using histone H1 and zz-Nup98(678–714) as readouts for CDK1 and PLK1 activity, respectively. Incorporation of 32P was analyzed by autoradiography.
First, we depleted PLK1 from the mitotic cell extract using PLK1-specific antibodies and analyzed the effect of depletion in the in vitro NPC disassembly system. Compared with the mock control, the PLK1-depleted extract was less efficient in triggering NPC disassembly. NE permeabilization was delayed by about 10 min, and the release of 2GFP-Nup58, a central FG Nup, from the NE was strongly retarded (Figures 1B–1F). Importantly, CDK1 activity of the mitotic extract was not affected by depletion of PLK1 as revealed by efficient in vitro phosphorylation of histone H1, an established readout for CDK1 activity (Brizuela et al., 1989). In contrast, phosphorylation of a PLK1 substrate, a peptide derived from Nup98 (see below and Figure S2), was impaired (Figure 1G). Collectively, these data suggest that the presence of PLK1 is required for timely NPC disassembly in vitro.
As a complementary approach, we inhibited PLK1 using the small-molecule inhibitor BI2536 (Steegmaier et al., 2007). Addition of BI2536 to the mitotic extract significantly inhibited NPC disassembly (Figures 2A–2D). Akin to PLK1 depletion, both dextran influx and dissociation of 2GFP-Nup58 from the NE were delayed. To confirm that inhibition of PLK1 accounted for the retardation of NPC disassembly, we added a 1.5-fold molar excess of recombinant PLK1 over BI2536, which restored the activity of the mitotic extract for in vitro phosphorylation of a PLK1 substrate. Importantly, the addition of excess PLK1 significantly enhanced both NE permeabilization and release of 2GFP-Nup58 from the NE compared with BI2536 addition alone. Histone H1 was equally efficiently phosphorylated in both control and PLK1-inhibited mitotic extracts (Figure 2E). Thus, PLK1 supports in vitro NPC disassembly without affecting the activity of CDK1.
Figure 2.

PLK1 Activity Is Required for Timely NPC Disassembly In Vitro
(A) Mitotic extracts used for NPC disassembly were supplemented with either DMSO, 500 nM of the PLK inhibitor BI2536, or BI2536 plus 750 nM purified PLK1. NPC disassembly was monitored by time-lapse microscopy. Scale bar, 10 μm.
(B) Quantification of dextran-positive nuclei over time. N = 3, n > 100 cells. Error bars, SEM.
(C) Quantification of 2GFP-Nup58 intensity at the NE. Error bars, SEM.
(D) Quantification of t50. Error bars, SD; ∗∗p < 0.005, unpaired t test, two-tailed.
(E) In vitro kinase assays monitoring PLK1 and CDK1 activities in the indicated mitotic extracts, as in Figure 1G.
PLK1 Localizes to the NE during Prophase
PLK1 is a key regulator of diverse mitotic processes (Archambault and Glover, 2009, Barr et al., 2004, Petronczki et al., 2008). It dynamically localizes to various intracellular structures in the course of mitotic progression, in part dictated by its interaction with numerous substrates (Schmucker and Sumara, 2014). When we examined the subcellular localization of human PLK1 by immunofluorescence, we observed that PLK1 was specifically enriched at the NE of cells during prophase (Figures 3A and 3C). The specificity of the immunolabeling was confirmed by small interfering RNA-mediated downregulation of PLK1 (Figures 3A and 3B). PLK1 localized to the NE in a punctate pattern, overlapping with GFP-Nup58-positive dots, indicative of NPC association (Figure 3C).
Figure 3.

PLK1 Co-localizes with NPCs in Prophase, and Its C-Terminal PBD Exerts a Dominant-Negative Effect on NPC Disassembly Kinetics In Vitro
(A) Confocal images of immunostaining of PLK1 and NPCs (mAb414) in combination with Hoechst (DNA) staining in HeLa cells. Note that PLK1 localizes to the nuclear rim specifically in prophase cells, as evident by the onset of chromatin condensation, but not in interphase cells (top row). Scale bar, 10 μm.
(B) Lysates of cells treated with control or PLK1 siRNAs were analyzed by immunoblotting with the indicated antibodies.
(C) Immunostaining of PLK1 in GFP-Nup58 expressing HeLa cells. DNA was visualized with Hoechst and prophase cells identified by the onset of chromatin condensation. Scale bar, 10 μm; inset 5 μm.
(D) Confocal images of HeLa cells expressing GFP-PLK, GFP-PBDWT, or GFP-PBDAA. Cells were immunostained with antibodies against Nup88 or mAb414, as indicated. Scale bar, 10 μm.
(E) Mitotic HeLa cell extracts were supplemented with either buffer, 2.5 μM recombinant PBDWT or a PBDAA mutant of reduced affinity for phospho-peptides, and used for in vitro NPC disassembly on semi-permeabilized, 2GFP-Nup58-expressing HeLa cells. Quantification of dextran-positive nuclei over time. Error bars, SEM.
(F) Quantification of 2GFP-Nup58 intensity at the NE. N = 3, n > 100 cells. Error bars, SEM.
(G) Quantification of t50. Error bars, SD; ∗p < 0.05, unpaired t test, two-tailed.
(H) In vitro kinase assay monitoring CDK1 activity in the supplemented mitotic extracts, as in Figure 1G.
The non-catalytic C-terminal domain of PLK1, the so-called polo-box domain (PBD), influences the dynamic subcellular localization of PLK1. The PBD recognizes phospho-epitopes on proteins that have been generated by so-called “priming” kinases, often CDK1. Mediated recruitment of PLK1 to these pre-phosphorylated or “primed” proteins then allows PLK1 to phosphorylate other sites in the vicinity, often on the same substrate (Elia et al., 2003, Reinhardt and Yaffe, 2013). To investigate whether targeting of PLK1 to the NE could involve binding to primed substrates, we tested if the PBD was sufficient for NE localization. Both full-length GFP-PLK1 and GFP-PBD were enriched at the NE in prophase cells (Figure 3D), similar to the endogenous kinase. In contrast, a GFP-PBDAA mutant harboring mutations in two residues critical for phospho-peptide binding, H538A and K540A (Cheng et al., 2003), did not localize to the NE. Thus, PBD targeting to the NE depends on phospho-peptide recognition, indicating that PLK1 may recognize a phosphorylated binding partner at the NE.
If targeting of PLK1 to NPCs by its PBD was functionally relevant for mitotic NPC disintegration, then the isolated PBD lacking the kinase domain should behave as a dominant-negative. Indeed, addition of recombinant GST-PBDWT to the mitotic extracts used in the in vitro NPC disassembly assay significantly delayed both nuclear influx of fluorescently labeled dextran and dissociation of GFP-Nup58 from the NE (Figures 3E–3G), akin to PLK1 depletion or chemical inhibition. In comparison, the GST-PBDAA mutant did not significantly impair the kinetics of NE permeabilization. Collectively, these results suggest a function of PLK1 in mitotic NPC disintegration, potentially guided by phospho-peptide recognition at the NPC mediated by the PBD.
Phosphorylation of Nup98 by CDK1 and PLK1 Contributes to Timely NE Permeabilization
Since interference with PLK1 activity delayed NE permeabilization, the gatekeeper nucleoporin Nup98 was a first candidate PLK1 substrate in mitotic NPC disassembly. Nup98 contains five sites resembling the PLK1 consensus motif [E/D]X[pS/pT][I/L/V/M]X[E] (Santamaria et al., 2011) within its C-terminal NPC targeting domain; two previously identified residues (S568 and S636) (Laurell et al., 2011) and three sites (T691, S692, and S697) that reside in a phosphorylation-sensitive epitope (Figures 4A and S2). Combined mutation of all five residues to alanines strongly impaired in vitro phosphorylation of the purified C-terminal domain of Nup98 (amino acids: 506–863) by PLK1 (Figure 4B).
Figure 4.

Nup98 Is a PLK1 Substrate, and Its Phosphorylation by Multiple Kinases Including PLK1 Promotes NPC Disassembly In Vitro
(A) Schematic representation of human Nup98 indicating the position of CDK1 and PLK1 consensus sites (referring to isoform P52948-2). GLFG, Gly-Leu-Phe-Gly repeat domain; GLEBS, Gle2-binding sequence motif; APD, autoproteolytic domain.
(B) The purified C-terminal domain (amino acids: 506–863) of Nup98 (WT) or a mutant derivative containing Ala substitutions at 5 PLK1 sites (5A) (Coomassie) were incubated with PLK1 in the presence of [γ-32P]ATP. Protein phosphorylation was analyzed by autoradiography.
(C) GFP-Nup98WT and the indicated phosphomimetic mutants were transiently expressed in HeLa cells and analyzed by confocal microscopy. Scale bar, 10 μm.
(D) Quantification of GFP intensity across the NE of the experiment in (C) by averaging line scans. n ≥ 50 cells per condition.
(E) Quantification of dextran-positive nuclei over time during NPC disassembly in semi-permeabilized cells expressing the indicated Nup98 derivatives. N = 3, n > 100 cells. Error bars, SEM.
(F) Quantification of t50. Error bars, SD; ∗p < 0.05, unpaired t test, two-tailed.
(G) Comparison of dextran influx during in vitro NPC disassembly in semi-permeabilized GFP-Nup98 and GFP-Nup58 expressing cells, in the absence or presence of BI2536. N = 4, n > 100 cells. Error bars, SEM.
(H) Comparison of GFP-Nup98 and GFP-Nup58 release in the absence or presence of BI2536. N = 4, n > 100 cells. Error bars, SEM.
(I) Quantification of t50. Error bars, SD; ∗∗p < 0.005, unpaired t test, two-tailed.
(J) In vitro kinase assay monitoring PLK1 activity in the mitotic extracts.
To examine whether phosphorylation of these sites by PLK1 could affect NPC association of Nup98, we analyzed the localization of a phosphomimetic GFP-Nup985E(PLK1) variant (Figures 4C and 4D). Compared with GFP-Nup98 wild-type (WT), GFP-Nup985E(PLK1) was targeted to the NE slightly less efficiently. Mislocalization was more pronounced for GFP-Nup988E(CDK1), a mutant in which all previously identified CDK1 sites in Nup98 (Laurell et al., 2011) were rendered to phosphomimetic Glu residues. Strikingly, the combined mutation of all CDK1 and PLK1 sites caused severe NPC targeting defects, as GFP-Nup9813E(CDK1/PLK1) was almost entirely displaced from the NE. This suggests that phosphorylation by both CDK1 and PLK1 may contribute to the mitotic dissociation of Nup98 from NPCs.
To investigate the relative impact of PLK1 and CDK1 phosphorylation of Nup98 on NE permeabilization, we generated inducible HeLa cell lines with GFP-Nup98 constructs harboring the corresponding phosphorylation-deficient alanine substitutions. To be able to remove endogenous Nup98 by RNAi, we also stably introduced an independent copy of full-length cypet-Nup96 (Figure S2H), as both Nups are normally synthesized from a common mRNA (Fontoura et al., 1999). Consistent with the tendency observed for NE localization of Nup98 phosphomimetic mutants, Nup985A(PLK1) delayed NE permeabilization only modestly, while GFP-Nup988A(CDK1) was slightly more inhibitory (Figure 4E). Only the combined mutation of both PLK1 and CDK1 significantly delayed the initiation of NPC disassembly as measured by dextran influx into nuclei (Figures 4E and 4F). Thus, our data demonstrate that phosphorylation of Nup98 by CDK1 and PLK1 contributes to timely mitotic NE permeabilization by phosphorylation of Nup98. Notably, NEK kinases also contribute (Laurell et al., 2011), suggesting that it is the concerted action of multiple protein kinases that aids disintegration of the NE permeability barrier at mitotic onset (see below, Figure 7).
Figure 7.

The Combined Activities of Recombinant Kinases Are Sufficient to Induce NPC Disassembly In Vitro
(A) In vitro kinase assays were used to adjust the activity of purified recombinant cyclinB1-CDK1, PLK1T210D, and NIMA to the respective activities in the mitotic extract (ME) used for in vitro NPC disassembly. Phosphorylation reactions were performed in buffer, ME, or in buffer supplemented with the indicated recombinant protein kinases using histone H1, zz-Nup98(678–714) and casein as readouts for CDK1, PLK1, and NIMA activity, respectively. Incorporation of 32P was analyzed by autoradiography. Coomassie-stained gel serves as loading control.
(B) Quantification of dextran influx into the nuclei of semi-permeabilized HeLa cells expressing 2GFP-Nup58 induced by the addition of the indicated recombinant mitotic protein kinases in buffer supplemented with an energy-regenerating system. N = 4, n > 100 cells. Error bars, SEM.
(C) Quantification of 2GFP-Nup58 intensity at the NE. Error bars, SEM. Note that upper and lower panels in (B) and (C) are derived from the same set of experiments.
(D) Representative confocal images derived form in vitro NPC disassembly experiments shown in (B) and (C), comparing buffer, the mixture of cyclinB1-CDK1, PLK1T210D, and NIMA, and ME.
Nup53 Is a Mitotic Binding Partner and Substrate of PLK1
Given the modest effect of the Nup98 PLK1 site mutant on NE permeabilization but the stronger PLK1 dependence of Nup58 dissociation from NPCs during NEBD (Figures 1 and 2), we directly compared the release of Nup98 and Nup58 from NPCs in the absence and presence of BI2536. PLK1 inhibition affected Nup58 more strongly than Nup98 (Figure 4H), suggesting that there exist additional PLK1 targets within the NPC. To identify additional PLK1 substrates among the Nups, we used the PBD of PLK1 for pull-down experiments with mitotic cell extracts. One particular nucleoporin, Nup53, was retrieved on GST-PBDWT but not on the GST-PBDAA mutant (Figures 5A and S3). Nup53 is a constituent of the IRC, where it functions as an essential linker Nup between the pore membrane and the other members of the IRC (Eisenhardt et al., 2014, Fischer et al., 2015, Hawryluk-Gara et al., 2005, Kosinski et al., 2016, Lin et al., 2016, Sachdev et al., 2012) (Figures 5B and S1). The IRC, in turn, anchors the Nup62/Nup58/Nup54 FG nucleoporin subcomplex within the central NPC (Schwartz, 2016). Notably, only Nup53 was isolated in the GST-PBD pull-down experiments, whereas subunits of the IRC (Nup93, Nup155, Nup188, and Nup205) and various other Nups were not retrieved under these conditions (Figures 5A and S3).
Figure 5.

Nup53 Is Phosphorylated by CDK1 and PLK1 in Mitosis
(A) GST pull-down experiment from mitotic HeLa cell extract using the indicated baits. Of the input and the eluates, 4% and 20%, respectively, were analyzed by immunoblotting with the indicated antibodies.
(B) Scheme of human Nup53 indicating its binding domains for partner Nups and mitotic phosphorylation sites. Sixteen sites matching the CDK1 consensus ([pS/pT]-[P]) are shown in black, and a site matching the PLK1 phosphorylation motif [E/D]X[pS/pT][I/L/V/M]X[E] is highlighted in red. Residues in green squares were confirmed by our mass spectrometric analysis of mitotic Nup53 (see Table S1). RRM, RNA recognition motif.
(C) Interphase or mitotic cell lysates from HeLa cell lines expressing GFP-Nup53WT or the indicated mutant variants were treated without or with alkaline phosphatase (CIP) and processed for immunoblotting with the indicated antibodies. Phosphorylation of histone H3 at S10 served as control for both the cell-cycle state of the extracts and phosphatase treatment. Actin served as control for equal loading. Note that we mutated S314 to a phosphodeficient cysteine as a change of S314 to alanine slightly affected NE localization of Nup53 (Figure S6), suggesting that the chemical features of this residue are critical for integration of Nup53 into NPCs.
(D) In vitro phosphorylation of GST-Nup53WT and GST-Nup5316A by cyclinB1-CDK1 using [γ-32P]ATP. Protein phosphorylation was analyzed by autoradiography.
(E) In vitro phosphorylation of GST-Nup53WT by cyclinB1-CDK1 or PLK1 using [γ-32P]ATP. Phosphorylated GST-Nup53WT was detected by autoradiography.
(F) Far-western analysis of the interaction between the PBD of PLK1 (bait) and GST-Nup53WT or GST-Nup5316A(CDK1) (preys), both pre-treated with cyclinB1-CDK1 in the presence of ATP before separation by SDS-PAGE and blotting onto a membrane.
(G) Sequential in vitro kinase assay. The indicated GST-Nup53 derivatives were first incubated with or without cyclinB1-CDK1 in the presence of unlabeled ATP. After heat inactivation of CDK1, samples were further incubated with [γ-32P]ATP in the absence or presence of PLK1. Phosphorylation of GST-Nup53 variants was analyzed by autoradiography.
Nup53 (also called Nup35 or MP-44) is known to be hyperphosphorylated during mitosis (Stukenberg et al., 1997). Hyperphosphorylation of Nup53 results in a strongly retarded electrophoretic mobility that is reverted by phosphatase treatment (Figure 5C). Altogether, Nup53 contains 16 sites matching the CDK1 consensus [pS/pT]-[P] and one site matching the PLK1 phosphorylation motif [E/D]X[pS/pT][I/L/V/M]X[E] (Santamaria et al., 2011) (Table S1). Mitosis-specific phosphorylation on most of these sites has been previously revealed by large-scale proteomic screens (Dephoure et al., 2008, Kettenbach et al., 2011, Olsen et al., 2010), and was confirmed by our own mass spectrometric analysis (Figure S4, Table S1). The 16 CDK1 sites are situated in presumably unstructured regions of Nup53 that are important for its interaction with the other members of the IRC (Figure 5B). The PLK1 phosphorylation site at S314 is located just upstream of an amphipathic α helix implicated in binding of the transmembrane nucleoporin NDC1 and membranes (Mansfeld et al., 2006, Vollmer et al., 2012).
To test whether the identified CDK1 and PLK1 sites contribute to mitotic hyperphosphorylation of Nup53, we expressed GFP-Nup53 or phosphodeficient mutants in HeLa cells. Whereas GFP-Nup53WT was efficiently hyperphosphorylated in mitosis, GFP-Nup5316A(CDK1) and GFP-Nup5316A/S314C(CDK1/PLK1) were not (Figure 5C). Both mutants migrated only slightly slower than dephosphorylated Nup53 in SDS-PAGE. This indicates that we have identified the majority of mitotic phosphorylation sites within Nup53, although some additional residues might be phosphorylated by other kinases (Table S1).
To confirm that Nup53 is a direct substrate for CDK1 and PLK1, we performed in vitro phosphorylation assays. CyclinB1-CDK1 efficiently phosphorylated recombinant GST-Nup53WT but not GST-Nup5316A(CDK1) (Figure 5D). Nup53 was also phosphorylated by PLK1, albeit very weakly (Figure 5E). Since CDK1 can act as a priming kinase for PLK1, we analyzed whether the PBD of PLK1 can interact with Nup53 depending on priming phosphorylation by cyclinB1-CDK1 using a far-western ligand-binding assay. Indeed, pre-phosphorylated GST-Nup53WT interacted with PBDWT but not with the PBDAA mutant (Figure 5F). In contrast, the GST-Nup5316A(CDK1) mutant did not bind the PBD.
As PLK1 localizes to NPCs in prophase cells (Figure 3), we next tested whether CDK1-mediated phosphorylation of Nup53 contributes to NE targeting of PLK1. RNAi-resistant transgenes encoding for GFP-Nup53WT or GFP-Nup5316A(CDK1) were expressed in HeLa cells in which endogenous Nup53 was simultaneously depleted by RNAi. Then, PLK1 localization was examined by immunofluorescence in late prophase cells, identified by the peripheral nuclear position of condensed chromatin (Figure S5). Indeed, PLK1 staining at the NE was reduced in cells expressing GFP-Nup5316A(CDK1). This indicates that phosphorylation of Nup53 by CDK1 supports PLK1 recruitment to the NE during late prophase, although other factors may contribute to NE targeting of PLK1 in addition.
To determine whether pre-phosphorylation by CDK1 can promote Nup53 phosphorylation by PLK1, we performed sequential kinase assays (Figure 5G). Recombinant GST-Nup53 variants were first incubated with or without cyclinB1-CDK1 in the presence of unlabeled ATP. Then, samples were heat inactivated, split, and further incubated with [γ-32P]ATP in the absence or presence of PLK1. PLK1-dependent phosphorylation of both GST-Nup53WT and GST-Nup5316A(CDK1) was minimal in the absence of prior phosphorylation by CDK1. In contrast, sequential exposure to CDK1 and PLK1 resulted in the efficient phosphorylation of GST-Nup53WT but not of GST-Nup5316A, confirming the inability of this mutant to provide docking sites for PLK1. Importantly, phosphorylation of GST-Nup53S314A(PLK1) by PLK1 was severely reduced compared with GST-Nup53WT, demonstrating that S314 is a major priming-dependent PLK1 phosphorylation site of Nup53.
CDK1- and PLK1-Mediated Phosphorylation of Nup53 Is Required for Its Efficient Release from the NPC
As several of the identified phosphorylation sites reside in regions critical for interaction of Nup53 with partner Nups (Figures 5B and S1), phosphorylation of Nup53 could be required for its release from the NPC during prophase. If so, then the respective phosphomimetic mutations in Nup53 should, in converse, impair NPC integration of Nup53 during interphase. GFP-Nup53WT and mutant variants were expressed in HeLa cells depleted of endogenous Nup53 by RNAi. Compared with the protein, GFP-Nup5316E(CDK1) was less efficiently recruited to the NE (Figures 6A and 6B). Similarly, mutation of the single PLK1 site at S314 in GFP-Nup53S314E(PLK1) affected NE localization. The combined mutation of the CDK1 and PLK1 sites to phosphomimetic residues almost completely abolished NPC integration of Nup53, indicating that hyperphosphorylation of Nup53 might be incompatible with its NPC association.
Figure 6.

CDK1- and PLK1-Mediated Phosphorylation of Nup53 Is Required for Its Efficient Release from the NPC
(A) Confocal images of HeLa cells depleted of endogenous Nup53 by RNAi and expressing siRNA-resistant GFP-Nup53WT and the indicated GFP-Nup53 variants. Scale bar, 10 μm.
(B) Quantification of GFP intensity across the NE of experiment in (A) by averaging line scans. n ≥ 50 cells per condition.
(C) Quantification of dextran influx in the course of in vitro NEBD reactions performed with semi-permeabilized HeLa cells depleted of endogenous Nup53 and expressing siRNA-resistant GFP-Nup53WT or GFP-Nup53 variants. WT(-TET); GFP-Nup53WT cells in the absence of tetracycline and RNAi. N = 4, n > 100 cells. Error bars, SEM.
(D) Quantification of release of GFP-Nup53WT or the indicated GFP-Nup53 variants from the NE in the course of NPC disassembly in vitro monitored by confocal microscopy. Error bars, SEM.
(E) Quantification of t50. Error bars, SD; ∗p < 0.05, ∗∗p < 0.005, unpaired t test, two-tailed.
(F) GST pull-down experiment from interphase HeLa cell extracts with either GST or the indicated Nup53 phosphomimetic mutants. Of the input and the eluates, 4% and 20%, respectively, were analyzed by immunoblotting with the indicated antibodies. Ponceau-stained membrane serves as loading control.
(G) GST, GST-Nup53WT or the indicated phosphomimetic GST-Nup53 variants (all with mutations F179E and W210E to decrease direct membrane binding), were incubated with lysates derived from E. coli overexpressing either the C-terminal membrane-embedded nucleoplasmic domain of Xenopus GP210 or mouse full-length NDC1. 25% of the eluates and 3% of the input were analyzed by immunoblotting using an anti-His6 antibody.
(H) Quantification of GP210 or NDC1 binding to the GST-Nup53 variants normalized to GST. Average of four independent experiments. Individual data points are shown.
Next, we addressed the importance of Nup53 phosphorylation for its mitotic release from NPCs by measuring the effect of phosphodeficient GFP-Nup53 variants in the in vitro NPC disassembly assay (Figures 6C–6E and S6). This analysis revealed that dextran influx into nuclei derived from GFP-Nup5316A(CDK1)-expressing cells was delayed by about 8 min compared with cells expressing GFP-Nup53WT, indicating that Nup53 phosphorylation by CDK1 contributes to NE permeabilization. GFP-Nup53S314C had no effect on NE permeabilization in the in vitro NPC disassembly assay, neither alone nor in combination with the mutated CDK1 sites (Figures 6C–6E). Importantly, however, GFP-Nup53S314C(PLK1) was retarded in its release from NPCs. Likewise, the dissociation of GFP-Nup5316A(CDK1) was delayed. The combined mutation of the CDK1 and PLK1 sites was not additive, consistent with the assumption that phosphorylation of S314 is promoted by priming phosphorylation of Nup53 by CDK1. Collectively, these data show that mitotic hyperphosphorylation of Nup53 by CDK1 and PLK1 contributes to its removal from NPCs.
Since Nup53 is a component of the IRC, which serves as the NPC anchor for the trimeric Nup62/Nup58/Nup54, we also tested whether phosphorylation of Nup53 might contribute to the release of Nup58 during mitotic NPC disassembly. Release of 2RFP-Nup58 from nuclei of GFP-Nup5316A(CDK1)-expressing cells was retarded compared with the GFP-Nup53WT control (Figure S7), indicating that phosphorylation of Nup53 promotes the liberation of the Nup62/Nup58/Nup54 subcomplex.
Nup53 Phosphorylation Diminishes the Interaction with Its Partner Nups
Mitotic hyperphosphorylation of Nup53 might break its contacts to the various neighboring Nups (Figure S1). To examine how modification of the identified phosphorylation sites might affect binding to other Nups, we performed pull-down experiments with recombinant phosphomimetic Nup53 variants from HeLa cell extracts. Strikingly, mutation of the 16 CDK1 sites in GST-Nup5316E(CDK1) impaired the interaction of Nup53 with IRC members Nup93 and Nup205 (Figure 6F), but still allowed binding to the transmembrane Nup NDC1, albeit with reduced efficiency. In contrast, mutation of the PLK1 phosphorylation site at S314, situated close to the C-terminal helix of Nup53, which is important for NDC1 binding (Mansfeld et al., 2006, Vollmer et al., 2012), only slightly decreased the interaction with Nup93 and Nup205, but impaired association with NDC1.
To further disentangle the role of S314 in NDC1 binding from interactions with the other partner Nups, we performed direct binding experiments. We expressed both mouse NDC1 and, as a negative control, a membrane-bound fragment of the pore membrane protein GP210 in Escherichia coli (Eisenhardt et al., 2014). Membrane proteins were then used for pull-down experiments with GST-Nup53 derivatives. To also separate NDC1 interaction of Nup53 from membrane binding, all Nup53 constructs harbored two mutations in the RNA recognition motif (RRM) domain that decrease its membrane association (Mansfeld et al., 2006, Vollmer et al., 2012). Strikingly, NDC1 interaction of the S314E mutant was reduced to background levels, whereas GST-Nup5316E(CDK1) bound NDC1 efficiently (Figures 6G and 6H). Taken together, these data identify the PLK1 phosphorylation site S314 of Nup53 as a critical residue for NDC1 interaction, thereby providing a molecular explanation as to why phosphorylation of this residue promotes the timely release of Nup53 from the NPC during NEBD.
Reconstitution of Initial Steps of Mitotic NPC Disassembly
In addition to CDK1 and the NEKs (Laurell et al., 2011), PLK1 is the third type of mitotic kinase that supports mitotic NPC disassembly (Figures 1, 2, 3, and 4). Conceptually, breakage of protein-protein interactions within the NPC by mitotic protein kinases might be a mechanism that could well explain the fast kinetics of NPC disassembly. To test this model and to provide further evidence for a direct role of these kinases in NPC disassembly, we analyzed whether they were sufficient as soluble factors to induce NPC disassembly on nuclei of semi-permeabilized cells in vitro. For these in vitro reconstitution experiments, we purified cyclinB1-CDK1 and a constitutively active variant of PLK1, PLK1T210D (Lee and Erikson, 1997), from baculovirus-infected insect cells. As a member of the NEK family, we used recombinant Aspergillus nidulans NIMA expressed in bacteria, guided by our previous observation that addition of NIMA to mitotic HeLa cell extracts stimulates NPC disassembly in vitro (Laurell et al., 2011). A further essential step in the reconstitution approach was the identification a buffer compatible with both the long-term stability of the nuclei in semi-permeabilized cells and the activities of the purified kinases (see the STAR Methods). Using this buffer, we adjusted the concentrations of the protein kinases on their respective substrates such that they corresponded to their activities in the mitotic HeLa cell extract used as positive control in the in vitro NEBD assay (Figure 7A).
We then studied both the individual and combinatorial effects of the recombinant mitotic kinases on nuclei of semi-permeabilized HeLa cells expressing 2GFP-Nup58 in the presence of a fluorescently labeled dextran and an energy mix containing ATP. Neither NIMA nor PLK1T210D alone had a significant effect on NE permeabilization and release of 2GFP-Nup58 from the NE (Figures 7B–7D). In contrast, cyclinB1-CDK1 triggered the onset of NE permeabilization, but with a long delay of more than 60 min, and with only a very minor impact on the dissociation of 2GFP-Nup58. Combined addition of PLK1T210D or NIMA and cyclinB1-CDK1 further promoted both dextran influx and Nup58 release. Importantly, the mixture of all three kinases was nearly as efficient as the mitotic extract in NE permeabilization, indicating that protein kinases are sufficient as soluble factors for the initiation of NPC disassembly. With respect to Nup58 dissociation, the kinase cocktail also accelerated the process considerably, although additional activities seem to stimulate the process in the mitotic extract in comparison. We conclude that PLK1 is an important player in mitotic NPC disassembly, but that it is the combined activities of cyclinB1-CDK1, PLK1, and NEK kinases that drive initial steps of NPC disassembly and promote the efficient disintegration of the central NPC scaffold.
Discussion
PLK1 Directly Contributes to Mitotic NPC Disassembly
This work demonstrates that protein kinases are major drivers of mitotic NPC disintegration, highlighting the key role of CDK1 and identifying a direct function of PLK1 in this process. Several previous studies performed in living cells or organisms had implicated PLK1 in the timely progression of cells into prometaphase (Lenart et al., 2007) or NEBD (Li et al., 2010, Rahman et al., 2015, Solc et al., 2015). However, whether this involved a direct role of PLK1 in NPC disassembly had been difficult to deduce. We provide three lines of evidence for a direct function of PLK1 in mitotic NPC disassembly. First, by exploiting a quantitative in vitro system that recapitulates NPC disassembly on nuclei of semi-permeabilized HeLa cells, we show that both chemical inhibition of PLK1 activity and PLK1 immunodepletion significantly delay NE permeabilization and the release of the central nucleoporin Nup58 from NPCs, without affecting the activity of CDK1 in the mitotic cell extracts used to induce NEBD. Second, PLK1 promotes NPC disintegration in a reconstituted system in which we added the purified protein kinases PLK1, cyclinB1-CDK1, and NIMA as the sole soluble components for in vitro NPC disassembly. And third, we identify mitotic PLK1 phosphorylation sites in two nucleoporins, Nup98 and Nup53, and demonstrate that phosphorylation of these sites contributes to NPC disintegration. Collectively, our molecular analysis extends previous studies on the role for CDK1 in NPC disassembly (Laurell et al., 2011, Mühlhäusser and Kutay, 2007, Turgay et al., 2014), and provides evidence for a direct function of PLK1 in NPC disintegration during NEBD in human cells. Notably, in a parallel study, Pintard and colleagues revealed that C. elegans PLK-1 is also recruited to NPCs in worm embryos to promote NEBD (Martino et al., 2017 [this issue of Developmental Cell]).
Mitotic Hyperphosphorylation of Key Multivalent Nups Contributes to NPC Disassembly
In vitro binding experiments and structural analyses have identified Nup98 and Nup53 as NPC constituents that interact with various other Nups, thereby serving as multivalent linker proteins within the NPC scaffold (Fischer et al., 2015, Hawryluk-Gara et al., 2005, Hawryluk-Gara et al., 2008, Kosinski et al., 2016, Lin et al., 2016, Stuwe et al., 2015). The C-terminal NPC targeting domain of Nup98 interacts with both the Y-complex, i.e., Nup96, and members of the IRC (Figure S1), thereby contributing to the connectivity between the peripheral and inner scaffold ring complexes. Nup53, in turn, serves as the core backbone for the assembly of the IRC, providing interaction surfaces for Nup155, Nup93, and Nup205 (Fischer et al., 2015, Hawryluk-Gara et al., 2005, Hawryluk-Gara et al., 2008, Kosinski et al., 2016, Lin et al., 2016, Stuwe et al., 2015). Importantly, Nup53 also links the IRC to the pore membrane by binding to both the transmembrane nucleoporin NDC1 and the lipid bilayer (Mansfeld et al., 2006, Vollmer et al., 2012).
Nup98 Hyperphosphorylation
Nup98 plays a decisive role in determining the NPC permeability characteristics, involving self-cohesive interactions of its N-terminal GLFG domain (Hülsmann et al., 2012). Consistent with this function, we had previously shown that hyperphosphorylation of Nup98 promotes timely NE permeabilization and its release from NPCs during mitotic entry (Laurell et al., 2011). Nup98 phosphorylation is mediated by several mitotic kinases, including CDK1, NEKs (Laurell et al., 2011) and, as shown here, PLK1.
The mitotic phosphorylation sites of Nup98 are spread over its entire C-terminal NPC targeting domain. Recent structural analysis of Nup145N, the yeast homolog of Nup98, revealed that this C-terminal domain engages into interactions with various other Nups, including Nup170 (human Nup155), Nup188 (human Nup188), and Nup192 (human Nup205) of the IRC, and Nup145C (human Nup96) of the Y-complex, thereby interconnecting distinct Nup subcomplexes within the NPC (Fischer et al., 2015, Kosinski et al., 2016, Lin et al., 2016). Our analyses demonstrate that multisite phosphorylation of Nup98 is needed to efficiently liberate Nup98 out of this protein interaction network to initiate NPC disassembly (Laurell et al., 2011, Lin et al., 2016). In support for a direct role of Nup98 phosphorylation in breaking Nup-Nup interaction, two phosphomimetic mutations in a loop of Nup98 contacting Nup155 perturb the interaction of both Nups in vitro (Laurell et al., 2011, Lin et al., 2016). Interestingly, the two residues seem not to directly contribute to Nup-Nup interaction (Laurell et al., 2011, Lin et al., 2016). Such configuration is ideally suited to explain how unzipping of Nup-Nup interactions by protein kinases could be initiated, since phosphorylation sites that trigger NPC disassembly should be readily accessible. Notably, we also identified a cluster of PLK1 sites (T691, S692, and S697) residing in a PLK1 phosphorylation-sensitive epitope in Nup98 fulfilling this criterion. The epitope is positioned in a potentially unstructured region of Nup98 preceding its C-terminal autoproteolytic domain, and is accessible to antibody recognition and phosphorylation by PLK1. Phosphorylation of this region could affect the overall configuration of the C-terminal domain of Nup98, perhaps by an allosteric mechanism. Notably, phosphorylation of Nup98's partner Nups may also contribute to its release from the NPC (see also below). In general, it can be expected that phosphorylation of various Nups situated at key interaction nodes within the NPC promotes the rapid disintegration of NPCs during NEBD.
Nup53 Hyperphosphorylation
Mitotic hyperphosphorylation of Nup53 is mainly driven by CDK1. The 16 CDK1 sites in human Nup53 are situated in the unstructured N- and C-terminal parts of Nup53 (flanking the central RRM domain), which are involved in the interaction with other IRC constituents. Consistently, we observed that a phosphomimetic Nup5316E(CDK1) mutant was compromised in binding to the IRC members Nup93 and Nup205. Yet, phosphorylation of Nup53 might not only disrupt protein-protein interactions within the IRC, but also weaken its association with the pore membrane, which involves binding to both the lipid bilayer and the transmembrane nucleoporin NDC1 (Mansfeld et al., 2006, Vollmer et al., 2012). Phosphorylation of two CDK1 sites within an N-terminal membrane-binding region of Xenopus laevis Nup53 (S94/T100 corresponding to S100/T106 in human Nup53) was shown to reduce Nup53's ability to bind to liposomes (Vollmer et al., 2012). Our analysis further indicates that dissociation of Nup53 from the pore membrane is supported by PLK1-mediated phosphorylation of S314. However, only the combined phosphomimetic substitution of the identified CDK1 and PLK1 sites impaired NPC incorporation of Nup53, indicating that multiple interactions contribute to the anchorage of Nup53 at NPCs.
Multisite phosphorylation of Nup53 is needed for breaking its multivalent interactions with partner nucleoporins and membranes in the context of NPC disassembly. Interestingly, the phosphodeficient Nup5316A(CDK1) mutant was not only retarded in its own release from the NE, but also caused a delay in both Nup58 dissociation and NE permeabilization. The latter could imply that phosphorylation of Nup53 contributes to the liberation of Nup98. Recent crosslinking analysis indeed suggests that Nup53 and Nup98 might be in close proximity (Kosinski et al., 2016). In addition, the IRC components Nup155 and Nup188/205 associate with both Nup53 and Nup98 (Lin et al., 2016), and their dissociation from Nup53 may support the timely release of Nup98 and thereby NE permeabilization.
Collectively, we have provided key insights into the mechanism of how the Nup53-Nup93 subcomplex is disintegrated, involving a sequence of phosphorylation events that promotes the unzipping of Nup-Nup interactions, culminating in the liberation of Nup53. According to such a model, initial CDK1-dependent phosphorylation of Nup53 would perturb its interaction with other scaffold Nups, i.e., Nup93, Nup205, and potentially Nup155, followed by the priming-dependent recruitment of PLK1 for phosphorylation of S314 to promote dissociation of Nup53 from NDC1. In this scenario, PLK1 could temporally couple Nup53 release from the NE to the rise in CDK1 activity accompanying prophase (Gavet and Pines, 2010) by “decoding” priming phosphorylation on Nup53.
Unzipping of Nup-Nup Interactions: Protein Kinases Execute Direct Control
Our reconstitution approach of NPC disassembly demonstrated that the protein kinases cyclinB1-CDK1, PLK1, and NIMA are together sufficient as soluble components to initiate mitotic NPC disassembly in vitro. Although it cannot be ruled out that other activities present in the semi-permeabilized cells also contribute, our data suggest that unzipping of Nup-Nup interactions by protein phosphorylation is an important principle underlying early steps of mitotic NPC disassembly. A phosphorylation-based mechanism of NPC disassembly would readily explain the rapid kinetics of the process and its reversibility during mitotic exit. Whereas a cocktail of mitotic kinases efficiently induced NE permeabilization, release of Nup58 by the purified kinases was slower compared with the mitotic cell extract, indicating that additional soluble factors may support the disassembly of the central NPC framework. Whether these are protein kinases or other factors awaits future investigation.
The great majority of the identified mitotic phosphorylation sites in Nups are CDK1 consensus sites, implicating a major role for this kinase in NPC disintegration. However, our reconstitution experiments showed that NPC disassembly required the concerted action of multiple kinases including PLK1 and NEKs. Thus, two mechanisms appear to safeguard that NPC disassembly is only occurring in cells fully committed to mitosis; the use of several mitotic kinases and, secondly, multisite phosphorylation which imposes an appropriate threshold of kinase activities (Salazar and Hofer, 2009). Three different types of kinases, namely CDK1, PLK1, and NEK6/NEK7 contribute to multisite phosphorylation of Nup98 and efficient NE permeabilization. The activation of these kinases is tightly interwoven. In mammalian cells, PLK1 has been suggested to act upstream of CDK1 activation (Gavet and Pines, 2010). CDK1 and PLK1, in turn, drive activation of NEK9, which promotes NEK6 and NEK7 activity (Bertran et al., 2011). The activity of CDK1 progressively increases during prophase, and NEBD occurs at a very high level of CDK1 activity compared with other processes accompanying mitotic entry (Gavet and Pines, 2010). In conclusion, hyperphosphorylation of multivalent interconnecting Nups by several protein kinases emerges as a key mechanism controlling and executing the rapid and complete disassembly of NPCs during mitotic entry.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-PLK1 antibody | This paper | N/A |
| Mouse monoclonal anti-actin | Sigma-Aldrich | cat# A1978 |
| Mouse monoclonal anti-tubulin | Sigma-Aldrich | cat# T5168 |
| Rabbit polyclonal anti-Nup88 antibody | This paper | N/A |
| Mouse monoclonal anti-mAb414 | Abcam | cat# ab24609 |
| Rabbit polyclonal anti-Nup53 antibody | This paper | N/A |
| Rabbit polyclonal anti-Nup98 antibody | Laurell et al., 2011 | N/A |
| Rabbit polyclonal anti-Nup188 antibody | This paper | N/A |
| Rabbit polyclonal anti-Nup155 antibody | This paper | N/A |
| Rabbit polyclonal anti-Nup93 antibody | This paper | N/A |
| Rabbit polyclonal anti-Ndc1 antibody | Mansfeld et al., 2006 | N/A |
| Goat polyclonal anti-GST antibody | Biacore | cat# BR-1002-23 |
| Rabbit polyclonal anti-GFP antibody | Turgay et al., 2014 | N/A |
| Rabbit polyclonal anti-pH3 antibody | Cell Signaling | cat# 9701S |
| Mouse monoclonal anti-mNup98 | Santa Cruz | cat# sc-74578; RRID: AB_2157953 |
| Rabbit polyclonal anti-Nup96 antibody | Laurell et al., 2011 | N/A |
| Rat monoclonal anti-Nup62 antibody | Abcam | cat# ab188413 |
| Rabbit polyclonal anti-Nup58 antibody | This paper | N/A |
| Rabbit polyclonal anti-Nup54 antibody | This paper | N/A |
| Rabbit polyclonal anti-Nup50 antibody | Abcam | cat# ab137092 |
| Mouse monoclonal anti-TPR | Hase et al., 2001 | N/A |
| Rabbit polyclonal anti-Nup160 antibody | This paper | N/A |
| Rabbit polyclonal anti-Nup133 antibody | This paper | N/A |
| Rabbit polyclonal anti-Nup107 antibody | Mansfeld et al., 2006 | N/A |
| Rabbit polyclonal anti-Nup205 antibody | Mansfeld et al., 2006 | N/A |
| Rabbit polyclonal anti-Nup58 antibody | This paper | N/A |
| Bacterial and Virus Strains | ||
| E.coli BL21 (DE3)-RIL (expression in bacteria) | Agilent technologies | N/A |
| Rosetta (expression in bacteria) | Novagen | N/A |
| BL21(DE3) (expression in bacteria) | NEB | N/A |
| DH10 | ThermoFisher | cat# 10361012 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| BI2536 | Selleckchem | cat# S1109 |
| Roscovitine | Calbiochem | cat# 557360 |
| CIP | NEB | cat# M0290S |
| Trypsin | Promega | cat# V5111 |
| Chymotrypsin | Pierce | cat# 90056 |
| Lysyl endopeptidase | Wako Chemicals | cat# 125-05061 |
| H1 | Calbiochem | cat# 382150 |
| TRITC-Dextran (155kDa) | Sigma | cat# T1287 |
| Casein | Sigma | cat# C4032 |
| γ-32P-ATP | Hartmann Analytic | cat# SRP301 |
| His-PLK1WT | This paper | N/A |
| His-PLK1T210D (constitutive active) | This paper | N/A |
| GST-PBDWT (aa326-603) | This paper | N/A |
| GST-PBDAA (aa326-603; H538A,K540A) | This paper | N/A |
| His-PBDWT (aa326-603) | This paper | N/A |
| His-PBDAA (aa326-603; H538A,K540A) | This paper | N/A |
| HiszzNup98 (aa678-714) | This paper | N/A |
| HisNup98WT (aa506-863) | This paper | N/A |
| His-Nup985A (PLK1 site mutant) | This paper | N/A |
| GST-Nup53WT | This paper | N/A |
| GST-Nup5316A (CDK1 mutant) | This paper | N/A |
| GST-Nup53S314A (PLK1 mutant) | This paper | N/A |
| GST-Nup5316A+S314A (CDK1 and PLK1 mutant) | This paper | N/A |
| His-cyclinB1 | Mühlhausser and Kutay, 2007 | N/A |
| His-CDK1 | Mühlhausser and Kutay, 2007 | N/A |
| His-NimA | Laurell et al., 2011 | N/A |
| GST-Nup53S314E (PLK1 mutant) | This paper | N/A |
| GST-Nup5316E (CDK1 mutant) | This paper | N/A |
| His-GP210 (TMR+NPD) | Eisenhardt et al., 2014) | N/A |
| His-Ndc1 (aa1-660) | Eisenhardt et al., 2014) | N/A |
| His-Nup98WT (aa 506-863) | This paper | N/A |
| His-Nup98S687A (aa 506-863) | This paper | N/A |
| His-Nup98S688A (aa 506-863) | This paper | N/A |
| His-Nup98T691A (aa 506-863) | This paper | N/A |
| His-Nup98S692A (aa 506-863) | This paper | N/A |
| His-Nup98S697A (aa 506-863) | This paper | N/A |
| Nup53 peptide: FAVEPQGPALGSEPMMLGC | This paper | N/A |
| Nup93 peptide: LENDGSRKPGVIDKFTSDTC | This paper | N/A |
| Nup155 peptide: FLDRNSQFAGGPLGNPNTTC | This paper | N/A |
| Nup188 peptide: CESQEPLIQLVQAFVRHMQR | This paper | N/A |
| Nup88 peptide: LREGLKNQSPTEAEKPASSC | This paper | N/A |
| Nup54 peptide: CPIIWEQAKVDNPDSEKLI | This paper | N/A |
| Nup58 peptide: DFSSSSDKKSDKTGTRPEDC | This paper | N/A |
| Nup160 peptide: ELSGAERERPRHFREFTVC | This paper | N/A |
| Nup133 peptide: CPYFEFVLKANYEYYVQGQI | This paper | N/A |
| Nup96 peptide: CSLHHPPDRTSDSTPDPQRV | Laurell et al., 2011 | N/A |
| Nup98 peptide: CNRDSENLASPSEYPENGER | Laurell et al., 2011 | N/A |
| Nup107 peptide: LLQASQDENFGNTTPRNQVC | Mansfeld et al., 2006 | N/A |
| NDC1 peptide: CQASAEHQKRLQQFLEFKE | Mansfeld et al., 2006 | N/A |
| Nup205 peptide: TPSLSETVNRDGPRQDTQAC | Mansfeld et al., 2006 | N/A |
| Experimental Models: Cell Lines | ||
| HeLa + 2GFPNup58 | Laurell et al., 2011 | N/A |
| HeLa T-Rex Flp-In | Hafner et al., 2014 | N/A |
| HeLa T-Rex Flp-In + GFP-PBDWT | This paper | N/A |
| HeLa T-Rex Flp-In + GFP-PBDAA | This paper | N/A |
| HeLa T-Rex Flp-In + GFP-Nup98WT + cypetNup96 | This paper | N/A |
| HeLa T-Rex Flp-In + GFP-Nup985A + cypetNup96 | This paper | N/A |
| HeLa T-Rex Flp-In + GFP-Nup988A + cypetNup96 | This paper | N/A |
| HeLa T-Rex Flp-In + GFP-Nup9813A + cypetNup96 | This paper | N/A |
| HeLa T-Rex Flp-In + GFP-Nup53WT | This paper | N/A |
| HeLa T-Rex Flp-In + GFP-Nup53S314E | This paper | N/A |
| HeLa T-Rex Flp-In + GFP-Nup5316E | This paper | N/A |
| HeLa T-Rex Flp-In + GFP-Nup5316E+S314E | This paper | N/A |
| HeLa T-Rex Flp-In + GFP-Nup53S314C | This paper | N/A |
| HeLa T-Rex Flp-In + GFP-Nup53S314A | This paper | N/A |
| HeLa T-Rex Flp-In + GFP-Nup5316A | This paper | N/A |
| HeLa T-Rex Flp-In + GFP-Nup5316A+S314C | This paper | N/A |
| HeLa T-Rex Flp-In + GFP-Nup53WT +2RFPNup58 | This paper | N/A |
| HeLa T-Rex Flp-In + GFP-Nup5316A +2RFPNup58 | This paper | N/A |
| Sf21 cells | Mühlhäusser and Kutay, 2007 | N/A |
| Oligonucleotides | ||
| siNup98/96: UCUGAAACAAAGACUCAUUdTdT | This paper | N/A |
| siPLK1: CGCGGGCAAGATTGTGCCTAAUUdTdT | This paper | N/A |
| siNup53: GGAAGUACUCCUAGGAUUUdTdT | This paper | N/A |
| Sicontrol: AllStars negative control | Badertscher et al., 2015 | N/A |
| PCR and cloning Primers | See Table S2 | N/A |
| Recombinant DNA | ||
| pOG44 (FLP recombinase) | ThermoFisher | cat# V600520 |
| pcDNA5/FRT/TO-GFP-PLK1 PBDWT | This paper | N/A |
| pcDNA5/FRT/TO-GFP-PLK1 PBDAA | This paper | N/A |
| pFastBac-His-PLK1 | This paper | N/A |
| pFastBacDual-His-PLK1T210D | This paper | N/A |
| pGEX-GST-PBDWT (aa326-603) | This paper | N/A |
| pGEX-GST-PBDAA (aa326-603; H538A,K540A) | This paper | N/A |
| pQE80-His-PBDWT (aa326-603) | This paper | N/A |
| pQE80-His-PBDAA (aa326-603; H538A,K540A) | This paper | N/A |
| pet28a-HiszzNup98 (aa678-714) | This paper | N/A |
| pet28a-HisNup98WT (aa506-863) | This paper | N/A |
| pet28a-His-Nup985A (PLK1 site mutant) | This paper | N/A |
| pGEX-GST-Nup53WT | This paper | N/A |
| pGEX-GST-Nup5316A (CDK1 mutant) | This paper | N/A |
| pGEX-GST-Nup53S314A (PLK1 mutant) | This paper | N/A |
| pGEX-GST-Nup5316A+S314A (CDK1 and PLK1 mutant) | This paper | N/A |
| pGEX-GST-Nup53WT | This paper | N/A |
| pGEX-GST-Nup5316E | This paper | N/A |
| pGEX-GST-Nup53S314E | This paper | N/A |
| pFastBac-His-cyclinB1 | Mühlhausser and Kutay, 2007 | N/A |
| pFastBac-His-CDK1 | Mühlhausser and Kutay, 2007 | N/A |
| His-NimA | Laurell et al., 2011 | N/A |
| pGEX-GST-Nup53S314E (PLK1 mutant) | This paper | N/A |
| pGEX-GST-Nup5316E (CDK1 mutant) | This paper | N/A |
| pet28a-MISTIC-GP210 (TMR+NPD) | Eisenhardt et al., 2014, Table S1 | N/A |
| pet28a MISTIC-Ndc1 (aa1-660) | Eisenhardt et al., 2014, Table S1 | N/A |
| pEGFPC1-Nup98 (aa 555-712) | This paper | N/A |
| pEGFPC1-Nup98 (aa 506-683) | This paper | N/A |
| pEGFPC1-Nup98 (aa 506-712) | This paper | N/A |
| pEGFPC1-Nup98 (aa 506-705) | This paper | N/A |
| pEGFPC1-Nup98S687E (aa 506-712) | This paper | N/A |
| pEGFPC1-Nup98S688E (aa 506-712) | This paper | N/A |
| pEGFPC1-Nup98T691E (aa 506-712) | This paper | N/A |
| pEGFPC1-Nup98S692E (aa 506-712) | This paper | N/A |
| pEGFPC1-Nup98S697E (aa 506-712) | This paper | N/A |
| pet28a-His-Nup98WT (aa 506-863) | This paper | N/A |
| pet28a-His-Nup98S687A (aa 506-863) | This paper | N/A |
| pet28a-His-Nup98S688A (aa 506-863) | This paper | N/A |
| pet28a-His-Nup98T691A (aa 506-863) | This paper | N/A |
| pet28a-His-Nup98S692A (aa 506-863) | This paper | N/A |
| pet28a-His-Nup98S697A (aa 506-863) | This paper | N/A |
| pcDNA5/FRT/TO-GFP- Nup98WT | This paper | N/A |
| pcDNA5/FRT/TO-GFP- Nup985E | This paper | N/A |
| pcDNA5/FRT/TO-GFP- Nup988E | This paper | N/A |
| pcDNA5/FRT/TO-GFP- Nup9813E | This paper | N/A |
| pcDNA5/FRT/TO-GFP- Nup985A | This paper | N/A |
| pcDNA5/FRT/TO-GFP- Nup988A | This paper | N/A |
| pcDNA5/FRT/TO-GFP- Nup9813A | This paper | N/A |
| pIRESNeo-cypetNup96 | This paper | N/A |
| pcDNA5/FRT/TO-GFP- Nup53WT | This paper | N/A |
| pcDNA5/FRT/TO-GFP- Nup53S314A | This paper | N/A |
| pcDNA5/FRT/TO-GFP- Nup53S314C | This paper | N/A |
| pcDNA5/FRT/TO-GFP- Nup5316A | This paper | N/A |
| pcDNA5/FRT/TO-GFP- Nup5316A+S314C | This paper | N/A |
| pGEX-GST-Nup5316E(CDK1 mutant) | This paper | N/A |
| pGEX-GST-Nup53S314E(CDK1 mutant) | This paper | N/A |
| pIRESNeo-2RFPNup58 | This paper | N/A |
| Software and Algorithms | ||
| Prism | GraphPad | Ver 5 |
| MATLAB | Mathworks | Ver R2011b |
| ImageJ | NIH | Ver 1.48g |
| Zen 2.3 | Zeiss | Ver 2.3 |
| Leica Application Suite LAS X | Leica | Ver 3.15.16308 |
| Huygens professional software 16.10 | SVI | Ver 16.10 |
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ulrike Kutay (ulrike.kutay@bc.biol.ethz.ch).
Experimental Model and Subject Details
Cell Lines
Cell lines used in this study are described in the Key Resources Table.
Cells were grown at 37°C in Dulbecco’s modified eagle medium (DMEM) with 10% tetracycline-free fetal bovine serum, 100 U/ml penicillin, and 100 U/ml streptomycin in a humidified incubator at 37°C and 5% CO2. Sf21 cells were grown at 27°C in insect medium SF900 with 10% fetal bovine serum. HeLa S3 cells were grown in RPMI medium supplemented with 10% fetal calf serum, 100 U/ml penicillin and 100 mg/ml streptomycin, and 1x ‘MEM Non-Essential Amino Acids’ in a humid atmosphere at 37°C and 5% CO2.
Method Details
Antibodies
All primary antibodies used in this study are listed in the Key Resources Table.
Horseradish-peroxidase-conjugated anti-rabbit or anti-mouse (Sigma) antibodies were used as secondary antibodies. Fluorescent secondary antibodies: goat anti-rabbit 568, goat anti-rabbit 488, goat anti-mouse 633 (all Life Technologies) and goat anti-mouse 568 (Thermo Fisher).
Molecular Cloning
The coding regions of human Nup98 (NM_016320), Nup53 (NM_138285) and PLK1 (NM_005030) were amplified from human cDNA and inserted into pcDNA5(GFP) FRT/TO for generation of stable, tetracycline-inducible HeLa cell lines or transient expression. Cypet-Nup96 and 2RFP-Nup58 were cloned into pIRESneo3. For generation of cell lines, see below. DNA fragments encoding for the PBD of PLK1 were cloned into pQE80 and pcDNA5(GFP) for expression in bacteria and HeLa cells, respectively. For insect cell expression, PLK1 was cloned into a modified pFastBacDUAL vector. Purification of Nup98(506-863), NIMA and cyclinB1-CDK1 have been described (Laurell et al., 2011, Mühlhäusser and Kutay, 2007). pET28a-Nup98(506-863) for protein expression in E. coli has been described (Laurell et al., 2011). A sequence coding for zz-Nup98(678–714) was cloned into pET28a. To express GST-Nup53 and GST in bacteria, PCR fragments were subcloned into pGEX. PLK1 and the constitutive active variant (PLK1T210D) were amplified by PCR and cloned for baculoviral expression into a modified pFastBacDUAL vector (Invitrogen), allowing simultaneous expression of EGFP and the His-fusion protein of interest.
Phosphodeficient or phosphomimetic mutations were introduced into Nup98 and Nup53 by either (1) ligation of primer-modified PCR products, (2) de novo gene synthesis (Geneart), or (3) QuikChange Site-Directed Mutagenesis (Agilent Technologies). RNAi-resistant Nup53 was generated using a synthetic DNA fragment (Geneart) of the Nup53 gene containing silent mutations in the binding sites for the Nup53 siRNA oligonucleotide. The constitutively active PLK1 mutant Thr210Asp and the PLK1 PBD mutant (His538A/Lys540A) deficient in phosphopeptide binding were generated by QuikChange. For interaction studies of GST-Nup53 with integral membrane proteins, mutations F179E and W210E, which prevent dimerization of the RRM domain and direct membrane interaction of Nup53 (Vollmer et al., 2012), were introduced by QuikChange.
To express M. musculus NDC1 and a C-terminal fragment of X. laevis GP210 (Eisenhardt et al., 2014) in bacteria, the respective coding sequences were cloned into pET28a with an N-terminal Mistic (membrane-integrating sequence for translation of integral membrane protein constructs) sequence to enhance expression in E. coli (Roosild et al., 2005). For NDC1, a synthetic DNA, codon-optimized for expression in E. coli encoding full-length Mus musculus NDC1 was cloned with a C-terminal His6-tag. A Xenopus laevis GP210 fragment comprising the transmembrane region and the nucleoplasmic part (Eisenhardt et al., 2014) was cloned into the same vector.
Oligonucleotide primers that have been used to generate constructs for this study are listed in Table S2.
Generation of Cell Lines
HeLa reporter cell lines carrying tetracycline-inducible GFP-Nup53 wild-type and variants were generated using a HeLa Flp-In T-REx cell line (Hafner et al., 2014). HeLa cells expressing HA-Strep-tagged Nup53 for affinity purification were produced by the same approach. HeLa cell lines stably expressing cypet-Nup96 and 2RFP-Nup58 were generated by random integration according to the manufacturer’s (Invitrogen) instructions. Stable cypet-Nup96 and 2RFP-Nup58 expressing clones were used for integration of GFP-Nup98WT/variants and GFP-Nup53WT/16A, respectively, at a predefined genomic FRT (flippase (Flp) recognition target) site. The HeLa cell line stably expressing 2GFP-Nup58 has been described previously (Laurell et al., 2011).
Treatment of Cell Lines
Expression of GFP-Nup98 and GFP-Nup53 was induced by addition of 0.1 μg/ml and 0.005 μg/ml tetracycline (Sigma), respectively. For the experiment shown in Figure S2A, HeLa cells were synchronized in mitosis by a single thymidine block, followed by release into fresh medium and a subsequent nocodazole arrest. Mitotic cells were collected by shake-off, and treated with DMSO or BI2536 (Selleckchem) at 100 nM for 1 hr at 37°C as indicated.
In Vitro NPC Disassembly
In vitro NPC disassembly on semi-permeabilized HeLa cells in presence of mitotic HeLa cell extract was performed as described (Marino et al., 2014). Briefly, HeLa cells expressing the indicated GFP-tagged nucleoporin were seeded into an Ibidi imaging chamber 12–24 hr before the NEBD assay. Cells were semi-permeabilized with 0.002% digitonin in ice-cold permeabilization buffer (PB: 20 mM HEPES-KOH pH 7.6, 110 mM KOAc, 5 mM Mg(OAc)2, 250 mM sucrose, and 0.5 mM EGTA) for 10 min followed by subsequent washing steps with PB. Nuclear disassembly was induced by addition of mitotic extract (ME) supplemented with a TRITC-labeled 155 kDa dextran and an energy regenerating system containing ATP and GTP. Progression of NEBD was monitored by time-lapse confocal microscopy.
For chemical inhibition of PLK1, the small-molecule inhibitor BI2536 (Selleckchem) was added directly to the ME to a final concentration of 500 nM, followed by pre-incubation at RT for 15 min. Recombinant PLK1 was added to the ME to a concentration of 750 nM. Depletion of PLK1 from ME was performed as described (Marino et al., 2014). Briefly, the PLK1 antibody was covalently coupled to protein A/G beads (9:1 mixture; GE Healthcare) using 2 mg of antibody per 1 ml protein A/G beads. PLK1 depletion from ME was performed directly prior to the NEBD assay with a 1:6 ratio of antibody-coupled A/G beads to ME in two subsequent rounds of depletion for 15 min at 4°C using Mobicol spin columns.
For reconstitution of NPC disassembly, HeLa cells expressing 2GFP-Nup58 were seeded at a density of 5000 per well (u-Slide 18-well, Ibidi) 24 hr before imaging, cells were semi-permeabilized with 0.002% digitonin in modified permeabilization buffer (mPB: 25 mM HEPES-KOH pH 7.6, 25 mM KOAc, 7.5 mM MgCl2, 250 mM sucrose, 0.5 mM DTT, and 0.5 mM EGTA). NEBD reactions were performed in mPB supplemented with 10 mg/ml BSA, TRITC–labeled 155 kDa dextran, 2 mM ATP, an energy generating system, 0.15 μM ocadaic acid and purified recombinant kinases. Time-lapse imaging was performed at 25°C at the indicated time intervals, acquiring z stacks on a laser scanning confocal microscope (LSM 710; 40x water immersion objective) (Marino et al., 2014).
siRNAs, Transfections, and Immunofluorescence
For siRNA target sequences, see the Key Resources Table.
siRNA transfections were performed with INTERFERin (Polyplus Transfection) at a final siRNA concentration of 10 nM for Nup96/98 and PLK1 for 30 hr. Depletion of Nup53 was performed by two consecutive transfection of siRNA (first 20 nM siRNA, second 10 nM siRNA after 60 hr) for a total of 5 days. AllStars (QIAGEN) was used as negative control. Transient plasmid transfections were done with X-tremeGene (Roche).
For immunofluorescence, cells were fixed with paraformaldehyde, washed with PBS and permeabilized for 5 min in 0.1% Triton X-100. Immunostaining was performed as described previously (Zemp et al., 2009). Briefly, cells were blocked with blocking solution (BS: 2% BSA in PBS) at RT for 1 hr or at 4°C o/n. All primary antibodies were diluted in BS and cells exposed to the antibody solution at RT for 1 hr. After three washing steps with BS, cells were stained with fluorescent secondary antibodies at a dilution of 1:300. After incubation at RT for 1 hr, cells were washed with BS and coverslips were mounted in Vectashield mounting medium on a glass slide. Confocal microscopy was performed with a Leica SP2 AOBS microscope using a 63×1.4NA, oil, HCX Plan-Apochromat objective or at a Zeiss LSM 880 upright microscope with a 63×1.4NA, oil, DIC Plan-Apochromat objective.
For NPC number determination in Figure S6, cells were seeded on high precision microscope cover glasses (Marienfeld, cat# 0117530). Cells were fixed and immunostained as described above. As secondary antibody an alexa goat anti-mouse 568 (Thermo Fisher cat# A11004) was used. Cover slips were mounted with ProLong® Diamond Antifade Mountant (Thermo Fisher cat# P36970). High-resolution imaging was performed on a STED microscope (Leica SP8 STED; 100x oil immersion objective, N.A. 1.4) and operated with the Leica LASX software. Deconvolution of raw images was performed using Huygens professional software 16.10. NPC numbers were counted per μm2 using ImageJ.
In Vitro Protein De/Phosphorylation
In vitro kinase reactions after NEBD used 1 μl of the respective NEBD reaction mix and were performed in PB in presence of 100 μM ATP, 1 μCi 32P-γ-ATP, and 2.5 μg H1 (Calbiochem), 10 μg casein (Sigma) or 2 μg purified recombinant zz-Nup98(678-714) in a 10 μl total sample volume at 30°C for 20 min. Kinase assays with recombinant kinases were done in kinase buffer KB (25 mM HEPES-KOH pH 7.5, 5 mM MgCl2, 5 mM EGTA and 1 mM DTT) using 2 μg protein substrate and 125 ng cyclinB1-CDK1 or 200 ng PLK1. Sequential kinase assays were performed as above except that 200 μM unlabeled ATP was used for the incubation step with or without cyclinB1-CDK1. Then, samples were heat-inactivated at 65°C for 5 min and further incubated in the presence of buffer or purified PLK1 in KB containing 1 μCi 32P-γ-ATP for 20 min at 30°C. Samples were analyzed by SDS-PAGE followed by autoradiography.
For phosphatase treatment, 5U calf intestinal phosphatase (CIP) was added to cell extracts in CIP buffer (25 mM Tris-HCl pH 8.0, 300 mM NaCl, 0.5% Triton X-100, 1 mM MgCl2, 1 mM DTT and protease inhibitors) and incubated for 1 hr at 37°C. The reaction was stopped by addition of SDS sample buffer.
Far-Western
Protein phosphorylation for Far-Western analysis was done with 1 mM unlabeled ATP. Samples were subjected to SDS-PAGE, transferred to nitrocellulose and incubated overnight with 2.5 μg/ml of His-PBDWT or His-PBDAA in binding buffer (50 mM Tris-HCl pH 7.4, 137 mM NaCl, 0.1% Tween 20, 1 mM DTT, 2 mM MgCl2 and 10% glycerol at 4°C. Bound PBD was detected with antibodies to PLK1.
Protein Expression and Purification from E. coli
His-fusion proteins were expressed in E. coli Rosetta (Novagen) at 18°C by induction with 0.5 mM IPTG. Cells were lysed by sonication in 25 mM Tris-HCl pH 7.5, 300 mM NaCl, 2 mM MgCl2, 0.25% Triton X-100, 5 mM imidazole and 10% glycerol. The lysate was cleared by ultracentrifugation, passed over Ni-NTA agarose, washed and eluted with 250 mM imidazole in elution buffer 25 mM Tris-HCl pH 7.5, 300 mM NaCl, 2 mM MgCl2, 10% Glycerol). Peak fractions were buffer exchanged to PB.
GST-fusion proteins were expressed in E. coli BL21(DE3)-RIL (Agilent Technologies) at 18°C by induction with 0.5 mM IPTG. GST-fusion proteins were lysed by sonication in 50 mM Tris-HCl pH 7.5, 300 mM NaCl, 5 mM MgCl2, 1mM EDTA, 0.25% Triton X-100 and 10% glycerol. The cleared lysate was incubated with equilibrated Glutathione Sepharose 4B (GE healthcare) for 2 hr at 4°C. GST-tagged proteins were eluted with 40 mM glutathione in 50 mM Tris-HCl and 300 mM NaCl, 5 mM MgCl2, 1 mM EDTA. Peak fractions were buffer exchanged to PB.
Integral membrane proteins were expressed in E. coli BL21(DE3) using auto-induction medium at 25°C overnight. Cell were harvested and lysed in 20 mM Tris-HCl pH 7.5, 150 mM NaCl, 1mM DTT, 1mM EDTA supplemented with protease inhibitors (2 μg/ml leupeptin, 1 μg/ml pepstatin, 2 μg/ml aprotinin). The lysate was cleared by centrifugation at 15,000xg for 15 min.
Protein Expression and Purification from Insect Cells
Recombinant baculoviruses coding for His-tagged PLK1WT, the constitutive active variant PLK1T210D, His-CDK1 and His-cyclinB1 were generated using the Bac-to-Bac Expression system (Invitrogen) and used to infect Sf21 insect cells, according to the manufacturer’s protocol. For complex formation His-CDK1 and His-cyclinB1 were co-expressed in baculovirus-infected Sf21 cells. After 3 days, (His-CDK1/His-cyclinB1) or after 2 days (His-PLK1), infected cells were harvested. For efficient lysis, cells were both dounced and passed through a 27 g needle in 300 mM NaCl, 20 mM Tris-HCl pH7.6, 1 mM DTT, 5 mM MgCl2, 5 mM imidazole, 10% gycerol, phosphatase and protease inhibitors on ice. The crude lysate was cleared by ultracentrifugation at 4°C followed by binding to equilibrated Ni-NTA agarose beads and incubated for 2 hr at 4°C on a rotating wheel. After extensive washing, His-tagged proteins were eluted with elution buffer (250 mM imidazole, 300 mM NaCl, 20 mM Tris-HCl pH7.6, 5 mM MgCl2, 1 mM DTT and 10% glycerol. Peak fractions were pooled and buffer exchanged to mPB (25 mM HEPES-KOH pH 7.6, 25 mM KOAc, 7.5 mM MgCl2, 250 mM sucrose, 0.5 mM DTT and 0.5 mM EGTA).
Pull-Down Experiments
For pull-down experiments, 15 μg of purified GST-fusion proteins were coupled to 15 μl glutathione sepharose. ME HeLa cell extracts were prepared as described (Marino et al., 2014). Pull-downs with GST-PBD(WT/AA) and with GST-Nup53 and mutant derivatives were performed from 50 μl ME (12 mg/ml) in 300 μl PB and from 300 μl interphase extract (7 mg/ml) in lysis buffer (25 mM Tris-HCl pH 7.6, 125 mM NaCl, 2 mM MgCl2, 1 mM DTT, 0.5% NP-40 and protease inhibitors), respectively, for 2 hr at 4°C. After washing, bound proteins were eluted with SDS sample buffer and resolved by electrophoresis. Interaction analyses of GST-Nup53 variants with NDC1 or a GP210 fragment expressed in E. coli were performed as described (Eisenhardt et al., 2014).
Identification of Nup53 Phosphorylation Sites
For identification of mitotic phosphorylation sites in Nup53, expression of GFP-Nup53 or HA-Strep-tagged Nup53 was induced in the respective HeLa cell lines by addition of tetracycline (0.1 μg/ml). Cells were synchronized in interphase by a double thymidine block or in mitosis by a single thymidine arrest followed by nocodazole treatment. Mitotic cells were collected by shake-off. After harvest, cells were resuspended in lysis buffer containing 25 mM Tris-HCl pH 7.6, 125 mM NaCl, 2 mM MgCl2, 1 mM DTT, 0.5% NP-40, protease and phosphatase inhibitors, followed by sonication. After clearance of the respective lysates by centrifugation (15’000 rpm, 30 min, 4°C), HA-Strep-Nup53 was purified by affinity chromatography with StrepTactin sepharose (IBA) for 30 min at 4°C, whereas GFP-Nup53 was immunoprecipated with an anti-GFP antibody coupled to a 9:1 mixture of protein A/G-sepharose beads for 2 h at 4°C. Beads were washed three times with lysis buffer and two times with buffer lacking NP-40. Bound proteins were eluted with SDS sample buffer, separated by SDS-PAGE followed by Coomassie blue staining. Excised gel fragments were processed for mass spectrometry.
Gel bands were cut in pieces and proteins digested in-gel as previously described (Shevchenko et al., 2006). To increase sequence coverage, proteins were separately digested by trypsin (Promega), lysyl endopeptidase (Wako Chemicals) and chymotrypsin (Pierce). Peptides were cleaned up and concentrated using 96-Well MiniSpin™ Plates (Nest Group) according to manufacturer’s instructions. Samples were measured on an EASY-nLC 1000 (Thermo Fisher) coupled to a Q Exactive Plus mass spectrometer (Thermo Fisher). Peptides were separated on a column (40 cm x 0.75 μm), packed in-house with reversed-phase ReproSil-Pur C18- AQ resin (1.9 μm, Dr. Maisch). Peptides were eluted for 80 min using a segmented linear gradient of 5% to 40% solvent B (99.9% acetonitrile, 0.1% formic acid) at a flow-rate of 300 nl/min. Survey full-scan mass spectra were acquired with mass range 350-1500 m/z, at a resolution of 70,000 at 200 m/z and the 20 most intense ions above an intensity of 3.6e4 were sequentially isolated, fragmented (HCD at 25 NCE) and measured at a resolution of 17,500 at 200 m/z. Peptides with a charge of +1 or with unassigned charge state were excluded from fragmentation for MS2, and a dynamic exclusion of 30 s was applied. Ions were accumulated to a target value of 3e6 for MS1 and of 1e5 for MS2.
Mass spectra were searched against a human database (Uniprot, downloaded at 13 November 2015) using Sequest HT. Carbamidomethylation of cysteine was set as a fixed modification and oxidation of methionine and phosphorylation of serine, threonine and tyrosine were allowed as a variable modification. Precursor mass tolerance was 10 ppm and fragment mass tolerance 0.02 Da. Chymotrypsin and trypsin were set as enzymes allowing for semi-tryptic cleavages and maximally 4 missed cleavages. Percolator was used to filter the results to 1% FDR at peptide level.
Quantification and Statistical Analysis
Image analysis of NPC disassembly was performed as described (Marino et al., 2014). The standard error of the mean (SEM) was calculated from at least 3 independent experiments. Statistical significance was tested using a two-tailed, unpaired Student’s t-test. For determination of t50 (time at which 50% of nuclei are dextran-positive), curves were fit using non-linear fitting with the Prism software.
Quantification of fluorescence intensity at the NE for Figures 4D, 6B, and S5B was done with ImageJ, generating 4 pixels-wide line profiles at a nuclear diameter of 90 pixels covering cytoplasmic regions on both nuclear sides of 30 pixels in length. Mean signal intensities of ≥ 50 cells per condition were plotted by using Graphpad Prism 5. Quantification of 2RFP-Nup58 for Figure S7 was performed as described previously using a MATLAB-based script (Ungricht et al., 2015).
Figure legends describe the applied statistical tests and statistical significance, with N = number of independent experiments, n = number of cells; SD, standard deviation; SEM, standard error of the mean.
Author Contributions
Conceptualization, U.K. with input from M.L., C.W., and J.M.; Methodology, M.L., M.K., P.B., M.W., C.W., J.M., P.P., W.A., and U.K.; Investigation, M.L., M.K., P.B., M.W., C.W., J.M., and W.A.; Writing – Original Draft, U.K., M.L., P.B., and W.A.; Writing – Review & Editing, all authors; Funding Acquisition, U.K.; Supervision, M.L., P.P., W.A., and U.K.
Acknowledgments
We thank Drs. L. Champion, S. Jonas, and R. Ungricht for helpful discussions and critical reading of the manuscript, R. Ungricht for help with illustration, L. Canelle and R. Aebersold, as well as J. Pfannstiel and B. Wuertz, for support in mass spectrometric analyses, V. Cordes for the anti-TPR antibody, and members of ScopeM for continuous support of microscopy. This work was supported by an ERC Advanced grant (NucEnv_322582) to U.K.
Published: October 23, 2017
Footnotes
Supplemental Information includes seven figures and two tables and can be found with this article online at https://doi.org/10.1016/j.devcel.2017.08.020.
Supporting Citations
The following references appear in the Supplemental Information: Beausoleil et al., 2006, Malik et al., 2008, Petrone et al., 2016, Sharma et al., 2014, von Appen et al., 2015.
Supplemental Information
References
- Archambault V., Glover D.M. Polo-like kinases: conservation and divergence in their functions and regulation. Nat. Rev. Mol. Cell Biol. 2009;10:265–275. doi: 10.1038/nrm2653. [DOI] [PubMed] [Google Scholar]
- Badertscher L., Wild T., Montellese C., Alexander L.T., Bammert L., Sarazova M., Stebler M., Csucs G., Mayer T.U., Zamboni N., Zemp I., Horvath P., Kutay U. Genome-wide RNAi screening identifies protein modules required for 40S subunit synthesis in human cells. Cell Rep. 2015;13:2879–2891. doi: 10.1016/j.celrep.2015.11.061. [DOI] [PubMed] [Google Scholar]
- Barr F.A., Sillje H.H., Nigg E.A. Polo-like kinases and the orchestration of cell division. Nat. Rev. Mol. Cell Biol. 2004;5:429–440. doi: 10.1038/nrm1401. [DOI] [PubMed] [Google Scholar]
- Bertran M.T., Sdelci S., Regue L., Avruch J., Caelles C., Roig J. Nek9 is a Plk1-activated kinase that controls early centrosome separation through Nek6/7 and Eg5. EMBO J. 2011;30:2634–2647. doi: 10.1038/emboj.2011.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beausoleil S.A., Villen J., Gerber S.A., Rush J., Gygi S.P. A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol. 2006;24:1285–1292. doi: 10.1038/nbt1240. [DOI] [PubMed] [Google Scholar]
- Brizuela L., Draetta G., Beach D. Activation of human CDC2 protein as a histone H1 kinase is associated with complex formation with the p62 subunit. Proc. Natl. Acad. Sci. USA. 1989;86:4362–4366. doi: 10.1073/pnas.86.12.4362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Champion L., Linder M.I., Kutay U. Cellular reorganization during mitotic entry. Trends Cell Biol. 2016;27:26–41. doi: 10.1016/j.tcb.2016.07.004. [DOI] [PubMed] [Google Scholar]
- Cheng K.Y., Lowe E.D., Sinclair J., Nigg E.A., Johnson L.N. The crystal structure of the human polo-like kinase-1 polo box domain and its phospho-peptide complex. EMBO J. 2003;22:5757–5768. doi: 10.1093/emboj/cdg558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dephoure N., Zhou C., Villen J., Beausoleil S.A., Bakalarski C.E., Elledge S.J., Gygi S.P. A quantitative atlas of mitotic phosphorylation. Proc. Natl. Acad. Sci. USA. 2008;105:10762–10767. doi: 10.1073/pnas.0805139105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dultz E., Zanin E., Wurzenberger C., Braun M., Rabut G., Sironi L., Ellenberg J. Systematic kinetic analysis of mitotic dis- and reassembly of the nuclear pore in living cells. J. Cell Biol. 2008;180:857–865. doi: 10.1083/jcb.200707026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenhardt N., Redolfi J., Antonin W. Interaction of Nup53 with Ndc1 and Nup155 is required for nuclear pore complex assembly. J. Cell Sci. 2014;127:908–921. doi: 10.1242/jcs.141739. [DOI] [PubMed] [Google Scholar]
- Elia A.E., Cantley L.C., Yaffe M.B. Proteomic screen finds pSer/pThr-binding domain localizing Plk1 to mitotic substrates. Science. 2003;299:1228–1231. doi: 10.1126/science.1079079. [DOI] [PubMed] [Google Scholar]
- Fischer J., Teimer R., Amlacher S., Kunze R., Hurt E. Linker Nups connect the nuclear pore complex inner ring with the outer ring and transport channel. Nat. Struct. Mol. Biol. 2015;22:774–781. doi: 10.1038/nsmb.3084. [DOI] [PubMed] [Google Scholar]
- Fontoura B.M., Blobel G., Matunis M.J. A conserved biogenesis pathway for nucleoporins: proteolytic processing of a 186-kilodalton precursor generates Nup98 and the novel nucleoporin, Nup96. J. Cell Biol. 1999;144:1097–1112. doi: 10.1083/jcb.144.6.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavet O., Pines J. Progressive activation of CyclinB1-Cdk1 coordinates entry to mitosis. Dev. Cell. 2010;18:533–543. doi: 10.1016/j.devcel.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner J., Mayr M.I., Mockel M.M., Mayer T.U. Pre-anaphase chromosome oscillations are regulated by the antagonistic activities of Cdk1 and PP1 on Kif18A. Nat. Commun. 2014;5:4397. doi: 10.1038/ncomms5397. [DOI] [PubMed] [Google Scholar]
- Hase M.E., Kuznetsov N.V., Cordes V.C. Amino acid substitutions of coiled-coil protein Tpr abrogate anchorage to the nuclear pore complex but not parallel, in-register homodimerization. Mol. Biol. Cell. 2001;12:2433–2452. doi: 10.1091/mbc.12.8.2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawryluk-Gara L.A., Platani M., Santarella R., Wozniak R.W., Mattaj I.W. Nup53 is required for nuclear envelope and nuclear pore complex assembly. Mol. Biol. Cell. 2008;19:1753–1762. doi: 10.1091/mbc.E07-08-0820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawryluk-Gara L.A., Shibuya E.K., Wozniak R.W. Vertebrate Nup53 interacts with the nuclear lamina and is required for the assembly of a Nup93-containing complex. Mol. Biol. Cell. 2005;16:2382–2394. doi: 10.1091/mbc.E04-10-0857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoelz A., Glavy J.S., Beck M. Toward the atomic structure of the nuclear pore complex: when top down meets bottom up. Nat. Struct. Mol. Biol. 2016;23:624–630. doi: 10.1038/nsmb.3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hülsmann B.B., Labokha A.A., Görlich D. The permeability of reconstituted nuclear pores provides direct evidence for the selective phase model. Cell. 2012;150:738–751. doi: 10.1016/j.cell.2012.07.019. [DOI] [PubMed] [Google Scholar]
- Kettenbach A.N., Schweppe D.K., Faherty B.K., Pechenick D., Pletnev A.A., Gerber S.A. Quantitative phosphoproteomics identifies substrates and functional modules of Aurora and Polo-like kinase activities in mitotic cells. Sci. Signal. 2011;4:rs5. doi: 10.1126/scisignal.2001497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knockenhauer K.E., Schwartz T.U. The nuclear pore complex as a flexible and dynamic gate. Cell. 2016;164:1162–1171. doi: 10.1016/j.cell.2016.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosinski J., Mosalaganti S., von Appen A., Teimer R., DiGuilio A.L., Wan W., Bui K.H., Hagen W.J., Briggs J.A., Glavy J.S. Molecular architecture of the inner ring scaffold of the human nuclear pore complex. Science. 2016;352:363–365. doi: 10.1126/science.aaf0643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurell E., Beck K., Krupina K., Theerthagiri G., Bodenmiller B., Horvath P., Aebersold R., Antonin W., Kutay U. Phosphorylation of Nup98 by multiple kinases is crucial for NPC disassembly during mitotic entry. Cell. 2011;144:539–550. doi: 10.1016/j.cell.2011.01.012. [DOI] [PubMed] [Google Scholar]
- Laurell E., Kutay U. Dismantling the NPC permeability barrier at the onset of mitosis. Cell Cycle. 2011;10:2243–2245. doi: 10.4161/cc.10.14.16195. [DOI] [PubMed] [Google Scholar]
- Lee K.S., Erikson R.L. Plk is a functional homolog of Saccharomyces cerevisiae Cdc5, and elevated Plk activity induces multiple septation structures. Mol. Cell. Biol. 1997;17:3408–3417. doi: 10.1128/mcb.17.6.3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenart P., Petronczki M., Steegmaier M., Di Fiore B., Lipp J.J., Hoffmann M., Rettig W.J., Kraut N., Peters J.M. The small-molecule inhibitor BI 2536 reveals novel insights into mitotic roles of polo-like kinase 1. Curr. Biol. 2007;17:304–315. doi: 10.1016/j.cub.2006.12.046. [DOI] [PubMed] [Google Scholar]
- Li H., Liu X.S., Yang X., Song B., Wang Y., Liu X. Polo-like kinase 1 phosphorylation of p150Glued facilitates nuclear envelope breakdown during prophase. Proc. Natl. Acad. Sci. USA. 2010;107:14633–14638. doi: 10.1073/pnas.1006615107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin D.H., Stuwe T., Schilbach S., Rundlet E.J., Perriches T., Mobbs G., Fan Y., Thierbach K., Huber F.M., Collins L.N. Architecture of the symmetric core of the nuclear pore. Science. 2016;352:aaf1015. doi: 10.1126/science.aaf1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindqvist A., Rodriguez-Bravo V., Medema R.H. The decision to enter mitosis: feedback and redundancy in the mitotic entry network. J. Cell Biol. 2009;185:193–202. doi: 10.1083/jcb.200812045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik R., Nigg E.A., Korner R. Comparative conservation analysis of the human mitotic phosphoproteome. Bioinformatics. 2008;24:1426–1432. doi: 10.1093/bioinformatics/btn197. [DOI] [PubMed] [Google Scholar]
- Mansfeld J., Guttinger S., Hawryluk-Gara L.A., Pante N., Mall M., Galy V., Haselmann U., Muhlhausser P., Wozniak R.W., Mattaj I.W. The conserved transmembrane nucleoporin NDC1 is required for nuclear pore complex assembly in vertebrate cells. Mol. Cell. 2006;22:93–103. doi: 10.1016/j.molcel.2006.02.015. [DOI] [PubMed] [Google Scholar]
- Marino J., Champion L., Wandke C., Horvath P., Mayr M.I., Kutay U. An in vitro system to study nuclear envelope breakdown. Methods Cell Biol. 2014;122:255–276. doi: 10.1016/B978-0-12-417160-2.00012-6. [DOI] [PubMed] [Google Scholar]
- Martino L., Morchoisne-Bolhy S., Cheerambathur D.K., Van Hove L., Dumont J., Joly N., Desai A., Doye V., Pintard L. Channel nucleoporins recruit the polo-like kinase PLK-1 to the nuclear pore complexes in prophase to direct nuclear envelope breakdown. Dev. Cell. 2017;43:157–171. doi: 10.1016/j.devcel.2017.09.019. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mühlhäusser P., Kutay U. An in vitro nuclear disassembly system reveals a role for the RanGTPase system and microtubule-dependent steps in nuclear envelope breakdown. J. Cell Biol. 2007;178:595–610. doi: 10.1083/jcb.200703002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen J.V., Vermeulen M., Santamaria A., Kumar C., Miller M.L., Jensen L.J., Gnad F., Cox J., Jensen T.S., Nigg E.A. Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci. Signal. 2010;3:ra3. doi: 10.1126/scisignal.2000475. [DOI] [PubMed] [Google Scholar]
- Petronczki M., Lenart P., Peters J.M. Polo on the rise-from mitotic entry to cytokinesis with Plk1. Dev. Cell. 2008;14:646–659. doi: 10.1016/j.devcel.2008.04.014. [DOI] [PubMed] [Google Scholar]
- Petrone A., Adamo M.E., Cheng C., Kettenbach A.N. Identification of candidate cyclin-dependent kinase 1 (Cdk1) substrates in mitosis by quantitative phosphoproteomics. Mol. Cell. Proteomics. 2016;15:2448–2461. doi: 10.1074/mcp.M116.059394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman M.M., Munzig M., Kaneshiro K., Lee B., Strome S., Muller-Reichert T., Cohen-Fix O. Caenorhabditis elegans polo-like kinase PLK-1 is required for merging parental genomes into a single nucleus. Mol. Biol. Cell. 2015;26:4718–4735. doi: 10.1091/mbc.E15-04-0244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhardt H.C., Yaffe M.B. Phospho-Ser/Thr-binding domains: navigating the cell cycle and DNA damage response. Nat. Rev. Mol. Cell Biol. 2013;14:563–580. doi: 10.1038/nrm3640. [DOI] [PubMed] [Google Scholar]
- Roosild T.P., Greenwald J., Vega M., Castronovo S., Riek R., Choe S. NMR structure of Mistic, a membrane-integrating protein for membrane protein expression. Science. 2005;307:1317–1321. doi: 10.1126/science.1106392. [DOI] [PubMed] [Google Scholar]
- Sachdev R., Sieverding C., Flotenmeyer M., Antonin W. The C-terminal domain of Nup93 is essential for assembly of the structural backbone of nuclear pore complexes. Mol. Biol. Cell. 2012;23:740–749. doi: 10.1091/mbc.E11-09-0761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar C., Hofer T. Multisite protein phosphorylation – from molecular mechanisms to kinetic models. FEBS J. 2009;276:3177–3198. doi: 10.1111/j.1742-4658.2009.07027.x. [DOI] [PubMed] [Google Scholar]
- Santamaria A., Wang B., Elowe S., Malik R., Zhang F., Bauer M., Schmidt A., Sillje H.H., Korner R., Nigg E.A. The Plk1-dependent phosphoproteome of the early mitotic spindle. Mol. Cell. Proteomics. 2011;10 doi: 10.1074/mcp.M110.004457. M110 004457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt H.B., Görlich D. Transport selectivity of nuclear pores, phase separation, and membraneless organelles. Trends Biochem. Sci. 2016;41:46–61. doi: 10.1016/j.tibs.2015.11.001. [DOI] [PubMed] [Google Scholar]
- Schmucker S., Sumara I. Molecular dynamics of PLK1 during mitosis. Mol. Cell Oncol. 2014;1:e954507. doi: 10.1080/23723548.2014.954507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz T.U. The structure inventory of the nuclear pore complex. J. Mol. Biol. 2016;428:1986–2000. doi: 10.1016/j.jmb.2016.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma K., D'Souza R.C., Tyanova S., Schaab C., Wisniewski J.R., Cox J., Mann M. Ultradeep human phosphoproteome reveals a distinct regulatory nature of Tyr and Ser/Thr-based signaling. Cell Rep. 2014;8:1583–1594. doi: 10.1016/j.celrep.2014.07.036. [DOI] [PubMed] [Google Scholar]
- Shevchenko A., Tomas H., Havlis J., Olsen J.V., Mann M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 2006;1:2856–2860. doi: 10.1038/nprot.2006.468. [DOI] [PubMed] [Google Scholar]
- Solc P., Kitajima T.S., Yoshida S., Brzakova A., Kaido M., Baran V., Mayer A., Samalova P., Motlik J., Ellenberg J. Multiple requirements of PLK1 during mouse oocyte maturation. PLoS One. 2015;10:e0116783. doi: 10.1371/journal.pone.0116783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steegmaier M., Hoffmann M., Baum A., Lenart P., Petronczki M., Krssak M., Gurtler U., Garin-Chesa P., Lieb S., Quant J. BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo. Curr. Biol. 2007;17:316–322. doi: 10.1016/j.cub.2006.12.037. [DOI] [PubMed] [Google Scholar]
- Stukenberg P.T., Lustig K.D., McGarry T.J., King R.W., Kuang J., Kirschner M.W. Systematic identification of mitotic phosphoproteins. Curr. Biol. 1997;7:338–348. doi: 10.1016/s0960-9822(06)00157-6. [DOI] [PubMed] [Google Scholar]
- Stuwe T., Bley C.J., Thierbach K., Petrovic S., Schilbach S., Mayo D.J., Perriches T., Rundlet E.J., Jeon Y.E., Collins L.N. Architecture of the fungal nuclear pore inner ring complex. Science. 2015;350:56–64. doi: 10.1126/science.aac9176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turgay Y., Champion L., Balazs C., Held M., Toso A., Gerlich D.W., Meraldi P., Kutay U. SUN proteins facilitate the removal of membranes from chromatin during nuclear envelope breakdown. J. Cell Biol. 2014;204:1099–1109. doi: 10.1083/jcb.201310116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungricht R., Klann M., Horvath P., Kutay U. Diffusion and retention are major determinants of protein targeting to the inner nuclear membrane. J. Cell Biol. 2015;209:687–703. doi: 10.1083/jcb.201409127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollmer B., Schooley A., Sachdev R., Eisenhardt N., Schneider A.M., Sieverding C., Madlung J., Gerken U., Macek B., Antonin W. Dimerization and direct membrane interaction of Nup53 contribute to nuclear pore complex assembly. EMBO J. 2012;31:4072–4084. doi: 10.1038/emboj.2012.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Appen A., Kosinski J., Sparks L., Ori A., DiGuilio A.L., Vollmer B., Mackmull M.T., Banterle N., Parca L., Kastritis P. In situ structural analysis of the human nuclear pore complex. Nature. 2015;526:140–143. doi: 10.1038/nature15381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemp I., Wild T., O'Donohue M.F., Wandrey F., Widmann B., Gleizes P.E., Kutay U. Distinct cytoplasmic maturation steps of 40S ribosomal subunit precursors require hRio2. J. Cell Biol. 2009;185:1167–1180. doi: 10.1083/jcb.200904048. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.