

ABSTRACT
Pseudomonas aeruginosa is a major causative agent of both acute and chronic infections. Although aminoglycoside antibiotics are very potent drugs against such infections, antibiotic failure is steadily increasing mainly because of increasing resistance of the bacteria. Many molecular mechanisms that determine resistance, such as acquisition of genes encoding aminoglycoside-inactivating enzymes or overexpression of efflux pumps, have been elucidated. However, there are additional, less well-described mechanisms of aminoglycoside resistance. In this study, we profiled a clinical tobramycin-resistant P. aeruginosa strain that exhibited a small-colony variant (SCV) phenotype. Both the resistance and colony morphology phenotypes were lost upon passage of the isolate under rich medium conditions. Transcriptional and mutational profiling revealed that the SCV harbored activating mutations in the two-component systems AmgRS and PmrAB. Introduction of these mutations individually into type strain PA14 conferred tobramycin and colistin resistance, respectively. However, their combined introduction had an additive effect on the tobramycin resistance phenotype. Activation of the AmgRS system slightly reduced the colony size of wild-type PA14, whereas the simultaneous overexpression of gacA, the response regulator of the GacSA two-component system, further reduced colony size. In conclusion, we uncovered combinatorial influences of two-component systems on clinically relevant phenotypes such as resistance and the expression of the SCV phenotype. Our results clearly demonstrate that the combined activation of P. aeruginosa two-component systems has pleiotropic effects with unforeseen consequences.
KEYWORDS: Pseudomonas aeruginosa, antibiotic resistance, small-colony variant, two-component systems
INTRODUCTION
Pseudomonas aeruginosa is one of the most important opportunistic pathogens and is a serious public health concern. Its high potential for intrinsic and acquired antimicrobial resistance is a major anticipated problem in hospitals (1–3). Aminoglycosides are highly potent broad-spectrum antibiotics. They are particularly used to treat P. aeruginosa pulmonary infections in cystic fibrosis patients, where the drugs are often inhaled to reach high local concentrations in the bronchial sections (4). Intravenously administered combination therapies, mainly with β-lactam antibiotics, are also common (5). However, as for most antimicrobial classes, higher rates of inappropriate empirical antimicrobial treatment are associated with increasing resistance to the antimicrobials in use.
Diverse mechanisms have been described to confer resistance to aminoglycosides on P. aeruginosa. The most prominent ones include drug inactivation through the activity of aminoglycoside-modifying enzymes and decreased drug accumulation inside the bacterial cell via active efflux or diminished cell wall permeability. Active efflux of aminoglycosides in P. aeruginosa can be achieved via upregulation of the MexXY efflux system (6) mainly caused by mutations in regulatory genes (7). Furthermore, adaptive expression of the MexXY efflux pump, e.g., in the presence of ribosome-targeting antibiotics, has been described (8). In cystic fibrosis isolates, active MexXY efflux is very common (9–11).
Besides these well-described strategies to resist aminoglycoside therapy, growth within biofilms (12), conversion to mucoidy (13), or the emergence of persister cells (14) can contribute to survival in the presence of antibiotics. Furthermore, slow-growing small-colony variant (SCV) populations have been associated with persistent infections and increased resistance to antibiotics, including aminoglycosides (15). It has also been shown that aminoglycoside exposure can induce the formation of SCVs in vivo and in vitro (16, 17). While the morphotypic switch to SCVs in Staphylococcus aureus is linked to electron transport deficiency in strains that are auxotroph for menadione or hemin or to thymidine auxotrophy (18), the underlying mechanisms of P. aeruginosa SCV generation seem to be more heterogeneous (19–22). In P. aeruginosa, the SCV phenotype is often, but not exclusively, associated with elevated intracellular levels of the second messenger cyclic di-GMP (12, 19, 23–25). Although the antibiotic susceptibility phenotype may vary a lot (26, 27), many clinical SCV isolates exhibit resistance to several antibiotics and express further phenotypic features like hyperpiliation or increased biofilm formation capabilities (25, 26). Overproduction of the exopolysaccharides Pel and Psl (28, 29) and, more recently, the involvement of a prophage genomic region (22) have also been linked to SCV formation.
In this report, we describe the underlying genetic determinants of clinical P. aeruginosa isolates that not only produce small colonies on agar plates but also exhibit an aminoglycoside resistance phenotype. Following passage under rich medium conditions, three clonally related SCVs produced revertants that exhibited larger colony morphologies and aminoglycoside susceptibility. By transcriptional and mutational profiling, we uncovered a combinatorial impact of the three two-component systems PmrAB, AmgRS, and GacSA on aminoglycoside resistance and the SCV morphotype.
RESULTS
Aminoglycoside resistance is linked to an SCV phenotype.
We have previously identified three clonally related tobramycin-resistant clinical P. aeruginosa isolates (30). The resistance phenotype of those isolates could not be explained by the presence of a gene encoding an aminoglycoside-modifying enzyme, nor did they express the mexXY multidrug efflux pump genes at highly elevated levels. (All three SCVs exhibited <2-fold greater mexX and mexY expression than the PA14 reference strain.) All three clinical isolates, however, were SCVs (Fig. 1). The formation of SCVs has been associated with persistent infections and increased resistance to aminoglycoside antibiotics (15). We therefore hypothesized that there might be a link between the phenotypic variation and aminoglycoside resistance of the three P. aeruginosa SCVs MHH8607, MHH9536, and MHH9604. As shown in Fig. 1, even among the closely related SCVs, differences in colony morphology were observed. MHH8607 seemed to exhibit an even smaller colony morphology than the other two SCV isolates. Via passage of the SCVs in rich medium, we generated stable revertants with larger-surface colonies. Morphological differences in the revertants were also observed. REV_MHH8607 formed shiny circular colonies, while the other two formed rather flat, dull colonies with a rough texture. Of note, all three revertants exhibited increased susceptibility to aminoglycosides (Table 1). However, only the revertant of MHH8607 (REV_MHH8607) had a tobramycin MIC that categorized it as susceptible.
FIG 1.
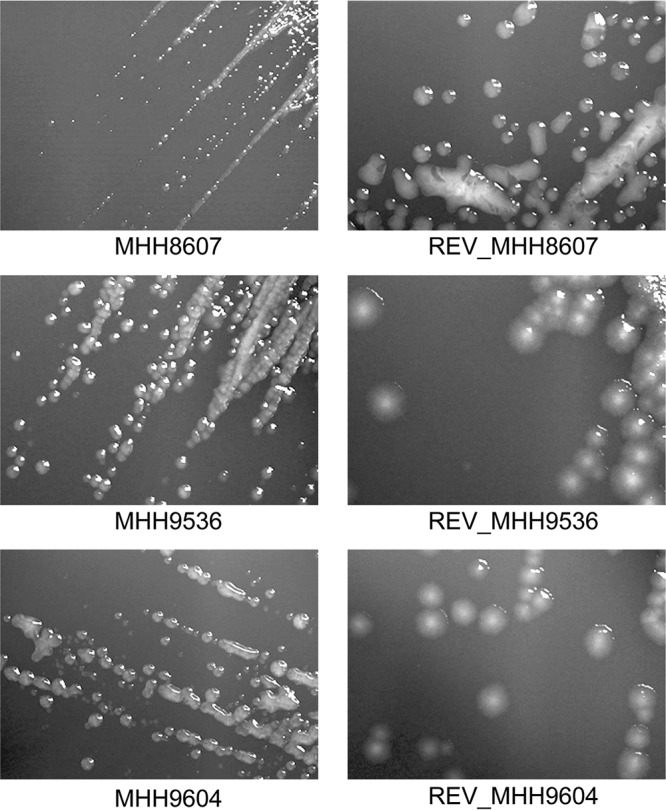
Colony morphology of three clonal SCVs and their respective revertants. Shown are SCVs MHH8607, MHH9536, and MHH9604 (left) and their respective revertants with larger and diverse colony surfaces (right) after 24 h of growth on Columbia blood agar plates.
TABLE 1.
Resistance profiles of P. aeruginosa SCV isolates and their respective revertants
| Isolate and colony morphology | MIC in μg/mla (phenotype)b |
|
|---|---|---|
| Tobramycin | Colistin | |
| MHH8607 | ||
| SCV | 32 (R) | 0.5 (S) |
| REVc | 2 (S) | 0.5 (S) |
| MHH9536 | ||
| SCV | 32 (R) | <0.25 (S) |
| REV | 8 (R) | <0.25 (S) |
| MHH9604 | ||
| SCV | 32 (R) | 0.5 (S) |
| REV | 16 (R) | 0.5 (S) |
MICs were determined by microdilution in cation-adjusted MH broth.
R, resistant; S, susceptible.
REV, revertant.
Transcriptional profiling reveals downregulation of the PmrAB two-component system in the revertants.
Transcriptional profiles of the three SCV isolates have been recorded before (31, 32) and were complemented in this study by RNA sequencing of the respective revertant strains. Gene expression analysis revealed a range of 64 to 84 genes that were differentially expressed between the SCVs and their respective revertants (Fig. 2; see Table S1 in the supplemental material). Among those genes, we found an overlap of 14 genes differentially regulated in all three SCV-revertant pairs (Table 2). Most of them (10 genes) belonged to the PmrAB two-component regulatory pathway.
FIG 2.
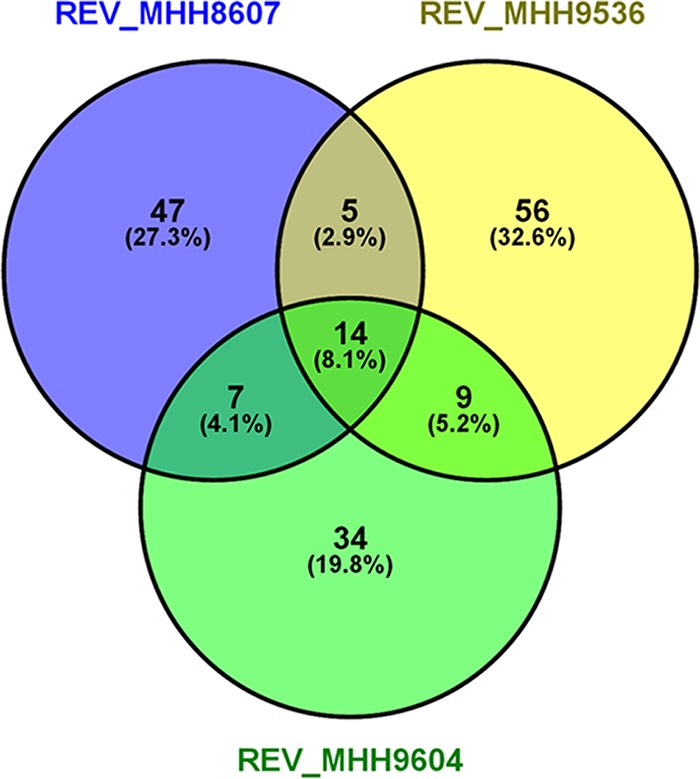
Differentially expressed genes in the SCV isolates in comparison with their respective revertants. A gene was categorized as differentially expressed when the absolute log2-fold change was ≥2 and the P value was ≤0.01. The Venn diagram shown was created with VENNY (60).
TABLE 2.
Genes differentially expressed in all three SCV-revertant pairs
| Locus | Gene | Product | Range of log2-fold changesa |
|---|---|---|---|
| PA14_18300 | Nucleotide sugar dehydrogenase | −5.11 to −7.02 | |
| PA14_18310 | arnF | LPS modification operon | −4.47 to −6.02 |
| PA14_18330 | arnT | −4.77 to −6.09 | |
| PA14_18340 | arnD | −4.77 to −6.85 | |
| PA14_18350 | arnA | −3.79 to 6.01 | |
| PA14_18360 | arnC | −3.61 to 5.38 | |
| PA14_28520 | Hypothetical protein | −3.32 to −4.72 | |
| PA14_44311 | Hypothetical protein | −3.70 to −5.90 | |
| PA14_63110 | speD | Spermidine biosynthesis | −7.48 to −10.08 |
| PA14_63120 | speE | −6.60 to −10.42 | |
| PA14_63130 | −5.66 to −6.69 | ||
| PA14_63150 | pmrA | Two-component response regulator | −4.62 to −5.63 |
| PA14_63160 | pmrB | Two-component sensor kinase | −4.63 to −5.28 |
| PA14_63220 | Hypothetical protein | −5.25 to −7.40 |
Range of log2-fold expression changes among the three SCV-revertant strain comparisons.
The sensor kinase-encoding gene pmrB and the response regulator-encoding gene pmrA were most strongly downregulated in all revertants. Furthermore, many of the genes in the downstream regulated gene operon arnBCADTEF (33) and the spermidine synthesis gene cluster speD (PA14_63110) and speE (PA14_63120) (34) were expressed at lower levels in the revertants. Expression of the arnBCADTEF operon is known to limit the interaction and self-promoted uptake of polycationic antibiotics (35), whereas spermidine is a polyamine and has been reported before to protect cells from antibiotic treatment and oxidative stress (34, 36).
Besides those commonly regulated genes, we found an individual set of differentially expressed genes in each revertant (Table S1). Closer inspection of isolate MHH8607 (where REV_MHH8607 reached aminoglycoside MICs in the susceptible range) revealed the downregulation of a further two-component system sensor kinase-encoding gene, amgS (PA14_68680, annotated as envZ in the PA14 reference genome) (log2-fold change of 3.67). The corresponding response regulator amgR (PA14_68700, annotated as ompR in the PA14 reference genome) also appeared to be downregulated with a log2-fold change of 2.31 (although a significant P value was not reached). The AmgRS system was reported before to be involved in aminoglycoside resistance, as it has been identified in a screening of a transposon mutant library for genes whose inactivation increased tobramycin sensitivity (37). The AmgRS system is a membrane stress-responsive two-component system involved in the transcriptional regulation of membrane proteases and other membrane proteins (38). In line with this, we found reduced expression of the protease HtpX (encoded by PA14_27480, log2-fold change of 3.99) and a membrane protein of unknown function (encoded by PA14_72930, log2-fold change of 3.97) in the REV_MHH8607 strain.
In conclusion, it seems that the downregulation of the PmrAB system in the revertants is involved in their regained aminoglycoside susceptibility, whereas the additional downregulation of AmgRS in REV_MHH8607 might account for the regained full tobramycin susceptibility of this particular strain.
The aminoglycoside-resistant SCVs exhibit increased pmrAB and amgRS expression levels.
We next explored whether the PmrAB and AmgRS two-component systems exhibit overall increased expression in the SCVs and thus might explain the aminoglycoside resistance phenotype. We therefore compared the expression levels of the respective genes with those of a P. aeruginosa type strain cultivated under identical conditions. Indeed, all three SCV isolates exhibited higher expression of the genes for both systems (up to 43-fold increased expression of pmrA and up to 5-fold increased expression of amgS; data not shown) than the PA14 reference strain. Also in comparison to the median expression level in 151 clinical isolates that have been sequenced before (32), we found higher expression of both two-component systems (data not shown).
The aminoglycoside-resistant SCVs exhibit gain-of-function mutations in the two-component systems.
As mutational activation of both the PmrAB and AmgRS systems has been described previously (39–41), the gene alleles of the SCV isolates were consequently screened for sequence alterations, which could be gain-of-function mutations. Indeed, in all three clonal SCV isolates, nonsynonymous sequence variations in comparison to the PA14 reference strain were discovered in both genes of the pmrAB operon, as well as in the sensor kinase-encoding gene amgS. The pmrA sequence variation caused an amino acid substitution in the corresponding protein (Leu71Arg) that is located within the signal receiver domain of the response regulator, which also contains the phosphorylation site of the protein. Within pmrB, two sequence variations were detected; one caused an amino acid exchange in the secretion signal of the protein (Thr4Ala), while the second is located in close proximity to the histidine kinase A and ATP binding domains (Leu323His) (40, 42). To explore whether these sequence variations act as activating mutations, we introduced them into the PA14 reference strain individually or in various combinations and analyzed the resulting phenotypes (Table 3).
TABLE 3.
Antibiotic susceptibilities of the PA14 reference strain mutants generated
| PA14 genotype | MIC in μg/mla (phenotype)b |
|
|---|---|---|
| Tobramycin | Colistin | |
| Wild type | 0.5 (S) | 1 (S) |
| amgS Ala28Glu | 1 (S) | 1 (S) |
| pmrA Leu71Arg | 0.5 (S) | 1 (S) |
| pmrB Thr4Ala | 0.5 (S) | 2 (S) |
| pmrB Thr4Ala Leu323His | 1 (S) | 64 (R) |
| pmrB Thr4Ala Leu323His + amgS Ala28Glu | 2 (S) | 64 (R) |
MICs were determined by microdilution in cation-adjusted MH broth.
R, resistant; S, susceptible.
Aminoglycoside susceptibility was slightly affected by the mutation in amgS, which led to a 2-fold increase in the tobramycin MIC (Table 3). The activating mutations within the PmrAB system did not influence the tobramycin MIC. However, the combined activation of AmgS and the PmrAB system led to a 4-fold increase in the tobramycin MIC. PmrAB is known to be involved in polymyxin resistance by activating the arnBCADTEF operon, which in turn modifies the lipopolysaccharide (LPS) structure. We therefore also tested the generated mutant strains for changes in colistin susceptibility. Single mutations in pmrA or pmrB led to no or only a slight decrease in colistin susceptibility, while both pmrB mutations in combination resulted in high-level resistance (MIC, 64 μg/ml). The aminoglycoside resistance-inducing mutation in amgS did not further enhance polymyxin susceptibility.
Intriguingly, the three clinical clonal SCV isolates investigated did not exhibit colistin resistance, although they carried the gain-of-function mutations in pmrAB and exhibited overexpression of the system and the downstream genes. There was also no difference in colistin susceptibility between the SCVs and their respective revertants, even though changes in arnBCADTEF expression occurred. We did not identify any insertions or deletions within the SCV arn genes. Nevertheless, we found 19 sequence variations overall (Table S2) within the arn genes of the SCVs compared to wild-type PA14. The possibility cannot be excluded that among them there is an inactivating mutation that might explain colistin susceptibility despite anr gene overexpression.
Susceptibility to colistin in the SCVs also could not be explained by the expression levels of the two-component system PhoPQ-OprH operon or ParRS. (We found up to 2.3-fold less phoQ, phoP, and oprH expression than in wild-type PA14 and up to 2.8-fold greater parS expression, but parR expression was not significantly changed [data not shown].)
Mutations that led to the switch to the revertant phenotype.
The PmrAB pathway has been previously reported to contribute to an antibiotic resistance phenotype. As this pathway was downregulated in all three revertants compared to their respective SCVs, we further explored whether this was due to mutational inactivation of the system in the revertants. We therefore used our RNA sequencing data set to identify mutations that have been acquired by the revertants during the course of their phenotypic switch. A range of polymorphic genes or regulatory pathways that were affected in more than one revertant strain were detected (Table 4). This included the PmrAB two-component system-encoding genes, in which all revertants had acquired a mutation in either the sensor kinase or the response regulator. Additionally, and in line with the transcriptional data, the two-component system AmgRS harbored a nonsense mutation in amgS in REV_MHH8607. Furthermore, the GacSA system harbored mutations in two of the three revertants (in REV_MHH8606 in gacS and in REV_MHH9536 44 bp upstream of gacA). Another mutational hot spot was the alternative sigma factor algU, where single-nucleotide polymorphisms occurred in close proximity in two revertant strains. The latter mutation is likely to be the cause of the observed morphological differences among the three revertants: Only REV_MHH8607 exhibited a shiny and more mucoid colony surface, while those with an acquired algU mutation and thus presumably lower alginate production formed dull, nonmucoid colonies, as described before (43).
TABLE 4.
Mutations in two-component systems acquired by the revertant strains during the course of their phenotypic switch
| Category and gene | Product | Function | Mutation |
||
|---|---|---|---|---|---|
| REV_MHH8607 | REV_MHH9536 | REV_MHH9604 | |||
| TCSsa | |||||
| PA14_63150_pmrA | TCS response regulator | LPS modification | Asp92Tyr | ||
| PA14_63160_pmrB | TCS sensor kinase | Ser420Arg | Gly423Cys | ||
| PA14_68700_amgR | TCS response regulator | Adaptive membrane stress response | |||
| PA14_68680_amgS | TCS sensor kinase | 28stop | |||
| PA14_52260_gacS | TCS sensor/response regulator hybrid | Phenotypic switch biofilm/motility | Gly474Val | ||
| PA14_30650_gacA | TCS response regulator | Intergenicb | |||
| Other | |||||
| PA14_54430_algU | RNA polymerase sigma factor | Envelope stress sigma factor | Ala54Asp | Arg66Cys | |
| PA14_60820_oprJ | Outer membrane protein | 173stop | |||
| PA14_08430 | ATPase | Leu71Ile | |||
| PA14_11980 | Hypothetical protein | Phe157Leu | |||
| PA14_55180_migA | Glycosyltransferase | LPS modification | Asp43Glu | ||
| PA14_72630 | ABC transporter permease | Trp26Cys | |||
TCSs, two-component systems.
44 bp 5′ gacA.
The combined activation of the AmgRS and GacSA systems in type strain PA14 produces an SCV phenotype.
The GacSA/RsmAZY signaling system is known to be involved in the phenotypic switch between chronic (persistence, SCV formation) and acute infectious lifestyles. Since the revertants of two of the clinical SCVs harbored mutations in the GacSA pathway, we overexpressed the gacA gene in wild-type PA14, as well as the PA14 strain harboring the gain-of-function mutations in pmrB and/or amgS to determine whether we could observe an influence on colony morphology. Overexpression of the gacA gene in PA14 or in the mutants overexpressing PmrB did not lead to alteration of colony morphology or the resistance phenotype (data not shown). However, in the mutant with an activation of the AmgRS two-component system, gacA overexpression led to an SCV phenotype. The resistance phenotype did not change in this strain background because of gacA overexpression (data not shown).
Our data thus indicate that the combined activation of the GacSA and AmgRS systems seems to explain the SCV phenotype in our clinical isolates, whereas the combined activation of the PmrAB and AmgRS systems confers enhanced aminoglycoside resistance.
DISCUSSION
In this study, we aimed to identify the molecular mechanisms underlying the expression of an aminoglycoside-resistant SCV phenotype in three clonally related clinical P. aeruginosa isolates. We found constitutive activation of the two two-component systems AmgRS and PmrAB in the SCV isolates. It has been reported previously that both systems can be activated not only by environmental triggers but also by gain-of-function mutations, especially within the sensor kinase-encoding gene sequence (39, 40). Amino acid substitutions at positions Arg182 and Val121 in AmgS have been shown to be activating mutations and to increase the tobramycin MIC 2- to 4-fold in clinical isolates (39). In this study, we identified a nonsynonymous sequence variation in amgS (Cys28Ala) in the SCV isolate. The introduction of this amgS sequence variation into the PA14 genetic background resulted in a 2-fold increase in the tobramycin MIC.
PmrAB is known to control polymyxin resistance in P. aeruginosa and other Gram-negative bacteria (41, 44). Polymyxins, such as colistin, are cationic antimicrobial peptides that interact with lipid A of the LPS of Gram-negative bacteria and lead to cell death and lysis (45). PmrAB becomes activated under divalent-cation-limiting conditions and stimulates the transcription of the arnBCADTEF operon, which drives resistance-conferring LPS modifications (41). An indirect involvement of PmrAB in aminoglycoside resistance might be due to its regulation of the spermidine synthesis-encoding genes PA14_63110 to PA14_63130. These are responsible for the production of polyamines that protect the bacterial membrane from antibiotic exposure and oxidative damage (46). Similar to AmgRS, mutations in pmrB (frequently in various combinations) have been shown to activate the system (40). In addition, mutations in pmrA seem to have an impact on polymyxin susceptibility (47). In this study, the combined introduction of two pmrB mutations resulted in high-level colistin resistance (MIC, 64 μg/ml), demonstrating that these mutations activate the two-component system. In contrast to what has been stated previously (48), activating mutations in pmrB alone can obviously confer high-level colistin resistance. However, no influence on the tobramycin MIC was observed.
The activating mutations in prmB clearly induced colistin resistance, and the activating mutation in amgS induced tobramycin resistance in type strain PA14. Interestingly, the combined activation of the PmrAB and AmgRS two-component systems further increased the tobramycin MIC.
There seems to be an association between LPS modifications as a result of an activated arnBCADTEF operon and the formation of an SCV phenotype following gentamicin exposure of a P. aeruginosa laboratory strain (21). Polymyxin resistance due to the activation of the arnBCADTEF operon is associated with an altered LPS structure and thus might contribute to fitness costs and/or growth retardation (41, 49–51), which may also affect colony morphology (40, 52). In some cases, reduced virulence was also observed (51, 53).
While this additive activity of AmgRS and PmrAB conferred the tobramycin resistance phenotype, the combined activation of the two systems was not sufficient to induce an SCV phenotype in the PA14 strain background. Only the additional activation of gacA in the AmgRS activating mutant background led to an SCV phenotype in type strain PA14. The expression of the GacAS system has been demonstrated previously to have an impact on the colony morphology of P. aeruginosa (54, 55). However, overexpression of gacA in the PA14 background did not change colony morphology. Only the gacA-overexpressing PA14 mutant with an activated AmgRS system developed an SCV phenotype. In the same line, the clinical SCVs analyzed in this study did not exhibit colistin resistance, although they carried the gain-of-function mutations in pmrAB and exhibited overexpression of the system and the downstream genes. It thus seems that the (combined) activation of the two-component systems PmrAB, AmgRS, and GacSA exhibits strong mutual influences whose pleiotropic effects on the resistance and SCV phenotypes are strongly impacted by the strain background.
MATERIALS AND METHODS
Bacterial strains, plasmids, and antibiotic susceptibility testing.
All of the isolates analyzed in this study were sampled at the Hannover Medical School (MHH). The three SCVs were isolated from the respiratory tract material of three individual patients. As a reference for differential gene expression and sequence variation analysis and for mutagenesis, the UCBPP-PA14 strain was used (56). For overexpression of gacA, the corresponding gene sequence (PA14_30650) was amplified from the PA14 chromosome with forward primer CCGGAATTCGTGATTAAGGTGCTGGTGGTCG and reverse primer CCCAAGCTTCTAGCTGGCGGCATCGAC and inserted between the EcoRI and HindIII restriction sites (underlined) of shuttle vector pUCP20 in accordance with standard molecular biology procedures (57). The PA3740-overexpressing strains were used for phenotypic assays.
Antibiotic susceptibility was determined by agar dilution as described previously (58). The classification of MIC breakpoints was performed according to the Clinical and Laboratory Standards Institute (CLSI) guidelines.
Generation of SCV revertants.
For the SCV isolates, a spontaneous variant with a larger-surface colony was generated. The SCV isolates were cultured in 10 ml of LB broth at 37°C, and when turbidity was visible, a 100-μl aliquot was again inoculated into 10 ml of LB broth. At the same time, an aliquot was spread on Columbia blood agar and examined for the occurrence of revertants exhibiting larger-surface colonies. This process was repeated until a stable revertant was generated (1 to 2 weeks).
RNA sequencing and data analysis.
Whole-transcriptome sequencing of the SCV clinical isolates was previously performed by our group by using a custom-made protocol with barcoded RNA libraries to enable pooled sequencing of several samples (31, 32). For this study, RNA sequencing of the revertant strains generated was performed in the same way. Sequencing data analysis was performed as described before (30).
Mutagenesis.
To insert point mutations into the PA14 reference strain, mutagenesis was carried out by homologous recombination with plasmid pEX18Ap (59) and the QuikChange II site-directed mutagenesis kit (Agilent Technologies). The mutagenic primers were designed with the web-based QuikChange Primer Design Program. The mutated plasmid was transformed into Escherichia coli DH5α cells, and the bacterial suspension was plated on LB agar plates containing 100 μg/ml ampicillin. Single colonies were screened for insertion of the point mutation by PCR and Sanger sequencing. The pEX18Ap constructs containing the desired mutation were transferred into P. aeruginosa by biparental mating with E. coli S17-1 as the donor strain.
Accession number(s).
All short-read data were uploaded at the Gene Expression Omnibus database repository and assigned accession number GSE99729.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by an ERC starter grant (RESISTOME 260276), the German Research Foundation (DFG SFB 900), and the Federal Ministry of Education and Research. M.S. and M.K. were funded by the Hannover Biomedical Research School. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
The Center for Clinical and Experimental Infection Research, Institute for Molecular Bacteriology, TWINCORE GmbH, is a joint venture of the Hannover Medical School and the Helmholtz Center for Infection Research.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.01178-17.
REFERENCES
- 1.Breidenstein EB, de la Fuente-Nunez C, Hancock RE. 2011. Pseudomonas aeruginosa: all roads lead to resistance. Trends Microbiol 19:419–426. doi: 10.1016/j.tim.2011.04.005. [DOI] [PubMed] [Google Scholar]
- 2.Michiels JE, Van den Bergh B, Verstraeten N, Fauvart M, Michiels J. 2016. In vitro emergence of high persistence upon periodic aminoglycoside challenge in the ESKAPE pathogens. Antimicrob Agents Chemother 60:4630–4637. doi: 10.1128/AAC.00757-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lister PD, Wolter DJ, Hanson ND. 2009. Antibacterial-resistant Pseudomonas aeruginosa: clinical impact and complex regulation of chromosomally encoded resistance mechanisms. Clin Microbiol Rev 22:582–610. doi: 10.1128/CMR.00040-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chiappini E, Taccetti G, de Martino M. 2014. Bacterial lung infections in cystic fibrosis patients: an update. Pediatr Infect Dis J 33:653–654. doi: 10.1097/INF.0000000000000347. [DOI] [PubMed] [Google Scholar]
- 5.Döring G, Conway SP, Heijerman HG, Hodson ME, Hoiby N, Smyth A, Touw DJ. 2000. Antibiotic therapy against Pseudomonas aeruginosa in cystic fibrosis: a European consensus. Eur Respir J 16:749–767. doi: 10.1034/j.1399-3003.2000.16d30.x. [DOI] [PubMed] [Google Scholar]
- 6.Poole K. 2001. Multidrug resistance in Gram-negative bacteria. Curr Opin Microbiol 4:500–508. doi: 10.1016/S1369-5274(00)00242-3. [DOI] [PubMed] [Google Scholar]
- 7.Matsuo Y, Eda S, Gotoh N, Yoshihara E, Nakae T. 2004. MexZ-mediated regulation of mexXY multidrug efflux pump expression in Pseudomonas aeruginosa by binding on the mexZ-mexX intergenic DNA. FEMS Microbiol Lett 238:23–28. doi: 10.1111/j.1574-6968.2004.tb09732.x. [DOI] [PubMed] [Google Scholar]
- 8.Morita Y, Sobel ML, Poole K. 2006. Antibiotic inducibility of the MexXY multidrug efflux system of Pseudomonas aeruginosa: involvement of the antibiotic-inducible PA5471 gene product. J Bacteriol 188:1847–1855. doi: 10.1128/JB.188.5.1847-1855.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sobel ML, McKay GA, Poole K. 2003. Contribution of the MexXY multidrug transporter to aminoglycoside resistance in Pseudomonas aeruginosa clinical isolates. Antimicrob Agents Chemother 47:3202–3207. doi: 10.1128/AAC.47.10.3202-3207.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morita Y, Tomida J, Kawamura Y. 2012. MexXY multidrug efflux system of Pseudomonas aeruginosa. Front Microbiol 3:408. doi: 10.3389/fmicb.2012.00408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Islam S, Jalal S, Wretlind B. 2004. Expression of the MexXY efflux pump in amikacin-resistant isolates of Pseudomonas aeruginosa. Clin Microbiol Infect 10:877–883. doi: 10.1111/j.1469-0691.2004.00991.x. [DOI] [PubMed] [Google Scholar]
- 12.Drenkard E, Ausubel FM. 2002. Pseudomonas biofilm formation and antibiotic resistance are linked to phenotypic variation. Nature 416:740–743. doi: 10.1038/416740a. [DOI] [PubMed] [Google Scholar]
- 13.Hatch RA, Schiller NL. 1998. Alginate lyase promotes diffusion of aminoglycosides through the extracellular polysaccharide of mucoid Pseudomonas aeruginosa. Antimicrob Agents Chemother 42:974–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Möker N, Dean CR, Tao J. 2010. Pseudomonas aeruginosa increases formation of multidrug-tolerant persister cells in response to quorum-sensing signaling molecules. J Bacteriol 192:1946–1955. doi: 10.1128/JB.01231-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Häussler S. 2004. Biofilm formation by the small colony variant phenotype of Pseudomonas aeruginosa. Environ Microbiol 6:546–551. doi: 10.1111/j.1462-2920.2004.00618.x. [DOI] [PubMed] [Google Scholar]
- 16.Gerber AU, Vastola AP, Brandel J, Craig WA. 1982. Selection of aminoglycoside-resistant variants of Pseudomonas aeruginosa in an in vivo model. J Infect Dis 146:691–697. doi: 10.1093/infdis/146.5.691. [DOI] [PubMed] [Google Scholar]
- 17.Langford PR, Anwar H, Gonda I, Brown MR. 1989. Outer membrane proteins of gentamicin induced small colony variants of Pseudomonas aeruginosa. FEMS Microbiol Lett 52:33–36. doi: 10.1111/j.1574-6968.1989.tb03547.x. [DOI] [PubMed] [Google Scholar]
- 18.Garcia LG, Lemaire S, Kahl BC, Becker K, Proctor RA, Denis O, Tulkens PM, Van Bambeke F. 2013. Antibiotic activity against small-colony variants of Staphylococcus aureus: review of in vitro, animal and clinical data. J Antimicrob Chemother 68:1455–1464. doi: 10.1093/jac/dkt072. [DOI] [PubMed] [Google Scholar]
- 19.Malone JG, Jaeger T, Manfredi P, Dötsch A, Blanka A, Bos R, Cornelis GR, Häussler S, Jenal U. 2012. The YfiBNR signal transduction mechanism reveals novel targets for the evolution of persistent Pseudomonas aeruginosa in cystic fibrosis airways. PLoS Pathog 8:e1002760. doi: 10.1371/journal.ppat.1002760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blanka A, Duvel J, Dötsch A, Klinkert B, Abraham WR, Kaever V, Ritter C, Narberhaus F, Häussler S. 2015. Constitutive production of c-di-GMP is associated with mutations in a variant of Pseudomonas aeruginosa with altered membrane composition. Sci Signal 8:ra36. doi: 10.1126/scisignal.2005943. [DOI] [PubMed] [Google Scholar]
- 21.Wei Q, Tarighi S, Dötsch A, Häussler S, Müsken M, Wright VJ, Camara M, Williams P, Haenen S, Boerjan B, Bogaerts A, Vierstraete E, Verleyen P, Schoofs L, Willaert R, De Groote VN, Michiels J, Vercammen K, Crabbe A, Cornelis P. 2011. Phenotypic and genome-wide analysis of an antibiotic-resistant small colony variant (SCV) of Pseudomonas aeruginosa. PLoS One 6:e29276. doi: 10.1371/journal.pone.0029276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lim WS, Phang KK, Tan AH, Li SF, Ow DS. 2016. Small colony variants and single nucleotide variations in Pf1 region of PB1 phage-resistant Pseudomonas aeruginosa. Front Microbiol 7:282. doi: 10.3389/fmicb.2016.00282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.D'Argenio DA, Calfee MW, Rainey PB, Pesci EC. 2002. Autolysis and autoaggregation in Pseudomonas aeruginosa colony morphology mutants. J Bacteriol 184:6481–6489. doi: 10.1128/JB.184.23.6481-6489.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hickman JW, Tifrea DF, Harwood CS. 2005. A chemosensory system that regulates biofilm formation through modulation of cyclic diguanylate levels. Proc Natl Acad Sci U S A 102:14422–14427. doi: 10.1073/pnas.0507170102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Häussler S, Tümmler B, Weissbrodt H, Rohde M, Steinmetz I. 1999. Small-colony variants of Pseudomonas aeruginosa in cystic fibrosis. Clin Infect Dis 29:621–625. doi: 10.1086/598644. [DOI] [PubMed] [Google Scholar]
- 26.Badenoch PR, Coster DJ. 1989. Selection of gentamicin-resistant variants of Pseudomonas aeruginosa in the rat cornea. J Ocul Pharmacol 5:19–25. doi: 10.1089/jop.1989.5.19. [DOI] [PubMed] [Google Scholar]
- 27.Kirisits MJ, Prost L, Starkey M, Parsek MR. 2005. Characterization of colony morphology variants isolated from Pseudomonas aeruginosa biofilms. Appl Environ Microbiol 71:4809–4821. doi: 10.1128/AEM.71.8.4809-4821.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Malone JG, Jaeger T, Spangler C, Ritz D, Spang A, Arrieumerlou C, Kaever V, Landmann R, Jenal U. 2010. YfiBNR mediates cyclic di-GMP dependent small colony variant formation and persistence in Pseudomonas aeruginosa. PLoS Pathog 6:e1000804. doi: 10.1371/journal.ppat.1000804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Starkey M, Hickman JH, Ma L, Zhang N, De Long S, Hinz A, Palacios S, Manoil C, Kirisits MJ, Starner TD, Wozniak DJ, Harwood CS, Parsek MR. 2009. Pseudomonas aeruginosa rugose small-colony variants have adaptations that likely promote persistence in the cystic fibrosis lung. J Bacteriol 191:3492–3503. doi: 10.1128/JB.00119-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Khaledi A, Schniederjans M, Pohl S, Rainer R, Bodenhofer U, Xia B, Klawonn F, Bruchmann S, Preusse M, Eckweiler D, Dötsch A, Häussler S. 2016. Transcriptome profiling of antimicrobial resistance in Pseudomonas aeruginosa. Antimicrob Agents Chemother 60:4722–4733. doi: 10.1128/AAC.00075-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dötsch A, Schniederjans M, Khaledi A, Hornischer K, Schulz S, Bielecka A, Eckweiler D, Pohl S, Häussler S. 2015. The Pseudomonas aeruginosa transcriptional landscape is shaped by environmental heterogeneity and genetic variation. mBio 6:e00749. doi: 10.1128/mBio.00749-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pohl S, Klockgether J, Eckweiler D, Khaledi A, Schniederjans M, Chouvarine P, Tummler B, Häussler S. 2014. The extensive set of accessory Pseudomonas aeruginosa genomic components. FEMS Microbiol Lett 356:235–241. doi: 10.1111/1574-6968.12445. [DOI] [PubMed] [Google Scholar]
- 33.McPhee JB, Lewenza S, Hancock RE. 2003. Cationic antimicrobial peptides activate a two-component regulatory system, PmrA-PmrB, that regulates resistance to polymyxin B and cationic antimicrobial peptides in Pseudomonas aeruginosa. Mol Microbiol 50:205–217. doi: 10.1046/j.1365-2958.2003.03673.x. [DOI] [PubMed] [Google Scholar]
- 34.Johnson L, Mulcahy H, Kanevets U, Shi Y, Lewenza S. 2012. Surface-localized spermidine protects the Pseudomonas aeruginosa outer membrane from antibiotic treatment and oxidative stress. J Bacteriol 194:813–826. doi: 10.1128/JB.05230-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fernández L, Gooderham WJ, Bains M, McPhee JB, Wiegand I, Hancock RE. 2010. Adaptive resistance to the “last hope” antibiotics polymyxin B and colistin in Pseudomonas aeruginosa is mediated by the novel two-component regulatory system ParR-ParS. Antimicrob Agents Chemother 54:3372–3382. doi: 10.1128/AAC.00242-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kwon DH, Lu CD. 2006. Polyamines induce resistance to cationic peptide, aminoglycoside, and quinolone antibiotics in Pseudomonas aeruginosa PAO1. Antimicrob Agents Chemother 50:1615–1622. doi: 10.1128/AAC.50.5.1615-1622.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee S, Hinz A, Bauerle E, Angermeyer A, Juhaszova K, Kaneko Y, Singh PK, Manoil C. 2009. Targeting a bacterial stress response to enhance antibiotic action. Proc Natl Acad Sci U S A 106:14570–14575. doi: 10.1073/pnas.0903619106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hinz A, Lee S, Jacoby K, Manoil C. 2011. Membrane proteases and aminoglycoside antibiotic resistance. J Bacteriol 193:4790–4797. doi: 10.1128/JB.05133-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lau CH, Fraud S, Jones M, Peterson SN, Poole K. 2013. Mutational activation of the AmgRS two-component system in aminoglycoside-resistant Pseudomonas aeruginosa. Antimicrob Agents Chemother 57:2243–2251. doi: 10.1128/AAC.00170-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moskowitz SM, Brannon MK, Dasgupta N, Pier M, Sgambati N, Miller AK, Selgrade SE, Miller SI, Denton M, Conway SP, Johansen HK, Hoiby N. 2012. PmrB mutations promote polymyxin resistance of Pseudomonas aeruginosa isolated from colistin-treated cystic fibrosis patients. Antimicrob Agents Chemother 56:1019–1030. doi: 10.1128/AAC.05829-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moskowitz SM, Ernst RK, Miller SI. 2004. PmrAB, a two-component regulatory system of Pseudomonas aeruginosa that modulates resistance to cationic antimicrobial peptides and addition of aminoarabinose to lipid A. J Bacteriol 186:575–579. doi: 10.1128/JB.186.2.575-579.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Olaitan AO, Morand S, Rolain JM. 2014. Mechanisms of polymyxin resistance: acquired and intrinsic resistance in bacteria. Front Microbiol 5:643. doi: 10.3389/fmicb.2014.00643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu H, Schurr MJ, Deretic V. 1995. Functional equivalence of Escherichia coli sigma E and Pseudomonas aeruginosa AlgU: E. coli rpoE restores mucoidy and reduces sensitivity to reactive oxygen intermediates in algU mutants of P. aeruginosa. J Bacteriol 177:3259–3268. doi: 10.1128/jb.177.11.3259-3268.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Adams MD, Nickel GC, Bajaksouzian S, Lavender H, Murthy AR, Jacobs MR, Bonomo RA. 2009. Resistance to colistin in Acinetobacter baumannii associated with mutations in the PmrAB two-component system. Antimicrob Agents Chemother 53:3628–3634. doi: 10.1128/AAC.00284-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yeaman MR, Yount NY. 2003. Mechanisms of antimicrobial peptide action and resistance. Pharmacol Rev 55:27–55. doi: 10.1124/pr.55.1.2. [DOI] [PubMed] [Google Scholar]
- 46.Lewenza S. 2013. Extracellular DNA-induced antimicrobial peptide resistance mechanisms in Pseudomonas aeruginosa. Front Microbiol 4:21. doi: 10.3389/fmicb.2013.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee JY, Ko KS. 2014. Mutations and expression of PmrAB and PhoPQ related with colistin resistance in Pseudomonas aeruginosa clinical isolates. Diagn Microbiol Infect Dis 78:271–276. doi: 10.1016/j.diagmicrobio.2013.11.027. [DOI] [PubMed] [Google Scholar]
- 48.Jochumsen N, Marvig RL, Damkiaer S, Jensen RL, Paulander W, Molin S, Jelsbak L, Folkesson A. 2016. The evolution of antimicrobial peptide resistance in Pseudomonas aeruginosa is shaped by strong epistatic interactions. Nat Commun 7:13002. doi: 10.1038/ncomms13002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pournaras S, Poulou A, Dafopoulou K, Chabane YN, Kristo I, Makris D, Hardouin J, Cosette P, Tsakris A, De E. 2014. Growth retardation, reduced invasiveness, and impaired colistin-mediated cell death associated with colistin resistance development in Acinetobacter baumannii. Antimicrob Agents Chemother 58:828–832. doi: 10.1128/AAC.01439-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sun S, Negrea A, Rhen M, Andersson DI. 2009. Genetic analysis of colistin resistance in Salmonella enterica serovar Typhimurium. Antimicrob Agents Chemother 53:2298–2305. doi: 10.1128/AAC.01016-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hraiech S, Roch A, Lepidi H, Atieh T, Audoly G, Rolain JM, Raoult D, Brunel JM, Papazian L, Bregeon F. 2013. Impaired virulence and fitness of a colistin-resistant clinical isolate of Acinetobacter baumannii in a rat model of pneumonia. Antimicrob Agents Chemother 57:5120–5121. doi: 10.1128/AAC.00700-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Snitkin ES, Zelazny AM, Gupta J, Program NCS, Palmore TN, Murray PR, Segre JA. 2013. Genomic insights into the fate of colistin resistance and Acinetobacter baumannii during patient treatment. Genome Res 23:1155–1162. doi: 10.1101/gr.154328.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.López-Rojas R, Dominguez-Herrera J, McConnell MJ, Docobo-Perez F, Smani Y, Fernández-Reyes M, Rivas L, Pachon J. 2011. Impaired virulence and in vivo fitness of colistin-resistant Acinetobacter baumannii. J Infect Dis 203:545–548. doi: 10.1093/infdis/jiq086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Davies JA, Harrison JJ, Marques LL, Foglia GR, Stremick CA, Storey DG, Turner RJ, Olson ME, Ceri H. 2007. The GacS sensor kinase controls phenotypic reversion of small colony variants isolated from biofilms of Pseudomonas aeruginosa PA14. FEMS Microbiol Ecol 59:32–46. doi: 10.1111/j.1574-6941.2006.00196.x. [DOI] [PubMed] [Google Scholar]
- 55.Malone JG. 2015. Role of small colony variants in persistence of Pseudomonas aeruginosa infections in cystic fibrosis lungs. Infect Drug Resist 8:237–247. doi: 10.2147/IDR.S68214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liberati NT, Urbach JM, Miyata S, Lee DG, Drenkard E, Wu G, Villanueva J, Wei T, Ausubel FM. 2006. An ordered, nonredundant library of Pseudomonas aeruginosa strain PA14 transposon insertion mutants. Proc Natl Acad Sci U S A 103:2833–2838. doi: 10.1073/pnas.0511100103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.West SE, Schweizer HP, Dall C, Sample AK, Runyen-Janecky LJ. 1994. Construction of improved Escherichia-Pseudomonas shuttle vectors derived from pUC18/19 and sequence of the region required for their replication in Pseudomonas aeruginosa. Gene 148:81–86. doi: 10.1016/0378-1119(94)90237-2. [DOI] [PubMed] [Google Scholar]
- 58.Wiegand I, Hilpert K, Hancock RE. 2008. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat Protoc 3:163–175. doi: 10.1038/nprot.2007.521. [DOI] [PubMed] [Google Scholar]
- 59.Hoang TT, Karkhoff-Schweizer RR, Kutchma AJ, Schweizer HP. 1998. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212:77–86. doi: 10.1016/S0378-1119(98)00130-9. [DOI] [PubMed] [Google Scholar]
- 60.Oliveros JC. 2007. VENNY. An interactive tool for comparing lists with Venn Diagrams. Spanish National Biotechnology Center, Madrid, Spain: http://bioinfogp.cnb.csic.es/tools/venny/index.html. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.