

Abstract
We have previously shown that monomeric globular αβ‐proteins can be designed de novo with considerable control over topology, size, and shape. In this paper, we investigate the design of cyclic homo‐oligomers from these starting points. We experimented with both keeping the original monomer backbones fixed during the cyclic docking and design process, and allowing the backbone of the monomer to conform to that of adjacent subunits in the homo‐oligomer. The latter flexible backbone protocol generated designs with shape complementarity approaching that of native homo‐oligomers, but experimental characterization showed that the fixed backbone designs were more stable and less aggregation prone. Designed C2 oligomers with β‐strand backbone interactions were structurally confirmed through x‐ray crystallography and small‐angle X‐ray scattering (SAXS). In contrast, C3‐C5 designed homo‐oligomers with primarily nonpolar residues at interfaces all formed a range of oligomeric states. Taken together, our results suggest that for homo‐oligomers formed from globular building blocks, improved structural specificity will be better achieved using monomers with increased shape complementarity and with more polar interfaces.
Keywords: computational protein design, de novo proteins, homo‐oligomer design, fixed and flexible backbone design
Short abstract
PDB Code(s): 4PWW
Introduction
Globular αβ‐ protein homo‐oligomers play important roles in molecular machines,1, 2 catalysis,3, 4 and regulation.5, 6 Considerable control over αβ‐ protein monomer topology and shape has been achieved with de novo protein design, but incorporating sequence features that specify a particular oligomerization state is a further challenge. The ability to design oligomers from scratch would enable the design of oligomeric functional proteins such as molecular machines without being limited to the set of naturally‐occurring oligomers. Previous homo‐oligomer design efforts have focused primarily on all α‐ proteins with nonglobular structures such as coiled coils7, 8, 9, 10, 11, 12, 13, 14 and repeat proteins,15 and disulfide‐linked oligomers.16, 17, 18, 19 Compared to oligomers made from elongated helical bundles, homo‐oligomeric structures made from globular building blocks have the advantage of multiple reconfigurable interfaces associated with subunit rotations along multiple axes.
The design of homo‐oligomers using αβ‐ proteins has been challenging, likely at least in part because a significant fraction of the interface will generally be involved. For example, Huang et al starting with protein G obtained a mixture of species.20 The best results have been obtained with strand‐strand interfaces as in a computationally designed β‐ sandwich homodimer.21 Here, we explore the design of a wide range of homo‐oligomeric geometries starting from de novo αβ‐ proteins.
Results
Starting with previously described de novo designed αβ‐ proteins,22, 23 we experimented with both fixed and flexible backbone methods for designing cyclic homo‐oligomers.
Fixed‐backbone oligomer design
De novo designed monomers were docked into C n oligomer conformations by repeatedly (1) applying a random rotation to the monomer, (2) sliding the monomers together until they come into contact, and (3) optimizing the identities and conformation of residues within 10Å of newly formed interfaces to minimize the binding energy using Rosetta Monte Carlo sequence design calculations (see Supporting Information).22 During the sequence design calculations, the energy is minimized with respect to the backbone and rigid body degrees of freedom but the changes in both are generally quite small. The thousands of alternative dock + designs generated using this procedure were ranked by shape complementarity,24 Rosetta binding energy,25 and the requirement that buried polar groups make hydrogen bonds (Fig. 1).8 The structural specificity of the top ranked designs was evaluated by carrying out multiple independent Rosetta symmetric docking calculations,26 and those with energy landscapes strongly funneling into the designed homo‐oligomer conformation were selected for experimental characterization.
Figure 1.
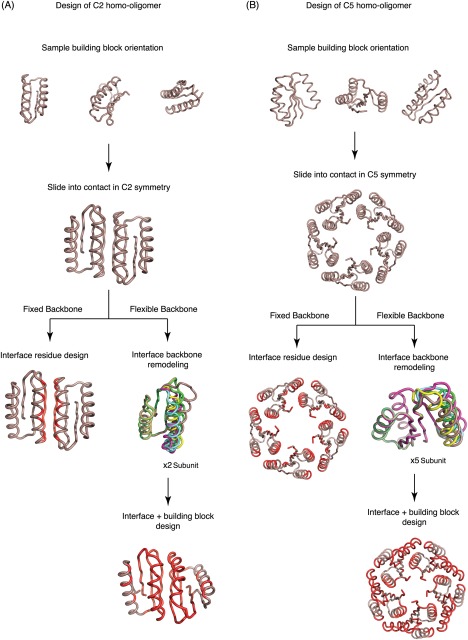
Design protocol. Schematic of fixed and flexible backbone design protocols for (Left panel) C2 homooligomers based on design Fd_7A and (Right panel) C5 homooligomers based on design Rsmn2x2_6
We used this fixed backbone protocol to design cyclic oligomers from de novo designed ferredoxin folds with a range of size and shapes.23 We obtained synthetic genes for eight designed homodimers built from Fd_7A, 6 designed trimers built from Fd_7A, and eight designed tetramers built Fd_9A (Supporting Information Tables S1–S3). The different oligomer designs built from the same monomer differ in sequence and in rigid body orientation. Synthetic genes were cloned into pET21 or pET29 E. coli expression vectors, and the designs were expressed, purified by immobilized metal affinity column (Ni‐NTA) and characterized by circular dichroism (CD) spectroscopy and size exclusion chromatography combined with multi‐angle light scattering (SEC‐MALS). We use the following naming convention: the oligomerization state (C2, C3, C4) is followed by the name of the monomer design and then by the number of the design in the series: for example, C2_Fd_7A_8 is the 8th dimer design built from the Fd_7A monomer.
Of the 22 designs, all but 3 had CD spectra expected for αβ‐proteins, suggesting that the many (20–26) amino acid residue changes made to create the designed interface did not disrupt the monomer fold. However, the SEC‐MALS results indicated that only 6 of 8 C2_Fd_7A designs had the molecular weights expected for the designed homo‐oligomers, and all of the C3_Fd_7A_v1 and C4_Fd_9A designs were polydisperse with multiple alternative oligomer conformations. Figure 2(B–E) shows experimental data for one of the best‐behaved designs from each of the three groups (C2, C3, C4); the data for the remaining designs are in Supporting Information Figures S2 and S3.
Figure 2.
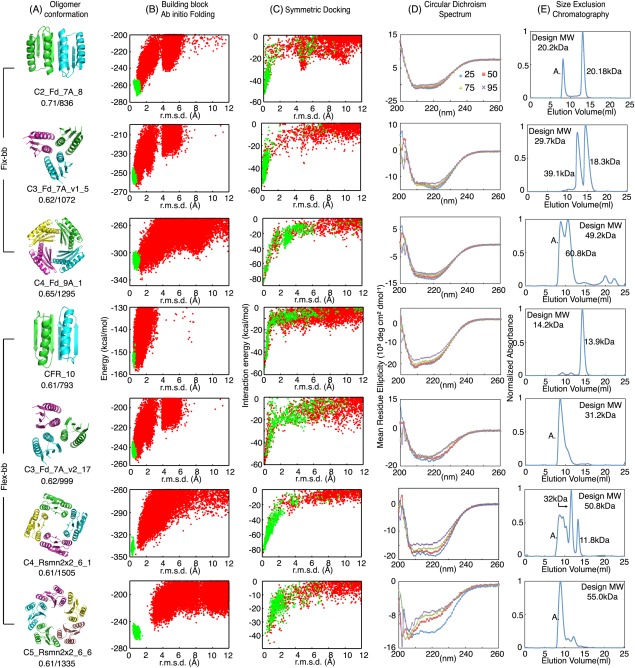
Characterization of computational designed oligomers. (A) Design models, with shape complementarity and interface area indicated below the names. (B) Energy landscapes obtained from Rosetta ab initio structure prediction simulations on Rosetta@home. Red points represent the lowest‐energy structures obtained in independent Monte Carlo structure prediction trajectories starting from an extended chain for each sequence; y axis, Rosetta all‐atom energy; x axis, Cα root mean square deviation (RMSD) from the design model. Green points represent the lowest‐energy structures obtained in trajectories starting from the design model. (C) Energy landscapes obtained from Rosetta symmetric docking. Red points represent the lowest‐energy docking conformations result from independent global sampling docking trajectories. X‐axis: Rosetta interaction energy. Y‐axis: Cα root mean square deviation (RMSD) from the designed oligomer conformation. Green points represent the lowest‐energy structures obtained from local sampling docking trajectories. (D) Far‐ultraviolet circular dichroism (CD) spectra at different temperatures. (E) Size‐exclusion chromatography spectra with molecular weight determined through multi‐angle light scattering (MALS)
We succeeded in solving the crystal structure of design C2_Fd_7A_8 (PDB:4PWW) at 1.47Å resolution. The crystal structure reveals a dimer very similar to the design model (backbone RMSD 1.2Å; Fig. 3, Supporting Information Table S8). The helices at the interface twist around each other in both design and crystal structure, but the extent of supercoiling is more significant in the crystal structure resulting in a more shape complementary interface. The extent of twisting of the helix brought about by the backbone minimization step during the design calculations is indicated in the comparison to the original design model on the right panel in Figure 3.
Figure 3.
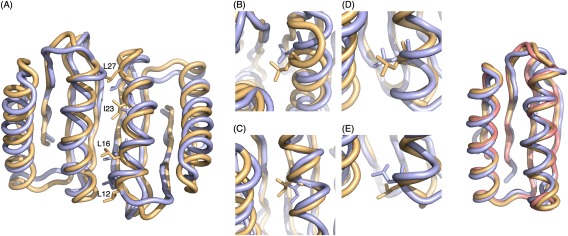
Crystal structure of design C2_Fd_7A_8. (Left panel) (A) Computational design model (yellow) aligned with crystal structure (light blue) with aliphatic residues at interface from one monomer shown. The helix at the interface was designed to bend toward the interacting helix in design model and the bending is even more significant in crystal structure. (B) (C) (D) (E) Close‐up view of L27, I23, L16, and L12 respectively. Due to the increase curvature, side chains are packed deeper into the interface core in crystal structure. (Right panel) Backbone superimposition of Fd_7A (pink), computational design model (yellow) and crystal structure (light blue) of C2_Fd_7A_8 chain A
Naturally‐occurring αβ‐protein oligomers have higher interface shape complementarity and area
An obvious limitation of fixed backbone approaches is that the shape complementarity between subunits is limited by the fixed backbone of the monomer (the changes in backbone conformation during the backbone relaxation step are quite modest). The increase in shape complementarity observed in the C2_Fd_7A_8 crystal structure suggested that the alternative oligomeric states observed in the C3 and C4 designs possibly resulted from insufficiently shape complementary interfaces. To determine whether the shape complementarity of the designs could be a contributor to the lack of success of the larger homo‐oligomers, we compared them to naturally‐occurring αβ‐ homo‐oligomers.
Analysis of a set of 31 naturally‐occurring αβ‐ cyclic homo‐oligomers from the PDB27 (see Supporting Information) showed that they generally have higher backbone (sequence independent) shape complementarity than the fixed backbone C3 and C4 homo‐oligomer docked configurations that the experimentally characterized designs were based on. In addition, the naturally‐occurring oligomers have larger interface areas because the subunits are generally larger than our de novo designed building blocks [Fig. 4(A–C)].
Figure 4.
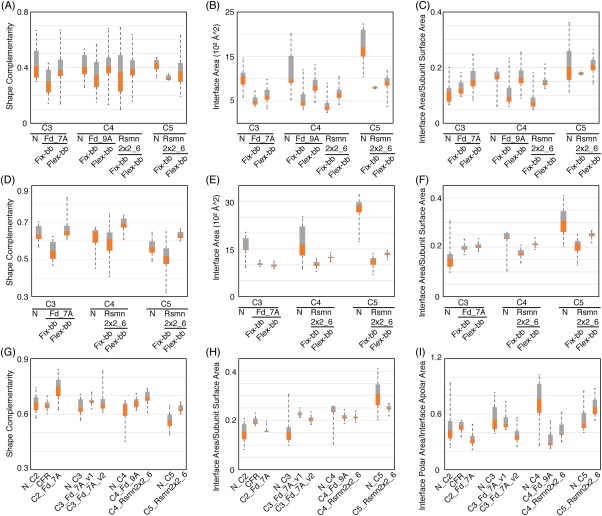
Comparison of interfaces in naturally occurring and fixed and flexible backbone designed homo‐oligomers. (A‐C) Evaluation of backbone complementarity, interface area and the ratio of interface area between neighboring subunits and total surface area per subunit between naturally‐occurring oligomers (N) and cyclic conformations sampled utilizing different de novo building blocks. Cyclic homo‐oligomer conformations were generated with either fixed‐backbone (fix‐bb) or flexible‐bb (flex‐bb) design method. Only backbone atoms were included in all calculations. Orange: first quartile to median. Gray: median to third quartile. (D–F) Same evaluation as (A–C) but including all atoms rather than just the backbone. (G,H) Same evaluation as D and F for all of the experimentally characterized designs. (I) Evaluation of interface polarity for all of the experimentally characterized designs
Based on the backbone movement resulting in the super‐helix‐like helical interface observed with crystal structure of C2_Fd_7A_8, we hypothesized that improved oligomeric interaction specificity could be achieved using backbone (N, NH, C, Cα, CO, and Cβ) remodeling to increase the surface area and shape complementarity24 of the designed interface.
Flexible‐backbone oligomer design
To more substantially remodel the backbone geometry of the monomers to increase shape complementarity between subunits, we used a flexible‐backbone design method combining Rosetta folding simulations22 with oligomer rigid body sampling. In the first step, fixed backbone Cn docking calculations were performed to identify potential oligomer interfaces. Segments of the monomer at the oligomer interface were then subjected to Rosetta remodeling in a Monte Carlo flexible docking trajectory in which small rigid body moves are alternated with broken chain remodeling of a randomly selected interface segment followed by loop closure. For example, the helices and flanking loops at the interfaces of the C2 and C5 oligomers in Figure 1 were selected for backbone remodeling.28, 29, 30 In the backbone remodeling step, the backbone torsion angles of randomly selected interface segments were replaced by fragments of the same length from the PDB (See Supporting Information) (Fig. 1). The flexible‐backbone protocol increased the sequence‐independent backbone shape complementarity and interface area significantly beyond the fixed backbone designs to close to that observed in the native complexes [Fig. 4 (A–C)].
We used the flexible‐backbone method to design C3 trimers based on Fd_7A (C3_Fd_7A_v2, building block: 2KL8) with β‐strands and alpha helices at the interface, C4 tetramers of Rsmn2x2_6 (C4_Rsmn2x2_6, building block: 2KPO), C5 pentamer of Rsmn2x2_6 (C5_Rsmn2x2_6, building block: 2KPO) and a de novo designed C2 dimer with an extended sheet interface as in the C2_Fd_7A_8 crystal structure (we call this design CFR because it resembles the structure of a C terminal fragment of Top7 protein).31 We chose C4_Rsmn2x2_6 over C4_Fd_9A for flexible‐backbone design because it has a larger core and is more stable23 and hence it can likely better maintain the overall fold even with substantial backbone remodeling. To generate C5 homo‐oligomers with high shape complementarity, we truncated α4 of 2KPO and remodeled α1 to interact with α2 and α3 of a neighboring subunit [C5_Rsmn2x2_6; Fig. 2(A)].
Oligomer conformations with backbone interface area larger then 240Å2 were selected for sequence design. Residues at the interface and within 8Å of remodeled segments were redesigned to optimize both monomer stability and interactions across the oligomer interface.20, 32 Overall, designs made with the flexible backbone protocol had higher backbone and sidechain shape complementarity and interface surface than those made with the fixed backbone protocol starting from the same building blocks [Fig. 4(D–F)].
For each design, to assess in silico the folding of the monomeric building block (perturbed more than in the fixed backbone case as both the sequence and the structure differ from the starting design), we performed multiple independent Rosetta ab initio structure prediction calculations33 starting from an extended chain. Designs with energy landscapes funneled into the remodeled monomer structure were then subjected to Rosetta symmetric docking calculations to assess the designed homo‐oligomeric interface.26
Genes encoding designs with docking energy landscapes strongly funneled into the design target conformation were obtained for experimental characterization; these include 10 designs for CFR, 18 for C3_Fd_7A_v2, 12 for C4_Rsmn2x2_6 and 8 for C5_Rsmn2x2_6 (Supporting Information Tables S4–S7). Solubly expressed designs were, as in the fixed backbone experiments, characterized with CD spectroscopy and SEC‐MALS after Ni‐NTA purification. The computational model and experimental results of the design with the highest thermal stability for each target oligomer conformation are shown in Figure 2. Half of the C3_Fd_7A_v2 and most of the C4_Rsmn2x2_6 designs were soluble and had the expected far‐UV CD spectra, but α4 truncation of 2KPO appeared to decrease tertiary structure stability as only two C5_Rsmn2x2_6 designs had αβ‐protein CD spectra at 25°C (Supporting Information Fig. S2). Unfortunately, the flexible backbone design protocol did not solve the polydispersity problem; multiple species were again observed for all the C3‐C5 solubly expressed designs.
More success was observed with the C2 flexible backbone design. Design 10 of CFR (CFR_10) had the CD spectrum of an αβ‐protein and had the expected molecular weight by SEC‐MALS (Fig. 2). We were unable to crystalize CFR_10 to compare with the design model, but SEC‐MALS indicated dimerization of CFR_10 and the solution small‐angle X‐ray scattering (SAXS)34, 35 profile was consistent with the design model ( χ = 1.64, Supporting Information Fig. S1).36, 37
Discussion
Taken together, we can draw several conclusions from the successes and failures described in this paper in designing assemblies of ∼100‐residue de novo αβ‐ protein with fixed‐ and flexible‐ backbone design methods and using αα‐, αβ‐, and ββ‐ interfaces.
First, the success with C2_Fd_7A and CFR highlights the robustness of designed interfaces with beta strand pairing to form an extended beta sheet, as found by Stranges and Kuhlman.21 Our recently described homo‐tetrameric TIM barrel also involves a beta sheet extending across the interface.38 Preorganized β‐ strands with exposed backbone amine and carbonyl groups allow strong association between subunits without introduction of large hydrophobic patches and hence partially circumvent the tradeoff between subunit solubility and interface stability. For interface geometries where backbone β strand pairing is not possible, extensive designed sidechain polar hydrogen bonding networks could increase structural specificity as observed for two ring helical bundles.8
Second, flexible backbone methods can generate assemblies with subunit‐subunit interfaces having shape complementarity in the range of native complexes, and with the monomers predicted to fold to the intended subunit structures. In contrast, the shape complementarity of assemblies for C3‐C5 generated using fixed backbone methods is generally quite a bit lower than that of native complexes.
Third, despite the good in silico metrics of the flexible backbone C3‐C5 designs, it is difficult to control the precise oligomerization state. The flexible backbone designs were readily expressed and purified, but were not monodisperse. To gain insight into the origins of these shortcomings, we compared our designs with naturally‐occurring homo‐oligomers and found that the latter generally have more polar interactions across the interface [Fig. 4(G–I)]. More extensive negative design could also improve success rates; backbone remodeling may result in flexibility that is consistent with oligomerization states beyond the design target.
Compared to the repeat proteins used as oligomer building blocks by Fallas et al., the de novo designed αβ‐ proteins have smaller hydrophobic cores stabilizing the individual subunits. Redesigning a large fraction of the surface residues for nonpolar subunit‐subunit interactions can disrupt the characteristic beta strand and alpha helical hydrophobic‐polar patterns and impact monomer folding and solubility, perhaps leading to the observed aggregates and multiple oligomeric states. One approach to maintain building block stability while designing protein–protein interaction would be addition of small extra elements for interaction as frequently observed in native homo‐oligomeric protein structures.39 Such inserted or appended structural elements can likely be optimized for interface shape complementarity without disturbing the stability of original building block.
In summary, for flexible backbone homo‐oligomer design, how to balance interactions across the homo‐oligomer interface with monomer foldability and stability remains an outstanding challenge. On the computational side, the flexible backbone interface design problem is closely related to the long studied and similarly challenging flexible backbone protein docking problem. The very large size of the joint search space (rigid body degrees of freedom X internal monomer degrees of freedom) make comprehensive sampling and robust identification of the global energy minimum quite challenging.
Materials and Methods
Computational homo‐oligomer design and experimental characterization methods are described in detail in Supporting Information.
Supporting information
Supporting Information
Acknowledgments
We thank the thousands of Rosetta@HOME volunteers for making this work possible. We thank L. Carter for assistance with SEC‐MALS. We thank the staff at the Advanced Light Source SIBYLS beamline at Lawrence Berkeley National Laboratory, including K. Burnett, G. Hura, M. Hammel, J. Tanamachi, and J. Tainer for the services provided through the mail‐in SAXS program, which is supported by the DOE Office of Biological and Environmental Research Integrated Diffraction Analysis program and the NIH project MINOS (Macromolecular Insights on Nucleic Acids Optimized by Scattering; grant no. RO1GM105404).
Statement of impact The sequences of protein homo‐oligomers must both encode the structure of the monomer and the monomer‐monomer interactions. We probe this balance by computationally designing and characterizing completely de novo protein homo‐oligomers.
References
- 1. Turner J, Anderson R, Guo J, Beraud C, Fletterick R, Sakowicz R (2001) Crystal structure of the mitotic spindle kinesin Eg5 reveals a novel conformation of the neck‐linker. J Biol Chem 276:25496–25502. [DOI] [PubMed] [Google Scholar]
- 2. Smith PC, Karpowich N, Millen L, Moody JE, Rosen J, Thomas PJ, Hunt JF (2002) ATP binding to the motor domain from an ABC transporter drives formation of a nucleotide sandwich dimer. Mol Cell 10:139–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Griffin MDW, Dobson RCJ, Pearce FG, Antonio L, Whitten AE, Liew CK, Mackay JP, Trewhella J, Jameson GB, Perugini MA, Gerrard JA (2008) Evolution of quaternary structure in a homotetrameric enzyme. J Mol Biol 380:691–703. [DOI] [PubMed] [Google Scholar]
- 4. Partanen ST, Novikov DK, Popov AN, Mursula AM, Hiltunen JK, Wierenga RK (2004) The 1.3 A crystal structure of human mitochondrial delta3‐delta2‐enoyl‐CoA isomerase shows a novel mode of binding for the fatty acyl group. J Mol Biol 342:1197–1208. [DOI] [PubMed] [Google Scholar]
- 5. Segura‐Peña D, Lichter J, Trani M, Konrad M, Lavie A, Lutz S (2007) Quaternary structure change as a mechanism for the regulation of thymidine kinase 1‐like enzymes. Structure 15:1555–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Stieglitz K, Stec B, Baker DP, Kantrowitz ER (2004) Monitoring the transition from the T to the R state in E. coli aspartate transcarbamoylase by X‐ray crystallography: crystal structures of the E50A mutant enzyme in four distinct allosteric states. J Mol Biol 341:853–868. [DOI] [PubMed] [Google Scholar]
- 7. Mou Y, Huang P‐S, Hsu F‐C, Huang S‐J, Mayo SL (2015) Computational design and experimental verification of a symmetric protein homodimer. Proc Natl Acad Sci USA 112:10714–10719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Boyken SE, Chen Z, Groves B, Langan RA, Oberdorfer G, Ford A, Gilmore JM, Xu C, DiMaio F, Pereira JH, Sankaran B, Seelig G, Zwart PH, Baker D (2016) De novo design of protein homo‐oligomers with modular hydrogen‐bond network‐mediated specificity. Science 352:680–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fletcher JM, Boyle AL, Bruning M, Barlett GJ, Vincent TL, Zaccai NR, Armstrong CT, Bromley EHC, Booth PJ, Brady RL, Thomson AR, Woolfson DN (2012) A basis set of de novo coiled‐coil peptide oligomers for rational protein design and synthetic biology. ACS Synth Biol 1:240–250. [DOI] [PubMed] [Google Scholar]
- 10. Dolphin GT (2006) A designed branched three‐helix bundle protein dimer. J Am Chem Soc 128:7287–7290. [DOI] [PubMed] [Google Scholar]
- 11. Egelman EH, Xu C, DiMaio F, Magnotti E, Modlin C, Yu X, Wright E, Baker D, Conticello VP (2015) Structural plasticity of helical nanotubes based on coiled‐coil assemblies. Structure 23:280–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hume J, Sun J, Jacquet R, Renfrew PD, Martin JA, Bonneau R, Gilchrist ML, Montclare JK (2014) Engineered coiled‐coil protein microfibers. Biomacromolecules 15:3503–3510. [DOI] [PubMed] [Google Scholar]
- 13. Barth P, Senes A (2016) Toward high‐resolution computational design of the structure and function of helical membrane proteins. Nat Struct Mol Biol 23:475–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Plecs JJ, Harbury PB, Kim PS, Alber T (2004) Structural test of the parameterized‐backbone method for protein design. J Mol Biol 342:289–297. [DOI] [PubMed] [Google Scholar]
- 15. Fallas JA, Ueda G, Sheffler W, Nguyen V, McNamara DE, Sankaran B, Pereira JH, Parmeggiani F, Brunette TJ, Cascio D, Yeates TR, Zwart P, Baker D (2016) Computational design of self‐assembling cyclic protein homo‐oligomers. Nat Chem 9:353–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Heinz DW, Matthews BW (1994) Rapid crystallization of T4 lysozyme by intermolecular disulfide cross‐linking. Protein Eng 7:301–307. [DOI] [PubMed] [Google Scholar]
- 17. Das M, Kobayashi M, Yamada Y, Sreeramulu S, Ramakrishnan C, Wakatsuki S, Kato R, Varadarajan R (2007) Design of disulfide‐linked thioredoxin dimers and multimers through analysis of crystal contacts. J Mol Biol 372:1278–1292. [DOI] [PubMed] [Google Scholar]
- 18. Meinke G, Phelan P, Fradet‐Turcotte A, Archambault J, Bullock PA (2011) Structure‐based design of disulfide‐linked oligomeric form of the simian virus 40 (sv40) large T antigen DNA‐binding domain. Acta Cryst D67:560–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kier BL, Anderson JM, Anderson NH (2015) Disulfide‐mediated β‐strand dimers: hyperstable β‐sheets lacking tertiary interactions and turns. J Am Chem Soc 137:5363–5371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Huang P‐S, Love JJ, Mayo SL (2007) A de novo designed protein–protein interface. Protein Sci 16:2770–2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stranges PB, Machius M, Miley MJ, Tripathy A, Kuhlman B (2011) Computational design of a symmetric homodimer using β‐strand assembly. Proc Natl Acad Sci USA 108:20562–20567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Koga N, Tatsumi‐Koga R, Liu G, Xiao R, Acton TB, Montelione GT, Baker D (2012) Principles for designing ideal protein structures. Nature 491:222–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lin Y‐R, Koga N, Tatsumi‐Koga R, Liu G, Clouser AF, Montelione GT, Baker D (2015) Control over overall shape and size in de novo designed proteins. Proc Natl Acad Sci USA 112:E5478–E5485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lawrence MC, Colman PM (1993) Shape complementarity at protein/protein interfaces. J Mol Biol 234:946–950. [DOI] [PubMed] [Google Scholar]
- 25. Karanicolas J, Kuhlman B (2009) Computational design of affinity and specificity at protein–protein interfaces. Curr Opin Struct Biol 19:458–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. André I, Bradley P, Wang C, Baker D (2007) Prediction of the structure of symmetrical protein assemblies. Proc Natl Acad Sci USA 104:17656–17661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TH, Weissig H, Shindyalov IN, Bourne PE (2000) The Protein Data Bank. Nucleic Acids Res 28:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Qian B, Raman S, Das R, Bradley P, McCoy AJ, Read RJ, Baker D (2007) High‐resolution structure prediction and the crystallographic phase problem. Nature 450:259–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Das R, Baker D (2008) Macromolecular modeling with rosetta. Ann Rev Biochem 77:363–382. [DOI] [PubMed] [Google Scholar]
- 30. Huang P‐S, Ban Y‐EA, Richter F, Andre I, Vernon R, Schief WR, Baker D (2011) RosettaRemodel: a generalized framework for flexible backbone protein design. PloS One 6:e24109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dantas G, Watters AL, Lunde B, Eletr Z, Isern NG, Roseman T, Lipfert J, Doniach S, Tompa M, Kuhlman B, Stoddard BL, Varani G, Baker D (2006) Mis‐translation of a computationally designed protein yields an exceptionally stable homodimer: implications for protein engineering and evolution. J Mol Biol 362:1004–1024. [DOI] [PubMed] [Google Scholar]
- 32. Kuhlman B, Dantas G, Ireton GC, Varani G, Stoddard BL, Baker D (2003) Design of a novel globular protein fold with atomic‐level accuracy. Science 302:1364–1368. [DOI] [PubMed] [Google Scholar]
- 33. Rohl CA, Strauss CEM, Misura KMS, Baker D (2004) Protein structure prediction using Rosetta. Methods Enzymol 383:66–93. [DOI] [PubMed] [Google Scholar]
- 34. Classen S, Hura GL, Holton JM, Rambo RP, Rodic I, McGuire PJ, Dyer K, Hammel M, Meigs G, Frankel KA, Tainer JA (2013) Implementation and performance of SIBYLS: a dual endstation small‐angle X‐ray scattering and macromolecular crystallography beamline at the Advanced Light Source. J Appl Cryst 46:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dyer KN, Hammel M, Rambo RP, Tsutakawa SE, Rodic I, Classen S, Tainer JA, Hura GL (2014) High‐throughput SAXS for the characterization of biomolecules in solution: a practical approach. Methods Mol Biol 1091:245–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schneidman‐Duhovny D, Hammel M, Tainer JA, Sali A (2013) Accurate SAXS profile computation and its assessment by contrast variation experiments. Biophys J 105:962–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schneidman‐Duhovny D, Hammel M, Tainer JA, Sali A (2016) FoXS, FoXSDock and MultiFoXS: single‐state and multi‐state structural modeling of proteins and their complexes based on SAXS profiles. Nucleic Acids Res 44:W424–W429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Huang P‐S, Feldmeier K, Parmeggiani F, Fernandez Velasco DA, Höcker B, Baker D (2016) De novo design of a four‐fold symmetric TIM‐barrel protein with atomic‐level accuracy. Nat Chem Biol 12:29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hashimoto K, Panchenko AR (2010) Mechanisms of protein oligomerization, the critical role of insertions and deletions in maintaining different oligomeric states. Proc Natl Acad Sci USA 107:20352–20357. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information