

ABSTRACT
Escherichia coli sequence type 131 (ST131), a pandemic clone responsible for the high incidence of extraintestinal pathogenic E. coli (ExPEC) infections, has been known widely for its contribution to the worldwide dissemination of multidrug resistance. Although other ExPEC-associated and extended-spectrum-β-lactamase (ESBL)-producing E. coli clones, such as ST38, ST405, and ST648 have been studied widely, no comparative genomic data with respect to other genotypes exist for ST131. In this study, comparative genomic analysis was performed for 99 ST131 E. coli strains with 40 genomes from three other STs, including ST38 (n = 12), ST405 (n = 10), and ST648 (n = 18), and functional studies were performed on five in-house strains corresponding to the four STs. Phylogenomic analysis results from this study corroborated with the sequence type-specific clonality. Results from the genome-wide resistance profiling confirmed that all strains were inherently multidrug resistant. ST131 genomes showed unique virulence profiles, and analysis of mobile genetic elements and their associated methyltransferases (MTases) has revealed that several of them were missing from the majority of the non-ST131 strains. Despite the fact that non-ST131 strains lacked few essential genes belonging to the serum resistome, the in-house strains representing all four STs demonstrated similar resistance levels to serum antibactericidal activity. Core genome analysis data revealed that non-ST131 strains usually lacked several ST131-defined genomic coordinates, and a significant number of genes were missing from the core of the ST131 genomes. Data from this study reinforce adaptive diversification of E. coli strains belonging to the ST131 lineage and provide new insights into the molecular mechanisms underlying clonal diversification of the ST131 lineage.
KEYWORDS: bacterial evolution, Escherichia coli, genomics, ST131 lineage, molecular epidemiology
IMPORTANCE
E. coli, particularly the ST131 extraintestinal pathogenic E. coli (ExPEC) lineage, is an important cause of community- and hospital-acquired infections, such as urinary tract infections, surgical site infections, bloodstream infections, and sepsis. The treatment of infections caused by ExPEC has become very challenging due to the emergence of resistance to the first-line as well as the last-resort antibiotics. This study analyzes E. coli ST131 against three other important and globally distributed ExPEC lineages (ST38, ST405, and ST648) that also produced extended-spectrum β-lactamase (ESBL). This is perhaps the first study that employs the high-throughput whole-genome sequence-based approach to compare and study the genomic features of these four ExPEC lineages in relation to their functional properties. Findings from this study highlight the differences in the genomic coordinates of ST131 with respect to the other STs considered here. Results from this comparative genomics study can help in advancing the understanding of ST131 evolution and also offer a framework towards future developments in pathogen identification and targeted therapeutics to prevent diseases caused by this pandemic E. coli ST131 clone.
INTRODUCTION
Extraintestinal pathogenic Escherichia coli (ExPEC) strains are a versatile variant of commensal E. coli demonstrating a complex genome plasticity and phylogeny (1). ExPEC strains can cause disease syndromes ranging from uncomplicated urinary tract infections to life-threatening septicemia in humans (1). Control of infections caused by ExPEC has been troublesome due to the emergence of antimicrobial resistance, particularly in the extended-spectrum β-lactamase (ESBL)-producing E. coli strains, which are linked with the increased rates of morbidity and mortality (2). Association of these multidrug-resistant (MDR) strains with a high rate of community- as well as hospital-acquired infections can be largely attributed to the evolution of a number of different clones with enhanced metabolic repertoires, along with increased levels of virulence and antibiotic resistance (3).
To understand the evolution of such clones, several classification schemes have been suggested, of which multilocus sequence typing (MLST) is considered to be the “gold standard” (4, 5). ESBL-producing E. coli ST131 was identified among thousands of sequence types (STs) or clones defined by MLST based on variations in seven housekeeping genes of E. coli (6, 7). Compared to other ExPEC clones, ST131 has been widely studied due to its predominant association with ExPEC infections, particularly urinary tract and bloodstream infections (8–10). This globally disseminated multidrug-resistant clone belonging to phylogenetic group B2 has also been associated with the worldwide emergence of a particular ESBL genotype, CTX-M-15 (11). Relatively higher virulence and metabolic capabilities along with resistance to wider classes of antibiotics can be attributed to widespread occurrence and persistence of this particular lineage (12, 13).
Recent reports suggest that a few other ExPEC-associated sequence types, such as ST38, ST405, and ST648, were also associated with global dissemination of CTX-M-producing E. coli (14). E. coli ST38 strains carrying CTX-M, NDM-1, and OXA-48 genes were earlier reported from several countries, such as Japan, Netherlands, South Korea, and Tanzania (15–21). Another study has reported that ST38 strains isolated from Mongolian birds were carrying the ESBL genes in their chromosomes instead of the plasmids (22). While, CTX-M-positive ST405 and ST648 were reported from different parts of the world (16, 23–28), NDM-producing ST405 and ST648 were reported from United Kingdom (29, 30). Genotype ST648 reported from companion animals was also reported to be usually associated with CTX-M (31). Comparative in vitro analysis of virulence determinants of strains belonging to ST38, ST131, ST405, and ST648 reported the presence of certain common virulence factor genes, such as sat, iutA, malX, usp, and ompT, which were associated with greater adaptability, competitiveness, and colonization capabilities (5, 32, 33).
Whole-genome-based comparative analysis provides insights into the variation in the genetic architecture within or among different species (34). While many ST131 whole-genome sequences have already been reported and analyzed worldwide, no comparative studies have yet been performed with ST38, ST405, and ST648 strains. An exhaustively comparative study on the genomic landscapes of ST131 versus other epidemiologically successful sequence types would be very pertinent to understand the evolutionary mechanisms of these STs; this therefore constituted the main focus of our study. This study included whole-genome sequencing of five ExPEC strains belonging to the four STs, which were previously obtained from western India (21). In addition, whole-genome-based comparative analysis of 139 genomes representative of strains belonging to ESBL-producing ST38, ST405, ST648, and ST131 as obtained from public as well as in-house databases was performed. Findings from this study provide an insight into the genetic-level differences in ST131 with respect to the other STs, which could be the potential basis for their global predominance in ExPEC infections.
RESULTS
Strain information and genome characteristics of in-house strains.
The assembly of NA023, NA081, NA090, NA101, and NA112 strains comprised 176, 223, 193, 175, and 168 contigs, respectively, with an approximate genome size of 5.2 Mb. The average G+C content was ~50%, and the average coding sequence (CDS) number of all the strains was found to be around ~5,000, amounting to a coding percentage of ~87%. The characteristics of the five genomes are presented in Table S1 in the supplemental material. While two strains, NA101 and NA112, belonged to ST131, strains NA023, NA081, and NA090 belonged to ST648, ST405, and ST38, respectively.
Background data and genome characteristics of the five in-house strains NA023, NA081, NA090, NA101, and NA112 belonging to four STs considered in this study. Download TABLE S1, PDF file, 0.2 MB (163.4KB, pdf) .
Copyright © 2017 Shaik et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Data set collection for the comparative analysis.
Apart from the five in-house strains, 134 strains belonging to the four STs were selected from the public databases. Thus, the data set included 99 ST131 strains (from all three clades A, B, and C), 12 ST38, 10 ST405, and 18 ST648 comprising 139 strains in total from different isolation sources such as stool, patients suffering from urinary tract infection (UTI) and bacteremia, etc. (see Table S2 in the supplemental material). These 139 strains were used for the in silico comparative analysis, and five in-house strains (NA023, NA081, NA090, NA101, and NA112) were characterized by various functional assays.
Information about 139 strains belonging to ST38, ST405, ST648, and ST131 considered for the comparative genomic analysis in this study. Download TABLE S2, PDF file, 0.2 MB (235.6KB, pdf) .
Copyright © 2017 Shaik et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Whole-genome-based phylogenetic analysis confirms clonality within the sequence types.
A core genome-based phylogenetic tree (see Fig. S1 in the supplemental material) showed four distinct clades emulating the MLST scheme depicting the clonality as well as distinctness at the whole-genome level. Three clades, namely, A (orange), B (pink), and C (red) as earlier reported (35, 36) were also observed within the ST131 data set. Clades of ST131 were observed to be more distant from the remaining STs, and also clades representing ST38 (purple) and ST405 (blue) were relatively closer than ST648 (green) (Fig. S1).
Whole-genome phylogenetic tree depicting individual STs as separate clades. Clades A, B, and C of ST131 were observed to be more distant than those of the remaining ST38, ST405, and ST648. Download FIG S1, TIF file, 2.7 MB (2.8MB, tif) .
Copyright © 2017 Shaik et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
STs exhibit similar plasmid and resistance profiles.
From the BLASTn (37) analysis of 139 strains against the downloaded Plasmid Finder database (38), it was observed that 98 out of 99 strains of ST131 harbored IncF plasmids. Among them, the in-house strains NA097 (39), NA101, and NA114 (40) harbored an IncF plasmid with pMLST profile F2:A1:B−, while NA112 had IncF with F4:A−:B− (see Table S3 in the supplemental material). These two pMLST profiles were earlier reported to be most common among the ST131 strains (41). IncF was also observed to be the major class within ST38 and ST405, where 11 out of 12 strains of ST38 and all 10 strains of ST405 harbored IncF. In the case of ST648, only 7 out of 18 genomes contained IncF. None of the strains from ST38, ST405, and ST648 had ST131-associated F2:A1:B− and F1:A2:B20 pMLST profiles. In addition to IncF, plasmid types IncHI2, IncI1, IncN, IncP, IncQ, IncB/O/K/Z, IncX1, IncX3, IncX4, and IncY were observed in lower prevalence in ST131, ST38, and ST405, while a few ST648 strains also harbored IncHI2 and IncN (Table S3).
Plasmid classes observed in each of the strains belonging to ST38, ST405, ST648, and ST131. A positive hit with identity of ≥95% and query coverage of ≥90% indicates the presence of that particular class in the strain. Download TABLE S3, PDF file, 0.5 MB (554.3KB, pdf) .
Copyright © 2017 Shaik et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
BLASTp (37) analysis of putative protein sequences of the 139 strains against the CARD (42) database (see Fig. S2A in the supplemental material) showed that the resistance profiles of strains belonging to all four STs were very similar. The majority of the strains from ST131 (44/99) and ST405 (8/10) carried the CTX-M-15 gene, while only 2 out of 12 strains of ST38 and 6 out of 18 strains of ST648 carried this gene. Similarly, variants of TEM, SHV, and OXA were also present in most of the strains from all four STs. Other resistance genes from different classes were also present in the majority of strains, but no ST-specific pattern was apparent. The five in-house strains NA023, NA081, NA090, NA101, and NA112 were tested in vitro for 18 different antimicrobials belonging to six different classes, namely, β-lactam antibiotics (penicillin and cephalosporin), fluoroquinolone, aminoglycoside, tetracycline, trimethoprim/sulfonamide, and macrolide. The four strains NA101, NA023, NA081, and NA090, belonging to different sequence types ST131, ST648, ST405, and ST38, were resistant to 10, 12, 12, and 8 antibiotics, respectively. The other strain of ST131, NA112, was resistant to only six antibiotics. In-house strains NA023, NA081, NA090, and NA101 harbored the CTX-M-15 gene. The complete resistome of in-house strains is represented in Fig. S2B.
(A) Heat map depicting the presence of CARD resistance genes in our data set containing 139 strains belonging to ST38, ST405, ST648, and ST131 (clades A, B, and C). Black indicates a positive hit with identity of ≥70% and query coverage of ≥75%. (B) Representation of in vitro analysis of five in-house strains tested for 18 different antimicrobials belonging to six different classes. Download FIG S2, TIF file, 1.4 MB (1.4MB, tif) .
Copyright © 2017 Shaik et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
ST131 exhibits a relatively robust virulence profile.
Previous in vitro virulence studies (5) have reported that certain virulence factors, such as sat (secreted autotransporter toxin) and iutA (aerobactin siderophore receptor), were significantly present in the four genotypes ST131, ST38, ST405, and ST648, whereas the pathogenicity island marker gene malX was present in all but ST38. Also, certain factors were reported to be specifically present in a single genotype, such as hra (heat-resistant agglutinin) and group 2 capsule variant K2 in ST38, kpsM-III group 3 capsule in ST405, and outer membrane protease T (ompT) in ST131 and ST648. sat, the uropathogenic-specific protein gene (usp), and the adhesion siderophore gene (iha) were significantly common in ST131. A similar pattern was also observed in our data set. Virulence factor kpsM-III group capsule was present in 4 out of 10 strains of ST405 and in a single strain of ST648 but was missing from all 99 strains of ST131. Also, hra, which was reported to be commonly present in ST38, was observed in 11 out of 12 strains and few strains from other STs. ompT, which was earlier reported in the strains of ST131 and ST648, was observed to be present in 17/18 strains of ST648 and all 99 strains of ST131 but was missing in the remaining STs. While usp was exclusively present in all 99 strains of ST131, cnf1 and iroN were present only in 14 and 8 ST131 strains, respectively. These three virulence factors (usp, cnf1, and iroN) were completely missing from ST38, ST405, and ST648. The pathogenicity island marker (malX) was present in all 139 strains irrespective of the STs.
Other than this, certain other observations were captured in the heat map (Fig. 1) that illustrates the frequency of the Virulence Factor Database (VFDB) genes (35) in all four STs. Adhesion- and invasion-related genes, such as upaG, ehaG, yfaL, ibeB, and ompA, were present in all 139 strains, but several autotransporter genes, serine protease genes (espP, epeA, pic, and EC55989_4660), and many uropathogenic E. coli (UPEC)-specific virulence genes were absent among ST38, ST405, and ST648 strains, while they were present in the majority of ST131 strains. Few autotransporter genes, such as ypjA, yejO, and ECIAI39_1904, were missing from ST131 but were present in the remaining STs (Fig. 1).
FIG 1 .
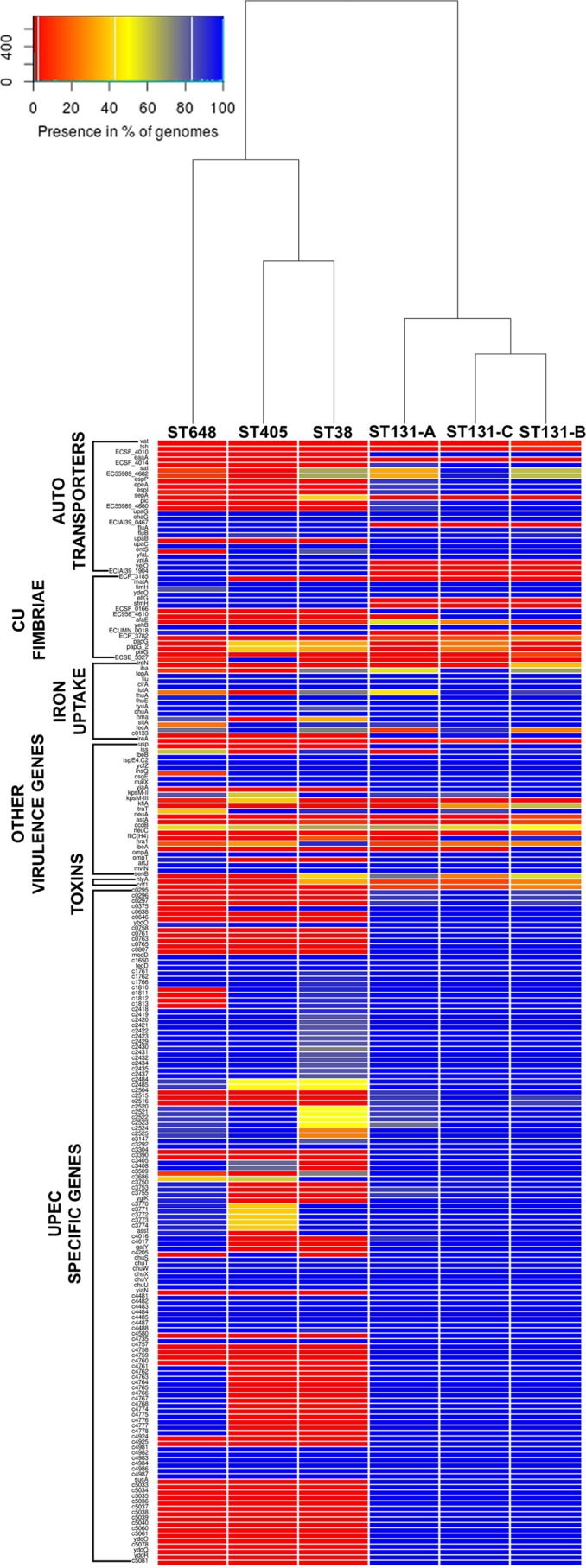
Gradient heat map depicting the frequency of virulence genes in ST38, ST405, ST648, and ST131 (clades A, B, and C). Frequency of each gene is calculated by the formula (presence in no. of strains of ST/total no. of strains in ST) × 100. Colors ranging toward red and blue depict lower and greater frequencies, respectively. Strains belonging to all three clades of ST131 harbored the majority of UPEC-specific genes, while the rest of the STs (ST38, ST405, and ST648) had them in low frequencies.
The in vitro adhesion and invasion assays revealed that all five strains were able to adhere and invade the bladder epithelial cell line T24 compared to the negative control, E. coli DH5α. Strains of all four sequence types were significantly more adherent (P < 0.001). There were certain strain-specific differences in adherence capabilities (Fig. 2A). Similarly, the strains were also significantly more invasive than the negative control (Fig. 2B). Thus, the strains demonstrated similar pathogenic capabilities in terms of adherence and invasion to epithelial cells, irrespective of sequence types.
FIG 2 .

In vitro assays performed on five in-house strains NA023 (ST648), NA081 (ST405), NA090 (ST38), NA101 (ST131), and NA112 (ST131) belonging to all four STs considered in this study. (A) Adhesion assay. (B) Invasion assay. All strains could significantly adhere to and invade T24 bladder cell lines compared to DH5α.
Mobile genetic elements indeed define ST131.
BLAST Ring Image Generator (BRIG [43]) analysis (Fig. 3A) of mobile genetic elements (MGEs) revealed that the genomic islands (GIs) GI-leuX, GI-pheV, and GI-thrW were conserved in the majority of the ST131 strains but were either missing partially or completely from non-ST131 strains. GI-thrW was observed to be missing in the two in-house ST131 strains NA101 and NA112. GI-pheV which did not follow any conservation pattern in case of ST131 strains was also completely or partially absent in all other STs. So was the case with the other mobile genetic elements. The 31,448-bp high-pathogenicity island (HPI) from EC958, which is 99% identical to that of Yersinia pestis (44), was found to be the only mobile genetic element in all strains of ST131, ST405, and ST648 and 10 out of 12 strains of ST38.
FIG 3 .

Mobile genetic elements and MTases in non-ST131 (ST38, ST405, and ST648) strains. (A) BLAST Ring Image Generator (BRIG) image showing the presence of mobile genetic elements reported in EC958 in our data set. (B) BRIG image with EC958 MTases as the reference. The majority of the mobile genetic elements and their associated MTases were found to be either missing or partially present in the non-ST131 genomes, while their presence was predominant in ST131 genomes.
A similar BRIG image (Fig. 3B) analyzing methyltransferases (MTases) revealed that chromosome-associated MTases such as M.EcoMDcm (EC958_2226), M.EcoMVI (EC958_3663), and M.EcoMDam (EC958_3778) were present in all strains of the data set. M.EcoMIII (EC958_0425), a type I restriction modification (R-M) system that is part of GI-thrW, was missing from all strains of ST38, ST405, and ST648 and also two ST131 strains (NA101 and NA112). Lack of GI-thrW in these strains as discussed above justifies the absence of this particular MTase. While M.EcoMIV (EC958_1101) encoded by prophage 2 was conserved in most of the strains of ST131 clade C strains, it was completely missing in the remaining strains. M.EcoMVII (EC958_4083) was observed to be present in only few strains of ST131 emulating the pattern of its associated mobile genetic element GI-selC. Other MTases, such as M1.EcoMI/M2.EcoMI (EC958_0008/EC958_0009), M.EcoMII (EC958_0078), M.EcoMV (EC958_1545), M.EcoMVIII (pEC958_A0009), McrBC (EC958_0011 and EC958_0012), and Mrr (EC958_0079) were present in almost all strains of clade C and a few other strains.
Comparison of essential gene sets of serum resistome.
All 139 strains were analyzed for the presence of 56 essential genes belonging to the serum resistome of EC958, which have been recently defined by transposon-directed insertion site sequencing (TraDIS [45]). It was observed from the BLASTp analysis (Fig. 4A) that a few genes were missing (hit identity of <70% and query coverage of <75%) from the complete data set of ST38, ST405, and ST648, which includes EC958_0460 (hyxA), EC958_0461 (hyxR), EC958_1112, EC958_1114, EC958_2371, EC958_2373, EC958_4029 (waaL), EC958_4030 (waaU [waaK]), EC958_4035 (waaB [rfaB]), EC958_4032 (waaY [rfaY]), EC958_4033 (waaJ [rfaJ]), and EC958_4034 (waaI [rfaI]) (Fig. 4B), while they were present in all ST131 strains. EC958_0858 (tolA), belonging to the Tol-Pal system of E. coli, was observed to be only partially present in these strains (129 bp out of 436 bp with identity of >90%).
FIG 4 .

Serum resistance analysis. (A) Heat map depicting the 56 essential serum resistome genes in our data set containing 139 strains belonging to ST38, ST405, ST648, and ST131 (clades A, B, and C). Black indicates a positive hit with identity of ≥70% and query coverage of ≥75%. (B) Serum resistance assay. Genome-wide screening of serum resistance genes revealed that few essential genes were missing from non-ST131 (ST38, ST405, and ST648) genomes, but the levels of phenotypic serum resistance were found to be comparable among all four STs.
In vitro serum resistance analysis of the in-house strains NA101, NA112, NA090, NA023, and NA081 belonging to all four sequence types revealed that all strains were found to be resistant to the bactericidal activity of human serum and showed significant growth in 50% human serum in comparison to the negative control (DH5α) (Fig. 4B), irrespective of their sequence types. NA114 and NA097 were also earlier reported to be resistant to human serum (39, 40).
Genes missing from ST131.
Core genome analysis was performed to identify the core gene content separately for each sequence type: ST38, ST131, ST405, and ST648. About 5,169, 8,189, 5,495, and 5,851 orthologous groups were identified in the cases of ST38, ST131, ST405, and ST648, respectively. Of these 4,101, 4,140, 4,362, and 4,279 belonged to their respective cores. The core protein sequences, including paralogs, were extracted from one representative strain of each ST, and another round of core genome analysis was performed using only the core protein sequences of individual STs. This analysis resulted in 4,328 orthologous groups, of which 3,472 belonged to the common core and 856 groups belonged to the accessory content.
Accessory content of the four cores, excluding the paralog clusters was further analyzed to identify any proteins that were specifically present or absent in ST131. We could not identify any ST131-specific proteins; however, 224 proteins that were specifically absent in the core of ST131 were identified. Another round of BLASTp analysis of these 224 protein sequences against the complete data set of 139 strains revealed that about 142 proteins (see Table S4 in the supplemental material) were indeed absent in all 99 strains of ST131 but were present in the remaining 40 strains belonging to ST38, ST405, and ST648.
Information about the list of proteins that are specifically missing in all 99 strains of ST131 while present in the remaining 40 strains belonging to ST38, ST405, and ST648. Presence represents a positive BLASTp hit with identity of ≥70%. Download TABLE S4, PDF file, 0.3 MB (280.5KB, pdf) .
Copyright © 2017 Shaik et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
The functional annotation of these 142 sequences revealed that 99 proteins had a Clusters of Orthologous Groups (COG) (46) domain. These 99 proteins included membrane proteins, transcriptional regulators, transporters, toxins and antitoxins, chaperones, fimbrial proteins, and those belonging to a wide range of COG categories (Table S4).
DISCUSSION
The enormous diversity of E. coli has made it the subject of innumerable comparative and phylogenetic studies in order to determine the mechanisms by which each of the subgroups/lineages has diversified and specialized. The recent emergence of the multidrug-resistant E. coli lineages (ST131, ST38, ST405, and ST648) as the globally dominant strains isolated from extraintestinal infections provides a new and important opportunity to understand how E. coli evolves and diversifies (14). Although the genomic architecture of ST131 has been widely studied, this is the first report on a genome-based comparison of ST131 strains together with three other emerging ESBL-producing ExPEC lineages: ST38, ST405, and ST648. Since E. coli is a very versatile organism, longitudinal studies on its genomics are very essential to understand the differences in the genomic coordinates and to understand its evolution. In this study, a data set of 5 in-house-sequenced strains along with 134 strains from the public databases belonging to these four STs was analyzed.
Clonality within the STs as depicted from the phylogenetic analysis (Fig. S1) suggests that these STs differ at the genetic level, although they are all responsible for ExPEC infections. Plasmid analysis has revealed that IncF plasmid is not just successful in ST131 as earlier reported but also most often found in the strains of the remaining STs analyzed in this study (35, 47, 48) (Table S3). The pMLST profiles F2:A1:B− and F1:A2:B20, which were commonly associated with IncF of ST131 strains (35), were not seen in any of the non-ST131 strains across the data set. Together with IncF, an intermixture of other plasmid classes, such as IncHI2, IncI1, IncN, IncP, IncQ, IncB/O/K/Z, IncX1, IncX3, IncX4, and IncY, was found among all the STs to a lesser extent. Similar to plasmid profiles, the antimicrobial resistance profiles as inferred from both genomic analysis of 139 strains and antimicrobial susceptibility assays on five in-house strains revealed that overall antimicrobial resistance was common and differed insignificantly between the four STs (Fig. S2). The presence of ESBL genes blaCTX-M, blaTEM, and blaSHV, together with several other resistance genes belonging to different classes, such as efflux pumps, pump regulators, and target inactivators, reflects the MDR genotypes of these STs (Fig. S2).
The presence of certain virulence-associated genes, such as hra (heat-resistant agglutinin), the kpsM-III group 3 capsule gene, ompT (outer membrane protease T), and malx (pathogenicity island marker) in these four STs was observed to emulate a previous study on in vitro virulence profiles of these four STs (5). Also, the presence of several serine proteases, autotransporters, and UPEC-specific virulence genes exclusively in the strains of ST131 was observed, which could be a reason for the predominance of ST131 strains in extraintestinal infections over any other single sequence type. The presence of genes related to adherence and invasion, such as ibeA, ibeB, fimH, papG, and afaE, among all the strains indicated comparable adhesion and invasion capabilities for the four STs. This supposition was validated by results of the adhesion and invasion assays on the in-house strains of all four STs (Fig. 2). Also, Wang et al. demonstrated that the components of the type 3 secretion system (T3SS) play roles in virulence and intracellular survival of an avian-pathogenic E. coli (APEC) strain in macrophages (49); thus, roles of such factors might also influence adhesion and invasion of ExPEC strains. In Gram-negative bacteria, the majority of serine proteases secreted by the autotransporter pathway are implicated in virulence (50). This could be the strategy of ST131 strains to enhance their virulence by consistently retaining the key serine protease autotransporters together with a unique set of virulence determinants.
Mobile genetic elements (MGEs) predominantly encode virulence and related factors (44, 51) that have been shown to play an important role in transforming UPEC strains from an acute infectious state to a chronic infectious one (52–54). An earlier study on comparative genomic analysis of ST131 strains has shown majority of the MGEs to be present in ST131 clade C (35). From our study, BRIG analysis of MGEs (previously defined in EC958 [44]) revealed that most of these regions were missing either completely or partially in ST38, ST405, and ST648 (Fig. 3A). Since EC958 belongs to ST131, it further indicates that ST131 strains have unique set of mobile genetic elements. Earlier in silico studies with ST131 strains (35) revealed that the presence of these regions was one of the major causes behind the diversity within the ST131 lineage. Also, MTases such as EcoMIII (EC958_0425), M.EcoMIV (EC958_1101), M.EcoMVII (EC958_4083), and M1.EcoMI/M2.EcoMI (EC958_0008/EC958_0009), which are associated with GI-thrW, Phi-2, GI-selC, and GI-leuX, respectively, were also found to be missing in strains lacking these MGEs (Fig. 3B). Three of the MTases mentioned above are part of restriction modification (R-M) systems, which are known to act as barriers for DNA exchange among species as well as within species. It would be possible that these MTases have a prominent role in segregating and shaping up the gene pool of ST131 (51). Lack of these MTases in remaining STs may indicate that they must be still open for gene exchange with other STs through HGT.
Serum resistance is one of the major survival mechanisms adopted by pathogenic E. coli to survive in the bloodstream of the host. BLASTp analysis of the 139 strains against 56 essential serum resistance genes of EC958 has revealed that 12 of these genes were missing from all 40 strains belonging to the non-ST131 strains but present in ST131 strains (Fig. 4A). Among these missing essential serum resistance genes, EC958_0460 (hyxA) and EC958_0461 (hyxR) were reported to have a role in regulating O-antigen chain length (45). Previous studies have also shown that EC958_0461 (hyxR) is linked to the suppression of nitrosactive stress response as well as important for intracellular survival within macrophages (55). EC958_1112 is one of the four glucose transferases found in the O-antigen, and the operons EC958_4029 (waaL), EC958_4030 (waaU [waaK]), and EC958_4035 (waaB [rfaB]) were linked to lipopolysaccharide (LPS) core biosynthesis. Mutants of all these genes except for EC958_1114 were reported to be relatively more sensitive to the serum than the wild-type genes (45). Although 12 essential serum resistance genes were missing from the non-ST131 strains, all of the five in-house strains representing the four STs exhibited similar resistance capabilities against the bactericidal activity of human serum (Fig. 4B). This may be because of the presence of uncharacterized genes in these STs, which may exhibit similar functional roles. The extra set (12 genes) of serum resistance genes identified exclusively in the ST131 lineage compared to the other three STs might help in circumventing the confounders in a natural infection state.
OrthoMCL (56) analysis of core proteins belonging to each of these four STs revealed that around 142 proteins were completely missing from all 99 strains of ST131 (Table S4) but were present in all 40 strains from ST38, ST405, and ST648. COG functional annotation of these proteins revealed that they represent a wide variety of COG functional classes, which include membrane proteins, transcriptional regulators, transcriptional chaperones, lyases, hydrogenases, etc. Also, two toxin-antitoxin modules, mazE-mazF and yafO-yafN, were found to be among the 142 missing genes of ST131. While mazE-mazF is known to cause programmed cell death under stress conditions, such as nutrient starvation and addition of antibiotics, yafO-yafN is involved in SOS response triggered by DNA damage (57). The gene coding for another important protein, diguanylate cyclase (yddV), involved in c-di-GMP signaling, was also among these 142 genes that were absent in the ST131 strains. It was reported earlier that this protein was found to be either truncated or completely deleted in many ExPEC strains. It was hypothesized that losing this protein would negatively regulate c-di-GMP, which may in turn enhance motility and also decrease expression of curli fibers to avoid local immune defenses (58). These observations indicate that strains of ST131 lineage utilize gene loss as a mechanism in order to fine-tune their genome for maximal fitness. The possibility of such a phenomenon has been described for some bacterial species (59).
In summary, the comparative genomic analysis of ST131 strains and other prominent non-ST131 strains (ST38, ST408, and ST648) spanning the extent of extraintestinal pathogenic E. coli strains provides novel insights on the evolutionary mechanisms of the ST131 lineage by revealing the molecular signatures that characterize their evolution. Our results suggest that adaptive strategies of ST131 E. coli strains were driven mainly by gene loss, genetic exchange, and coevolution with an antimicrobial resistance repertoire. We provide an inventory of both ST131-specific genes and those that are completely lacking in the E. coli ST131 lineage compared to E. coli genomes of other three STs (ST38, ST408, and ST648). These data will help in advancing the understanding of ST131 evolution and also offer a framework to advance future developments in pathogen identification and targeted therapeutics to prevent diseases caused by this pandemic E. coli ST131 clone.
MATERIALS AND METHODS
Strain selection and whole-genome sequencing.
The five strains NA023, NA081, NA090, NA101, and NA112, belonging to ST38, ST405, ST648, and ST131 from the collection described in reference 21, were sequenced using an Illumina Miseq sequencer with an insert size of 400 to 500 bp.
Assembly and annotation of in-house strains.
High-quality reads obtained from NGS QC Toolkit (60) were used for de novo assembly of contigs using Velvet (61). The thus-generated contigs were then ordered and scaffolded using C-L-Authenticator (62), and the final draft genomes were obtained by merging these scaffolds using a series of N’s. These draft genomes were then submitted to the RAST server (63) for annotation, and the genome statistics from the resulting file were extracted using ARTEMIS (64). The sequence type (ST) of each of these strains was determined by submitting the de novo contigs to https://cge.cbs.dtu.dk/services/MLST/.
Data set collection for comparative genomics. (i) Collection of 99 ST131 strains.
Apart from the two strains from the present study, 97 whole-genome sequences, including NA097, NA114, JJ1886, and EC958 from the NCBI and other ST131 genomes described by Petty et al. (35), were considered for the study.
(ii) Collection of ST38, ST405, and ST648 strains.
As there are not many studies performed at the genomic level of these STs, an in-house pipeline was devised to extract the genomic data from NCBI. As of 8 June 2015, genomic data (both complete and incomplete) belonging to 3,051 E. coli strains were downloaded, and in the case of strains with incomplete data, contigs that were >500 in number were eliminated. In each genome, in-house scripts were used to extract the sequences of the seven housekeeping genes (fum, adk, gyr, icd, pur, mdh, and rec) and an ST number was assigned based on the combination of allelic numbers of these genes (supported by the database data downloaded from http://mlst.warwick.ac.uk/mlst/dbs/Ecoli/Downloads_HTML). Among all of the STs, 12 strains from ST38, 10 strains from ST405, and 18 strains from ST648 were separated out. The ST of each of these strains was further verified by submitting the contigs to https://cge.cbs.dtu.dk/services/MLST/. Information about all the strains used in this study is provided in Table S3.
Phylogenomic analysis.
A core-genome-based phylogenetic tree of all 139 strains belonging to the four STs was constructed using Harvest (65), and the resulting tree was visualized using FigTree (http://tree.bio.ed.ac.uk/software/figtree/).
Detection of plasmid classes and resistance profiles.
In case of each of these strains, BLASTn of the contigs/whole genome was performed against the data downloaded from Plasmid Finder (38). A positive hit (90% query coverage and 95% identity) indicated the presence of a particular plasmid class in respective strains.
For all of the genomes, protein sequences were predicted using GeneMarkS (66). Resistance profiles of each of these strains were obtained by performing a BLASTp analysis of the predicted protein sequences against the Comprehensive Antibiotic Resistance Database (CARD). Only the genes that had hits with 70% identity and 75% query coverage were given the status of being present. A heat map was generated using R to depict the positive hits (represented in black) in each of these genomes.
Determination of virulence gene profile.
Virulence profiles of each of these strains were obtained by performing a BLASTn analysis of 139 genomes against the database of selected VFDB (35, 67) virulence genes. Only the genes that had hits with 70% identity and query coverage of either ≥50% or ≥800 bp were given the status of being present. The frequency of each of these virulence genes at the ST level was calculated using the formula (presence in no. of strains of ST/total no. of strains in ST) × 100. The frequencies of all of the virulence genes in each of these STs are represented in the form of a heat map using R.
Genomic islands, MTases, and serum resistome analysis.
BRIG analysis (Fig. 3A) was performed with the data set containing 12, 10, and 18 strains belonging to ST38, ST405, and ST648 using the MGEs and MTases reported in the ST131 representative strain EC958 (44, 51) as a reference. Only 20 strains from ST131 data set belonging to all three clades A, B, and C were selected for this analysis.
A BLASTp analysis was performed to detect the presence of 56 essential serum resistance genes that were earlier reported in EC958 (45). Positive hits were defined and represented in the form of a heat map, as discussed earlier in the section “Detection of plasmid classes and resistance profiles.”
Comparative core-accessory genome analysis.
Due to the differences in the sample sizes of each sequence type, core genes of individual sequence types were determined first. Core orthologous clusters of individual STs were identified using OrthoMCL (56, 68). The parameters for deciding orthologs such as identity and E value cutoff were set to 70% and 0.00001, respectively. The genes with less than 50 amino acids were excluded from the analysis. The clusters that contained orthologs in at least 90% of the sample size constituted the core. Genes belonging to the core orthologous groups of ST38, ST131, ST405, and ST648 were extracted from one respective representative strain.
Core genes (including recent paralogs) from the orthologous clusters of the four STs were then considered for further inter-ST core-accessory genome analysis. To identify genes that were specifically present or missing in ST131, orthologous clusters of all four ST core genes were once again determined using OrthoMCL. Orthologous clusters that had missing genes only from the ST131 core were further analyzed at the complete data set level using BLASTp as described above. A final list of genes that were present in all 40 strains belonging to ST38, ST405, and ST648 but missing from all 99 strains of ST131 was identified. COG classification and their functional annotations of all the genes of interest were determined using batch CD-search (69) against the COG database.
In vitro phenotypic characterization of the strains.
All five representative strains of the four different sequence types were characterized for the phenotypic properties, such as resistance to 18 different antibiotics, ESBL production, adhesion and invasion capabilities toward human bladder epithelial (T24) cell lines, and ability to resist bactericidal activity of human serum. Resistance toward antimicrobials was determined by standard Kirby Bauer disk diffusion technique using ICOSA UTI strips (Himedia, India) as per the guidelines of CLSI, and ESBL production was tested by a double-disk synergy test (12). The adhesion and invasion assays were performed as per the standard protocols described previously (70). Briefly, T24 cells were grown to monolayer in 24-well plates and infected at a multiplicity of infection (MOI) of 10 with different strains in triplicates. After 3 h of incubation at 37°C in a CO2 incubator, the cells were washed three times with 1× phosphate-buffered saline (PBS) and then lysed using 0.1% Triton X-100. The lysates were collected and plated after serial dilutions. For invasion, after 3 h of incubation cells were additionally incubated for 1.5 h with medium containing 100 µg/ml of gentamicin. After incubation, cells were lysed as in adhesion step and plated and CFU counts determined for all strains. The serum resistance assay was performed in sterile 50% human serum (Pan Biotech, Germany) for 3 h as per the protocols described elsewhere (70). The growth in serum was observed in terms of CFU. The adhesion and invasion assays were performed three times, while the serum resistance assay was performed twice in triplicates. For serum resistance and the adhesion assay, the Mann-Whitney test was employed to get P values against the negative control (DH5α), while for the invasion assay, P values were obtained by employing Wilcoxon’s signed-rank test against the negative control.
Accession number(s).
The GenBank accession numbers of the five genomes sequenced for this study are JSXK00000000 (NA023), JSXM00000000 (NA081), MVIO00000000 (NA090), JSXN00000000 (NA101), and JSXO00000000 (NA112).
ACKNOWLEDGMENTS
We acknowledge funding support from the Department of Biotechnology, Government of India (Ref. No. BT/HRD/NBA/34/01/2011(ix)). Also, the Indo-German International Research Training Group, Internationales Graduiertenkolleg (GRK1673)-functional molecular infection epidemiology, an initiative of the German Research Foundation (DFG) and the University of Hyderabad (India), of which N.A. and L.H.W. served as speakers, is gratefully acknowledged. N.A. is an Adjunct Professor of the Academy of Scientific and Innovative Research (AcSIR), India. icddr,b acknowledges with gratitude the Governments of Bangladesh, Canada, Sweden, and the United Kingdom for providing core/unrestricted support. The authors would also like to thank Microsoft Corporation for the “Microsoft Azure for Research Award” to N.A.
Footnotes
Citation Shaik S, Ranjan A, Tiwari SK, Hussain A, Nandanwar N, Kumar N, Jadhav S, Semmler T, Baddam R, Islam MA, Alam M, Wieler LH, Watanabe H, Ahmed N. 2017. Comparative genomic analysis of globally dominant ST131 clone with other epidemiologically successful extraintestinal pathogenic Escherichia coli (ExPEC) lineages. mBio 8:e01596-17. https://doi.org/10.1128/mBio.01596-17.
REFERENCES
- 1.Kaper JB, Nataro JP, Mobley HL. 2004. Pathogenic Escherichia coli. Nat Rev Microbiol 2:123–140. doi: 10.1038/nrmicro818. [DOI] [PubMed] [Google Scholar]
- 2.Tumbarello M, Sanguinetti M, Montuori E, Trecarichi EM, Posteraro B, Fiori B, Citton R, D’Inzeo T, Fadda G, Cauda R, Spanu T. 2007. Predictors of mortality in patients with bloodstream infections caused by extended-spectrum-beta-lactamase-producing Enterobacteriaceae: importance of inadequate initial antimicrobial treatment. Antimicrob Agents Chemother 51:1987–1994. doi: 10.1128/AAC.01509-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dobrindt U. 2005. (Patho-)genomics of Escherichia coli. Int J Med Microbiol 295:357–371. doi: 10.1016/j.ijmm.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 4.Sullivan CB, Diggle MA, Clarke SC. 2005. Multilocus sequence typing: data analysis in clinical microbiology and public health. Mol Biotechnol 29:245–254. doi: 10.1385/MB:29:3:245. [DOI] [PubMed] [Google Scholar]
- 5.Van der Bij AK, Peirano G, Pitondo-Silva A, Pitout JD. 2012. The presence of genes encoding for different virulence factors in clonally related Escherichia coli that produce CTX-Ms. Diagn Microbiol Infect Dis 72:297–302. doi: 10.1016/j.diagmicrobio.2011.12.011. [DOI] [PubMed] [Google Scholar]
- 6.Peirano G, Pitout JD. 2010. Molecular epidemiology of Escherichia coli producing CTX-M beta-lactamases: the worldwide emergence of clone ST131 O25:H4. Int J Antimicrob Agents 35:316–321. doi: 10.1016/j.ijantimicag.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 7.Wirth T, Falush D, Lan R, Colles F, Mensa P, Wieler LH, Karch H, Reeves PR, Maiden MC, Ochman H, Achtman M. 2006. Sex and virulence in Escherichia coli: an evolutionary perspective. Mol Microbiol 60:1136–1151. doi: 10.1111/j.1365-2958.2006.05172.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nicolas-Chanoine MH, Blanco J, Leflon-Guibout V, Demarty R, Alonso MP, Caniça MM, Park YJ, Lavigne JP, Pitout J, Johnson JR. 2008. Intercontinental emergence of Escherichia coli clone O25:H4-ST131 producing CTX-M-15. J Antimicrob Chemother 61:273–281. doi: 10.1093/jac/dkm464. [DOI] [PubMed] [Google Scholar]
- 9.Hussain A, Ewers C, Nandanwar N, Guenther S, Jadhav S, Wieler LH, Ahmed N. 2012. Multiresistant uropathogenic Escherichia coli from a region in India where urinary tract infections are endemic: genotypic and phenotypic characteristics of sequence type 131 isolates of the CTX-M-15 extended-spectrum-β-lactamase-producing lineage. Antimicrob Agents Chemother 56:6358–6365. doi: 10.1128/AAC.01099-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jadhav S, Hussain A, Devi S, Kumar A, Parveen S, Gandham N, Wieler LH, Ewers C, Ahmed N. 2011. Virulence characteristics and genetic affinities of multiple drug resistant uropathogenic Escherichia coli from a semi urban locality in India. PLoS One 6:e18063. doi: 10.1371/journal.pone.0018063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schembri MA, Zakour NL, Phan MD, Forde BM, Stanton-Cook M, Beatson SA. 2015. Molecular characterization of the multidrug resistant Escherichia coli ST131 clone. Pathogens 4:422–430. doi: 10.3390/pathogens4030422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hussain A, Ranjan A, Nandanwar N, Babbar A, Jadhav S, Ahmed N. 2014. Genotypic and phenotypic profiles of Escherichia coli isolates belonging to clinical sequence type 131 (ST131), clinical non-ST131, and fecal non-ST131 lineages from India. Antimicrob Agents Chemother 58:7240–7249. doi: 10.1128/AAC.03320-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vimont S, Boyd A, Bleibtreu A, Bens M, Goujon JM, Garry L, Clermont O, Denamur E, Arlet G, Vandewalle A. 2012. The CTX-M-15-producing Escherichia coli clone O25b:H4-ST131 has high intestine colonization and urinary tract infection abilities. PLoS One 7:e46547. doi: 10.1371/journal.pone.0046547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pitout JD. 2012. Extraintestinal pathogenic Escherichia coli: a combination of virulence with antibiotic resistance. Front Microbiol 3:9. doi: 10.3389/fmicb.2012.00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Suzuki S, Shibata N, Yamane K, Wachino J, Ito K, Arakawa Y. 2009. Change in the prevalence of extended-spectrum-beta-lactamase-producing Escherichia coli in Japan by clonal spread. J Antimicrob Chemother 63:72–79. doi: 10.1093/jac/dkn463. [DOI] [PubMed] [Google Scholar]
- 16.van der Bij AK, Peirano G, Goessens WH, van der Vorm ER, van Westreenen M, Pitout JD. 2011. Clinical and molecular characteristics of extended-spectrum-beta-lactamase-producing Escherichia coli causing bacteremia in the Rotterdam area, Netherlands. Antimicrob Agents Chemother 55:3576–3578. doi: 10.1128/AAC.00074-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim J, Bae IK, Jeong SH, Chang CL, Lee CH, Lee K. 2011. Characterization of IncF plasmids carrying the blaCTX-M-14 gene in clinical isolates of Escherichia coli from Korea. J Antimicrob Chemother 66:1263–1268. doi: 10.1093/jac/dkr106. [DOI] [PubMed] [Google Scholar]
- 18.Mshana SE, Imirzalioglu C, Hain T, Domann E, Lyamuya EF, Chakraborty T. 2011. Multiple ST clonal complexes, with a predominance of ST131, of Escherichia coli harbouring blaCTX-M-15 in a tertiary hospital in Tanzania. Clin Microbiol Infect 17:1279–1282. doi: 10.1111/j.1469-0691.2011.03518.x. [DOI] [PubMed] [Google Scholar]
- 19.Poirel L, Bernabeu S, Fortineau N, Podglajen I, Lawrence C, Nordmann P. 2011. Emergence of OXA-48-producing Escherichia coli clone ST38 in France. Antimicrob Agents Chemother 55:4937–4938. doi: 10.1128/AAC.00413-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamamoto T, Takano T, Iwao Y, Hishinuma A. 2011. Emergence of NDM-1-positive capsulated Escherichia coli with high resistance to serum killing in Japan. J Infect Chemother 17:435–439. doi: 10.1007/s10156-011-0232-3. [DOI] [PubMed] [Google Scholar]
- 21.Ranjan A, Shaik S, Mondal A, Nandanwar N, Hussain A, Semmler T, Kumar N, Tiwari SK, Jadhav S, Wieler LH, Ahmed N. 2016. Molecular epidemiology and genome dynamics of New Delhi metallo-β-lactamase-producing extraintestinal pathogenic Escherichia coli strains from India. Antimicrob Agents Chemother 60:6795–6805. doi: 10.1128/AAC.01345-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guenther S, Semmler T, Stubbe A, Stubbe M, Wieler LH, Schaufler K. 2017. Chromosomally encoded ESBL genes in Escherichia coli of ST38 from Mongolian wild birds. J Antimicrob Chemother 72:1310–1313. doi: 10.1093/jac/dkx006. [DOI] [PubMed] [Google Scholar]
- 23.Coque TM, Novais A, Carattoli A, Poirel L, Pitout J, Peixe L, Baquero F, Cantón R, Nordmann P. 2008. Dissemination of clonally related Escherichia coli strains expressing extended-spectrum beta-lactamase CTX-M-15. Emerg Infect Dis 14:195–200. doi: 10.3201/eid1402.070350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jones GL, Warren RE, Skidmore SJ, Davies VA, Gibreel T, Upton M. 2008. Prevalence and distribution of plasmid-mediated quinolone resistance genes in clinical isolates of Escherichia coli lacking extended-spectrum beta-lactamases. J Antimicrob Chemother 62:1245–1251. doi: 10.1093/jac/dkn406. [DOI] [PubMed] [Google Scholar]
- 25.Mihaila L, Wyplosz B, Clermont O, Garry L, Hipeaux MC, Vittecoq D, Dussaix E, Denamur E, Branger C. 2010. Probable intrafamily transmission of a highly virulent CTX-M-3-producing Escherichia coli belonging to the emerging phylogenetic subgroup D2 O102-ST405 clone. J Antimicrob Chemother 65:1537–1539. doi: 10.1093/jac/dkq155. [DOI] [PubMed] [Google Scholar]
- 26.Smet A, Martel A, Persoons D, Dewulf J, Heyndrickx M, Claeys G, Lontie M, Van Meensel B, Herman L, Haesebrouck F, Butaye P. 2010. Characterization of extended-spectrum beta-lactamases produced by Escherichia coli isolated from hospitalized and nonhospitalized patients: emergence of CTX-M-15-producing strains causing urinary tract infections. Microb Drug Resist 16:129–134. doi: 10.1089/mdr.2009.0132. [DOI] [PubMed] [Google Scholar]
- 27.Cortés P, Blanc V, Mora A, Dahbi G, Blanco JE, Blanco M, López C, Andreu A, Navarro F, Alonso MP, Bou G, Blanco J, Llagostera M. 2010. Isolation and characterization of potentially pathogenic antimicrobial-resistant Escherichia coli strains from chicken and pig farms in Spain. Appl Environ Microbiol 76:2799–2805. doi: 10.1128/AEM.02421-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zong Z, Yu R. 2010. Escherichia coli carrying the blaCTX-M-15 gene of ST648. J Med Microbiol 59:1536–1537. doi: 10.1099/jmm.0.022459-0. [DOI] [PubMed] [Google Scholar]
- 29.Hornsey M, Phee L, Wareham DW. 2011. A novel variant, NDM-5, of the New Delhi metallo-β-lactamase in a multidrug-resistant Escherichia coli ST648 isolate recovered from a patient in the United Kingdom. Antimicrob Agents Chemother 55:5952–5954. doi: 10.1128/AAC.05108-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mushtaq S, Irfan S, Sarma JB, Doumith M, Pike R, Pitout J, Livermore DM, Woodford N. 2011. Phylogenetic diversity of Escherichia coli strains producing NDM-type carbapenemases. J Antimicrob Chemother 66:2002–2005. doi: 10.1093/jac/dkr226. [DOI] [PubMed] [Google Scholar]
- 31.Ewers C, Bethe A, Stamm I, Grobbel M, Kopp PA, Guerra B, Stubbe M, Doi Y, Zong Z, Kola A, Schaufler K, Semmler T, Fruth A, Wieler LH, Guenther S. 2014. CTX-M-15-D-ST648 Escherichia coli from companion animals and horses: another pandemic clone combining multiresistance and extraintestinal virulence? J Antimicrob Chemother 69:1224–1230. doi: 10.1093/jac/dkt516. [DOI] [PubMed] [Google Scholar]
- 32.Peirano G, Mulvey GL, Armstrong GD, Pitout JD. 2013. Virulence potential and adherence properties of Escherichia coli that produce CTX-M and NDM β-lactamases. J Med Microbiol 62:525–530. doi: 10.1099/jmm.0.048983-0. [DOI] [PubMed] [Google Scholar]
- 33.Johnson JR, Johnston B, Clabots C, Kuskowski MA, Castanheira M. 2010. Escherichia coli sequence type ST131 as the major cause of serious multidrug-resistant E. coli infections in the United States. Clin Infect Dis 51:286–294. doi: 10.1086/653932. [DOI] [PubMed] [Google Scholar]
- 34.Ahmed N. 2009. A flood of microbial genomes—do we need more? PLoS One 4:e5831. doi: 10.1371/journal.pone.0005831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Petty NK, Ben Zakour NL, Stanton-Cook M, Skippington E, Totsika M, Forde BM, Phan MD, Gomes Moriel D, Peters KM, Davies M, Rogers BA, Dougan G, Rodriguez-Baño J, Pascual A, Pitout JD, Upton M, Paterson DL, Walsh TR, Schembri MA, Beatson SA. 2014. Global dissemination of a multidrug resistant Escherichia coli clone. Proc Natl Acad Sci U S A 111:5694–5699. doi: 10.1073/pnas.1322678111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McNally A, Oren Y, Kelly D, Pascoe B, Dunn S, Sreecharan T, Vehkala M, Välimäki N, Prentice MB, Ashour A, Avram O, Pupko T, Dobrindt U, Literak I, Guenther S, Schaufler K, Wieler LH, Zhiyong Z, Sheppard SK, McInerney JO, Corander J. 2016. Combined analysis of variation in core, accessory and regulatory genome regions provides a super-resolution view into the evolution of bacterial populations. PLoS Genet 12:e1006280. doi: 10.1371/journal.pgen.1006280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 38.Carattoli A, Zankari E, García-Fernández A, Voldby Larsen M, Lund O, Villa L, Møller Aarestrup F, Hasman H. 2014. In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob Agents Chemother 58:3895–3903. doi: 10.1128/AAC.02412-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ranjan A, Shaik S, Hussain A, Nandanwar N, Semmler T, Jadhav S, Wieler LH, Ahmed N. 2015. Genomic and functional portrait of a highly virulent, CTX-M-15-producing H30-Rx subclone of Escherichia coli sequence type 131. Antimicrob Agents Chemother 59:6087–6095. doi: 10.1128/AAC.01447-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Avasthi TS, Kumar N, Baddam R, Hussain A, Nandanwar N, Jadhav S, Ahmed N. 2011. Genome of multidrug-resistant uropathogenic Escherichia coli strain NA114 from India. J Bacteriol 193:4272–4273. doi: 10.1128/JB.05413-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Phan MD, Forde BM, Peters KM, Sarkar S, Hancock S, Stanton-Cook M, Ben Zakour NL, Upton M, Beatson SA, Schembri MA. 2015. Molecular characterization of a multidrug resistance IncF plasmid from the globally disseminated Escherichia coli ST131 clone. PLoS One 10:e0122369. doi: 10.1371/journal.pone.0122369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McArthur AG, Waglechner N, Nizam F, Yan A, Azad MA, Baylay AJ, Bhullar K, Canova MJ, De Pascale G, Ejim L, Kalan L, King AM, Koteva K, Morar M, Mulvey MR, O’Brien JS, Pawlowski AC, Piddock LJ, Spanogiannopoulos P, Sutherland AD, Tang I, Taylor PL, Thaker M, Wang W, Yan M, Yu T, Wright GD. 2013. The comprehensive antibiotic resistance database. Antimicrob Agents Chemother 57:3348–3357. doi: 10.1128/AAC.00419-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Alikhan NF, Petty NK, Ben Zakour NL, Beatson SA. 2011. BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 12:402. doi: 10.1186/1471-2164-12-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Totsika M, Beatson SA, Sarkar S, Phan MD, Petty NK, Bachmann N, Szubert M, Sidjabat HE, Paterson DL, Upton M, Schembri MA. 2011. Insights into a multidrug resistant Escherichia coli pathogen of the globally disseminated ST131 lineage: genome analysis and virulence mechanisms. PLoS One 6:e26578. doi: 10.1371/journal.pone.0026578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Phan MD, Peters KM, Sarkar S, Lukowski SW, Allsopp LP, Gomes Moriel D, Achard ME, Totsika M, Marshall VM, Upton M, Beatson SA, Schembri MA. 2013. The serum resistome of a globally disseminated multidrug resistant uropathogenic Escherichia coli clone. PLoS Genet 9:e1003834. doi: 10.1371/journal.pgen.1003834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tatusov RL, Galperin MY, Natale DA, Koonin EV. 2000. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res 28:33–36. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lanza VF, de Toro M, Garcillán-Barcia MP, Mora A, Blanco J, Coque TM, de la Cruz F. 2014. Plasmid flux in Escherichia coli ST131 sublineages, analyzed by plasmid constellation network (PLACNET), a new method for plasmid reconstruction from whole genome sequences. PLoS Genet 10:e1004766. doi: 10.1371/journal.pgen.1004766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nicolas-Chanoine MH, Bertrand X, Madec JY. 2014. Escherichia coli ST131, an intriguing clonal group. Clin Microbiol Rev 27:543–574. doi: 10.1128/CMR.00125-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang S, Liu X, Xu X, Yang D, Wang D, Han X, Shi Y, Tian M, Ding C, Peng D, Yu S. 2016. Escherichia coli type III secretion system 2 ATPase EivC is involved in the motility and virulence of avian pathogenic Escherichia coli. Front Microbiol 7:1387. doi: 10.3389/fmicb.2016.01387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pallen MJ, Chaudhuri RR, Henderson IR. 2003. Genomic analysis of secretion systems. Curr Opin Microbiol 6:519–527. doi: 10.1016/j.mib.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 51.Forde BM, Phan MD, Gawthorne JA, Ashcroft MM, Stanton-Cook M, Sarkar S, Peters KM, Chan KG, Chong TM, Yin WF, Upton M, Schembri MA, Beatson SA. 2015. Lineage-specific methyltransferases define the methylome of the globally disseminated Escherichia coli ST131 clone. mBio 6:e01602-15. doi: 10.1128/mBio.01602-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Luo C, Hu GQ, Zhu H. 2009. Genome reannotation of Escherichia coli CFT073 with new insights into virulence. BMC Genomics 10:552. doi: 10.1186/1471-2164-10-552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hochhut B, Wilde C, Balling G, Middendorf B, Dobrindt U, Brzuszkiewicz E, Gottschalk G, Carniel E, Hacker J. 2006. Role of pathogenicity island-associated integrases in the genome plasticity of uropathogenic Escherichia coli strain 536. Mol Microbiol 61:584–595. doi: 10.1111/j.1365-2958.2006.05255.x. [DOI] [PubMed] [Google Scholar]
- 54.Blum G, Ott M, Lischewski A, Ritter A, Imrich H, Tschäpe H, Hacker J. 1994. Excision of large DNA regions termed pathogenicity islands from tRNA-specific loci in the chromosome of an Escherichia coli wild-type pathogen. Infect Immun 62:606–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bateman SL, Seed PC. 2012. Epigenetic regulation of the nitrosative stress response and intracellular macrophage survival by extraintestinal pathogenic Escherichia coli. Mol Microbiol 83:908–925. doi: 10.1111/j.1365-2958.2012.07977.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li L, Stoeckert CJ, Roos DS. 2003. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res 13:2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yamaguchi Y, Inouye M. 2011. Regulation of growth and death in Escherichia coli by toxin-antitoxin systems. Nat Rev Microbiol 9:779–790. doi: 10.1038/nrmicro2651. [DOI] [PubMed] [Google Scholar]
- 58.Povolotsky TL, Hengge R. 2015. Genome-based comparison of cyclic di-GMP signaling in pathogenic and commensal Escherichia coli strains. J Bacteriol 198:111–126. doi: 10.1128/JB.00520-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Maurelli AT, Fernández RE, Bloch CA, Rode CK, Fasano A. 1998. ‘Black holes’ and bacterial pathogenicity: a large genomic deletion that enhances the virulence of Shigella spp. and enteroinvasive Escherichia coli. Proc Natl Acad Sci U S A 95:3943–3948. doi: 10.1073/pnas.95.7.3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Patel RK, Jain M. 2012. NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PLoS One 7:e30619. doi: 10.1371/journal.pone.0030619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shaik S, Kumar N, Lankapalli AK, Tiwari SK, Baddam R, Ahmed N. 2016. Contig-layout-authenticator (CLA): a combinatorial approach to ordering and scaffolding of bacterial contigs for comparative genomics and molecular epidemiology. PLoS One 11:e0155459. doi: 10.1371/journal.pone.0155459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M, Meyer F, Olsen GJ, Olson R, Osterman AL, Overbeek RA, McNeil LK, Paarmann D, Paczian T, Parrello B, Pusch GD, Reich C, Stevens R, Vassieva O, Vonstein V, Wilke A, Zagnitko O. 2008. The RAST Server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream MA, Barrell B. 2000. Artemis: sequence visualization and annotation. Bioinformatics 16:944–945. doi: 10.1093/bioinformatics/16.10.944. [DOI] [PubMed] [Google Scholar]
- 65.Treangen TJ, Ondov BD, Koren S, Phillippy AM. 2014. The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol 15:524. doi: 10.1186/PREACCEPT-2573980311437212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Besemer J, Lomsadze A, Borodovsky M. 2001. GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res 29:2607–2618. doi: 10.1093/nar/29.12.2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chen L, Yang J, Yu J, Yao Z, Sun L, Shen Y, Jin Q. 2005. VFDB: a reference database for bacterial virulence factors. Nucleic Acids Res 33:D325–D328. doi: 10.1093/nar/gki008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Qumar S, Majid M, Kumar N, Tiwari SK, Semmler T, Devi S, Baddam R, Hussain A, Shaik S, Ahmed N. 2017. Genome dynamics and molecular infection epidemiology of multidrug-resistant Helicobacter pullorum isolates obtained from broiler and free-range chickens in India. Appl Environ Microbiol 83. doi: 10.1128/AEM.02305-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Marchler-Bauer A, Bryant SH. 2004. CD-Search: protein domain annotations on the fly. Nucleic Acids Res 32:W327–W331. doi: 10.1093/nar/gkh454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nandanwar N, Janssen T, Kühl M, Ahmed N, Ewers C, Wieler LH. 2014. Extraintestinal pathogenic Escherichia coli (ExPEC) of human and avian origin belonging to sequence type complex 95 (STC95) portray indistinguishable virulence features. Int J Med Microbiol 304:835–842. doi: 10.1016/j.ijmm.2014.06.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Background data and genome characteristics of the five in-house strains NA023, NA081, NA090, NA101, and NA112 belonging to four STs considered in this study. Download TABLE S1, PDF file, 0.2 MB (163.4KB, pdf) .
Copyright © 2017 Shaik et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Information about 139 strains belonging to ST38, ST405, ST648, and ST131 considered for the comparative genomic analysis in this study. Download TABLE S2, PDF file, 0.2 MB (235.6KB, pdf) .
Copyright © 2017 Shaik et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Whole-genome phylogenetic tree depicting individual STs as separate clades. Clades A, B, and C of ST131 were observed to be more distant than those of the remaining ST38, ST405, and ST648. Download FIG S1, TIF file, 2.7 MB (2.8MB, tif) .
Copyright © 2017 Shaik et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Plasmid classes observed in each of the strains belonging to ST38, ST405, ST648, and ST131. A positive hit with identity of ≥95% and query coverage of ≥90% indicates the presence of that particular class in the strain. Download TABLE S3, PDF file, 0.5 MB (554.3KB, pdf) .
Copyright © 2017 Shaik et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
(A) Heat map depicting the presence of CARD resistance genes in our data set containing 139 strains belonging to ST38, ST405, ST648, and ST131 (clades A, B, and C). Black indicates a positive hit with identity of ≥70% and query coverage of ≥75%. (B) Representation of in vitro analysis of five in-house strains tested for 18 different antimicrobials belonging to six different classes. Download FIG S2, TIF file, 1.4 MB (1.4MB, tif) .
Copyright © 2017 Shaik et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Information about the list of proteins that are specifically missing in all 99 strains of ST131 while present in the remaining 40 strains belonging to ST38, ST405, and ST648. Presence represents a positive BLASTp hit with identity of ≥70%. Download TABLE S4, PDF file, 0.3 MB (280.5KB, pdf) .
Copyright © 2017 Shaik et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.