

ABSTRACT
The number of infections caused by Gram-negative pathogens carrying carbapenemases is increasing, and the group of carbapenem-hydrolyzing class D β-lactamases (CHDLs) is especially problematic. Several clinically important CHDLs have been identified in Acinetobacter baumannii, including OXA-23, OXA-24/40, OXA-58, OXA-143, OXA-235, and the chromosomally encoded OXA-51. The selection and dissemination of carbapenem-resistant A. baumannii strains constitutes a serious global threat. Carbapenems have been successfully utilized as last-resort antibiotics for the treatment of multidrug-resistant A. baumannii infections. However, the spread of OXA carbapenemases is compromising the continued use of these antimicrobials. In response to this clinical issue, it is necessary and urgent to design and develop new specific inhibitors with efficacy against these enzymes. The aim of this work was to characterize the inhibitory activity of LN-1-255 (a 6-alkylidene-2-substituted penicillin sulfone) and compare it to that of two established inhibitors (avibactam and tazobactam) against the most relevant enzymes of each group of class D carbapenemases in A. baumannii. The β-lactamase inhibitor LN-1-255 demonstrated excellent microbiological synergy and inhibition kinetics parameters against all tested CHDLs and a significantly higher activity than tazobactam and avibactam. A combination of carbapenems and LN-1-255 was effective against A. baumannii class D carbapenemases. Docking assays confirmed the affinity of LN-1-255 for the active site of these enzymes. LN-1-255 represents a potential new β-lactamase inhibitor that may have a significant role in eradicating infections caused by A. baumannii isolates carrying CHDLs.
KEYWORDS: β-lactamase inhibitors, Acinetobacter baumannii, antimicrobial resistance, carbapenem-hydrolyzing class D β-lactamases
INTRODUCTION
The Gram-negative Acinetobacter baumannii may be the pathogen that has evolved the most drug resistance in recent decades. While A. baumannii was an uncommon infectious agent in the 1970s and 1980s, presently this human pathogen is associated with a broad spectrum of infectious diseases, including pneumonia and bloodstream infections (1, 2). Most clinical strains of A. baumannii are multidrug resistant (3, 4), which dramatically reduces the available treatment options.
In the available clinical arsenal of antimicrobials, the β-lactams comprise more than 60% of the world's antibiotics (5, 6). The dissemination and expression of β-lactamase enzymes is the most relevant mechanism of resistance in Gram-negative bacteria, including A. baumannii. β-Lactamases confer resistance to β-lactams via the cleavage of the β-lactam ring and the release of an inactive product, preventing the binding of drugs to the target penicillin-binding proteins.
Four molecular classes of β-lactamases are recognized based on their amino acid sequences. Three of them, classes A, C, and D, are active-site serine enzymes, and the other one, class B, contains zinc-dependent enzymes (7). Class D (also known as the OXA-type β-lactamases) is the fastest-growing class of β-lactamases, with more than 500 reported enzymes (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA313047). The OXA-like enzymes are divided into three categories based on their substrate specificities: narrow-spectrum enzymes, such as OXA-1 or OXA-10, extended-spectrum β-lactamases (ESBLs), such as OXA-13 or OXA-17, and carbapenem-hydrolyzing class D β-lactamases (named CHDLs), such as OXA-23 or OXA-24/40 (8). From the first description of OXA-23 (9), the number of CHDLs discovered worldwide has dramatically increased in problematic Gram-negative pathogens, such as Pseudomonas aeruginosa or Enterobacteriaceae, and mostly in A. baumannii (10).
Until 1 decade ago, the most clinically relevant β-lactamases were the ESBLs and the AmpC β-lactamases, which are unable to hydrolyze carbapenems. However, in recent years there has been an increase in the number of β-lactamase carbapenemases, of which the group of CHLDs is especially problematic. Several CHDLs have been identified in A. baumannii, including the mostly acquired OXA-23, OXA-24/40, OXA-58, OXA-143, and OXA-235 and the naturally occurring OXA-51 (11–16). These enzymes were the first CHDLs described in each of the six families of CHDLs in A. baumannii and constitute representative examples of the OXA-23-like, the OXA24/40-like, the OXA-58-like, the OXA-143-like, the OXA-235-like, and the OXA-51-like enzymes (16–18). These carbapenem-hydrolyzing class D β-lactamases are the main enzymes responsible for the majority of infections caused by carbapenem-resistant A. baumannii in clinics around the world (19, 20), with OXA-23 being the major source of carbapenem resistance in this pathogen (21). The OXA-51 enzyme is regarded as a weak carbapenemase; however, it can confer resistance to carbapenems when its gene acquires a strong promoter by upstream insertion of an insertion sequence (IS) or becomes located in a plasmid, resulting in increased expression rates. These OXA-51-related enzymes are intrinsic to A. baumannii and are naturally found on its chromosome (18, 22).
Clinical selection and dissemination of carbapenem-resistant A. baumannii strains constitutes a serious threat associated with severe adverse clinical outcomes, including increased mortality (23, 24). The prevalence and plasmid-mediated dissemination of carbapenemase genes is an important clinical challenge. The carbapenems have been successfully utilized as last-resort antibiotics for the treatment of multidrug-resistant Acinetobacter infections; however, the spread of OXA carbapenemases in the last decade is seriously compromising the use of these antimicrobials, which is a major obstacle in the preservation of carbapenem efficacy against A. baumannii. In the United States, the percentage of A. baumannii isolates resistant to imipenem has increased from an average of 10% between 1999 and 2005 to 48% in 2008 (25). In response to this clinical issue, two strategies may preserve the utility of β-lactams: (i) the design of new β-lactam antibiotics, which are able to evade the inactivation conferred by β-lactamases, and (ii) the design of specific inhibitors against β-lactamases (26).
In clinical therapy, β-lactamase inhibitors are routinely successfully used in combination with β-lactam antibiotics (i.e., amoxicillin-clavulanic acid, piperacillin-tazobactam, ampicillin-sulbactam, etc.). However, these inhibitors mainly have affinity for class A β-lactamases and are ineffective in infections caused by pathogens carrying class D carbapenemases (27). Avibactam, a new inhibitor of class A and C β-lactamases, is able to inhibit some OXA enzymes, but it has not been effective in any CHDL-producing A. baumannii strains tested so far (28). Thus, the development of efficient inhibitors of A. baumannii class D carbapenemases is urgently needed to preserve the efficacy of carbapenem antimicrobials (29).
Buynak et al. (30, 31) designed a group of 6-alkylidene-2-substituted penicillin sulfones that are able to inhibit β-lactamases, and the compound LN-1-255 demonstrated significant potency (Fig. 1). LN-1-255 is able to inhibit class A β-lactamases, such as SHV-1 and -2 (31), and class D non-carbapenem-hydrolyzing enzymes, such as OXA-10, -14, and -17 (32), and even has proven activity against the CHDLs OXA-24/40. In 2010, the hydrophobic bridge of OXA-24/40 (composed of Tyr-112 and Met-223) was shown to be important in conferring inhibition by LN-1-255. However, this bridge is not a universal feature of CHDLs; for example, OXA-48 does not possess it. Nevertheless, we have observed that LN-1-255 also confers significant inhibition against this carbapenemase in Enterobacteriaceae (33).
FIG 1.
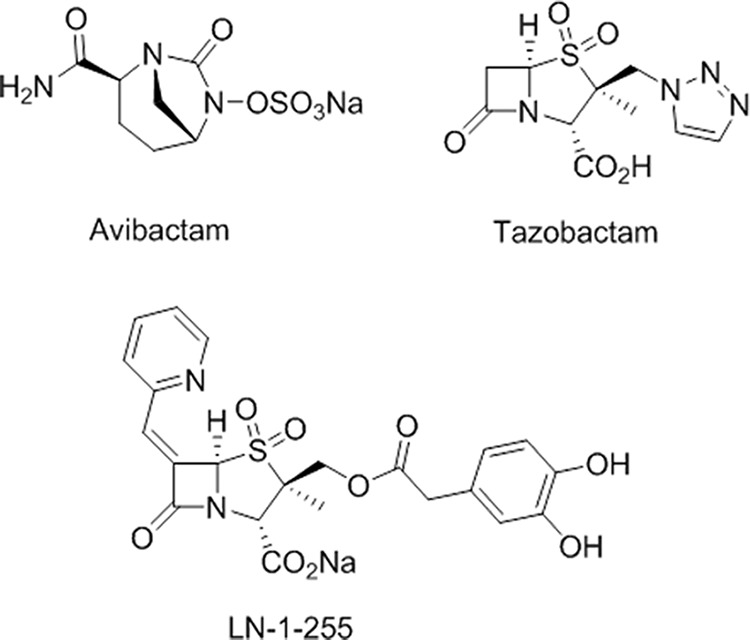
β-Lactamase inhibitors used in this study.
The aim of this work was to study the activity of inhibition of this compound in the major representative enzymes of all families of CHDLs described to date in A. baumannii. We compared the already demonstrated activity of this inhibitor against OXA-24/40 with its activity against the other OXA carbapenemases in A. baumannii. Microbiological, kinetic, and molecular docking studies were performed in order to demonstrate the synergy of LN-1-255 with carbapenems, as well as its inhibition activity against all of the CHDLs of A. baumannii.
RESULTS
Microbiological experiments.
MIC assays revealed differences between clinical isolates and the isogenic strains expressing each CHDL protein tested (Table 1). High resistance levels to carbapenems were observed in the ATCC 17978 strain containing β-lactamases, from basal MICs of 0.5 mg/liter to 4 to 128 mg/liter, except for the transformant carrying OXA-235, which had the lowest MIC (2 mg/liter). The highest MICs of carbapenems observed both in clinical isolates and ATCC 17978 transformants were in those carrying OXA-24/40 and OXA-143. The hyperexpression of OXA-51-like in the ATCC 17978 strain increased the MICs of carbapenems 8-fold.
TABLE 1.
β-Lactam MIC values with A. baumannii ATCC 17978 and clinical isolates producing CHDLs in the presence and absence of β-lactamase inhibitors
| A. baumannii strain | MICs (mg/liter) fora: |
||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IP | Inhibitors at 16 mg/liter |
Inhibitors at 4 mg/liter |
MP | Inhibitors at 16 mg/liter |
Inhibitors at 4 mg/liter |
AP | Inhibitors at 16 mg/liter |
Inhibitors at 4 mg/liter |
|||||||||||||
| IP + TZ | IP + AV | IP + LN | IP + TZ | IP + AV | IP + LN | MP + TZ | MP + AV | MP + LN | MP + TZ | MP + AV | MP + LN | AP + TZ | AP + AV | AP + LN | AP + TZ | AP + AV | AP + LN | ||||
| ATCC 17978 (OXA-23) | 16 | 8 | 8 | 0.5 | 16 | 8 | 2 | 8 | 8 | 4 | 0.5 | 8 | 8 | 1 | 8,192 | 8,192 | 512 | 32 | 8,192 | 2,048 | 128 |
| Clinical isolate (OXA-23) | 32 | 16 | 8 | 1 | 16 | 16 | 4 | 16 | 8 | 8 | 1 | 16 | 16 | 4 | 16,384 | 16,384 | 1,024 | 128 | 16,384 | 4,096 | 1,024 |
| ATCC 17978 (OXA-24) | 64 | 64 | 32 | 0.5 | 64 | 32 | 2 | 64 | 64 | 16 | 0.25 | 64 | 64 | 2 | 16,384 | 8,192 | 1,024 | 32 | 8,192 | 4,096 | 128 |
| Clinical isolate (OXA-24) | 256 | 256 | 128 | 8 | 256 | 256 | 128 | 256 | 256 | 128 | 8 | 256 | 256 | 128 | >16,384 | >16,384 | 8,192 | 2,048 | >16,384 | >16,384 | 16,384 |
| ATCC 17978 (OXA-58) | 32 | 16 | 4 | 0.5 | 16 | 16 | 2 | 8 | 4 | 2 | 0.5 | 4 | 4 | 1 | 8,192 | 8,192 | 1,024 | 32 | 8,192 | 4,096 | 256 |
| Clinical isolate (OXA-58) | 8 | 8 | 4 | 1 | 8 | 4 | 2 | 4 | 4 | 1 | 0.5 | 4 | 4 | 1 | 8,192 | 4,096 | 1,024 | 128 | 8,192 | 4,096 | 256 |
| ATCC 17978 (OXA-51 like) | 4 | 4 | 2 | 1 | 4 | 4 | 2 | 4 | 2 | 2 | 0.5 | 4 | 2 | 1 | 8,192 | 4,096 | 1,024 | 32 | 8,192 | 4,096 | 256 |
| ATCC 17978 (OXA-143) | 64 | 32 | 8 | 1 | 64 | 32 | 4 | 128 | 128 | 16 | 1 | 128 | 64 | 16 | 16,384 | 8,192 | 512 | 32 | 16,384 | 2,048 | 256 |
| Clinical isolate (OXA-143) | 32 | 16 | 4 | 2 | 32 | 16 | 8 | 128 | 128 | 32 | 4 | 128 | 64 | 32 | 16,384 | 16,384 | 1,024 | 256 | 16,384 | 4,096 | 512 |
| ATCC 17978 (OXA-235) | 2 | 2 | 2 | 0.5 | 2 | 2 | 1 | 2 | 1 | 1 | 0.5 | 1 | 1 | 0.5 | 2,048 | 512 | 128 | 32 | 2,048 | 256 | 64 |
| Clinical isolate (OXA-235) | 8 | 4 | 2 | 1 | 8 | 2 | 2 | 4 | 2 | 0.5 | 0.5 | 2 | 2 | 1 | 16,384 | 8,192 | 128 | 128 | 16,384 | 1,024 | 1,024 |
| ATCC 17978 pET-RA | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 64 | 32 | 32 | 32 | 64 | 32 | 32 |
IP, imipenem; MP, meropenem; AM, ampicillin; TZ, tazobactam; AV, avibactam; LN, LN-1-255. Data represent the means from three independent experiments.
To determine the synergy of LN-1-255 with the CHDLs of A. baumannii, in vitro susceptibility to imipenem–LN-1-255, meropenem–LN-1-255, and ampicillin–LN-1-255 were compared in combination with the β-lactamase inhibitors tazobactam and avibactam. The most significant inhibitory results were obtained when inhibitors were used at a concentration of 16 mg/liter. Tazobactam did not significantly decrease the MIC of carbapenems in any ATCC 17978 transformant expressing an OXA enzyme (0- to 1-fold). Avibactam decreased the MIC of both carbapenems of OXA-143 by 8-fold, OXA-58 decreased the MIC of imipenem and meropenem by 8- and 4-fold, respectively, and OXA-24/40 decreased the MIC of meropenem by 4-fold. In the other combinations tested, avibactam did not decrease the MIC against carbapenems. In contrast, LN-1-255 at the same concentration significantly decreased the MICs of carbapenems, with LN-1-255-cotreated transformants displaying the basal level of susceptibility to carbapenems (Table 1, Fig. 2). ATCC 17978 transformants expressing OXA-51-like and OXA-235 had low MICs of carbapenems. The MIC of OXA-51-like to both imipenem and meropenem was 4 mg/liter, while the MIC of OXA-235 was 2 mg/liter. Therefore, the MIC of ampicillin was also tested to better observe the inhibition of the MICs. The MIC of OXA-51-like against ampicillin was 8,192 mg/liter, which decreased to 4,096 mg/liter, 1,024 mg/liter, and 32 mg/liter in the presence of tazobactam, avibactam, and LN-1-255, respectively. Similarly, the MIC of ampicillin in OXA-235 was 2,048 mg/liter, which decreased to 512 mg/liter, 128 mg/liter, and 32 mg/liter in the presence of tazobactam, avibactam, and LN-1-255, respectively. In these two weak carbapenemases, we observed a significantly better inhibitory activity with LN-1-255 than with the other two inhibitors. When inhibitors were used at 4 mg/liter, LN-1-255 also had better inhibitory activity than avibactam and tazobactam; however, the decrease in MIC was less than that observed at 16 mg/liter.
FIG 2.

Assessment of synergy between inhibitors. Pictures represent checkerboard assays using imipenem and inhibitors with cultures of A. baumannii ATCC 17978 and clinical isolate Ab1 (both carrying OXA-23). Pink wells indicate growth and blue wells indicate growth inhibition.
Similar results were obtained with the A. baumannii clinical isolates tested, and although LN-1-255 was able to increase the efficacy of β-lactams, the MICs were slightly higher than those observed in the ATCC 17978 transformants, mainly in the clinical isolate carrying OXA-24/40. It is likely that other minor resistance mechanisms are present in these clinical isolates. Nevertheless, following the EUCAST criteria, in the presence of LN-1-255 all clinical isolates and transformants were susceptible to both carbapenems (≤2 mg/liter), except the clinical isolate carrying OXA-24/40 (intermediate susceptibility to imipenem and meropenem) and the clinical isolate carrying OXA-143 (intermediate susceptibility to meropenem) (34). The remaining data are shown in Table 1.
The antimicrobial activity of the three inhibitors was also tested, and tazobactam was the only one with intrinsic antimicrobial activity (MICs of 32 mg/liter for ATCC 17978 carrying the empty vector pET-RA and 32 to 128 mg/liter for the ATCC 17978 transformants carrying the OXA enzymes). Conversely, LN-1-255 and avibactam did not show any intrinsic antimicrobial activity, with MICs of ≥1,024 mg/liter.
Checkerboard assays were also used to determine the activity of LN-1-255 in both OXA-23 strains tested, the ATCC 17978 transformant and the clinical isolate. The MIC of imipenem alone in the ATCC 17978 transformant carrying OXA-23 was 16 mg/liter, while the addition of LN-1-255 at 8 mg/liter reduced the MIC of imipenem to 0.5 mg/liter, thus totally inhibiting the carbapenemase; however, the MIC of imipenem decreased to 8 mg/liter with avibactam, and no decrease in MIC was detected in the presence of tazobactam. Similarly, in the clinical isolate carrying OXA-23, the MIC of imipenem alone was 32 mg/liter, which decreased to 2, 8, and 8 mg/liter in the presence of LN-1-255, avibactam, and tazobactam (8 mg/liter), respectively (Fig. 2 reflects a representative assay performed with OXA-23). The checkerboard assays performed with the remaining A. baumannii clinical isolates and transformants included in this study were also in agreement with the MIC assays (Table 1 and data not shown).
Kinetic parameters of CHDL enzymes.
Class D carbapenemases were purified, and their steady-state kinetic parameters were measured. All purified enzymes could hydrolyze nitrocefin at a high rate, which has been previously reported (16, 35–37) (Table 2). Nitrocefin hydrolysis rates were used as a reference ([S] > Km, where [S] is the concentration of the substrate) to perform the inhibition kinetics assays, with nitrocefin as a reporter substrate (Table 3).
TABLE 2.
Nitrocefin hydrolysis kinetics of CHDLsb
| CDHL | kcat (s−1) | Km (μM) | kcat/Km (μM−1 s−1) | [S]a (μM) |
|---|---|---|---|---|
| OXA-23 | 177 ± 52 | 170 ± 45 | 1.04 | 350 |
| OXA-24/40 | 27.0 ± 15.1 | 29.1 ± 7.7 | 0.93 | 100 |
| OXA-58 | 50.9 ± 25.7 | 30.2 ± 5.6 | 1.68 | 100 |
| OXA-143 | 20.3 ± 4.9 | 41.4 ± 15.4 | 0.49 | 150 |
| OXA-235 | 130 ± 73 | 59.9 ± 11.6 | 2.17 | 150 |
| OXA-51 | 123 ± 8 | 248 ± 44 | 0.50 | 350 |
Concentration of nitrocefin, which was used as a reporter substrate.
Data represent the means from three independent experiments.
TABLE 3.
Inhibition kinetics of A. baumannii CHDLa
| β-Lactamase and substrate | IC50 (μM) | Ki app (μM) | k2/K (M−1 s−1) | KI (μM) | kinact (s−1) | kinact/KI (M−1 s−1) | tn |
|---|---|---|---|---|---|---|---|
| OXA-23 | |||||||
| Avibactam | 8.93 ± 0.99 | 105.56 ± 3.08 | 101.37 ± 15.96 | 203.93 ± 22.6 | 0.057 ± 0.009 | 286.60 ± 61.78 | 150 |
| Tazobactam | 2.13 ± 0.80 | 11.39 ± 2.49 | 193.53 ± 39.10 | 19.73 ± 4.71 | 0.023 ± 0.004 | 1180 ± 77.48 | 51 |
| LN-1-255 | 0.012 ± 0.008 | 0.088 ± 0.006 | (2.90 ± 0.03) × 104 | 0.30 ± 0.08 | 0.041 ± 0.005 | (1.39 ± 0.29) × 105 | 2 |
| OXA-24 | |||||||
| Avibactam | 22.32 ± 2.79 | 154.25 ± 19.93 | 22.12 ± 5.53 | 113.93 ± 35.93 | 0.018 ± 0.007 | 159.92 ± 40.63 | 202 |
| Tazobactam | 27.91 ± 2.55 | 78.07 ± 30.05 | 28.65 ± 8.19 | 12.38 ± 3.14 | 0.007 ± 0.002 | 724.48 ± 364.13 | 4,180 |
| LN-1-255 | 0.015 ± 0.004 | 0.289 ± 0.054 | (2.05 ± 0.74) × 104 | 0.24 ± 0.05 | 0.022 ± 0.002 | (9.82 ± 3.21) × 104 | 6 |
| OXA-51 | |||||||
| Avibactam | 34.9 ± 17.7 | >500 | 2.77 ± 1.20 | 430.94 ± 122.43 | 0.004 ± 0.0008 | 9.28 ± 0.93 | 9,600 |
| Tazobactam | >5000 | >3000 | NDb | >5000 | ND | ND | >20,000 |
| LN-1-255 | 1.70 ± 0.37 | 56.80 ± 27.76 | 17.67 ± 4.41 | 29.82 ± 7.81 | 0.0022 ± 0.0005 | 74.9 ± 3.8 | 520 |
| OXA-58 | |||||||
| Avibactam | 6.06 ± 2.56 | 71.04 ± 10.95 | 119.40 ± 39.80 | 62.5 ± 21.43 | 0.022 ± 0.006 | 388.99 ± 162.73 | 53 |
| Tazobactam | 22.62 ± 3.77 | 171.61 ± 15.22 | 16.00 ± 4.00 | 88.75 ± 25.53 | 0.018 ± 0.005 | 206.47 ± 51.12 | 314 |
| LN-1-255 | 0.041 ± 0.009 | 0.350 ± 0.052 | (8.76 ± 1.44) × 103 | 0.30 ± 0.09 | 0.022 ± 0.008 | (7.52 ± 2.1) × 104 | 4 |
| OXA-143 | |||||||
| Avibactam | 11.94 ± 2.43 | 50.44 ± 11.25 | 194.92 ± 77.97 | 107.33 ± 40.15 | 0.072 ± 0.028 | 681.13 ± 8.13 | 110 |
| Tazobactam | 5.90 ± 1.86 | 21.36 ± 0.68 | 181.93 ± 81.15 | 20.69 ± 10.14 | 0.017 ± 0.006 | 955.37 ± 385.40 | 237 |
| LN-1-255 | 0.014 ± 0.003 | 0.166 ± 0.016 | (3.98 ± 0.47) × 104 | 0.09 ± 0.03 | 0.018 ± 0.003 | (1.21 ± 0.83) × 105 | 3 |
| OXA-235 | |||||||
| Avibactam | 1.44 ± 0.34 | 9.33 ± 1.41 | 290.32 ± 17.34 | 5.73 ± 1.01 | 0.016 ± 0.006 | 2,808.64 ± 585.87 | 17 |
| Tazobactam | 4.88 ± 1.32 | 60.23 ± 10.28 | 7.77 ± 1.92 | 266.22 ± 50.05 | 0.050 ± 0.017 | 197.45 ± 83.02 | 306 |
| LN-1-255 | 0.008 ± 0.003 | 0.054 ± 0.010 | (1.76 ± 0.80) × 104 | 0.044 ± 0.025 | 0.024 ± 0.007 | (7.58 ± 5.17) × 105 | 3 |
Data represent the means from three independent experiments.
ND, not determined, due to very low inhibition.
All enzymes, except OXA-51-like enzyme, demonstrated a Ki for LN-1-255 in the nanomolar range (54 to 289 nM). Avibactam and tazobactam were different, with a Ki of 9 to 154 μM and 11 to 172 μM, respectively, approximately 3 logs higher than the affinity of LN-1-255. The OXA-51-like enzyme had a low affinity for inhibitors, with a Ki of 56 μM, >500 μM, and >3,000 μM for LN-1-255, avibactam, and tazobactam, respectively. The OXA enzymes with the greatest affinity for LN-1-255 were OXA-23 and OXA-235, which had a Ki of 88 and 54 nM, respectively.
Similarly, the 50% inhibitory concentration (IC50) of LN-1-255 was in the nanomolar range (8 to 40 nM) while it was in the micromolar range for avibactam (1 to 35 μM) and tazobactam (2 to 28 μM) for all tested enzymes except the OXA-51-like enzyme. The OXA-51-like enzyme had an IC50 of 1.7 μM, 35 μM, and >5,000 μM for LN-1-255, avibactam, and tazobactam, respectively. The best IC50s for LN-1-255 were the same as those for OXA-23 (12 nM) and OXA-235 (8 nM).
Inactivation efficiencies (kinact/KI) were determined with the three compounds. While kinact values were similar between LN-1255, tazobactam, and avibactam, large differences were observed in the affinity (KI) of the three inhibitors, with the affinity of LN-1-255 being 65- to 6,045-fold higher than that of tazobactam and 14- to 1,126-fold higher than that of avibactam. As a consequence, the inactivation efficiencies were also higher for LN-1-255 than for tazobactam (117- to 3,847-fold) and avibactam (8- to 614-fold). Similarly, apparent onset of acylation (k2/K) values were higher for LN-1-255 than for avibactam and tazobactam, being 73- to 927-fold and 150- to 3,194-fold higher, respectively.
The turnover number is the time-dependent partitioning of the initial enzyme/inhibitor complex between hydrolysis and enzyme inactivation and is represented by the equation tn = kcat/kinact. In the case of LN-1-255, the tn was very similar for all CHDLs tested, between 2 and 6, except for OXA-51-like enzyme, which showed a tn number of 520. The turnover numbers for avibactam and tazobactam were significantly higher, with 17 to 202 for avibactam and 51 to 4,180 for tazobactam, and even higher for OXA-51-like enzyme, 9,600 and 20,000, respectively (Fig. 3). The tn of OXA-24/40 is remarkably high for tazobactam, 13- to 84-fold higher than that of the other CHDLs. This result was in agreement with the MICs observed for OXA-24/40 in the presence of tazobactam, which was inactivated at the lowest concentration out of all the carbapenemases tested.
FIG 3.

Plots of turnover numbers (tn). Ratio of inhibitor concentration to enzyme concentration necessary to decrease the enzyme activity by 90%.
Computational studies of activity of LN-1-255 against CHDLs enzymes.
Reasoning that the differences in inhibitory activity observed between the β-lactamase inhibitors evaluated in this study against the CHDLs enzymes are due to the binding mode of these ligands in the serine active site of the enzymes, molecular docking studies were performed. These studies were carried out using the program GOLD version 5.2 (http://www.ccdc.cam.ac.uk/solutions/csd-discovery/components/gold/) and the enzyme geometries of OXA-23 (2.14 Å; PDB code 4JF4) (38) and OXA-58 (2.6 Å; PDB code 4Y0U) (39), which are covalently modified by meropenem and 6α-hydroxymethylpenicillanate, respectively. In addition, atomic structure determinations of OXA-143 and OXA-235 are not available, so their corresponding homology models were constructed by using the SWISS-MODEL homology modeling web server (40). The resulting OXA-143 and OXA-235 structures had 88% and 61% sequence identity with the templates of OXA-24/40 (PDB code 3PAG) and OXA-51 (PDB code 4ZDX), respectively.
Comparison of the available crystal structures of OXA-23 (2.14 Å; PDB code 4JF4) (38), OXA-24/40 (2.14 Å; PDB code 3G4P) (35), OXA-51 (1.61 Å; PDB code 5KZH) (41), and OXA-58 (1.8 Å; PDB code 5BOH) (42) enzymes and the constructed three-dimensional structures of OXA-143 and OXA-235 reveals (i) similarities in their overall structure and (ii) a highly conserved active site and cleft located between the two domains that are involved in hydrolysis (Fig. 4). It has been proposed that the ability of these enzymes to hydrolyze carbapenems is provided by a tunnel-like entrance to the active site formed by the side chains of a tyrosine or phenylalanine and a methionine, with the exception of OXA-51, in which the latter is replaced by tryptophan (43). The presence of a Trp residue makes the active site of OXA-51 particularly small and inaccessible (see Fig. S1 in the supplemental material), which might explain its reduced carbapenemase activity. The Tyr/Phe and Met residues are crucial for the architecture of the tunnel-like active site that permits the specific accommodation of carbapenems. In general, this tunnel-like structure of CHDL enzymes forms a hydrophobic barrier that controls the access to the active site by only certain substrates and remains mainly unchanged after ligand binding (44). Despite the similarities found in all of the CHDL enzymes, important differences in (i) the arrangement and sequence of the Ω-loop, which is located near the tunnel entrance, and (ii) the strength of the intramolecular interactions that constitute the tunnel-like structure were identified (Fig. 4A and 5B). The differences in the Ω-loop might cause significant variations in the plasticity of these loops and therefore in the efficacy of the ligands. In addition, for OXA-24/40 and OXA-143 enzymes and with the exception of OXA-51-like, the presence of a Tyr residue in the tunnel-like structure provides a more closed active site than OXA-23, OXA-58, and OXA-235 (Fig. 4C and 5D), which might also be relevant for activity.
FIG 4.

(A) Comparison of the OXA-type carbapenemases employed in this study, OXA-23 (gray), OXA-24 (yellow), OXA-51 (green), OXA-58 (pink), OXA-143 (blue), and OXA-235 (cyan), highlighting the side chain residues responsible of the major structural differences. The side chain of the catalytic serine is also shown. (B) Detailed view of the Ω-loop and neighbor turn involving β1-strand and α1-helix. Note the relevant differences in the arrangement and sequence of the Ω-loop among the enzymes studied. (C and D) Selected views of the tunnel-like entrance to the active site of OXA-24 (C) and OXA-23 (D) enzymes. The position of the Tyr/Phe and Met residues that are involved in this entrance are highlighted in green and red, respectively. The catalytic serine is shown in orange. Note how the active site of OXA-24 containing a Try residue is more closed than the OXA-23 one having a Phe residue.
FIG 5.

Selected view of inactivation of OXA-23 (A and B) and OXA-58 (C and D) from A. baumannii by LN-1-255 (A and C) and avibactam (B and D) obtained by molecular docking studies. Hydrogen bonding and electrostatic interactions between the ligands and the enzymes are shown as dashed blue lines. Relevant side chain residues are shown and labeled.
Docking studies carried out in the active site of the relevant carbapenemase, OXA-23 with LN-1-255, showed that the indolizine adduct obtained after covalent modification of the catalytic Ser-79 by LN-1-255 would establish strong polar interactions with residues Arg-259, Lys-216, Ser-126, and Thr-217 (Fig. S2). Specifically, the sulfinate group would be anchored to the active site via an electrostatic interaction with the guanidinium group of Arg-259 as well as hydrogen bonding with Thr-217. In addition, the NH group would establish hydrogen bonding with the side chain of Ser-126. The indolizine moiety would be embedded in the apolar pocket near the tunnel-like entrance involving Val-128 and Leu-166. Remarkably, the large pocket close to the active site would be partially occupied by the catechol moiety. The catechol moiety would be quite flexible but would interact by hydrogen bonding with the ε-amino group of Lys-216. The avibactam adduct would also establish strong polar interactions with residues Arg-259, Lys-216, Ser-126, and Thr-217 (Fig. 5B). The contacts between the catechol moiety of LN-1-255 with the large pocket close to the active site would be absent. Moreover, the predicted binding mode of the studied ligands, either Michaelis complexes or ligand adducts, in the active site of OXA-58 (Fig. 5C and D), OXA-143, and OXA-235 proved to be rather similar to that observed for OXA-23.
DISCUSSION
In the present study, we have described the inhibitory activity of a member of the family of C2/C3 substitute penicillin sulfones and compared it to the inhibitory activities of tazobactam and a new β-lactamase inhibitor, avibactam, against the most clinically relevant class D carbapenemases of A. baumannii. The CHDLs studied were selected because they were the first isolated and constitute the main examples of each family of CHDLs in A. baumannii (18).
These β-lactamase inhibitors must penetrate the Gram-negative membrane to effectively decrease the MICs of β-lactam antimicrobials. The molecule LN-1-255, which was previously described, coopts the microbial iron uptake system to traverse the outer membrane, increasing the penetration rate (30, 31). LN-1-255 has previously proven activity against OXA-1, OXA-10, and the CHDLs OXA-24/40 of A. baumannii and OXA-48 of Klebsiella pneumoniae (26, 33, 35). In the present study, we have demonstrated the activity of LN-1-255 as a paninhibitor of all A. baumannii CHDLs. Our microbiological data confirmed that LN-1-255 can penetrate the membrane at a high enough concentration to be effective and thus was able to inhibit the main A. baumannii CDHLs. The kinetic data presented here are in agreement with the microbiological data and, overall, indicated that this compound inhibits the OXA carbapenemases with Ki values that were significantly lower than those of avibactam and tazobactam. Susceptibility testing showed that a combination treatment with imipenem–LN-1-255 or meropenem–LN-1-255 significantly lowered the MICs against carbapenem-resistant A. baumannii strains carrying CHLDs. The new compound did not show intrinsic antibiotic activity against any of the clinical isolates and ATCC strains tested. However, a dose-dependent effect was observed for this compound, as the observed MICs decreased more at a concentration of 16 mg/liter than 4 mg/liter. Checkerboard assays indicated that a clinical breakpoint of 2 mg/liter (susceptible) was reached at a concentration of 8 mg/liter of LN-1-255, which is a reasonable concentration to reach in plasma during treatment with other β-lactamase inhibitors, such as avibactam or sulbactam (45, 46). The microbiological and kinetics results described in the present study are in good agreement with previous inhibition studies performed with these three inhibitors against class D β-lactamases OXA-24/40 (28, 32, 35) and with avibactam against OXA-23 (47).
Class D β-lactamases have a broader profile of substrates in A. baumannii than other classes of β-lactamases. Some OXAs have limited substrate affinity and only hydrolyze penicillins and first-generation cephalosporins (e.g., OXA-1), while others have extended their specificity to extended-spectrum cephalosporins (e.g., OXA-144, which is an OXA-2-like enzyme) and carbapenems such as the OXAs included in this study and OXA-48, which are the most clinically worrisome. The potent activity of LN-1-255 against all tested OXAs β-lactamases is evident (32, 33, 35), and combinations of antimicrobials such as ceftazidime or imipenem with this inhibitor could be good options for treatment of CHDL-carrying A. baumannii.
Molecular docking studies were performed to ascertain if the binding mode of these ligands in the serine active site of the enzymes could account for the observed differences in inhibitory activity observed between the β-lactamase inhibitors evaluated in this study against CHDL enzymes. Comparison of the predicted binding mode of the OXA–LN-1-255 and OXA–avibactam adducts obtained by molecular docking revealed that both adducts would have similar polar (electrostatic and hydrogen-bonding) interactions involving the groups that most contribute to anchoring the ligands in the active site, the sulfate and sulfinate/carboxylate groups, with mostly the same active-site residues, specifically, Arg-259, Thr-217, and Ser-126 (numbering for OXA-23). However, significant differences were identified in the interactions of the indolizine and catechol moieties with two nearby pockets, specifically (i) the tunnel-like entrance and (ii) the catalytic serine (Fig. 5). Those interactions involving residues Val-128, Leu-166, and Lys-216 (numbering for OXA-23) would be absent from the OXA-avibactam adducts, which might account for the higher efficiency of LN-1-255.
The search for new inhibitors against β-lactamases is a key step in the fight against bacterial resistance; however, while compounds that inhibit different classes of β-lactamases are being developed, few of them exhibit activity against the class D enzymes. Avibactam is a non-β-lactam β-lactamase inhibitor. It has good inhibitory properties against clinically relevant enzymes, such as class A extended-spectrum β-lactamases (ESBL) and some carbapenemases, as well as class C β-lactamases (47). It can also inhibit certain class D β-lactamases; however, while it is able to inhibit OXA-48 enzymes, it does not have good inhibition kinetics against OXA-24/40 (28). In the present study, avibactam did not show any inhibitory activity against OXA-24/40 or any of the other CHDLs tested, and the average measured k2/K for all enzymes was approximately 170-fold lower for avibactam than that for LN-1-255. OXA-235 was strongly inhibited by avibactam, which is in agreement with the microbiological data, where avibactam mainly decreased the MICs to ampicillin.
Few other molecules are able to inhibit the class D β-lactamases. Methylene penems such as BRL 42715 and other new penem inhibitors have Kis in the nanomolar range against OXA-1; however, against the carbapenemase OXA-24/40 they acted like substrates, with no inhibition of this enzyme under mild conditions (26, 48–50). Methylene penems, such as the penicillin sulfones (including LN-1-255), are derived from β-lactams, and some non-β-lactam derivatives (such as avibactam) are also being developed against class D β-lactamases, such as the phosphonates and boronic acids (10). The phosphonates are also inhibitors with activity against OXA β-lactamases and have affinity in the nanomolar range against OXA-1 and OXA-10 (26, 51, 52). When they were tested against OXA-24/40, the thiophenyl oxime-derived phosphonates had Ki values in the micromolar range (53). Finally, a boronic acid (4,7-dichloro-1-benzothien-2-yl-sufonyl-aminomethyl boronic acid) also had an IC50 against OXA-24/40 in the micromolar range and thus is the first boronic acid-based class D β-lactamase inhibitor (54). Polycarboxylic acids could be a good scaffold for novel lipophilic inhibitors. Designed through fragment-based lead discovery, these polycarboxylic acids showed binding affinity against OXA-10 in the micromolar range. They are also promising candidates in the search for a new group of class D β-lactamase inhibitors (55–57).
Inhibition studies with the compound LN-1-255 also have been performed with other classes of β-lactamases. Activity was observed against class A β-lactamases (TEM-1, SHV-1, IMI-1, and others) and class C β-lactamases (P99, MIR-1, and others) (30, 31, 58), thus showing a potent and broad-spectrum activity. However, no studies have been published with 6-alkylidene-2-substituted penicillin sulfones against class B β-lactamases. The profound differences in enzyme mechanisms and active-site geometry between serine- and metallo-β-lactamases greatly hamper the design of common inhibitors for the four Ambler classes of β-lactamases.
In summary, in the present study we describe the inhibitory activity of the compound LN-1-255 against the class D carbapenemases of A. baumannii. Carbapenems in combination with LN-1-255 were effective against these carbapenem-resistant A. baumannii strains carrying β-lactamases, and biochemical assays demonstrated that LN-1-255 has better inhibitory efficiency than tazobactam or avibactam. Therefore, LN-1-255 represents a potential new therapeutic option in combination with carbapenems against resistant A. baumannii strains. Additional preclinical studies in infectious animal models are necessary to further develop this compound for clinical applications.
MATERIALS AND METHODS
Bacterial strains and media.
The recipient strain for OXA enzymes was an antibiotic-susceptible A. baumannii ATCC 17978 type strain, and blaOXA genes were isolated from previously studied clinical strains of A. baumannii (12, 15, 16, 59, 60). The OXA-51-like gene used in this study was OXA-95, which is the chromosomal blaOXA gene of the type strain ATCC 17978. Escherichia coli TG1 was used as the recipient strain for cloning experiments, and E. coli BL21was used for expression experiments. All strains used in this study are listed in Table 4.
TABLE 4.
Laboratory strains and clinical isolates used in this work
| Strain | blaOXA genea | Strain description | Reference or source |
|---|---|---|---|
| E. coli BL21 | NA | F− ompT hsdSB(rB− mB−) gal dcm | ATCC |
| E. coli TG1 | NA | supE hsdΔ5 thi Δ(lac-proAB) F′ [traD36 proAB+ lacIqΔM15] | ATCC |
| A. baumannii Ab1 | OXA-23 | A. baumannii clinical isolate, isolated from bronchial aspirate from Spain | 60 |
| A. baumannii RYC 52763/97 | OXA-24/40 | A. baumannii clinical isolate, isolated from bronchial aspirate from Spain, first description of OXA-24/40 | 12 |
| A. baumannii ATCC17978 | OXA-95 (OXA-51-like) | A. baumannii type strain, OXA-51 like variant OXA-95, completely sequenced reference strain | ATCC |
| A. baumannii HRS1 | OXA-58 | A. baumannii clinical isolate from Spain | 59 |
| A. baumannii 135040 | OXA-143 | A. baumannii clinical isolate, isolated from blood culture from Brazil, first description of OXA-143 | 15 |
| A. baumannii AF 401 | OXA-235 | A. baumannii clinical isolate, isolated from small intestine from Mexico, first description of OXA-235 | 16 |
NA, not available.
Bacterial strains were frozen in Luria-Bertani broth (LB) with 10% glycerol and were maintained at −80°C until analysis. Strains of E. coli and A. baumannii were grown at 37°C in LB medium. When necessary, LB medium was supplemented with ampicillin (20 mg/liter) or kanamycin (50 mg/liter) (Sigma-Genosys Ltd., United Kingdom). MICs and checkerboard assays were performed in Mueller-Hinton II broth (Becton, Dickinson and Company, Sparks, MD).
Susceptibility testing of antibiotics and inhibitors.
When the blaOXA genes were codified in a plasmid (blaOXA-23, blaOXA-58, and blaOXA-143), the plasmid DNA was purified using the GeneJET plasmid miniprep kit (Thermo Fisher Scientific, MA) according to the manufacturer's instructions. In the cases of blaOXA-24/40, blaOXA-51-like, and blaOXA-235, which are chromosomal genes, genomic DNA was obtained using the Wizard genomic DNA purification kit (Promega, Madison, WI). The blaOXA genes were amplified using PrimeSTAR HS DNA polymerase (TaKaRa, Berkeley, CA) and a primer pair containing recognition sites for the restriction enzyme XbaI (Fermentas, Glen Burnie, MD) (Table 5). The amplified DNA fragments were purified with a GeneJET PCR purification kit (Thermo Fisher Scientific, Waltham, MA, USA), digested with XbaI, and ligated into the pET-RA plasmid previously digested with XbaI (61) using T4 DNA ligase (Thermo Fisher Scientific, Waltham, MA, USA). The expression of the enzymes was under the control of the blaCTX-M-14 gene promoter previously cloned into the pET-RA plasmid, which also carried a kanamycin resistance gene. These plasmid constructs (pET-RA-OXA-23, pET-RA-OXA-24/40, pET-RA-OXA-51, pET-RA-OXA-58, pET-RA-OXA-143, and pET-RA-OXA-235) were used to transform E. coli TG1 for subcloning and then transformed into the ATCC 17978 A. baumannii wild-type strain by selection in LB plates containing 50 mg/liter of kanamycin.
TABLE 5.
Primers used in this work
| blaOXA gene | Primer sequencea |
|
|---|---|---|
| Amplification and sequencing of blaOXA genes | Amplification and purification of OXA enzymes | |
| OXA-23 | OXA-23fw, GCTCTAGAGCATGAATAAATATTTTACTTGCT | OXA-23fw, CGCGGATCCTTAATAAATGAAACCCCGAGTC |
| OXA-23rv, GCTCTAGAGCTCAGATTATAAAAGGCCCAT | OXA-23rv, CCGGAATTCTTAAATAATATTCAGCTGTTTTAATG | |
| OXA-24 | OXA-24fw, GCTCTAGAGCATGAAAAAATTTATACTTCC | OXA-24fw, AAGGATCCTCTATTAAAACTAAATCTGAAG |
| OXA-24rv, GCTCTAGAGCTTAAATGATTCCAAGATTTT | OXA-24rv, AAAGAATTCTTAAATGATTCCAAGATTTTC | |
| OXA-95 (OXA-51-like) | OXA-51fw, GCTCTAGAGCATGAACATTAAAGCACTCTT | OXA-51fw, CGCGGATCCAATCCAAATCACAGCGCTTCAAA |
| OXA-51rv, GCTCTAGAGCCTATAAAATACCTAATTGTT | OXA-51rv, CCGGAATTCCTATAAAATACCTAATTGTTCTAAA | |
| OXA-58 | OXA-58fw, GCTCTAGAGCATGAAATTATTAAAAATATT | OXA-58fw, CGCGGATCCGAGCATAGTATGAGTCGAGCA |
| OXA-58rv, GCTCTAGAGCTTATAAATAATGAAAAACAC | OXA-58rv, CCGGAATTCTTATAAATAATGAAAAACACCCAAC | |
| OXA-143 | OXA-143fw, GCTCTAGAGCATGAAAAAATTTATACTTCC | OXA-143fw, CGCGGATCCTGCTCATCTATTCAAACTAAATTT |
| OXA-143rv, GCTCTAGAGCTTATATAATCCCTAAATTCT | OXA143rv, TCCCCCGGGTTATATAATCCCTAAATTCTCTAAT | |
| OXA-235 | OXA-235fw, GCTCTAGAGCATGAAAACTCTTATTTTGTTGC | OXA-235fw, CCGGAATTCTTGCCTGTTTCAAATTCGTCC |
| OXA-235rv, GCTCTAGAGCTTACCCTTCAGCTTTCGGAT | OXA-235rv, CCGCTCGAGTTACCCTTCAGCTTTCGGATA | |
The underlined sequences represent sites for restriction enzymes.
MICs of ampicillin (Sigma-Genosys, Dorset, United Kingdom), imipenem (Actavis, Cork, Ireland), and meropenem (Accordpharma, Middlesex, United Kingdom) were determined by broth microdilution, in the presence and absence of inhibitors, according to CLSI criteria (62). The inhibitors used were tazobactam (Sigma-Genosys, Dorset, United Kingdom), avibactam (Astrazeneca, London, United Kingdom), and LN-1-255, each at 4 and 16 mg/liter (Fig. 1). MICs of inhibitors were also determined. LN-1-255 was prepared at the Center for Research in Biological Chemistry and Molecular Materials (CIQUS, University of Santiago of Compostela, Spain), as described previously (58). Turbidity was recorded in 96-well plates after 20 to 22 h of incubation at 37°C in Mueller-Hinton II broth. The MICs presented here are the means from three independent replicates.
The activity of inhibitors in combination with imipenem was also assessed in checkerboard assays (63). After 20 h of incubation, 10 μl of alamarBlue reagent (Thermo-Scientific, Waltham, MA, USA) was added to all wells and incubated for 4 h at 37°C to identify those wells containing viable bacteria.
Purification of OXA carbapenemases.
To purify the CHDL enzymes, the blaOXA genes were amplified and DNA fragments were cloned into the pGEX-6P-1 vector using BamHI and EcoRI sites, except in the cases of blaOXA-143 and blaOXA-235, where BamHI/XmaI and BamHI/XhoI restriction sites, respectively, were used. This genetic construction led to a fusion protein containing glutathione S-transferase (GST) and the target enzymes. The signal peptide of the CHDLs was not included in the fusion proteins. Cloning procedures were similar to those described above, using the primers described in Table 5. The genetic constructs were introduced into E. coli BL21 by electroporation and selection in LB plates containing 50 mg/liter of ampicillin. The recombinant β-lactamases were then purified to homogeneity using the GST gene fusion system (Amersham Pharmacia Biotech, Munich, Germany) according to the manufacturer's instructions. After SDS-PAGE, the purified proteins appeared as a band of approximately 29 kDa (≥95% purity). Spots with proteins were excised from gels, washed, digested with trypsin, and identified by matrix-assisted laser desorption ionization-tandem time of flight using the software ProteinPilot4.0.
Kinetic parameters.
All experiments were performed in triplicate at 25°C in 50 mM sodium phosphate (pH 7.0) with 20 mM sodium bicarbonate (64), in 1.0-cm-path-length cuvettes, using purified proteins under steady-state conditions on an Epoch 2 microplate spectrophotometer (Biotek, VT, USA) as previously described (33).
Measurements of hydrolysis were performed with nitrocefin (NCF) (Oxoid, Hampshire, United Kingdom) at a wavelength of 482 nm (extinction coefficient, 15,900 M−1 cm−1). The apparent kcat, Vmax, and Km were determined by measuring the initial reaction velocities at different nitrocefin concentrations. The data were fit to the Michaelis-Menten equation (nonlinear least-square fit) (equation 1) (65).
| (1) |
For inhibition kinetics, the IC50 was calculated as the inhibitor concentration resulting in 50% reduction of nitrocefin hydrolysis after 10 min of preincubation of the enzyme and inhibitor at 25°C, as previously described (35).
The apparent Ki was obtained as a competitive inhibition constant (Ki app) in the presence of nitrocefin as previously described (66). Increasing concentrations of each inhibitor were added to the enzyme and nitrocefin mixtures, and the initial velocities (vi) were measured. The nitrocefin concentrations used were 350 μM for OXA-23 and OXA-51, 150 μM for OXA-143 and OXA-235, and 100 μM for OXA-24/40 and OXA-58 (2- to 3-fold over the Km) (Table 2). Inverse initial velocities (1/v0) were plotted against inhibitor concentrations ([I]), and the data were fit to a linear equation where the y intercept divided by the slope of the line was defined as the Ki app observed. The initial velocity (v0) was determined by equation 2. Ki app observed then was corrected for nitrocefin affinity using equation 3.
| (2) |
| (3) |
The inhibitor complex inactivation rate (kinact) in the presence of nitrocefin was measured and the KI determined as previously described (65, 67). Nitrocefin was used as a reporter substrate with the same concentration described above and increasing concentrations of inhibitors over a 15-min time course (45 min for OXA-51-like enzyme). The kobs values were determined using nonlinear least-squares fit of the data, employing GraphPad software (La Jolla, CA, USA) and equation 4.
| (4) |
Here, A is absorbance, v0 is initial velocity, vf is the final velocity, and x is time. The kobs values were plotted against inhibitor concentrations to determine kinact and KI using equation 5. The KI value was corrected using equation 3.
| (5) |
The apparent onset of acylation (k2/K) was determined as the slope of the line obtained by plotting [I] against kobs (33). This value was corrected for nitrocefin affinity by following equation 6.
| (6) |
The turnover numbers (tn), defined as the inhibitor/enzyme ratio (I:E) necessary for >90% inhibition of nitrocefin hydrolysis, were determined after 20 h of incubation with increasing concentrations of inhibitors and 10 nM enzymes, with various molar I:E ratios (35). Incubations were performed in a final reaction volume of 200 μl, and nitrocefin was added immediately before measuring in order to determine the residual enzyme activity for 180 s.
Binding studies using molecular docking.
The binding mode of LN-1-255 and avibactam in the active site of the major representative enzymes of all families of CHDLs described to date in A. baumannii were analyzed by docking studies by using the available crystal structures. As no three-dimensional structures are available for OXA-143 and OXA-235, their corresponding homology models were constructed as indicated below.
Building of the wild-type OXA-143 and OXA-235 models.
The SWISS-Model homology modeling web server was used to model the three-dimensional structures of OXA-143 and OXA-235 (40). The coordinates of the crystallographically determined OXA-24/40 (2.25 Å; PDB code 3PAG) (44) and OXA-51 (2.0 Å; PDB code 4ZDX) (22) were chosen as the main templates for building the two models.
Docking studies.
The docking studies were carried out using GOLD 5.2 (http://www.ccdc.cam.ac.uk/solutions/csd-discovery/components/gold/), and the enzyme geometries of OXA-23 (2.14 Å; PDB code 4JF4) (38) and OXA-58 (2.6 Å; PDB code 4Y0U) (39) and the homology models of OXA-143 and OXA-235 were created. The crystal structures of PDB codes 4JF4 and 4Y0U were chosen because the position of the modified ligand is defined. Ligand geometries were minimized using AM1 Hamiltonian as implemented in the program Gaussian 09 (68) and used as MOL2 files. Each ligand was docked in 25 independent genetic algorithm (GA) runs, and for each of these a maximum number of 100,000 GA operations were performed on a single population of 50 individuals. Operator weights for crossover, mutation, and migration in the entry box were used as default parameters (95, 95, and 10, respectively), as well as the hydrogen bonding (4.0 Å) and van der Waals (2.5 Å) parameters. For OXA-23 and OXA-58, the position of the adducts present in the above-mentioned crystal structures were used to define the active site, whereas for the homology models, the position of the catalytic serine was employed. For all cases, the radius was set to 10 Å. The “flip ring corners” flag was switched on, while all other flags were off. The GOLD scoring function was used to rank the ligands in order of fitness. Figures depicting structures were prepared using PYMOL (69).
Supplementary Material
ACKNOWLEDGMENTS
This work was financially supported by Planes Nacionales de I+D+i 2008-2011/2013-2016 and Instituto de Salud Carlos III, Subdirección General de Redes y Centros de Investigación Cooperativa, Ministerio de Economía y Competitividad, Spanish Network for Research in Infectious Diseases (REIPI RD12/0015/0014 and REIPI RD16/0016/006), cofinanced by the European Development Regional Fund “A Way to Achieve Europe” and the operative program Intelligent Growth 2014-2020. This work was also supported by the Galician Innovation Agency (IN607A 2016/22) and the ISCIII-General Subdirection of Assessment and Promotion of the Research (PI15/00860 to G.B. and PI14/00059 to M.P. and A.B.). This study was also supported by the Spanish Ministry of Economy and Competiveness (SAF2016-75638-R), the Xunta de Galicia (Centro Singular de Investigación de Galicia Accreditation 2016-2019, EDG431G/09), and the European Union (European Regional Development Fund-ERDF) to C.G.-B. J.C.V.-U. and A.B. were financially supported by the Miguel Servet Programme ISCIII-FEDER (CP13/00226). M.M. thanks the Spanish Ministry of Science and Innovation for her FPU fellowship. M.M.G. was financially supported by the grant “Clara Roy” from the Spanish Society of Clinical Microbiology and Infectious Diseases. Research reported in this publication was supported in part by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award numbers R01AI100560, R01AI063517, and R01AI072219 (to R.A.B.) and T32-GM-7250 to Case Western Reserve University, MLW. This study was supported in part by funds and/or facilities provided by the Cleveland Department of Veterans Affairs, award number 1I01BX001974, to R.A.B., and by the Biomedical Laboratory Research & Development Service of the VA Office of Research and Development and the Geriatric Research Education and Clinical Center VISN 10 to R.A.B.
The contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health or the Department of Veterans Affairs.
We have no conflicts of interest to declare.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.01172-17.
REFERENCES
- 1.Peleg AY, Seifert H, Paterson DL. 2008. Acinetobacter baumannii: emergence of a successful pathogen. Clin Microbiol Rev 21:538–582. doi: 10.1128/CMR.00058-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dijkshoorn L, Nemec A, Seifert H. 2007. An increasing threat in hospitals: multidrug-resistant Acinetobacter baumannii. Nat Rev Microbiol 5:939–951. doi: 10.1038/nrmicro1789. [DOI] [PubMed] [Google Scholar]
- 3.Kallen AJ, Hidron AI, Patel J, Srinivasan A. 2010. Multidrug resistance among gram-negative pathogens that caused healthcare-associated infections reported to the National Healthcare Safety Network, 2006-2008. Infect Control Hosp Epidemiol 31:528–531. doi: 10.1086/652152. [DOI] [PubMed] [Google Scholar]
- 4.Rosenthal VD, Maki DG, Jamulitrat S, Medeiros EA, Todi SK, Gomez DY, Leblebicioglu H, Abu Khader I, Miranda Novales MG, Berba R, Ramírez Wong FM, Barkat A, Pino OR, Dueñas L, Mitrev Z, Bijie H, Gurskis V, Kanj SS, Mapp T, Hidalgo RF, Ben Jaballah N, Raka L, Gikas A, Ahmed A, Thu TA, Guzmán Siritt ME, INICC Members 2010. International Nosocomial Infection Control Consortium (INICC) report, data summary for 2003-2008, issued June 2009. Am J Infect Control 38:95–104.e2. doi: 10.1016/j.ajic.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 5.Toth M, Antunes NT, Stewart NK, Frase H, Bhattacharya M, Smith CA, Vakulenko SB. 2016. Class D β-lactamases do exist in Gram-positive bacteria. Nat Chem Biol 12:9–14. doi: 10.1038/nchembio.1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Poole K. 2004. Resistance to beta-lactam antibiotics. Cell Mol Life Sci 61:2200–2223. doi: 10.1007/s00018-004-4060-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Massova I, Mobashery S. 1998. Kinship and diversification of bacterial penicillin-binding proteins and beta-lactamases. Antimicrob Agents Chemother 42:1–17. doi: 10.1093/jac/42.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaitany KC, Klinger NV, June CM, Ramey ME, Bonomo RA, Powers RA, Leonard DA. 2013. Structures of the class D carbapenemases OXA-23 and OXA-146: mechanistic basis of activity against carbapenems, extended-spectrum cephalosporins, and aztreonam. Antimicrob Agents Chemother 57:4848–4855. doi: 10.1128/AAC.00762-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Paton R, Miles RS, Hood J, Amyes SG. 1993. ARI 1: beta-lactamase-mediated imipenem resistance in Acinetobacter baumannii. Int J Antimicrob Agents 2:81–87. doi: 10.1016/0924-8579(93)90045-7. [DOI] [PubMed] [Google Scholar]
- 10.Leonard DA, Bonomo RA, Powers RA. 2013. Class D β-lactamases: a reappraisal after five decades. Acc Chem Res 46:2407–2415. doi: 10.1021/ar300327a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Afzal-Shah M, Woodford N, Livermore DM. 2001. Characterization of OXA-25, OXA-26, and OXA-27, molecular class D beta-lactamases associated with carbapenem resistance in clinical isolates of Acinetobacter baumannii. Antimicrob Agents Chemother 45:583–588. doi: 10.1128/AAC.45.2.583-588.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bou G, Oliver A, Martínez-Beltrán J. 2000. OXA-24, a novel class D beta-lactamase with carbapenemase activity in an Acinetobacter baumannii clinical strain. Antimicrob Agents Chemother 44:1556–1561. doi: 10.1128/AAC.44.6.1556-1561.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Corvec S, Poirel L, Naas T, Drugeon H, Nordmann P. 2007. Genetics and expression of the carbapenem-hydrolyzing oxacillinase gene blaOXA-23 in Acinetobacter baumannii. Antimicrob Agents Chemother 51:1530–1533. doi: 10.1128/AAC.01132-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Poirel L, Marqué S, Héritier C, Segonds C, Chabanon G, Nordmann P. 2005. OXA-58, a novel class D β-lactamase involved in resistance to carbapenems in Acinetobacter baumannii. Antimicrob Agents Chemother 49:202–208. doi: 10.1128/AAC.49.1.202-208.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Higgins PG, Poirel L, Lehmann M, Nordmann P, Seifert H. 2009. OXA-143, a novel carbapenem-hydrolyzing class D beta-lactamase in Acinetobacter baumannii. Antimicrob Agents Chemother 53:5035–5038. doi: 10.1128/AAC.00856-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Higgins PG, Pérez-Llarena FJ, Zander E, Fernández A, Bou G, Seifert H. 2013. OXA-235, a novel class D β-lactamase involved in resistance to carbapenems in Acinetobacter baumannii. Antimicrob Agents Chemother 57:2121–2126. doi: 10.1128/AAC.02413-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Poirel L, Naas T, Nordmann P. 2010. Diversity, epidemiology, and genetics of class D beta-lactamases. Antimicrob Agents Chemother 54:24–38. doi: 10.1128/AAC.01512-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Evans BA, Amyes SG. 2014. OXA β-lactamases. Clin Microbiol Rev 27:241–263. doi: 10.1128/CMR.00117-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Queenan AM, Bush K. 2007. Carbapenemases: the versatile beta-lactamases. Clin Microbiol Rev 20:440–458. doi: 10.1128/CMR.00001-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walther-Rasmussen J, Hoiby N. 2006. OXA-type carbapenemases. J Antimicrob Chemother 57:373–383. doi: 10.1093/jac/dki482. [DOI] [PubMed] [Google Scholar]
- 21.Mugnier PD, Poirel L, Naas T, Nordmann P. 2010. Worldwide dissemination of the blaOXA-23 carbapenemase gene of Acinetobacter baumannii. Emerg Infect Dis 16:35–40. doi: 10.3201/eid1601.090852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith CA, Antunes NT, Stewart NK, Frase H, Toth M, Kantardjieff KA, Vakulenko S. 2015. Structural basis for enhancement of carbapenemase activity in the OXA-51 family of class D β-lactamases. ACS Chem Biol 10:1791–1796. doi: 10.1021/acschembio.5b00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abbo A, Carmeli Y, Navon-Venezia S, Siegman-Igra Y, Schwaber MJ. 2007. Impact of multi-drug-resistant Acinetobacter baumannii on clinical outcomes. Eur J Clin Microbiol Infect Dis 26:793–800. doi: 10.1007/s10096-007-0371-8. [DOI] [PubMed] [Google Scholar]
- 24.Livermore DM, Hill RL, Thomson H, Charlett A, Turton JF, Pike R, Patel BC, Manuel R, Gillespie S, Balakrishnan I, Barrett SP, Cumberland N, Twagira M, C-MRAB Study Group 2010. Antimicrobial treatment and clinical outcome for infections with carbapenem- and multiply-resistant Acinetobacter baumannii around London. Int J Antimicrob Agents 35:19–24. doi: 10.1016/j.ijantimicag.2009.09.014. [DOI] [PubMed] [Google Scholar]
- 25.Rhomberg PR, Jones RN. 2009. Summary trends for the Meropenem Yearly Susceptibility Test Information Collection Program: a 10-year experience in the United States (1999-2008). Diagn Microbiol Infect Dis 65:414–426. doi: 10.1016/j.diagmicrobio.2009.08.020. [DOI] [PubMed] [Google Scholar]
- 26.Drawz SM, Bonomo RA. 2010. Three decades of beta-lactamase inhibitors. Clin Microbiol Rev 23:160–201. doi: 10.1128/CMR.00037-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bush K, Jacoby GA. 2010. Updated functional classification of beta-lactamases. Antimicrob Agents Chemother 54:969–976. doi: 10.1128/AAC.01009-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lahiri SD, Mangani S, Jahic H, Benvenuti M, Durand-Reville TF, De Luca F, Ehmann DE, Rossolini GM, Alm RA, Docquier JD. 2015. Molecular basis of selective inhibition and slow reversibility of avibactam against class D carbapenemases: a structure-guided study of OXA-24 and OXA-48. ACS Chem Biol 10:591–600. doi: 10.1021/cb500703p. [DOI] [PubMed] [Google Scholar]
- 29.Perez-Llarena FJ, Bou G. 2009. Beta-lactamase inhibitors: the story so far. Curr Med Chem 16:3740–3765. doi: 10.2174/092986709789104957. [DOI] [PubMed] [Google Scholar]
- 30.Buynak JD, Rao AS, Doppalapudi VR, Adam G, Petersen PJ, Nidamarthy SD. 1999. The synthesis and evaluation of 6-alkylidene-2′beta-substituted penam sulfones as beta-lactamase inhibitors. Bioorg Med Chem Lett 9:1997–2002. doi: 10.1016/S0960-894X(99)00325-X. [DOI] [PubMed] [Google Scholar]
- 31.Pattanaik P, Bethel CR, Hujer AM, Hujer KM, Distler AM, Taracila M, Anderson VE, Fritsche TR, Jones RN, Pagadala SR, van den Akker F, Buynak JD, Bonomo RA. 2009. Strategic design of an effective beta-lactamase inhibitor: LN-1-255, a 6-alkylidene-2′-substituted penicillin sulfone. J Biol Chem 284:945–953. doi: 10.1074/jbc.M806833200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Drawz SM, Bethel CR, Doppalapudi VR, Sheri A, Pagadala SR, Hujer AM, Skalweit MJ, Anderson VE, Chen SG, Buynak JD, Bonomo RA. 2010. Penicillin sulfone inhibitors of class D beta-lactamases. Antimicrob Agents Chemother 54:1414–1424. doi: 10.1128/AAC.00743-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vallejo JA, Martínez-Guitián M, Vázquez-Ucha JC, González-Bello C, Poza M, Buynak JD, Bethel CR, Bonomo RA, Bou G, Beceiro A. 2016. LN-1-255, a penicillanic acid sulfone able to inhibit the class D carbapenemase OXA-48. J Antimicrob Chemother 71:2171–2180. doi: 10.1093/jac/dkw105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.European Committee on Antimicrobial Susceptibility Testing. 2015. Breakpoint tables for interpretation of MICs and zone diameters, version 5.0. http://www.eucast.org/clinical_breakpoints.
- 35.Bou G, Santillana E, Sheri A, Beceiro A, Sampson JM, Kalp M, Bethel CR, Distler AM, Drawz SM, Pagadala SR, van den Akker F, Bonomo RA, Romero A, Buynak JD. 2010. Design, synthesis, and crystal structures of 6-alkylidene-2′-substituted penicillanic acid sulfones as potent inhibitors of Acinetobacter baumannii OXA-24 carbapenemase. J Am Chem Soc 132:13320–13331. doi: 10.1021/ja104092z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Torol S, Kasap M. 2013. Purification and characterization of OXA-23 from Acinetobacter baumannii. J Enzyme Inhib Med Chem 28:836–842. doi: 10.3109/14756366.2012.689296. [DOI] [PubMed] [Google Scholar]
- 37.Verma V, Testero SA, Amini K, Wei W, Liu J, Balachandran N, Monoharan T, Stynes S, Kotra LP, Golemi-Kotra D. 2011. Hydrolytic mechanism of OXA-58 enzyme, a carbapenem-hydrolyzing class D β-lactamase from Acinetobacter baumannii. J Biol Chem 286:37292–37303. doi: 10.1074/jbc.M111.280115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smith CA, Antunes NT, Stewart NK, Toth M, Kumarasiri M, Chang M, Mobashery S, Vakulenko SB. 2013. Structural basis for carbapenemase activity of the OXA-23 β-lactamase from Acinetobacter baumannii. Chem Biol 20:1107–1115. doi: 10.1016/j.chembiol.2013.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pratap S, Katiki M, Gill P, Kumar P, Golemi-Kotra D. 2015. Active-site plasticity is essential to carbapenem hydrolysis by OXA-58 class D β-lactamase of Acinetobacter baumannii. Antimicrob Agents Chemother 60:75–86. doi: 10.1128/AAC.01393-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, Kiefer F, Gallo Cassarino T, Bertoni M, Bordoli L, Schwede T. 2014. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res 42:W252–W258. doi: 10.1093/nar/gku340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.June CM, Muckenthaler TJ, Schroder EC, Klamer ZL, Wawrzak Z, Powers RA, Szarecka A, Leonard DA. 2016. The structure of a doripenem-bound OXA-51 class D β-lactamase variant with enhanced carbapenemase activity. Protein Sci 25:2152–2163. doi: 10.1002/pro.3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Saino H, Sugiyabu T, Ueno G, Yamamoto M, Ishii Y, Miyano M. 2015. Crystal structure of OXA-58 with the substrate-binding cleft in a closed state: insights into the mobility and stability of the OXA-58 structure. PLoS One 10:e0145869. doi: 10.1371/journal.pone.0145869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Santillana E, Beceiro A, Bou G, Romero A. 2007. Crystal structure of the carbapenemase OXA-24 reveals insights into the mechanism of carbapenem hydrolysis. Proc Natl Acad Sci U S A 104:5354–5359. doi: 10.1073/pnas.0607557104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schneider KD, Ortega CJ, Renck NA, Bonomo RA, Powers RA, Leonard DA. 2011. Structures of the class D carbapenemase OXA-24 from Acinetobacter baumannii in complex with doripenem. J Mol Biol 406:583–594. doi: 10.1016/j.jmb.2010.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Soto E, Shoji S, Muto C, Tomono Y, Marshall S. 2014. Population pharmacokinetics of ampicillin and sulbactam in patients with community-acquired pneumonia: evaluation of the impact of renal impairment. Br J Clin Pharmacol 77:509–521. doi: 10.1111/bcp.12232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Das S, Li J, Armstrong J, Learoyd M, Edeki T. 2015. Randomized pharmacokinetic and drug-drug interaction studies of ceftazidime, avibactam, and metronidazole in healthy subjects. Pharmacol Res Perspect 3:e00172. doi: 10.1002/prp2.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ehmann DE, Jahic H, Ross PL, Gu RF, Hu J, Durand-Réville TF, Lahiri S, Thresher J, Livchak S, Gao N, Palmer T, Walkup GK, Fisher SL. 2013. Kinetics of avibactam inhibition against class A, C, and D β-lactamases. J Biol Chem 288:27960–27971. doi: 10.1074/jbc.M113.485979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bethel CR, Distler AM, Ruszczycky MW, Carey MP, Carey PR, Hujer AM, Taracila M, Helfand MS, Thomson JM, Kalp M, Anderson VE, Leonard DA, Hujer KM, Abe T, Venkatesan AM, Mansour TS, Bonomo RA. 2008. Inhibition of OXA-1 beta-lactamase by penems. Antimicrob Agents Chemother 52:3135–3143. doi: 10.1128/AAC.01677-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Che T, Bethel CR, Pusztai-Carey M, Bonomo RA, Carey PR. 2014. The different inhibition mechanisms of OXA-1 and OXA-24 β-lactamases are determined by the stability of active site carboxylated lysine. J Biol Chem 289:6152–6164. doi: 10.1074/jbc.M113.533562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weiss WJ, Petersen PJ, Murphy TM, Tardio L, Yang Y, Bradford PA, Venkatesan AM, Abe T, Isoda T, Mihira A, Ushirogochi H, Takasake T, Projan S, O'Connell J, Mansour TS. 2004. In vitro and in vivo activities of novel 6-methylidene penems as beta-lactamase inhibitors. Antimicrob Agents Chemother 48:4589–4596. doi: 10.1128/AAC.48.12.4589-4596.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Adediran SA, Nukaga M, Baurin S, Frère JM, Pratt RF. 2005. Inhibition of class D beta-lactamases by acyl phosphates and phosphonates. Antimicrob Agents Chemother 49:4410–4412. doi: 10.1128/AAC.49.10.4410-4412.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Majumdar S, Adediran SA, Nukaga M, Pratt RF. 2005. Inhibition of class D beta-lactamases by diaroyl phosphates. Biochemistry 44:16121–16129. doi: 10.1021/bi051719s. [DOI] [PubMed] [Google Scholar]
- 53.Tan Q, Ogawa AM, Raghoobar SL, Wisniewski D, Colwell L, Park YW, Young K, Hermes JD, Dininno FP, Hammond ML. 2011. Thiophenyl oxime-derived phosphonates as nano-molar class C beta-lactamase inhibitors reducing MIC of imipenem against Pseudomonas aeruginosa and Acinetobacter baumannii. Bioorg Med Chem Lett 21:4363–4365. doi: 10.1016/j.bmcl.2011.04.122. [DOI] [PubMed] [Google Scholar]
- 54.Tan Q, Ogawa AM, Painter RE, Park YW, Young K, DiNinno FP. 2010. 4,7-Dichloro benzothien-2-yl sulfonylaminomethyl boronic acid: first boronic acid-derived beta-lactamase inhibitor with class A, C, and D activity. Bioorg Med Chem Lett 20:2622–2624. doi: 10.1016/j.bmcl.2010.02.065. [DOI] [PubMed] [Google Scholar]
- 55.Nichols DA, Renslo AR, Chen Y. 2014. Fragment-based inhibitor discovery against β-lactamase. Future Med Chem 6:413–427. doi: 10.4155/fmc.14.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Beck J, Vercheval L, Bebrone C, Herteg-Fernea A, Lassaux P, Marchand-Brynaert J. 2009. Discovery of novel lipophilic inhibitors of OXA-10 enzyme (class D beta-lactamase) by screening amino analogs and homologs of citrate and isocitrate. Bioorg Med Chem Lett 19:3593–3597. doi: 10.1016/j.bmcl.2009.04.149. [DOI] [PubMed] [Google Scholar]
- 57.Docquier JD, Benvenuti M, Calderone V, Giuliani F, Kapetis D, De Luca F, Rossolini GM, Mangani S. 2010. Crystal structure of the narrow-spectrum OXA-46 class D beta-lactamase: relationship between active-site lysine carbamylation and inhibition by polycarboxylates. Antimicrob Agents Chemother 54:2167–2174. doi: 10.1128/AAC.01517-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Buynak JD. 2004. The discovery and development of modified penicillin- and cephalosporin-derived beta-lactamase inhibitors. Curr Med Chem 11:1951–1964. doi: 10.2174/0929867043364847. [DOI] [PubMed] [Google Scholar]
- 59.Ruiz M, Marti S, Fernandez-Cuenca F, Pascual A, Vila J. 2007. High prevalence of carbapenem-hydrolysing oxacillinases in epidemiologically related and unrelated Acinetobacter baumannii clinical isolates in Spain. Clin Microbiol Infect 13:1192–1198. doi: 10.1111/j.1469-0691.2007.01825.x. [DOI] [PubMed] [Google Scholar]
- 60.Merino M, Poza M, Roca I, Barba MJ, Sousa MD, Vila J, Bou G. 2014. Nosocomial outbreak of a multiresistant Acinetobacter baumannii expressing OXA-23 carbapenemase in Spain. Microb Drug Resist 20:259–263. doi: 10.1089/mdr.2013.0127. [DOI] [PubMed] [Google Scholar]
- 61.Aranda J, Poza M, Pardo BG, Rumbo S, Rumbo C, Parreira JR, Rodríguez-Velo P, Bou G. 2010. A rapid and simple method for constructing stable mutants of Acinetobacter baumannii. BMC Microbiol 10:279. doi: 10.1186/1471-2180-10-279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Clinical and Laboratory Standards Institute. 2012. Performance standards for antimicrobial susceptibility testing: 17th informational supplement M07–A9. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 63.Hsieh MH, Yu CM, Yu VL, Chow JW. 1993. Synergy assessed by checkerboard. A critical analysis. Diagn Microbiol Infect Dis 16:343–349. doi: 10.1016/0732-8893(93)90087-N. [DOI] [PubMed] [Google Scholar]
- 64.Golemi D, Maveyraud L, Vakulenko S, Samama JP, Mobashery S. 2001. Critical involvement of a carbamylated lysine in catalytic function of class D beta-lactamases. Proc Natl Acad Sci U S A 98:14280–14285. doi: 10.1073/pnas.241442898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Winkler ML, Papp-Wallace KM, Bonomo RA. 2015. Activity of ceftazidime/avibactam against isogenic strains of Escherichia coli containing KPC and SHV β-lactamases with single amino acid substitutions in the Ω-loop. J Antimicrob Chemother 70:2279–2286. doi: 10.1093/jac/dkv094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Papp-Wallace KM, Winkler ML, Gatta JA, Taracila MA, Chilakala S, Xu Y, Johnson JK, Bonomo RA. 2014. Reclaiming the efficacy of β-lactam-β-lactamase inhibitor combinations: avibactam restores the susceptibility of CMY-2-producing Escherichia coli to ceftazidime. Antimicrob Agents Chemother 58:4290–4297. doi: 10.1128/AAC.02625-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bethel CR, Taracila M, Shyr T, Thomson JM, Distler AM, Hujer KM, Hujer AM, Endimiani A, Papp-Wallace K, Bonnet R, Bonomo RA. 2011. Exploring the inhibition of CTX-M-9 by beta-lactamase inhibitors and carbapenems. Antimicrob Agents Chemother 55:3465–3475. doi: 10.1128/AAC.00089-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas ¨O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. 2009. Gaussian 09, revision D.01. Gaussian, Inc, Wallingford, CT. [Google Scholar]
- 69.DeLano WL. 2008. The PyMOL molecular graphics system. DeLano Scientific LLC, Palo Alto, CA. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.