

Abstract
Dedicated computing resources are expensive to develop, maintain, and administrate. Frequently, research groups require bursts of computing power, during which progress is still limited by available computing resources. One way to alleviate this bottleneck would be to use additional computing resources. Today, many computing devices remain idle most of the time. Passive volunteer computing exploits this unemployed reserve of computing power by allowing device‐owners to donate computing time on their own devices. Another complementary way to alleviate bottlenecks in computing resources is to use more efficient algorithms. Engaging volunteer computing employs human intuition to help solve challenging problems for which efficient algorithms are difficult to develop or unavailable. Designing engaging volunteer computing projects is challenging but can result in high‐quality solutions. Here, we highlight four examples.
Keywords: distributed computing, volunteer computing, crowd sourcing, protein structure prediction, interaction networks, protein‐ligand docking
Abbreviations
- C2D
Connect to Decode
- BOINC
Berkeley Open Infrastructure for Network Computing
- D@H
Docking@Home
- ExSciTech
Explore Science, Technology, and Health
- FLOPS
floating point operations per second
- GPU
graphics processing unit
- IPW
interactome pathway
- M‐PMV
Mason Pfizer monkey virus
- SSNIc
Species Specific Network Inference challenge
- Mtb
Mycobacterium tuberculosis
- VC
volunteer computing
- VMD
Visual Molecular Dynamics.
Introduction
Crowd sourcing, volunteer computing (VC), citizen science—these terms refer to the use of idle computing resources to solve problems. In general, such computing resources can be both of a biological and silicon variety. Here, we use the VC term and differentiate between two types of VC based on the role of volunteers. If the volunteers use their human intuition to actively participate in finding the solution to a problem, this is termed engaging VC. If the volunteers only donate processing cycles on their computer devices, this is termed passive VC. Several passive VC projects, especially within astronomy,1, 2 have been widely used over the years. The Folding@Home3 and Docking@Home4 projects were developed as passive VC projects to explore protein and protein‐ligand complex structures. However, it is more challenging to successfully develop an engaging VC project. Therefore, only a select few engaging VC projects have been developed for any discipline and specifically for protein‐related science. The transition between passive and engaging VC projects requires engagement to be introduced at various levels (Fig. 1). In fact, one key challenge of designing engaging VC projects is to properly focus the effort of participants on those parts of a problem where they can provide the greatest impact.
Figure 1.
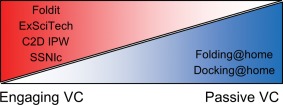
Gradient of volunteer computing (VC). VC is a scale with engaging projects on one side and passive projects on the other side and a smooth transition between them. Only a handful of projects have taken advantage of VC
Below we present a review of engaging VC projects in protein science made possible through crowd sourcing. The review is organized as follows. First, we present BOINC, a framework enabling volunteer computing and crowd sourcing projects to be easily developed by researchers. We then provide four case‐studies describing examples of volunteer computing and crowd sourcing that take advantage of human intelligence to help solve difficult problems involving protein‐related science. The review concludes by identifying open avenues for future research to realize the full potential of volunteer computing and crowd‐sourcing scientific problems.
Boinc
The Berkeley Open Infrastructure for Network Computing (BOINC) provides a middleware software solution for distributing compute jobs across a network of volunteer‐donated computers. Currently, almost 200,000 volunteers provide over 1 million computers resulting in over 16 petaFLOPS of computing power. This is enough computing power to place the BOINC VC network among the top five super computers in the world. BOINC was designed to be manageable for an average research group to set‐up a project within about a week. In addition, BOINC allows separate and diverse projects to share the same pool of resources and reward participants. Each project is entirely setup and maintained by the group conducting a project. BOINC provides tools to set up the server and configure the software to handle the data and schedule the flow between the project and its participants. Participants need only to register on a given project's website and download the BOINC client software.
Enabling volunteer distributed computing presented challenges that BOINC needed to overcome. First, sending out computing jobs to unknown computers could result in calculation errors or malicious users. To overcome these challenges, BOINC submits redundant jobs to multiple, different participants. This allows the project to analyze the results and determine which ones are correct based on multiple samples. Also, results from malicious users will be just one of N redundant results and therefore can be appropriately filtered. Second, the server infrastructure of a BOINC project is typically very small compared to the computers provided by participants such that the server could get overloaded if all participants interacted with the server simultaneously. Therefore, BOINC has built‐in checks to limit the interaction of participants' computers with the project server to keep the server running smoothly. Third, participants are highly motivated by a reward system. BOINC developed a fair scoring system that provides credit for donated time, which is difficult to cheat by participants. For long computing jobs, participants still want to receive credit without waiting until the computing job is entirely finished. So, BOINC provides methods for the project to provide credit as the computation proceeds.
To foster strong community support, BOINC provides features for participants such as options for creating teams, profiles, messaging, and screensaver graphics. BOINC also allows the use of GPU accelerated computing. Due to its flexibility and design such that any research group can create a project, BOINC is the go‐to solution for developing volunteer computing projects for protein chemistry research such as ligand‐protein docking4, 5, 6, 7, 8 and protein structure prediction.3, 9, 10 These projects are passive VC projects as they only use the computing resources of volunteers. BOINC can also be used for engaging volunteer computing, as highlighted below for the Foldit11 and ExSciTech12 projects.
C2d Ipw
Mycobacterium tuberculosis (Mtb) is the pathogen causing tuberculosis.13 To develop new pharmaceuticals against multi‐drug‐resistant strains, a high quality interactome was needed to identify potential new therapeutic targets. Importantly, much protein interaction data existed in the literature but had not been curated into databases from which all the components of the biological system could be simultaneously considered. Therefore, the authors developed an approach to enable the community to annotate the Mtb genome in an accurate, complete, and detailed way (Fig. 2). This crowd sourcing method was termed “Connect to Decode” (C2D) and resulted in the “interactome pathway” (IPW) for Mtb. Participants reviewed over 10,000 papers, with an average of 3–4 papers being required to have enough information to annotate a protein. In total, it was estimated that 300 years of manual labor were condensed into four months.
Figure 2.
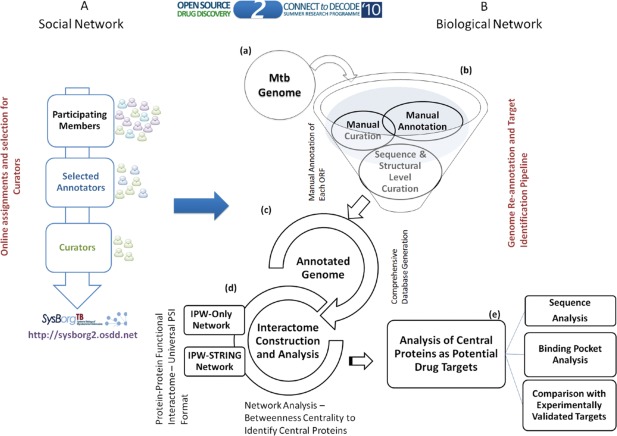
Overview of the workflow for C2D IPW to obtain the protein interaction map for Mtb (Figure reproduced from Ref. 13). Without focusing on the details in the content of the figure, the reader can appreciate the amount of organization, planning, and development that came together in order to successfully integrate all of the volunteer effort into a focused scientific outcome. The organizers built‐in multiple feedback loops in order to ensure high‐quality results
The annotation method employed one of two different standard operating protocols depending on whether literature was available relating to the protein. If no data was available regarding a protein specifically within Mtb, information from other closely related organisms was used. Literature was identified based on relevant keywords in standard databases (e.g., PubMed). Prospective participants underwent training and evaluation to ensure appropriate capability. After filtering under‐performing candidates, over 100 researchers and graduate and undergraduate students were selected from an initial registration of over 800. For proteins where no literature was available, information from the closest available homolog was used, with this being highlighted to distinguish homologous annotations from direct literature data. Participants were given a presentation so they could curate the annotation data gathered in the IPW. The presentation consisted of suggested background reading and instructions for editing, correcting, and color coding data by status. The IPW data were contained in a shared Google Document spreadsheet to allow users to easily edit the content.
The IPW covers 87% of the Mtb genome, which is a 67% increase over the coverage of previous Mtb genome annotations. The IPW also contains data from multiple interaction databases to complement the manually curated data. IPW annotates 71% of the proteins thought to comprise the Mtb proteome. IPW added 1762 interactions and 29 proteins above those found in an entirely computationally derived database. The authors performed network analysis to identify potential drug targets. The analysis identified several proteins that were both previously identified and validated as drug targets and also novel potential drug targets.
ExSciTech
The Explore Science, Technology, and Health (ExSciTech) project combines both active and passive user engagement to advance protein‐ligand docking studies.12 ExSciTech provides several games to train users in concepts needed to understand protein‐ligand docking. ExSciTech also promotes user participation in protein‐ligand docking studies through games to exploit human intuition such that the large potential search space can be reduced. ExSciTech is formulated around Docking@Home (D@H), which is an established VC project for searching for ligands effective against breast cancer and HIV. D@H is solely a passive crowd‐sourcing platform, with users just donating free computer cycles. ExSciTech augments D@H by actively engaging volunteers during the cross‐docking phase of drug identification, whereby interactions of ligands with closely related proteins are studied to investigate possible non‐specific binding of a ligand. This has implications for potential side‐effects caused by a molecule. Users contribute to this process by reducing the number of ligands and protein binding pockets that need to be explored by the docking algorithm.
ExSciTech uses gaming to educate and engage users in the scientific process. This allows users to be assessed for their capability and to improve, thereby identifying appropriate tasks. Thus, actively engaged users will provide more resources to the project. ExSciTech can be divided into two stages according to the user: a learning stage and an engaged stage. During the learning stage, volunteers learn about the scientific process and protein‐ligand docking. Users are assessed as Novice, Amateur, or Professional Chemist at the end of the learning stage. The game for the learning stage consists of identifying and classifying properties of molecules as they fall across the screen one‐at‐a‐time. Hints are provided to guide a player to the correct answer. This can be described as a “flashcard game”. After the learning stage, participants can apply their knowledge to develop jobs for distribution through the D@H VC network. The game developed for the engaging stage was termed “Drag'n Dock”. Here, users are given a protein and they identify the binding pocket, then use a spaceship to drag a ligand into the binding pocket to obtain an initial docking structure. Once satisfied with the ligand position, the user then flies the spaceship through a portal and the job is submitted to the D@H network. Users are rewarded based on the quality of the results they generate as indicated by the energy of the protein‐ligand complex. Providing computer time for other users' jobs is also rewarded. Because of the modular design, additional games can easily be added into ExSciTech. ExSciTech takes advantage of established software such as BOINC and Visual Molecular Dynamics (VMD). Using BOINC provides a heavily used and well established infrastructure for handling the master‐worker relationship needed to distribute jobs to users. VMD provides high quality representations.
ExSciTech was tested to identify its effect on learning in the game environment had compared to a traditional learning approach. Twenty‐four graduate or undergraduate students without training in biochemistry were given a brief introduction to the properties of peptides, carbohydrates, lipids, and nucleotides. The students were then asked to classify a set of molecules. The students were split into two groups: one group was presented with the molecules on paper; the other group was presented with the molecules through the ExSciTech game. Three metrics were compared between the two groups: score on the task, enjoyment of the task, and time to complete the task. The ExSciTech group indicated a higher level of enjoyment with smaller variance than the traditional group. The ExSciTech group also featured a steeper learning curve as indicated by a reduction in score during the first eight test molecules. The ExSciTech group further showed an overall lower score than the traditional group. The authors attributed the difference to several aspects of ExSciTech. One is the speed at which the molecule was presented and disappeared to the user. Allowing the user to control the speed, or adapting the speed to the user's performance could result in a higher score. Also, ExSciTech users could not go back to reexamine their previous answers, but this was an option for the traditional group. Further, the 3D representation provided by ExSciTech can obfuscate aspects of molecules that are easily identifiable from a 2D representation, as presented to the traditional group. Finally, the representations presented to the ExSciTech group could not render double bonds and instead indicated double bonds as single bonds. The authors state that they plan to improve upon these shortcomings as ExSciTech matures.
Foldit
The protein folding problem arises from the fact that the amino acid sequence determines the three dimensional topology of a protein structure, but the number of degrees of freedom the amino acids allow results in a vast search space that must be traversed to locate the lowest energy and native conformation. Further complicating the goal of predicting structure from sequence are the many local minima of the energy landscape. The current state‐of‐the‐art program in protein structure prediction software is the Rosetta program.14 Rosetta's energy function can properly identify a native conformation as having the lowest energy.15 Rosetta uses a combination of large and small, random and deterministic structural perturbations in an attempt to sample the native conformation, but its ability to sample the native conformation is its bottleneck.16
Foldit is a game developed to use human intuition to facilitate the prediction of high‐quality protein structure models.11 The portion of the protein folding problem Foldit targets for human improvement over algorithmic methods are the large‐scale, stochastic perturbations. Foldit is based on Rosetta and exposes Rosetta functionality through a game‐friendly interface (Fig. 3). Players participate in challenges that are posted online with the goal being to achieve the highest score for that protein structure. Players are able to directly manipulate the protein structure and perform specific tasks such as perturbing helix or β‐sheet conformations. Players are also able to perform automatic Rosetta functions such as side‐chain repacking, fragment insertion, or gradient‐based minimization. They also can affect the automatic perturbations by, for example, preventing portions of a protein from being moved or restraining portions to move together. Foldit provides visual cues to the users indicating specific protein properties such as hydrophobicity, clashes, cavities, and energetic favorability along the backbone as calculated by the Rosetta energy function. These cues allow players to focus on where to improve the protein structure. Foldit provides beginning puzzles for users not familiar with protein chemistry to learn about the important aspects of the visual interface and functionality for manipulating the protein structure.
Figure 3.
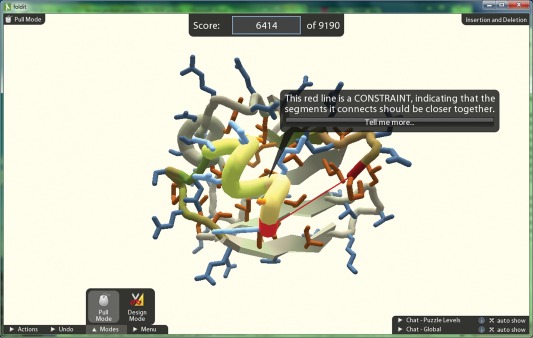
Screenshot of Foldit during one of the learning tutorials. Foldit uses color cues and structural simplification to provide important information to players without overwhelming them. The tutorials provide text‐box clues with information about the game in order to familiarize players with the mechanics. In Foldit, players are tasked with achieving the highest score, which corresponds to the most favorable three‐dimensional conformation of a protein
Ten proteins were used to test players' abilities to find correct protein structures. None of the protein structures were available during the duration of the challenges. Players were given starting structures derived from initial Rosetta protein structure prediction results and asked to improve the structures. The authors found players outperformed Rosetta in scenarios where a cascade of changes is required in order to reach a preferred conformation. Under such conditions, the stochastic search process of Rosetta is unlikely to sample a series of steps which may require energetically unfavorable intermediate states. Additionally, given the choice of multiple initial conformations, players were able to select the conformation which would allow them to most easily reach the native conformation.11
Players used a wide variety of exploration strategies to drive the proteins into native conformations. As a result, Foldit was extended to allow users to codify their strategies as “recipes”. These could then be shared and distributed throughout the Foldit community for other players to use and build upon. Over 5000 recipes were created, with twenty‐six being run more than 1000 times. Different recipes are used at different stages of the model building process. Interestingly, all of the recipes rely upon player input. Therefore, the recipes did not replace human involvement but complemented it. The Foldit community independently discovered a fine‐tuning optimization routine that outperformed previously published Rosetta methods in terms of speed and model improvement. Further, this routine also could more quickly reach a superior energy compared to an unpublished routine the researchers had developed.17
Foldit was applied to several structural and crystallographic problems. In particular, the Mason Pfizer monkey virus (M‐PMV) is a target for preventing simian AIDS. Crystals of M‐PMV were available, but the structure was not solved for over ten years. Foldit players were tasked with developing a model suitable for molecular replacement based on NMR data. These players were able to improve the NMR models such that, within just days after conclusion of the puzzle, a final refined structure was made available through the use of standard molecular replacement tools.18
Foldit was extended to allow players to build models directly into electron density maps. The extension included visualization of the electron density as an iso‐surface whose representation the players could control to remove extraneous electron density. The Foldit score was updated to include an electron density agreement score to incentivize players to consider the electron density. Their ability to correctly determine a structure from electron density was tested via a competition between Foldit players, automated methods, trained crystallographers, and undergraduate students taking an introductory crystallography course. The best structure produced in the competition was a structure from Foldit, which showed improved side‐chain conformations compared to the best structure produced by the trained crystallographers. The result of this contest allowed the protein to be identified as a member of the histidine triad protein family.19
Foldit was also used for protein design. Players were asked to improve the activity of a computationally designed enzyme that catalyzes the Diels‐Alder reaction. The task was broken down into three puzzles that allowed an iterative cycle between the Foldit players and the scientists. The Foldit players were able to achieve more than an 18‐fold improvement in enzyme activity compared to the starting protein. Further, the Foldit players could predict the structure of their designed protein with high accuracy.20
SSNIc
The Species Specific Network Inference challenge (SSNIc) was created to develop a protein interaction network specific to humans and rats.21 Participants were given phosphoprotein, gene expression, and cytokine data, and an initial reference network from which they could remove or add edges between nodes (Fig. 4). The goal of the challenge was to improve the transferability of drugs developed in rats such that they would also be safe and effective when tested in humans. New technologies allow the activity of thousands of genes to be simultaneously measured, but no computational method has arisen to meet the data influx, with different methods having various strengths and weaknesses. SSNIc was set‐up to take advantage of the massive reasoning skills available through crowd‐sourcing.
Figure 4.
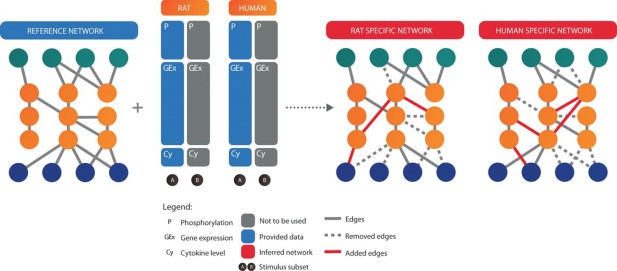
Schematic description of the Species‐Specific Network Inference challenge (SSNIc). The figure is reproduced from Ref. 21. Participants were tasked with updating a reference network to differentiate between human and rat interactions by removing or adding edges to the graph based on available experimental data
One challenge was devising a method for evaluating the participant‐generated networks. The authors decided to use a method that combined multiple predictions from orthogonal methods to rank and evaluate the networks. One scoring method consisted of an automatically generated network, used as what was termed the “silver standard”. A second scoring method involved a blind assessment by three independent judges. The judgment was based on the description provided by the participants as to the method they used to generate their network. Several criteria were used including, for example, rigor, originality, and ability to implement the methodology computationally. Judges provided scores between one and five, with five being the best, that then were averaged.
The final consensus network was derived from all the participants' results. This was accomplished by considering the number of times an edge was indicated to be an edge by participants and deriving the probability the indicated connection was in‐fact correct. The initial reference network had 220 nodes and 501 edges. Participants could only add or remove edges, not nodes, from the reference network. In the end, for the human and rat networks the median number of edges were 406 and 429, respectively. The lower number of edges in the participant networks compared to the reference network indicates that the reference network over estimated the activity of pathways leading to false positives. Evaluation scores showed little variation between the reviewers. The final score for each participant was simply the average of the results from each scoring method (agreement with the “silver network” and evaluation by reviewers). The best teams according to the consensus network were also the best determined by the total scoring system. The authors calculated that about eight votes by teams were needed to affirm the existence of an edge.
Comparing the consensus networks for human and rat, the authors found differences in pathways at strictly local and isolated locations, but at higher levels of organization the networks were similar, as would be expected. Because participants who performed well based on multiple evaluation criteria also performed well when compared to the consensus, simply using the consensus as a scoring criterion could be one possibility in the future. This would have the advantage that the bias of the “silver standard” due to reliance on a particular algorithm would be removed. Consensus scoring relies upon having little correlation between predictions in the ensemble. The number of participants could have been larger and would have enabled the identification of additional edges, so in this instance, the number of participants was a limiting factor in the results obtained. Overall the consensus networks highlight a few differences in interactions between human and rat pathways that could be taken into account during drug testing and development. Participants used a broad array of methods to develop their networks ranging from network to statistical to heuristic methods. Even when using the same method, sharp disparities in performance can result from the many choices needed to set up a given method. This further supports the utility of crowd‐sourcing to enable the investigation of not only a breadth of different methods but also variations within methods.
Discussion
In order for a crowd‐sourced project to succeed, several pieces must come together from the participants' perspective.22 First, project organizers must make sure the goal is clearly articulated to participants. Second, participants must be provided feedback to facilitate their development of successful strategies and properly focus their efforts. Third, the interface provided should ideally incorporate an iterative feedback loop between the participants and the project creators to continuously improve how participants interact with the task.23 Finally, the project must be advertised to attract the attention of the VC community.
Project organizers must carefully consider the design of the project. If the task cannot be parallelized across many participants, passive or engaging VC may not be the best solution. Project developers must design the computational task for participants to maximize the speed‐up achieved by distributing it over many computers.3 Developing a project for engaging VC introduces significant challenges beyond a passive VC project, because designers need to frame the task in a manner that attracts participants' interest and participation. Simultaneously, the project must also be designed to address a scientific problem. Typically, passive VC projects are designed so that the calculations do not interfere with the participants' usage of their computing device. With engaging VC, both the participants and their computing devices can be engaged in the project.
With the growing ubiquity of personal computing devices featuring increased computational power and efficiency, the potential for VC continues to expand. Although designing a popular VC project may be challenging, examples reported here demonstrate that the initial effort can be well rewarded by the efforts volunteers donate to solve difficult scientific problems in protein chemistry. Continued improvements in middleware such as BOINC and programming tools to develop user interfaces will further reduce the effort needed to set‐up a VC project. As the amount of experimental data expands with the development of new techniques, VC provides an avenue for analyzing the data at low cost and in ways not conventionally accessible by standard algorithms.
Disclosure Statements
None.
Authors Contribution
N.S.A. conceived the manuscript. N.S.A. and K.P. wrote the manuscript.
Acknowledgments
We thank Dr. Leslie T. Webster, Jr. for helpful comments on this manuscript. This work was supported by funding from the National Institutes of Health (EY009339 and EY027283 to K.P.), (EY025007 to N.S.A.). K.P. is the John H. Hord Professor of Pharmacology.
Contributor Information
Nathan S. Alexander, Email: nsa36@case.edu
Krzysztof Palczewski, Email: kxp65@case.edu.
References
- 1. net NEWS (1998) … and a Search for Alien Life. Science 282:839–839. [Google Scholar]
- 2. Lintott CJ, Schawinski K, Slosar A, Land K, Bamford S, Thomas D, Raddick MJ, Nichol RC, Szalay A, Andreescu D, Murray P, Vandenberg J (2008) Galaxy Zoo: morphologies derived from visual inspection of galaxies from the Sloan Digital Sky Survey*. Monthly Notices Royal Astron Soc 389:1179–1189. [Google Scholar]
- 3. Shirts M, Pande VS (2000) Screen savers of the world unite! Science 290:1903. [DOI] [PubMed]
- 4. Estrada T, Armen R, Taufer M (2010) Automatic selection of near‐native protein‐ligand conformations using a hierarchical clustering and volunteer computing. Proceedings of the First ACM International Conference on Bioinformatics and Computational Biology. Niagara Falls, New York: ACM; p 204–213.
- 5. Zhang B, Estrada T, Cicotti P, Balaji P, Taufer M (2017) Enabling scalable and accurate clustering of distributed ligand geometries on supercomputer. Parallel Comput 63:38–60. [Google Scholar]
- 6. Estrada T, Zhang B, Cicotti P, Armen RS, Taufer M (2012) A scalable and accurate method for classifying protein‐ligand binding geometries using a MapReduce approach. Comput Biol Med 42:758–771. [DOI] [PubMed] [Google Scholar]
- 7. Pradeep P, Struble C, Neumann T, Sem DS, Merrill SJ (2015) A novel scoring based distributed protein docking application to improve enrichment. Ieee‐Acm Trans Comput Biol Bioinform 12:1464–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gaudreault F, Najmanovich RJ (2015) FlexAID: Revisiting docking on non‐native‐complex structures. J Chem Inform Model 55:1323–1336. [DOI] [PubMed] [Google Scholar]
- 9. Strunk T, Wolf M, Brieg M, Klenin K, Biewer A, Tristram F, Ernst M, Kleine PJ, Heilmann N, Kondov I, Wenzel W (2012) SIMONA 1.0: An efficient and versatile framework for stochastic simulations of molecular and nanoscale systems. J Comput Chem 33:2602–2613. [DOI] [PubMed] [Google Scholar]
- 10. Strunk T, Wolf M, Wenzel W (2012) Peptide structure prediction using distributed volunteer computing networks. J Math Chem 50:421–428. [Google Scholar]
- 11. Cooper S, Khatib F, Treuille A, Barbero J, Lee J, Beenen M, Leaver‐Fay A, Baker D, Popovic Z, Players F (2010) Predicting protein structures with a multiplayer online game. Nature 466:756–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Matheny M, Schlachter S, Crouse LM, Kimmel ET, Estrada T, Schumann M, Armen R, Zoppetti G, Taufer M (2012) ExSciTecH: Expanding volunteer computing to Explore Science, Technology, and Health. October 8–12, p 1–8.
- 13. Vashisht R, Mondal AK, Jain A, Shah A, Vishnoi P, Priyadarshini P, Bhattacharyya K, Rohira H, Bhat AG, Passi A, Mukherjee K, Choudhary KS, Kumar V, Arora A, Munusamy P, Subramanian A, Venkatachalam A, Raj S, Chitra V, Verma K, Zaheer S, Gurusamy M, Razeeth M, Raja I, Thandapani M, Mevada V, Soni R, Rana S, Ramanna GM, Raghavan S, Subramanya SN, Kholia T, Patel R, Bhavnani V, Chiranjeevi L, Sengupta S, Singh PK, Atra N, Gandhi S, Avasthi TS, Nisthar S, Anurag M, Sharma P, Hasija Y, Dash D, Sharma A, Scaria V, Thomas Z, Consortium O, Chandra N, Brahmachari SK, Bhardwaj A (2012) Crowd sourcing a new paradigm for interactome driven drug target identification in Mycobacterium tuberculosis. PLoS One 7:e39808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Simons KT, Kooperberg C, Huang E, Baker D (1997) Assembly of protein tertiary structures from fragments with similar local sequences using simulated annealing and Bayesian scoring functions. J Mol Biol 268:209–225. [DOI] [PubMed] [Google Scholar]
- 15. Bradley P, Misura KM, Baker D (2005) Toward high‐resolution de novo structure prediction for small proteins. Science 309:1868–1871. [DOI] [PubMed] [Google Scholar]
- 16. Misura KMS, Baker D (2005) Progress and challenges in high‐resolution refinement of protein structure models. Proteins 59:15–29. [DOI] [PubMed] [Google Scholar]
- 17. Khatib F, Cooper S, Tyka MD, Xu K, Makedon I, Popović Z, Baker D, Players F (2011) Algorithm discovery by protein folding game players. Proc Natl Acad Sci USA 108:18949–18953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Khatib F, DiMaio F, Cooper S, Kazmierczyk M, Gilski M, Krzywda S, Zabranska H, Pichova I, Thompson J, Popovic Z, Jaskolski M, Baker D (2012) Crystal structure of a monomeric retroviral protease solved by protein folding game players. Nat Struct Mol Biol 19:364–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Horowitz S, Koepnick B, Martin R, Tymieniecki A, Winburn AA, Cooper S, Flatten J, Rogawski DS, Koropatkin NM, Hailu TT, Jain N, Koldewey P, Ahlstrom LS, Chapman MR, Sikkema AP, Skiba MA, Maloney FP, Beinlich FRM, Popovic Z, Baker D, Khatib F, Bardwell JCA (2016) Determining crystal structures through crowdsourcing and coursework. Nat Commun 7:12549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Eiben CB, Siegel JB, Bale JB, Cooper S, Khatib F, Shen BW, Players F, Stoddard BL, Popovic Z, Baker D (2012) Increased Diels‐Alderase activity through backbone remodeling guided by Foldit players. Nat Biotech 30:190–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bilal E, Sakellaropoulos T, Participants C, Melas IN, Messinis DE, Belcastro V, Rhrissorrakrai K, Meyer P, Norel R, Iskandar A, Blaese E, Rice JJ, Peitsch MC, Hoeng J, Stolovitzky G, Alexopoulos LG, Poussin C (2015) A crowd‐sourcing approach for the construction of species‐specific cell signaling networks. Bioinformatics 31:484–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lessl M, Bryans JS, Richards D, Asadullah K (2011) Crowd sourcing in drug discovery. Nat Rev Drug Discov 10:241–242. [DOI] [PubMed] [Google Scholar]
- 23. Cooper S, Treuille A, Barbero J, Leaver‐Fay A, Tuite K, Khatib F, Snyder AC, Beenen M, Salesin D, Baker D, Popovi Z (2010) The challenge of designing scientific discovery games. Proceedings of the Fifth International Conference on the Foundations of Digital Games. Monterey, California: ACM p 40–47. [Google Scholar]