

ABSTRACT
The agr system of Staphylococcus aureus promotes invasion of host tissues, and as expected, agents that block agr quorum sensing have anti-infective properties. Paradoxically, agr-defective mutants are frequently recovered from patients, especially those persistently infected with S. aureus. We found that an agr deficiency increased survival of cultured bacteria during severe stress, such as treatment with gentamicin, ciprofloxacin, heat, or low pH. With daptomycin, deletion of agr decreased survival. Therefore, agr activity can be either detrimental or protective, depending on the type of lethal stress. Deletion of agr had no effect on the ability of the antimicrobials to block bacterial growth, indicating that agr effects are limited to lethal action. Thus, the effect of an agr deletion is on bacterial tolerance, not resistance. For gentamicin and daptomycin, activity can be altered by agr-regulated secreted factors. For ciprofloxacin, a detrimental function was downregulation of glutathione peroxidase (bsaA), an enzyme responsible for defense against oxidative stress. Deficiencies in agr and bsaA were epistatic for survival, consistent with agr having a destructive role mediated by reactive oxygen species. Enhanced susceptibility to lethal stress by wild-type agr, particularly antimicrobial stress, helps explain why inactivating mutations in S. aureus agr commonly occur in hospitalized patients during infection. Moreover, the agr quorum-sensing system of S. aureus provides a clinically relevant example in which a single-step change in the response to severe stress alters the evolutionary path of a pathogen during infection.
KEYWORDS: Antibiotics, Staphylococcus aureus, agr, ciprofloxacin, daptomycin, gentamicin, resistance, tolerance
IMPORTANCE
When phenotypes produced in response to an environmental stress are inadequate to buffer against that stress, changes that do buffer may become genetically encoded by natural selection. A clinically relevant example is seen with S. aureus mutants that are deficient in the key virulence regulator agr. Paradoxically, defects in agr are selected during serious hospital infection and have been associated with worse outcome. The current work helps resolve this paradox: agr mutants are often less readily killed by lethal stressors without affecting MIC, a phenomenon known as tolerance. Our results indicate that tolerance, which would not be detected as resistance, can be selected in clinical settings. The data also support the ideas that (i) S. aureus broadly hedges against environmental change and stress through genome plasticity, (ii) reactive oxygen can be involved in the self-destructive response in bacteria, and (iii) therapeutic targeting of agr and virulence can be counterproductive.
INTRODUCTION
Experimental (1–3) and observational (4–7) work suggests that mutation of global regulators drives adaptive leaps made by microbes. In Staphylococcus aureus, the agr quorum-sensing transcriptional regulator is likely to be such a system. The agr regulon governs the expression of secreted virulence factors that appear to enhance acute infection and bacterial dissemination among healthy hosts. However, these factors are not needed, or are needed less, for pathogen persistence inside human tissues (8–12). Indeed, the prototypical within-host adaptation during S. aureus infection results in partial or complete inactivation of agr (13–17). While this regulator, which controls ~200 genes in vitro (18), is critical for pathogenesis in a variety of contexts (19–22), agr-defective mutants arise and are enriched during human infection when treated with antimicrobials (23–26). The result is poor clinical outcome (27, 28). Thus, the S. aureus agr system provides an opportunity to study how inactivation of a virulence regulon shifts the pathogen to a more persistent state. Moreover, understanding the enrichment of global regulator mutants during antimicrobial treatment is central to managing infections by S. aureus and other pathogens.
The agr locus consists of two divergent transcription units driven by promoters P2 and P3 (reviewed in reference 29). The P2 operon encodes the signaling module, which contains four genes—agrB, -D, -C, and -A—each of which is required for transcriptional activation of the agr regulon. AgrC is the receptor-histidine kinase, and AgrA is the response regulator. AgrD is the autoinducing, secreted peptide that is derived from a propeptide processed by AgrB. The P3 transcript is a regulatory RNA (RNAIII) that also contains the structural gene for delta-hemolysin. Regulation of target genes by agr occurs through two pathways: (i) an RNAIII-dependent regulation of virulence genes and (ii) an RNAIII-independent, AgrA-mediated regulation of metabolic genes and small cytolytic toxins known as phenol-soluble modulins (PSMs) (18). The regulatory connection between these processes links virulence to metabolism.
To study agr-stressor effects, it is important to distinguish growth-related phenotypes from those specific to survival. For example, treatment with an antimicrobial leads to damage that is specific to the test agent. This primary damage halts growth, which is measured as the MIC. The MIC reflects drug uptake, efflux, and target affinity; high MIC values are associated with antimicrobial resistance. Some forms of primary damage also kill cells, with much of the lethal process arising from a self-destructive, secondary bacterial response to the primary damage (reviewed in references 30 to 32). To focus experimental measurements on the lethal response, lethal drug concentrations are normalized to the MIC. It is also important to recognize that lethal stress may be transient. For example, reactive oxygen species (ROS) can accelerate killing without increasing the extent of killing (33). Consequently, overnight killing assays, such as those commonly used to measure minimal bactericidal concentration (MBC), may take too much time to detect changes in killing rate and may therefore be uninformative with respect to the stress response (33).
Highly lethal antimicrobials are important probes for studying bacterial responses to lethal stress, particularly for responses involving the accumulation of toxic ROS (30–32, 34–36). The present work used a range of both drug concentrations and treatment times to probe effects of agr status on the response of S. aureus to lethal stress. We found that an agr defect increased S. aureus survival by an order of magnitude following treatment with some antimicrobials (gentamicin) but that it conferred hyperlethality on others (daptomycin). agr-deficiency-mediated protection operated through a variety of mechanisms that depended on the underlying lethal stress. The data lead to a framework for interpreting the phenotypes of known and newly emergent infection-adaptive mutations. Broadly speaking, mutation can create, in a single step, complex traits that explain how loss of a seemingly important facilitator of virulence can be adaptive during infection by suppressing the lethal effects of stressors.
RESULTS
Transcription from agr promoters during growth in culture.
Because agr is a quorum-sensing regulon (29, 37), differences in antimicrobial-mediated killing between the wild type and Δagr mutants must be interpreted within the context of growth phase and agr induction. Using a β-lactamase reporter fused to the principal agr promoter, P3, we confirmed, in laboratory strain S. aureus Newman, that maximal agr activity is seen in late exponential growth phase and is followed by a plateau or decrease (see Fig. S1A in the supplemental material), as reported previously (38). All subsequent experiments were performed at late exponential phase.
Tests of agrP3 promoter activity. S. aureus Newman cells containing agrP3-blaZ (SaPI1 attC::agrP3-blaZ; strain BS983) were grown in TSB (A) or TSB in the presence of 20% (vol/vol) human serum (B). agrP3 activity (β-lactamase units/culture OD600) was assayed at the indicated times (see Materials and Methods). Symbols: broken line, growth; empty circles, agrP3 activity in TSB; filled circles, agrP3 activity in medium containing serum. Data represented the means from biological replicates ± standard deviations (n = 3). Download FIG S1, TIF file, 0.1 MB (108.4KB, tif) .
Copyright © 2017 Kumar et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Survey of lethal agents affected by deletion of agr.
agr-defective strains have been associated with the development of vancomycin tolerance (39, 40); however, agr dysfunction is associated with only small reductions in killing that are apparent only with long-term, time-kill experiments (41, 42). Consequently, we focused on several agents known to be rapid, concentration-dependent killers of S. aureus (gentamicin, ciprofloxacin, and daptomycin). For these agents, a deficiency in agr (agr::tetM deletion) had no effect on bacteriostatic activity (MIC) compared with a wild-type strain (Table 1). Thus, agr has no effect on the initial, bacteriostatic lesion created by the drugs; lethal activity is likely a stress response to those lesions.
TABLE 1 .
Antimicrobial susceptibilityb
| Antibiotic or compound | Strain | TSB MIC (μg/ml for antibiotics, mM for compounds) for strain: |
Serum MIC (μg/ml) for strain: |
||
|---|---|---|---|---|---|
| WT | Δagr mutant | WT | Δagr mutant | ||
| Antibiotics | |||||
| Cipro | Newman | 0.5 | 0.5 | 1 | 1 |
| ATCC 25923 | 0.5 | 0.5 | 1 | 1 | |
| LAC | 0.5 | 0.5 | 1 | 1 | |
| Gent | Newman | 6 | 6 | 12 | 12 |
| ATCC 25923 | 6 | 6 | 12 | 12 | |
| LAC | 6 | 6 | 12 | 12 | |
| 126a | 3 | 3 | 6 | 6 | |
| 127b | 3 | ND | 6 | ND | |
| BS39 | 1.5 | ND | 3 | ND | |
| BS40 | 1.5 | ND | 3 | ND | |
| Oxa | Newman | 0.25 | 0.25 | ND | ND |
| Daptoa | Newman | 4 | 4 | ND | ND |
| LAC | 2 | 2 | ND | ND | |
| Compounds | |||||
| H2O2 | Newman | 0.258 | 0.258 | ||
| 2,2′-Bipyridyl | Newman | 8 | 8 | ||
| Thiourea | Newman | 400 | 400 | ||
Daptomycin MICs were determined in Mueller-Hinton broth supplemented with 50 μg/ml Ca2+ and 25 μg/ml Mg2+.
Abbreviations: Cipro, ciprofloxacin; Dapto, daptomycin; Gent, gentamicin; ND, not determined; Oxa, oxacillin; WT, wild type.
When we compared survival rates of Δagr mutant and wild-type cells, we found that the mutation increased survival by about 10-fold for gentamicin and approximately 3-fold for ciprofloxacin when drug concentration was varied (Fig. 1A and B). A similar result was obtained using various treatment times at a fixed drug concentration (Fig. 1C and D). Since the presence of serum may alter antimicrobial activity (43) and agr functionality (44), we also examined lethal activity in 20% (vol/vol) human serum, the highest concentration of serum that failed to detectably affect bacterial growth. Inclusion of serum reduced agrP3-blaZ expression by severalfold (Fig. S1B), but protection conferred by the agr deficiency was greater against gentamicin and similar against ciprofloxacin relative to that observed in the absence of serum (Fig. S2A to D). Collectively, these data show that rapid lethal activity responds differently to an agr defect than bacteriostatic action, as if wild-type agr specifically stimulates a lethal response to the primary damage.
FIG 1 .
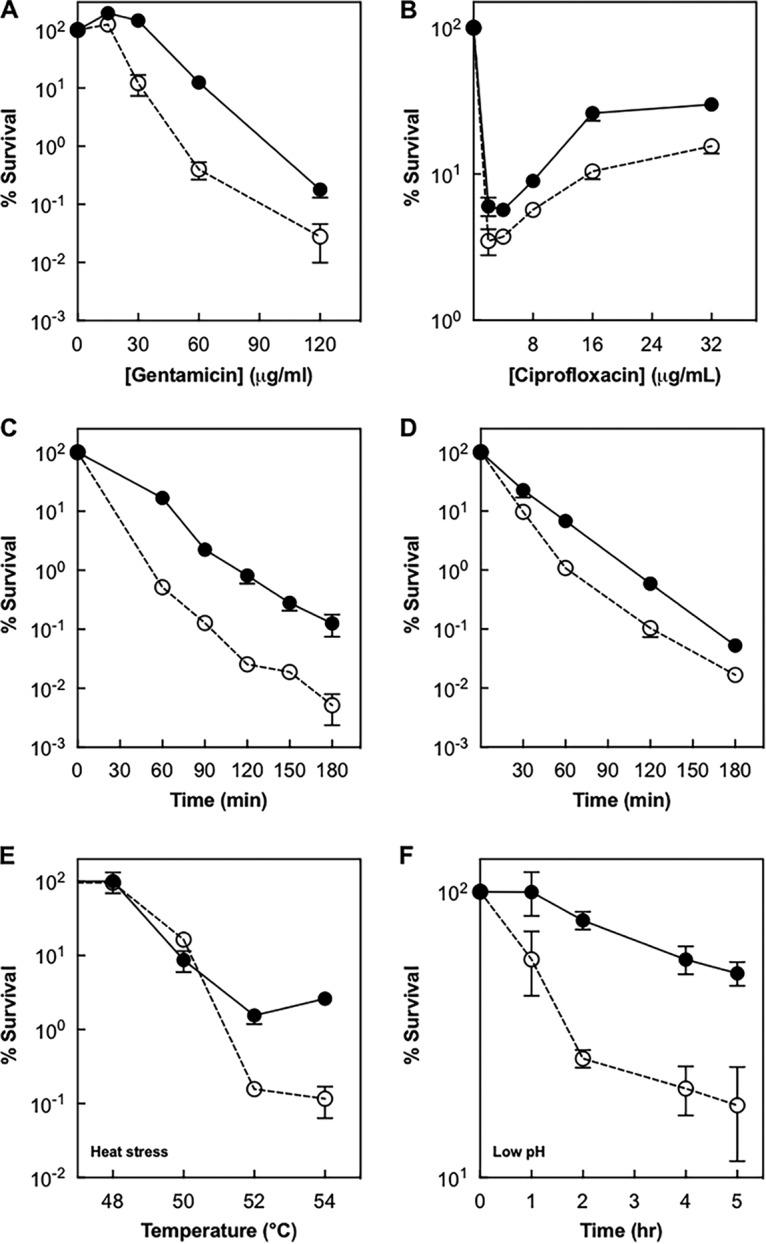
agr deficiency increases bacterial survival following exposure to antimicrobial and environmental stresses. Wild-type S. aureus Newman (BS12) and the Δagr strain (BS13) were grown to late log phase in TSB and treated with the indicated concentrations of gentamicin for 60 min (A) or with 60 μg/ml of gentamicin for the times indicated (C). Likewise, cultures were treated with indicated concentrations of ciprofloxacin for 60 min (B) or with 10 μg/ml of ciprofloxacin for the times indicated (D). At the end of treatment, aliquots were removed, serially diluted, and plated for determination of viable counts, from which percent survival was calculated relative to a sample taken at the time of drug addition. Similarly, cells grown to late log phase in TSB were incubated at the indicated temperatures for 10 min (E) or in acidic TSB (pH 3.0) for indicated times (F). Symbols: filled circles, Δagr strain; empty circles, wild type. Data represent means from biological replicates ± standard deviations (n = 3).
Serum enhances agr-mediated differences in antimicrobial killing. Wild-type S. aureus Newman (BS12) and Δagr mutant (BS13) were grown to late log phase in TSB with serum and treated with the indicated concentrations of gentamicin for 60 min (A) or with 60 μg/ml of gentamicin for the times indicated (C). Likewise, cultures were treated with indicated concentrations of ciprofloxacin for 60 min (B) or with 10 μg/ml of ciprofloxacin for the times indicated (D). Symbols: filled circles, Δagr strain; empty circles, wild type. Data represent means of biological replicates ± standard deviations (n = 3). Download FIG S2, TIF file, 0.1 MB (144.8KB, tif) .
Copyright © 2017 Kumar et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
As expected, agr-mediated differences in killing correlated with growth phase, with maximal effects occurring in late exponential phase when agr was induced (Fig. 2A). Complementation tests employing chromosomally integrated, wild-type agr confirmed that the agr deletion was responsible for protection from killing by gentamicin and ciprofloxacin (Fig. 2B and C). A protective effect of the agr mutation was also observed when cells were treated with heat or low pH (Fig. 1E and F), suggesting that a destructive, wild-type agr-mediated response occurs with a variety of severe stressors.
FIG 2 .

Antimicrobial-mediated killing is growth phase and agr specific. Cells were grown in serum for 2, 4, 6, or 24 h as indicated and treated with 15 μg/ml of gentamicin for 60 min (A). To determine whether the observed difference in killing was due to agr, wild-type (WT; BS12), Δagr mutant (BS13), and complemented Δagr mutant (BS519) cells were treated with 15 μg/ml of gentamicin (B) or 2.5 μg/ml of ciprofloxacin (C) for 60 min. Survival was determined as described in the legend to Fig. 1. Significance was examined by unpaired two-tailed t test (P < 0.05). ***, P < 0.01; ****, P < 0.001. Data represent the means from biological replicates ± standard deviations (n = 3).
Results with the other two antimicrobials differed from those described above. Killing by oxacillin was unaffected by deletion of agr (Fig. 3A). Oxacillin exhibits low to moderate, time-dependent killing. As with killing with other cell wall agents, such as vancomycin, killing by oxacillin likely reflects processes that are different from those occurring with agents that kill more rapidly, such as ciprofloxacin and gentamicin. Daptomycin, a rapid-killing, cell-membrane-targeting agent, showed increased killing in the agr-deficient mutant (Fig. 3B). Killing by the environmental stressor hydrogen peroxide was also increased by the agr deficiency (Fig. 3C). For these two stressors, wild-type agr exhibited a protective role. Thus, inactivation of agr protects cells from killing by some types of stress, while it has little effect on or enhances the lethal action of others.
FIG 3 .

Effect of an agr deficiency on the lethal activity of oxacillin, daptomycin, and exogenous H2O2. Wild-type cells (BS12, empty circles) or agr-deficient mutant cells (BS13, filled circles) were grown to late log phase and then treated with the indicated concentrations of oxacillin for 3 h (A), daptomycin for 90 min (B), or H2O2 for 60 min (C). Survival was determined as described in the legend to Fig. 1. Growth medium was TSB (oxacillin and H2O2) or Ca2+-supplemented Mueller-Hinton broth (daptomycin). Data represent the means from biological replicates ± standard deviations (n = 3).
Effect of agr deficiency on response to gentamicin.
To better understand the destructive effect of wild-type agr, we first asked whether the protective effect of an agr deletion against gentamicin-mediated killing acts through RNAIII. We observed no effect (Fig. 4A), indicating that destruction is agrA dependent.
FIG 4 .

agrA-mediated protection from gentamicin-mediated killing is largely independent of PMF. (A) Survival following gentamicin treatment. Late-log-phase cultures were grown in TSB plus 20% (vol/vol) human serum and treated with 15 μg/ml gentamicin for 60 min (wild-type [WT] strain BS12 [white bar], ΔRNAIII BS669 [black bar], and Δagr mutant BS13 [gray bar]). (B) Effect of pretreatment with CCCP. Cells were grown as described for panel A; they were pretreated with CCCP for 5 min and subsequently with 15 μg/ml of gentamicin for 60 min. (C) Effect of alkaline pH. Cells were grown as described for panel A, late-log-phase cultures were concentrated by centrifugation, and cells were resuspended in serum-containing TSB medium with pH adjusted to 8.0. They were then treated with 15 μg/ml of gentamicin for 60 min. Symbols: white bars, wild-type strain BS12; black bars, agr mutant BS13. (D) Effect of Δagr or saeS::bursa single mutation (BS13 and BS984) and double mutation (BS985) on exoproteins. Exoproteins were extracted from late-log-phase cultures as described in Materials and Methods and separated by electrophoresis in a 15% polyacrylamide gel containing SDS, and protein bands were stained with Coomassie blue. Lane M, molecular mass markers. (E) Effect of Δagr or saeS::bursa single mutations and double mutation on survival during treatment with gentamicin. Cells from late-log-phase cultures were treated with 15 μg/ml of gentamicin for 60 min, and then survival was determined. (F) Mixed-culture killing. Wild-type cells (BS12) and an agr mutant (BS13) were mixed in equal amounts and grown together in TSB to late log phase; cultures were then treated with 60 μg/ml of gentamicin for 60 min. Percent survival in mixed-culture kill assays was calculated by enumerating survivor colonies grown on sheep blood agar. Unpaired two-tailed t test was used to evaluate the significance (P < 0.05). ns, not significant (P > 0.05); ***, P < 0.01. Data represent the means from biological replicates ± standard deviations (n = 3).
Aminoglycosides, such as gentamicin, require the proton motive force (PMF) of the bacterial membrane for penetration into cells (45). The PMF consists of a transmembrane pH gradient and a transmembrane electrical potential. Thus, inhibitors that eliminate the proton gradient, such as carbonyl cyanide m-chlorophenylhydrazone (CCCP), inhibit aminoglycoside uptake and thereby activity (46), while increasing the pH of the medium enhances gentamicin uptake (47). When CCCP was used with gentamicin, a protective effect was still observed with the Δagr mutant, and the effect of CCCP was muted (Fig. 4B). When pH was raised, gentamicin became more lethal; the agr deficiency remained protective, but less so (Fig. 4C). These partial effects of an agr deficiency suggested that wild-type agr stimulates antimicrobial lethality for gentamicin through both PMF-dependent and PMF-independent pathways.
S. aureus Newman, the strain employed to generate many of the results described above, has a naturally occurring upregulating mutation in the two-component signaling system sae. Unlike agr, sae senses environmental signals (48), rather than a quorum-sensing peptide, to tailor the production of S. aureus virulence factors. The upregulating mutation in sae results in constitutive activation of numerous genes that contribute to the exoproteome of Newman strains, even when agr is absent (49, 50). To test the possibility that sae lies on the pathway leading to protection from the lethal activity of gentamicin, we performed killing assays employing an engineered strain deficient in both agr and saeS (BS985). The double mutant demonstrated an almost complete lack of exoprotein secretion (Fig. 4D), and it showed survival of gentamicin treatment comparable to that of wild-type cells (Fig. 4E). Thus, the saeS deficiency reversed the protective effect of Δagr. As expected, a sae-complemented strain was killed to the same extent as the Δagr strain (Fig. 4E). These data suggest that exoproteins are a source of protection from gentamicin-mediated killing of S. aureus afforded by agr-inactivating mutations. To explore this possibility, we examined whether differences seen between agr-positive and agr-defective strains in monoculture are eliminated in coculture. In coculture with equal starting inocula, the difference between the wild-type and agr mutant strains was much smaller with respect to killing by gentamicin, consistent with complementation in trans through a shared, extracellular factor (Fig. 4F).
S. aureus strains vary in protection from lethal stress by an agr deficiency.
We next examined the effect of agr functionality with the methicillin-susceptible strain ATCC 25923 and with the prototype community-acquired methicillin-resistant strain LAC. The protective effect of an agr deletion on killing by gentamicin was observed with strain ATCC 25923 but was marginal in LAC (Fig. 5A and B). Thus, the effect of an agr deficiency on the response to lethal stress is strain dependent, but it is not lineage specific.
FIG 5 .

agr-mediated phenotypes among diverse S. aureus strains. (A to D) Wild-type (WT) laboratory strains ATCC 25923 (BS902) (A) and LAC (BS819) (B) and clinical isolates 127b (C) and BS40 (D) with naturally occurring agr mutations (indicated in panels) were grown to late log phase and treated with indicated concentrations of gentamicin for 60 min. Symbols: open circles, wild-type cells; filled symbols, Δagr mutants. (E) Effect of agr group-specific differences on survival with gentamicin. Newman and Δagr strains and isogenic variants with the indicated agr allele (I to IV; BS519 to BS522) were grown to late log phase in TSB and treated with 15 μg/ml of gentamicin for 60 min. The asterisk denotes the strain containing the agr-III allele (BS519), which has a mutation in agrC that attenuates agr function (3) (see text). Data represent the means from biological replicates ± standard deviations (n = 3).
Differences observed in assays utilizing laboratory-constructed mutants may be obscured during infection by changes elsewhere in the genome. Accordingly, we assayed clinical agr-defective strains using a small set of genotypically diverse agr-defective MRSA clones initially obtained from mixed cultures containing agr-positive and agr-negative cells (51). Strains were previously genotyped to confirm that variants within a single specimen were otherwise isogenic (51). That work also traced the basis of agr dysfunction to inactivating mutations in agrA or agrC. Of the 4 clinical isolates in our collection that were susceptible to gentamicin, 2 genotypically distinct clones (see Table S1 in the supplemental material) exhibited 10-fold protection from killing by gentamicin (Fig. 5C and D). Thus, the data are consistent with data from laboratory strains indicating that agr inactivation reveals cryptic genetic variation among strains in their response to lethal stress. Moreover, variation in intrinsic (wild-type) tolerance was also observed. The mechanisms underlying strain-dependent differences in agr-mediated and intrinsic tolerance to lethal stress are unknown. Future work will investigate to what extent they are stress specific.
Previous work shows allelic variation in the S. aureus agr genes, identified as four specificity groups based on induction timing and strength. Induction is the earliest and strongest with agr-IV and -I, agr-II is intermediate, and induction with agr-III is delayed and weak (38). Weak agr-III induction levels result from a single-nucleotide polymorphism that changes amino acid 55 of AgrC (G55R) (10). This substitution is found in hospital-associated MRSA (HA-MRSA) clonal complex 30 lineage clones associated with poor outcome in bacteremic patients (16, 52). When we examined group-specific differences in agr autoinduction and virulence gene regulation using previously characterized isogenic variants of strain Newman (28), each harboring an S. aureus agr allele, we found an agr-III-specific attenuation of protection to gentamicin (Fig. 5E). Thus, partial loss-of-function mutations of agr may tune levels of signaling to balance virulence and antimicrobial tolerance.
Effect of agr deficiency on stress response to ciprofloxacin.
As with gentamicin, the protective effect of an agr deletion against ciprofloxacin-mediated killing does not act through RNAIII—the ΔRNAIII mutant demonstrated no effect on killing for ciprofloxacin, indicating that protection is agrA dependent (Fig. 6A).
FIG 6 .

agrA-mediated protection from ciprofloxacin-mediated killing involves ROS-dependent and ROS-independent pathways. (A to C) Effect of RNAIII and bsaA on survival during treatment with ciprofloxacin or gentamicin. Wild-type (WT) strain Newman (BS12), Δagr mutant (BS13), and ΔRNAIII mutant (BS669) (A) or wild-type strain Newman, Δagr mutant, and Δagr ΔbsaA double mutant (BS982) (B) were grown to late log phase and treated with 2.5 μg/ml of ciprofloxacin. Wild-type strain Newman, the Δagr mutant, and the Δagr bsaA::bursa double mutant were treated with 60 μg/ml gentamicin (C). (D and E) Effect of ROS quenchers on killing. Cells were grown to late log phase in TSB and treated with 10 μg/ml of ciprofloxacin (D) or 60 μg/ml of gentamicin (E) in the presence of 2,2′-bipyridyl and thiourea (BT), each at 0.5× MIC, for 60 min. Significance was determined by unpaired two-tailed t test (P < 0.05). ns, not significant; **, P < 0.05; ***, P < 0.01. Data represent the means from biological replicates ± standard deviations (n = 3).
The DNA-binding domain of AgrA contains an intramolecular disulfide switch as part of an oxidation-sensing mechanism. Oxidation leads to dissociation of AgrA from DNA, which prevents AgrA-mediated downregulation of glutathione peroxidase (BsaA), an enzyme that detoxifies ROS (53). Accordingly, inactivating mutations in agr elevate the expression of bsaA, which is expected to reduce oxidation-mediated lethality arising from treatment with antimicrobials such as fluoroquinolones (33). A Δagr bsaA::bursa double mutation lowered survival following treatment with ciprofloxacin but not with gentamicin (Fig. 6B and C), indicating that agr acts differently on the effects of the two drugs.
Since bsaA is involved in detoxification of ROS, we tested for elimination of Δagr-mediated protection by an ROS scavenger and iron chelator. When wild-type and Δagr mutant cells were pretreated with subinhibitory (0.5× MIC) concentrations of thiourea plus 2,2′-bipyridyl to block hydroxyl radical accumulation, the lethal action of ciprofloxacin, but not gentamicin, was reduced in both strains, and the Δagr-mediated protection for ciprofloxacin was eliminated (Fig. 6D and E). These data indicate that wild-type agr stimulates ciprofloxacin lethality largely through an ROS-dependent pathway. These data are consistent with the observation that ciprofloxacin MBCs were identical for wild-type and Δagr strains (1 μg/ml): MBC reflects killing extent, while ROS-mediated effects are seen as rate changes (32, 33). The observed lack of an effect of Δagr on peroxide-mediated killing is consistent with previous work indicating that the action of exogenous hydrogen peroxide overshadows endogenous-ROS-mediated effects (54).
Overall, agr deficiency-dependent escape from lethal stress is common among S. aureus strains and correlates with reduction in the activity of some antimicrobials (gentamicin) and interference in the lethal response to others (ciprofloxacin). Thus, representative stressors illustrate how mutations in agr reveal a general mechanism of adaptive evolution through attenuation of lethal stress. Complexity was uncovered by daptomycin having the opposite effect, as described below in the Discussion.
DISCUSSION
The work described above addresses the general issue of adaptive leaps made by bacterial pathogens, using as an example the paradoxical finding that defects in the agr virulence regulon are associated with poor patient outcome from staphylococcal infection, particularly during antimicrobial treatment (27, 28). The major observation is that wild-type agr stimulates the lethal action of several stressors, including gentamicin and ciprofloxacin; thus, defective mutants will tend to persist under stressful conditions rather than being killed by stressors that may include synthetic antimicrobials and host defenses such as neutrophil-generated ROS. In the case of gentamicin, agr-mediated stimulation of lethal activity relies on the bacterial production of exoproteins; for ciprofloxacin, agr normally downregulates a protein that protects from ROS. How agr stimulates killing by heat and low pH is not yet known. The lethal action of these diverse stressors may apply to many other challenges experienced by S. aureus during persistent infection and thereby help explain the selective enrichment of agr-deficient mutants. Since stimulation of killing by agr is not universal, as shown by daptomycin being more lethal with the agr mutant, determining whether agr has a positive or negative effect on the lethality of a particular stressor will be important for combining anti-Agr agents with antimicrobials.
A striking observation was the absence of an agr effect on bacteriostatic activity. Previous distinctions between bacteriostatic and lethal activity with fluoroquinolones (55) led to the idea that some forms of lethal activity, in particular those involving ROS, are a cellular response to a primary lesion (34). The current work strongly supports separation of bacteriostatic and bactericidal effects, thereby emphasizing the need to normalize survival data to MIC when considering how stress kills bacterial cells (32). Our conclusion that lethal activity exerts selective pressure independently of bacteriostatic activity also emphasizes the importance of considering antimicrobial lethality during bacterial infection (current treatment, diagnosis, and surveillance are based largely on bacteriostatic activity [MIC]).
The ability of bacteria to survive lethal stress that still blocks growth is a form of tolerance: paradoxical enrichment of agr-deficient mutants is a clinical example of tolerance that would not be detected by standard susceptibility testing for resistance. Our results support the growing body of evidence that mutations in global regulators constitute a prominent mechanism underlying tolerance (56–60). From a clinical perspective, tolerance to severe stress presents a major challenge: in contrast to the specificity of resistance, tolerance can confer a survival advantage against a broad spectrum of selective pressures that ultimately lead to antimicrobial resistance (59) and to altered host-pathogen interactions that favor persistent infection. Thus, understanding tolerance is critical for addressing the decreasing efficacy of antibiotics.
Within our sample of stressors, daptomycin was unusual in exhibiting greater lethality with the agr-deficient mutant. Test conditions are important, as indicated by consideration of previous work in which the opposite result was obtained with nongrowing S. aureus in deep stationary phase, long after induction of agr and expression of agr transcripts (61). Daptomycin causes the release of membrane phospholipids that bind to and inactivate the antibiotic (61); agrA triggers secretion of phenol-soluble modulins (PSMs) that bind to phospholipids and prevent daptomycin inactivation. Our experiments were performed in late exponential phase when PSM levels may be lower and less protective (18). The complex relationship between daptomycin lethality, agr status, and bacterial physiological state illustrates the importance of understanding agr biology before applying novel therapies that target agr (62).
Secreted factors that bind to drugs or block their uptake are expected to affect the MIC. Since no difference in MIC was observed for agr alleles (Table 1) (61), the protective mechanism induced by daptomycin, described by Pader et al. (61), likely involves cell damage and release of phospholipids occurring at drug concentrations above the MIC. As with daptomycin, gentamicin interacts strongly with anionic sites in the plasma membrane and in particular phospholipids. We reason that concentrations above MIC are required to trigger leakage of bacterial components, explaining the lack of agr-mediated perturbation of MIC for gentamicin.
Ciprofloxacin-mediated killing merits additional comment because a role for ROS still remains controversial (63–65). agr normally downregulates bsaA, a gene encoding glutathione peroxidase, which detoxifies ROS (53). The Δagr defect allows expression of a protective protein, thereby explaining the drop in ciprofloxacin-mediated killing. Thus, the present work is most readily explained by a contribution of ROS to killing by quinolones and helps resolve a controversy (31, 32, 35). Indeed, with S. aureus ciprofloxacin is more likely to exhibit ROS-mediated lethality than are more potent fluoroquinolones that tend to kill by an ROS-independent mechanism, as deduced from studies of Escherichia coli (66, 67).
Previous reports indicate that inactivation of RNAIII is associated with a growth advantage for Δagr mutants in the presence of subinhibitory concentrations of several antibiotics (ciprofloxacin, mupirocin, and rifampin) (25). These data, plus the present work, lead to the conclusion that two distinct subsets of agr antimicrobial fitness exist: an RNAIII-independent one that impacts antimicrobial lethality and an RNAIII-dependent form that controls antimicrobial-associated fitness for growth. The mechanism underlying agr dysfunction among strains derived from clinical isolates is almost always traced to inactivating mutations in agrC and agrA, the sensor component and response regulator, respectively, of the agr system (14–17). Since selection for agr-defective strains occurs in mixtures with agr-positive parental strains, inactivation of agrD or agrB does not silence agr (autoinducing peptide is produced in trans by the agr-positive strain). However, this scenario does not explain why RNAIII is not targeted by selection for loss of agr function. Identification of the role of agrA in protection from the lethal response to antimicrobial-mediated stress resolves the dilemma, since inactivating mutations in agrCA will inactivate both agrP2-agrA and agrP3-RNAIII operons.
In summary, comparison of clinical strains entering hospitals with those emerging from patients provides insight into how infection remodels pathogens with respect to a major regulator. Additional lethality screening is needed to determine the frequency and specificity with which agr inactivation results in tolerance to specific stresses among clinical agr-defective mutants. We expect that additional lethality screening will identify other bacterial regulators having activities that can be either destructive or protective, depending on the type and level of lethal stress. Understanding the basis for such antimicrobial tolerance can be clinically significant when it informs efforts to personalize antimicrobial management through strain-specific pathogen characteristics. For example, use of anti-agr agents or therapeutic vaccines (62) may be ill advised for applications in which the absence of agr reduces antimicrobial lethality. Identifying other adaptations that erode the lethal activities of antimicrobials could lead to novel strategies for selectively bolstering antimicrobial effectiveness (68–70).
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
S. aureus strains and plasmids used in the study are described in Table S1 in the supplemental material. Cells were cultured in tryptic soy broth (TSB) with constant aeration (rotary shaking at 250 rpm) or on tryptic soy agar (TSA) plates. In some cases, TSB was supplemented with 20% (vol/vol) human serum. Incubation was at 37°C. Phages 80α and Φ11 were used to transduce marker-disrupted alleles (71); transductants were selected on TSA plates containing the appropriate antimicrobial.
Strains. Download TABLE S1, DOCX file, 0.03 MB (37.2KB, docx) .
Copyright © 2017 Kumar et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Antimicrobials and chemicals.
Antimicrobials and off-the-clot human serum were obtained from Sigma-Aldrich (St. Louis, MO) and SeraCare (Milford, MA), respectively. Chemicals and reagents were obtained from Sigma-Aldrich and Fisher (Fair Lawn, NJ).
Construction of mutants.
S. aureus Newman agr::tet and RNAIII::cd were generated by transducing the disrupted alleles from RN6911 and BS640, respectively, using phage 80α. The Δagr bsaA::bursa double mutant (strain BS982) was generated by transducing bsaA::bursa, obtained from the University of Nebraska transposon mutant (ΦNE) library, into agr::tet Newman using phage 80α. An S. aureus Newman Δagr saeS::bursa double mutant was obtained by moving saeS::bursa from strain VJT12.22 (51) to agr::tet Newman using phage 80α-mediated transduction. For saeS complementation, plasmid Plgt-saeSpOS1, expressing saeS under the control of the constitutive promoter hprK, was introduced into the double mutant strain Newman agr::tet saeS::bursa by bacterial transduction (51).
Reporter assays.
We employed an agrP3-blaZ reporter cassette integrated into the S. aureus chromosome at the SaPI1 attC site (72). Overnight cultures were diluted to an optical density at 600 nm (OD600) of 0.05 in TSB with or without 20% (vol/vol) human serum and incubated at 37°C with shaking. Cultures were collected at various times; normalized β-lactamase activity (Vmax/OD600) was determined using the nitrocefin method as described previously (38). Briefly, 50 μl cells was mixed with 50 μl of nitrocefin solution (119 μg/ml prepared in 100 mM sodium phosphate buffer, pH 5.8); OD490 and OD600 were measured using a Synergy H1 hybrid microplate reader (BioTek).
Exoprotein analysis.
Exoproteins were extracted as described previously (26). Briefly, cells were grown overnight in hydrolysate-yeast extract-containing medium (CCY) (3% [wt/vol] yeast extract, 2% Bacto Casamino Acids, 2.3% sodium pyruvate, 0.63% Na2HPO4, and 0.041% KH2PO4 [pH 6.7]). Overnight cultures were diluted and grown to late log phase (~5 h) in fresh CCY, and 1.5-ml aliquots were centrifuged to remove bacteria. Culture supernatants were treated with an equal volume of ice-cold 20% trichloroacetic acid, and the precipitate was collected by centrifugation. The precipitated proteins were separated and analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (73).
Susceptibility and survival measurements.
Inhibition of growth (MIC values, Table 1) was determined by agar or broth dilution. For the latter, about 105 cells were applied to a series of broth cultures containing antimicrobials at various concentrations (2-fold dilutions). Turbid growth and optical density (OD600) were detected after 1 day. The MIC was taken as the minimal concentration that blocked growth of liquid cultures.
To measure lethal action, overnight cultures were diluted 50-fold in TSB or TSB plus serum and grown with shaking to late log phase, a condition in which agr is maximally activated. Cultures (~3 × 108 CFU/ml) were exposed to antimicrobials under aerobic conditions, diluted in drug-free medium, plated on drug-free agar, and incubated overnight at 37°C. Percent survival was estimated by colony formation relative to that of an untreated control sampled at the time of antimicrobial addition. To measure the effect of alkaline pH on gentamicin-mediated lethality, the pH of serum was adjusted with 6 N NaOH to 8.5. CCCP was added to cultures 5 min prior to the addition of gentamicin. To test the effect of ROS quenchers on gentamicin- and ciprofloxacin-mediated lethality, cultures grown in TSB were treated with 0.5× MIC of 2,2′-bipyridyl and thiourea 5 min prior to the addition of the antimicrobial. Since daptomycin requires Ca2+ for activity, late-log-phase cultures were supplemented with 50 μg/ml Ca2+ and 25 μg/ml Mg2+ when treated with various concentrations of daptomycin for 90 min in Mueller-Hinton broth. To measure the lethal effects of high-temperature stress, cells were grown in TSB to ~3 × 108 CFU/ml, incubated at various temperatures for 10 min in a PCR thermocycler (Eppendorf, Hamburg, Germany), serially diluted, and plated on drug-free agar for determination of viable colony numbers. To measure the lethal effect of low pH, cells from late-log-phase cultures were concentrated by centrifugation, resuspended in TSB adjusted to various values of pH with HCl, and incubated at 37°C under aerobic conditions for various times. For H2O2 treatment, late-log-phase cultures (~107 CFU/ml) were treated with various concentrations of peroxide for 1 h at 37°C under aerobic conditions. All experiments were repeated at least three times; similar results were obtained from the biological replicates.
Statistical analysis.
For killing assays, comparisons were made using an unpaired two-tailed t test (P < 0.05). P values of <0.05 were considered statistically significant.
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grants R01 AI103268 and NIAID HHS N272201400019C.
B.S. is a consultant for Regeneron Pharmaceuticals.
We thank Marila Gennaro, Richard Novick, Jeffrey Weiser, and Xilin Zhao for critical comments on the manuscript.
Footnotes
Citation Kumar K, Chen J, Drlica K, Shopsin B. 2017. Tuning of the lethal response to multiple stressors with a single-site mutation during clinical infection by Staphylococcus aureus. mBio 8:e01476-17. https://doi.org/10.1128/mBio.01476-17.
REFERENCES
- 1.Quan S, Ray JC, Kwota Z, Duong T, Balázsi G, Cooper TF, Monds RD. 2012. Adaptive evolution of the lactose utilization network in experimentally evolved populations of Escherichia coli. PLoS Genet 8:e1002444. doi: 10.1371/journal.pgen.1002444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Saxer G, Krepps MD, Merkley ED, Ansong C, Deatherage Kaiser BL, Valovska MT, Ristic N, Yeh PT, Prakash VP, Leiser OP, Nakhleh L, Gibbons HS, Kreuzer HW, Shamoo Y. 2014. Mutations in global regulators lead to metabolic selection during adaptation to complex environments. PLoS Genet 10:e1004872. doi: 10.1371/journal.pgen.1004872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Spencer CC, Bertrand M, Travisano M, Doebeli M. 2007. Adaptive diversification in genes that regulate resource use in Escherichia coli. PLoS Genet 3:e15. doi: 10.1371/journal.pgen.0030015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carter MQ, Parker CT, Louie JW, Huynh S, Fagerquist CK, Mandrell RE. 2012. RcsB contributes to the distinct stress fitness among Escherichia coli O157:H7 curli variants of the 1993 hamburger-associated outbreak strains. Appl Environ Microbiol 78:7706–7719. doi: 10.1128/AEM.02157-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Das S, Lindemann C, Young BC, Muller J, Österreich B, Ternette N, Winkler AC, Paprotka K, Reinhardt R, Förstner KU, Allen E, Flaxman A, Yamaguchi Y, Rollier CS, van Diemen P, Blättner S, Remmele CW, Selle M, Dittrich M, Müller T, Vogel J, Ohlsen K, Crook DW, Massey R, Wilson DJ, Rudel T, Wyllie DH, Fraunholz MJ. 2016. Natural mutations in a Staphylococcus aureus virulence regulator attenuate cytotoxicity but permit bacteremia and abscess formation. Proc Natl Acad Sci U S A 113:E3101–E3110. doi: 10.1073/pnas.1520255113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kisiela DI, Radey M, Paul S, Porter S, Polukhina K, Tchesnokova V, Shevchenko S, Chan D, Aziz M, Johnson TJ, Price LB, Johnson JR, Sokurenko EV. 2017. Inactivation of transcriptional regulators during within-household evolution of Escherichia coli. J Bacteriol 199:e00036-17. doi: 10.1128/JB.00036-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith EE, Buckley DG, Wu Z, Saenphimmachak C, Hoffman LR, D’Argenio DA, Miller SI, Ramsey BW, Speert DP, Moskowitz SM, Burns JL, Kaul R, Olson MV. 2006. Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients. Proc Natl Acad Sci U S A 103:8487–8492. doi: 10.1073/pnas.0602138103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baines SL, Holt KE, Schultz MB, Seemann T, Howden BO, Jensen SO, van Hal SJ, Coombs GW, Firth N, Powell DR, Stinear TP, Howden BP. 2015. Convergent adaptation in the dominant global hospital clone ST239 of methicillin-resistant Staphylococcus aureus. mBio 6:e00080-15. doi: 10.1128/mBio.00080-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheung GY, Kretschmer D, Duong AC, Yeh AJ, Ho TV, Chen Y, Joo HS, Kreiswirth BN, Peschel A, Otto M. 2014. Production of an attenuated phenol-soluble modulin variant unique to the MRSA clonal complex 30 increases severity of bloodstream infection. PLoS Pathog 10:e1004298. doi: 10.1371/journal.ppat.1004298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DeLeo FR, Kennedy AD, Chen L, Bubeck Wardenburg J, Kobayashi SD, Mathema B, Braughton KR, Whitney AR, Villaruz AE, Martens CA, Porcella SF, McGavin MJ, Otto M, Musser JM, Kreiswirth BN. 2011. Molecular differentiation of historic phage-type 80/81 and contemporary epidemic Staphylococcus aureus. Proc Natl Acad Sci U S A 108:18091–18096. doi: 10.1073/pnas.1111084108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Laabei M, Uhlemann AC, Lowy FD, Austin ED, Yokoyama M, Ouadi K, Feil E, Thorpe HA, Williams B, Perkins M, Peacock SJ, Clarke SR, Dordel J, Holden M, Votintseva AA, Bowden R, Crook DW, Young BC, Wilson DJ, Recker M, Massey RC. 2015. Evolutionary trade-offs underlie the multi-faceted virulence of Staphylococcus aureus. PLoS Biol 13:e1002229. doi: 10.1371/journal.pbio.1002229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.López-Collazo E, Jurado T, de Dios Caballero J, Pérez-Vázquez M, Vindel A, Hernández-Jiménez E, Tamames J, Cubillos-Zapata C, Manrique M, Tobes R, Máiz L, Cantón R, Baquero F, Del Campo R. 2015. In vivo attenuation and genetic evolution of a ST247-SCCmecI MRSA clone after 13 years of pathogenic bronchopulmonary colonization in a patient with cystic fibrosis: implications of the innate immune response. Mucosal Immunol 8:362–371. doi: 10.1038/mi.2014.73. [DOI] [PubMed] [Google Scholar]
- 13.Painter KL, Krishna A, Wigneshweraraj S, Edwards AM. 2014. What role does the quorum-sensing accessory gene regulator system play during Staphylococcus aureus bacteremia? Trends Microbiol 22:676–685. doi: 10.1016/j.tim.2014.09.002. [DOI] [PubMed] [Google Scholar]
- 14.Shopsin B, Drlica-Wagner A, Mathema B, Adhikari RP, Kreiswirth BN, Novick RP. 2008. Prevalence of agr dysfunction among colonizing Staphylococcus aureus strains. J Infect Dis 198:1171–1174. doi: 10.1086/592051. [DOI] [PubMed] [Google Scholar]
- 15.Shopsin B, Eaton C, Wasserman GA, Mathema B, Adhikari RP, Agolory S, Altman DR, Holzman RS, Kreiswirth BN, Novick RP. 2010. Mutations in agr do not persist in natural populations of methicillin-resistant Staphylococcus aureus. J Infect Dis 202:1593–1599. doi: 10.1086/656915. [DOI] [PubMed] [Google Scholar]
- 16.Smyth DS, Kafer JM, Wasserman GA, Velickovic L, Mathema B, Holzman RS, Knipe TA, Becker K, von Eiff C, Peters G, Chen L, Kreiswirth BN, Novick RP, Shopsin B. 2012. Nasal carriage as a source of agr-defective Staphylococcus aureus bacteremia. J Infect Dis 206:1168–1177. doi: 10.1093/infdis/jis483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Traber KE, Lee E, Benson S, Corrigan R, Cantera M, Shopsin B, Novick RP. 2008. agr function in clinical Staphylococcus aureus isolates. Microbiology 154:2265–2274. doi: 10.1099/mic.0.2007/011874-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Queck SY, Jameson-Lee M, Villaruz AE, Bach TH, Khan BA, Sturdevant DE, Ricklefs SM, Li M, Otto M. 2008. RNAIII-independent target gene control by the agr quorum-sensing system: insight into the evolution of virulence regulation in Staphylococcus aureus. Mol Cell 32:150–158. doi: 10.1016/j.molcel.2008.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abdelnour A, Arvidson S, Bremell T, Rydén C, Tarkowski A. 1993. The accessory gene regulator (agr) controls Staphylococcus aureus virulence in a murine arthritis model. Infect Immun 61:3879–3885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheung AL, Eberhardt KJ, Chung E, Yeaman MR, Sullam PM, Ramos M, Bayer AS. 1994. Diminished virulence of a sar-/agr- mutant of Staphylococcus aureus in the rabbit model of endocarditis. J Clin Invest 94:1815–1822. doi: 10.1172/JCI117530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gillaspy AF, Hickmon SG, Skinner RA, Thomas JR, Nelson CL, Smeltzer MS. 1995. Role of the accessory gene regulator (agr) in pathogenesis of staphylococcal osteomyelitis. Infect Immun 63:3373–3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wright JS III, Jin R, Novick RP. 2005. Transient interference with staphylococcal quorum sensing blocks abscess formation. Proc Natl Acad Sci U S A 102:1691–1696. doi: 10.1073/pnas.0407661102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Butterfield JM, Tsuji BT, Brown J, Dodds Ashley ED, Hardy D, Brown K, Forrest A, Lodise TP. 2011. Predictors of agr dysfunction in methicillin-resistant Staphylococcus aureus (MRSA) isolates among patients with MRSA bloodstream infections. Antimicrob Agents Chemother 55:5433–5437. doi: 10.1128/AAC.00407-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mwangi MM, Wu SW, Zhou Y, Sieradzki K, de Lencastre H, Richardson P, Bruce D, Rubin E, Myers E, Siggia ED, Tomasz A. 2007. Tracking the in vivo evolution of multidrug resistance in Staphylococcus aureus by whole-genome sequencing. Proc Natl Acad Sci U S A 104:9451–9456. doi: 10.1073/pnas.0609839104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Paulander W, Nissen Varming A, Bæk KT, Haaber J, Frees D, Ingmer H. 2013. Antibiotic-mediated selection of quorum-sensing-negative Staphylococcus aureus. mBio 3:e00459-12. doi: 10.1128/mBio.00459-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Traber K, Novick R. 2006. A slipped-mispairing mutation in AgrA of laboratory strains and clinical isolates results in delayed activation of agr and failure to translate delta- and alpha-haemolysins. Mol Microbiol 59:1519–1530. doi: 10.1111/j.1365-2958.2006.04986.x. [DOI] [PubMed] [Google Scholar]
- 27.Fowler VG Jr, Sakoulas G, McIntyre LM, Meka VG, Arbeit RD, Cabell CH, Stryjewski ME, Eliopoulos GM, Reller LB, Corey GR, Jones T, Lucindo N, Yeaman MR, Bayer AS. 2004. Persistent bacteremia due to methicillin-resistant Staphylococcus aureus infection is associated with agr dysfunction and low-level in vitro resistance to thrombin-induced platelet microbicidal protein. J Infect Dis 190:1140–1149. doi: 10.1086/423145. [DOI] [PubMed] [Google Scholar]
- 28.Schweizer ML, Furuno JP, Sakoulas G, Johnson JK, Harris AD, Shardell MD, McGregor JC, Thom KA, Perencevich EN. 2011. Increased mortality with accessory gene regulator (agr) dysfunction in Staphylococcus aureus among bacteremic patients. Antimicrob Agents Chemother 55:1082–1087. doi: 10.1128/AAC.00918-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Novick RP. 2003. Autoinduction and signal transduction in the regulation of staphylococcal virulence. Mol Microbiol 48:1429–1449. doi: 10.1046/j.1365-2958.2003.03526.x. [DOI] [PubMed] [Google Scholar]
- 30.Dwyer DJ, Collins JJ, Walker GC. 2015. Unraveling the physiological complexities of antibiotic lethality. Annu Rev Pharmacol Toxicol 55:313–332. doi: 10.1146/annurev-pharmtox-010814-124712. [DOI] [PubMed] [Google Scholar]
- 31.Zhao X, Drlica K. 2014. Reactive oxygen species and the bacterial response to lethal stress. Curr Opin Microbiol 21:1–6. doi: 10.1016/j.mib.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao X, Hong Y, Drlica K. 2015. Moving forward with reactive oxygen species involvement in antimicrobial lethality. J Antimicrob Chemother 70:639–642. doi: 10.1093/jac/dku463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu Y, Liu X, Qu Y, Wang X, Li L, Zhao X. 2012. Inhibitors of reactive oxygen species accumulation delay and/or reduce the lethality of several antistaphylococcal agents. Antimicrob Agents Chemother 56:6048–6050. doi: 10.1128/AAC.00754-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dorsey-Oresto A, Lu T, Mosel M, Wang X, Salz T, Drlica K, Zhao X. 2013. YihE kinase is a central regulator of programmed cell death in bacteria. Cell Rep 3:528–537. doi: 10.1016/j.celrep.2013.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dwyer DJ, Belenky PA, Yang JH, MacDonald IC, Martell JD, Takahashi N, Chan CT, Lobritz MA, Braff D, Schwarz EG, Ye JD, Pati M, Vercruysse M, Ralifo PS, Allison KR, Khalil AS, Ting AY, Walker GC, Collins JJ. 2014. Antibiotics induce redox-related physiological alterations as part of their lethality. Proc Natl Acad Sci U S A 111:E2100–E2109. doi: 10.1073/pnas.1401876111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kohanski MA, Dwyer DJ, Hayete B, Lawrence CA, Collins JJ. 2007. A common mechanism of cellular death induced by bactericidal antibiotics. Cell 130:797–810. doi: 10.1016/j.cell.2007.06.049. [DOI] [PubMed] [Google Scholar]
- 37.Novick RP, Geisinger E. 2008. Quorum sensing in staphylococci. Annu Rev Genet 42:541–564. doi: 10.1146/annurev.genet.42.110807.091640. [DOI] [PubMed] [Google Scholar]
- 38.Geisinger E, Chen J, Novick RP. 2012. Allele-dependent differences in quorum-sensing dynamics result in variant expression of virulence genes in Staphylococcus aureus. J Bacteriol 194:2854–2864. doi: 10.1128/JB.06685-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dai Y, Chang W, Zhao C, Peng J, Xu L, Lu H, Zhou S, Ma X. 2017. VraR binding to the promoter region of agr inhibits its function in vancomycin-intermediate Staphylococcus aureus (VISA) and heterogeneous VISA. Antimicrob Agents Chemother 61:e02740-16. doi: 10.1128/AAC.02740-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sakoulas G, Moellering RC Jr, Eliopoulos GM. 2006. Adaptation of methicillin-resistant Staphylococcus aureus in the face of vancomycin therapy. Clin Infect Dis 42(Suppl 1):S40–S50. doi: 10.1086/491713. [DOI] [PubMed] [Google Scholar]
- 41.Johnson PJ, Levin BR. 2013. Pharmacodynamics, population dynamics, and the evolution of persistence in Staphylococcus aureus. PLoS Genet 9:e1003123. doi: 10.1371/journal.pgen.1003123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sakoulas G, Eliopoulos GM, Moellering RC Jr, Wennersten C, Venkataraman L, Novick RP, Gold HS. 2002. Accessory gene regulator (agr) locus in geographically diverse Staphylococcus aureus isolates with reduced susceptibility to vancomycin. Antimicrob Agents Chemother 46:1492–1502. doi: 10.1128/AAC.46.5.1492-1502.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kavanaugh JS, Horswill AR. 2016. Impact of environmental cues on staphylococcal quorum sensing and biofilm development. J Biol Chem 291:12556–12564. doi: 10.1074/jbc.R116.722710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hall PR, Elmore BO, Spang CH, Alexander SM, Manifold-Wheeler BC, Castleman MJ, Daly SM, Peterson MM, Sully EK, Femling JK, Otto M, Horswill AR, Timmins GS, Gresham HD. 2013. Nox2 modification of LDL is essential for optimal apolipoprotein B-mediated control of agr type III Staphylococcus aureus quorum-sensing. PLoS Pathog 9:e1003166. doi: 10.1371/journal.ppat.1003166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Taber HW, Mueller JP, Miller PF, Arrow AS. 1987. Bacterial uptake of aminoglycoside antibiotics. Microbiol Rev 51:439–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Allison KR, Brynildsen MP, Collins JJ. 2011. Metabolite-enabled eradication of bacterial persisters by aminoglycosides. Nature 473:216–220. doi: 10.1038/nature10069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lebeaux D, Chauhan A, Létoffé S, Fischer F, de Reuse H, Beloin C, Ghigo JM. 2014. pH-mediated potentiation of aminoglycosides kills bacterial persisters and eradicates in vivo biofilms. J Infect Dis 210:1357–1366. doi: 10.1093/infdis/jiu286. [DOI] [PubMed] [Google Scholar]
- 48.Novick RP, Jiang D. 2003. The staphylococcal saeRS system coordinates environmental signals with agr quorum sensing. Microbiology 149:2709–2717. doi: 10.1099/mic.0.26575-0. [DOI] [PubMed] [Google Scholar]
- 49.Adhikari RP, Novick RP. 2008. Regulatory organization of the staphylococcal sae locus. Microbiology 154:949–959. doi: 10.1099/mic.0.2007/012245-0. [DOI] [PubMed] [Google Scholar]
- 50.Geiger T, Goerke C, Mainiero M, Kraus D, Wolz C. 2008. The virulence regulator Sae of Staphylococcus aureus: promoter activities and response to phagocytosis-related signals. J Bacteriol 190:3419–3428. doi: 10.1128/JB.01927-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Benson MA, Lilo S, Wasserman GA, Thoendel M, Smith A, Horswill AR, Fraser J, Novick RP, Shopsin B, Torres VJ. 2011. Staphylococcus aureus regulates the expression and production of the staphylococcal superantigen-like secreted proteins in a Rot-dependent manner. Mol Microbiol 81:659–675. doi: 10.1111/j.1365-2958.2011.07720.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fowler VG Jr, Nelson CL, McIntyre LM, Kreiswirth BN, Monk A, Archer GL, Federspiel J, Naidich S, Remortel B, Rude T, Brown P, Reller LB, Corey GR, Gill SR. 2007. Potential associations between hematogenous complications and bacterial genotype in Staphylococcus aureus infection. J Infect Dis 196:738–747. doi: 10.1086/520088. [DOI] [PubMed] [Google Scholar]
- 53.Sun F, Liang H, Kong X, Xie S, Cho H, Deng X, Ji Q, Zhang H, Alvarez S, Hicks LM, Bae T, Luo C, Jiang H, He C. 2012. Quorum-sensing agr mediates bacterial oxidation response via an intramolecular disulfide redox switch in the response regulator AgrA. Proc Natl Acad Sci U S A 109:9095–9100. doi: 10.1073/pnas.1200603109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li L, Hong Y, Luan G, Mosel M, Malik M, Drlica K, Zhao X. 2014. Ribosomal elongation factor 4 promotes cell death associated with lethal stress. mBio 5:e01708-14. doi: 10.1128/mBio.01708-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Drlica K, Malik M, Kerns RJ, Zhao X. 2008. Quinolone-mediated bacterial death. Antimicrob Agents Chemother 52:385–392. doi: 10.1128/AAC.01617-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Girgis HS, Harris K, Tavazoie S. 2012. Large mutational target size for rapid emergence of bacterial persistence. Proc Natl Acad Sci U S A 109:12740–12745. doi: 10.1073/pnas.1205124109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hansen S, Lewis K, Vulić M. 2008. Role of global regulators and nucleotide metabolism in antibiotic tolerance in Escherichia coli. Antimicrob Agents Chemother 52:2718–2726. doi: 10.1128/AAC.00144-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Honsa ES, Cooper VS, Mhaissen MN, Frank M, Shaker J, Iverson A, Rubnitz J, Hayden RT, Lee RE, Rock CO, Tuomanen EI, Wolf J, Rosch JW. 2017. RelA mutant Enterococcus faecium with multiantibiotic tolerance arising in an immunocompromised host. mBio 8:e02124-16. doi: 10.1128/mBio.02124-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Levin-Reisman I, Ronin I, Gefen O, Braniss I, Shoresh N, Balaban NQ. 2017. Antibiotic tolerance facilitates the evolution of resistance. Science 355:826–830. doi: 10.1126/science.aaj2191. [DOI] [PubMed] [Google Scholar]
- 60.Mok WW, Orman MA, Brynildsen MP. 2015. Impacts of global transcriptional regulators on persister metabolism. Antimicrob Agents Chemother 59:2713–2719. doi: 10.1128/AAC.04908-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pader V, Hakim S, Painter KL, Wigneshweraraj S, Clarke TB, Edwards AM. 2016. Staphylococcus aureus inactivates daptomycin by releasing membrane phospholipids. Nat Microbiol 2:16194. doi: 10.1038/nmicrobiol.2016.194. [DOI] [PubMed] [Google Scholar]
- 62.Khan BA, Yeh AJ, Cheung GY, Otto M. 2015. Investigational therapies targeting quorum-sensing for the treatment of Staphylococcus aureus infections. Expert Opin Invest Drugs 24:689–704. doi: 10.1517/13543784.2015.1019062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Imlay JA. 2015. Diagnosing oxidative stress in bacteria: not as easy as you might think. Curr Opin Microbiol 24:124–131. doi: 10.1016/j.mib.2015.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Keren I, Wu Y, Inocencio J, Mulcahy LR, Lewis K. 2013. Killing by bactericidal antibiotics does not depend on reactive oxygen species. Science 339:1213–1216. doi: 10.1126/science.1232688. [DOI] [PubMed] [Google Scholar]
- 65.Liu Y, Imlay JA. 2013. Cell death from antibiotics without the involvement of reactive oxygen species. Science 339:1210–1213. doi: 10.1126/science.1232751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Malik M, Hussain S, Drlica K. 2007. Effect of anaerobic growth on quinolone lethality with Escherichia coli. Antimicrob Agents Chemother 51:28–34. doi: 10.1128/AAC.00739-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang X, Zhao X, Malik M, Drlica K. 2010. Contribution of reactive oxygen species to pathways of quinolone-mediated bacterial cell death. J Antimicrob Chemother 65:520–524. doi: 10.1093/jac/dkp486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gusarov I, Shatalin K, Starodubtseva M, Nudler E. 2009. Endogenous nitric oxide protects bacteria against a wide spectrum of antibiotics. Science 325:1380–1384. doi: 10.1126/science.1175439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liu Y, Zhou J, Qu Y, Yang X, Shi G, Wang X, Hong Y, Drlica K, Zhao X. 2016. Resveratrol antagonizes antimicrobial lethality and stimulates recovery of bacterial mutants. PLoS One 11:e0153023. doi: 10.1371/journal.pone.0153023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shatalin K, Shatalina E, Mironov A, Nudler E. 2011. H2S: a universal defense against antibiotics in bacteria. Science 334:986–990. doi: 10.1126/science.1209855. [DOI] [PubMed] [Google Scholar]
- 71.Novick RP. 1991. Genetic systems in staphylococci. Methods Enzymol 204:587–636. [DOI] [PubMed] [Google Scholar]
- 72.Chen J, Yoong P, Ram G, Torres VJ, Novick RP. 2014. Single-copy vectors for integration at the SaPI1 attachment site for Staphylococcus aureus. Plasmid 76:1–7. doi: 10.1016/j.plasmid.2014.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tests of agrP3 promoter activity. S. aureus Newman cells containing agrP3-blaZ (SaPI1 attC::agrP3-blaZ; strain BS983) were grown in TSB (A) or TSB in the presence of 20% (vol/vol) human serum (B). agrP3 activity (β-lactamase units/culture OD600) was assayed at the indicated times (see Materials and Methods). Symbols: broken line, growth; empty circles, agrP3 activity in TSB; filled circles, agrP3 activity in medium containing serum. Data represented the means from biological replicates ± standard deviations (n = 3). Download FIG S1, TIF file, 0.1 MB (108.4KB, tif) .
Copyright © 2017 Kumar et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Serum enhances agr-mediated differences in antimicrobial killing. Wild-type S. aureus Newman (BS12) and Δagr mutant (BS13) were grown to late log phase in TSB with serum and treated with the indicated concentrations of gentamicin for 60 min (A) or with 60 μg/ml of gentamicin for the times indicated (C). Likewise, cultures were treated with indicated concentrations of ciprofloxacin for 60 min (B) or with 10 μg/ml of ciprofloxacin for the times indicated (D). Symbols: filled circles, Δagr strain; empty circles, wild type. Data represent means of biological replicates ± standard deviations (n = 3). Download FIG S2, TIF file, 0.1 MB (144.8KB, tif) .
Copyright © 2017 Kumar et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Strains. Download TABLE S1, DOCX file, 0.03 MB (37.2KB, docx) .
Copyright © 2017 Kumar et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.