

ABSTRACT
In a multiple-dose-ranging trial, we previously evaluated higher doses of rifampin in patients for 2 weeks. The objectives of the current study were to administer higher doses of rifampin for a longer period to compare the pharmacokinetics, safety/tolerability, and bacteriological activity of such regimens. In a double-blind, randomized, placebo-controlled, phase II clinical trial, 150 Tanzanian patients with tuberculosis (TB) were randomized to receive either 600 mg (approximately 10 mg/kg of body weight), 900 mg, or 1,200 mg rifampin combined with standard doses of isoniazid, pyrazinamide, and ethambutol administered daily for 2 months. Intensive pharmacokinetic sampling occurred in 63 patients after 6 weeks of treatment, and safety/tolerability was assessed. The bacteriological response was assessed by culture conversion in liquid and solid media. Geometric mean total exposures (area under the concentration-versus-time curve up to 24 h after the dose) were 24.6, 50.8, and 76.1 mg · h/liter in the 600-mg, 900-mg, and 1,200-mg groups, respectively, reflecting a nonlinear increase in exposure with the dose (P < 0.001). Grade 3 adverse events occurred in only 2 patients in the 600-mg arm, 4 patients in the 900-mg arm, and 5 patients in the 1,200-mg arm. No significant differences in the bacteriological response were observed. Higher daily doses of rifampin (900 and 1,200 mg) resulted in a more than proportional increase in rifampin exposure in plasma and were safe and well tolerated when combined with other first-line anti-TB drugs for 2 months, but they did not result in improved bacteriological responses in patients with pulmonary TB. These findings have warranted evaluation of even higher doses of rifampin in follow-up trials. (This study has been registered at ClinicalTrials.gov under identifier NCT00760149.)
KEYWORDS: drug safety, pharmacokinetics, rifampin, tuberculosis
INTRODUCTION
There is an urgent need to improve the treatment of tuberculosis (TB), considering that approximately 10.4 million people develop active TB and 1.8 million people die from it each year (1). Since its introduction in the 1960s, rifampin has been considered the cornerstone in TB treatment (2). It is active against Mycobacterium tuberculosis in the exponential growth phase, and it possesses activity against nonreplicating persistent bacilli. It is applied in a daily dose of 10 mg/kg of body weight, which corresponds to 600 mg in most populations. The evidence underpinning the selection of this recommended dose of rifampin is scant and based on the idea that effective rifampin plasma concentrations were achieved with this dose. This was coupled with a fear of adverse effects at higher doses and cost considerations for programmatic implementation at the time that rifampin was introduced (in the 1960s and 1970s) (3).
Accumulating evidence suggests that higher doses of rifampin may be more effective and, thus, could lead to shortening of the duration of TB treatment. More specifically, in vitro experiments in a pharmacokinetic-pharmacodynamic model of TB showed that higher levels of exposure to rifampin kill the mycobacteria more rapidly and prevent the emergence of resistance to this drug (4). Experiments in mice have also shown that the current 10-mg/kg dose of rifampin is at the lower end of the dose-response curve and that an increase in the dose leads to enhanced sterilizing activity and a decrease in the duration of treatment for pulmonary TB in mice (5–8). A systematic review identified 14 randomized controlled clinical trials that evaluated doses of rifampin of up to 1,200 mg (20 mg/kg) administered at different dosing intervals for the treatment of pulmonary TB (9). Most of the trials were performed before 1980, and only four of them were considered to be of high quality according to published guidelines, yet several of them suggested an advantage in terms of the likelihood of culture conversion among patients receiving at least 900 mg of rifampin (9, 10). These combined findings and the results of more recent clinical studies on the concept of high-dose rifampin (11, 12) encouraged us to develop a series of clinical trials to thoroughly investigate higher doses of rifampin for pulmonary TB. We have previously reported on our initial multiple-dose-ranging trial in which we evaluated higher doses of rifampin in patients with pulmonary TB for a short period of 2 weeks (the Pan African Consortium for the Evaluation of Antituberculosis Antibiotics [PanACEA] HIGHRIF study 1) (13). Here we report on the findings of our second trial (PanACEA HIGHRIF study 2; ClinicalTrials.gov identifier NCT00760149), which was initiated at the same time as the PanACEA HIGHRIF study 1. In PanACEA HIGHRIF study 2, we evaluated higher doses of rifampin for a longer period of 2 months.
The objectives of this clinical trial were to determine the effect of a higher than standard dose of rifampin (900 and 1,200 mg of rifampin compared to 600 mg) on the pharmacokinetics of rifampin, on the occurrence of adverse events, and on the bacteriological response of M. tuberculosis when these higher doses of rifampin were combined with other first-line anti-TB drugs for 2 months.
RESULTS
Patients.
A total of 150 smear-positive pulmonary tuberculosis patients, predominantly male patients (n = 135, 90%), were randomized. The patient characteristics were found to be similar across the three arms of the study (Table 1).
TABLE 1.
Demographic and baseline characteristics at inclusiona
| Characteristic | Value(s) for subjects receiving: |
|||
|---|---|---|---|---|
| All subjects | 600 mg rifampin | 900 mg rifampin | 1,200 mg rifampin | |
| Total no. of subjects randomized | 150 | 50 | 50 | 50 |
| No. (%) of subjects by gender | ||||
| Maleb | 135 (90) | 44 (88) | 46 (92) | 45 (90) |
| Female | 15 (10) | 6 (12) | 4 (8) | 5 (10) |
| Median (IQR) age (yr) | 33.5 (27.0–40.0) | 35.0 (28.0–40.0) | 33.0 (27.0–41.0) | 33.5 (27.0–41.0) |
| Median (IQR) wt (kg) | 55.5 (52.0–59.3) | 55.0 (51.0–59.3) | 55.0 (52.0–59.3) | 56.0 (53.0–60.0) |
| No. (%) of subjects who were outpatients | 43 (29) | 13 (26) | 16 (32) | 14 (28) |
| No. (%) of HIV-positive subjects | 15 (10) | 4 (12) | 5 (10) | 6 (12) |
| No. (%) of subjects with the following MGIT resultc: | ||||
| Positive | 143 (95) | 48 (96) | 48 (96) | 47 (94) |
| Contaminated | 1 (1) | 0 | 0 | 1 (2) |
| Missing | 6 (4) | 2 (4) | 2 (4) | 2 (4) |
| No. (%) of subjects with the following result on LJc: | ||||
| Negative | 12 (8) | 3 (6) | 6 (12) | 3 (6) |
| 1+ | 40 (27) | 17 (34) | 12 (24) | 11 (22) |
| 2+ | 49 (33) | 12 (24) | 16 (32) | 21 (42) |
| 3+ | 9 (6) | 4 (8) | 3 (6) | 2 (4) |
| 4+ | 21 (14) | 6 (12) | 8 (16) | 7 (14) |
| Missing | 19 (13) | 8 (16) | 5 (10) | 6 (12) |
| No. (%) of subjects with the following ZN staining resultc: | ||||
| Negative | 0 | 0 | 0 | 0 |
| 1+ | 15 (10) | 4 (8) | 6 (12) | 5 (10) |
| 2+ | 51 (34) | 18 (36) | 18 (36) | 15 (30) |
| 3+ | 55 (37) | 18 (36) | 18 (36) | 19 (38) |
| 4+ | 24 (16) | 7 (14) | 7 (14) | 10 (20) |
| Missing | 5 (3) | 3 (6) | 1 (2) | 1 (2) |
| Median (IQR) TTP for MGIT (days)c | 5.13 (3.96, 6.71) | 5.10 (4.13, 6.58) | 4.92 (3.83, 6.71) | 5.13 (3.71, 6.79) |
| Median (IQR) no. of log CFUc | 5.25 (4.04, 6.79) | 5.25 (4.21, 6.79) | 5.29 (4.04, 6.75) | 5.13 (3.71, 6.79) |
HIV, human immunodeficiency virus; IQR, interquartile range; LJ, Lowenstein-Jensen medium; MGIT, mycobacterial growth indicator tube; TTP, time to positivity; ZN, Ziehl-Neelsen.
In Tanzania, there are generally more male than females admitted for the treatment of tuberculosis because of a benefit in access to health care. In previous studies in Tanzania, we observed that men were less likely to be under community-based directly observed treatment (DOT) (3) and relatively more men were hospitalized (4).
The result for the earliest positive culture within 14 days of randomization.
Pharmacokinetics.
Pharmacokinetic data were available for 23, 21, and 19 patients in the 600-, 900-, and 1,200-mg dose groups, respectively. The majority of patients in these groups (91%, 91%, and 90%, respectively) were male; the median ages were 35, 36, and 31 years, respectively, and the median weights were 57, 54, and 56 kg, respectively, similar to the median values for the patients in the whole group.
Upon doubling of the dose of rifampin from 600 mg to 1,200 daily, the geometric mean area under the concentration-versus-time curve (AUC) up to 24 h after the dose (AUC0–24) of rifampin increased 3-fold (from 24.6 to 76.1 mg · h/liter), which reflects a more than dose-proportional increase in the level of exposure with the dose (Table 2; Fig. 1). More than dose-proportional increases in the average maximum plasma concentration (Cmax) of rifampin were also observed. In line with this finding, one-way analysis of variance (ANOVA) showed statistically significant differences in the rifampin exposure measures AUC0–24 (P < 0.001) and Cmax (P < 0.001) and in apparent clearance (CL/F) (P = 0.013) between each of the three dose groups. In addition, there was a significant increase in the elimination half-life (P = 0.02) with higher doses compared to that with the 600-mg dose, suggesting the saturation of elimination processes. Of note, large interindividual variability in AUC0–24 and Cmax occurred (Table 2). However, the minimum exposure recorded in each group (in terms of either AUC0–24 or Cmax) still increased with the dose administered.
TABLE 2.
Doses and pharmacokinetics of TB drugsa
| Drug | Pharmacokinetic parameter | Values for subjects receiving: |
P valuec | ||
|---|---|---|---|---|---|
| 600 mg rifampin (n = 23)b | 900 mg rifampin (n = 21) | 1,200 mg rifampin (n = 19) | |||
| Rifampin | Dose (mg/kg) | 10.7 (8.3–12.0) | 16.7 (14.1–17.7) | 21.4 (17.1–23.5) | |
| AUC0–24 (mg · h/liter) | 23.9 (9.1–118.5) | 50.8 (18.9−153.6) | 76.1 (43.5–167.0) | <0.001 | |
| Cmax (mg/liter) | 5.3 (2.0–23.3) | 9.1 (4.9–15.4) | 14.1 (8.1–29.0) | <0.001 | |
| Tmax (h) | 4.0 (2.0–6.1) | 4.0 (2.0–6.1) | 4.0 (2.5–6.2) | 0.879 | |
| CL/F (liters/h) | 24.4 (5.1–65.6) | 17.2 (5.9–47.7) | 15.8 (7.2–27.6) | 0.013 | |
| V/F (liters) | 77.0 (17.6–212.7) | 70.4 (41.8–130.6) | 54.8 (34.0–97.0) | 0.1 | |
| t1/2 (h) | 1.9 (1.1–4.5) | 2.8 (1.4–7.2) | 2.4 (1.4–3.4) | 0.02 | |
| Isoniazid | Dose (mg/kg) | 5.4 (4.2–6.0) | 5.6 (4.7–5.9) | 5.4 (4.3–5.9) | |
| AUC0–24 (mg · h/liter) | 12.4 (4.6–30.9) | 9.6 (2.8–22.3) | 11.5 (2.0–28.1) | 0.333 | |
| Cmax (mg/liter) | 2.7 (1.4–6.9) | 2.2 (1.2–4.6) | 2.6 (0.74–4.9) | 0.253 | |
| Tmax (h) | 2.5 (1.0–4.1) | 3.0 (1.0–4.1) | 2.6 (1.0–4.0) | 0.365 | |
| CL/F (liters/h) | 24.1 (9.7–65.7) | 31.1 (13.5–108.1) | 26.0 (10.7–147.7) | 0.333 | |
| V/F (liters) | 108.1 (41.6–1034.1) | 113.3 (54.3–373.8) | 109.5 (42.5–350.1) | 0.952 | |
| t1/2 (h) | 3.1 (1.5−10.9) | 2.5 (1.4–4.7) | 2.9 (1.2–16.6) | 0.415 | |
| Pyrazinamide | Dose (mg/kg) | 28.6 (22.2–32.0) | 29.6 (25.0–31.4) | 28.6 (22.9–31.4) | |
| AUC0–24 (mg · h/liter) | 332.2 (246.2–586.4) | 306.4 (188.7–419.5) | 284.2 (190.0–452.3) | 0.098 | |
| Cmax (mg/liter) | 33.5 (23.8–54.7) | 33.1 (23.7–50.3) | 32.6 (27.6–43.2) | 0.884 | |
| Tmax (h) | 3.0 (1.0–6.1) | 3.95 (1.0–6.0) | 2.5 (2.0–4.0) | 0.293 | |
| CL/F (liters/h) | 4.8 (2.7–6.5) | 5.2 (3.8–8.5) | 5.6 (3.5–8.4) | 0.098 | |
| V/F (liters) | 51.0 (37.1–228.4) | 43.9 (30.2–51.9) | 47.2 (19.0–164.3) | 0.346 | |
| t1/2 (h) | 7.3 (5.1–46.2) | 5.8 (3.8–7.7) | 5.8 (1.6–32.2) | 0.157 | |
Pharmacokinetic parameters after daily administration of 600, 900, or 1,200 mg rifampin combined with 300 mg isoniazid, 1,600 mg pyrazinamide, and 1,100 mg ethambutol, recorded at week 6 of treatment. Data represent the geometric mean (range) for all parameters except dose and Tmax, for which they represent the median (range). Abbreviations: AUC0–24 = area under the concentration-versus-time curve up to 24 h after the dose; Cmax, maximum plasma concentration; Tmax, time to the maximum concentration; CL/F, clearance; V/F, apparent volume of distribution; F, bioavailability; t1/2, elimination half-life.
In the 600-mg rifampin group, data are for 23 patients who received rifampin but 22 patients who received isoniazid, pyrazinamide, and ethambutol.
P values were determined by one-way ANOVA on log-transformed pharmacokinetic parameters (all parameters except Tmax) or the Kruskal-Wallis test (for Tmax).
FIG 1.
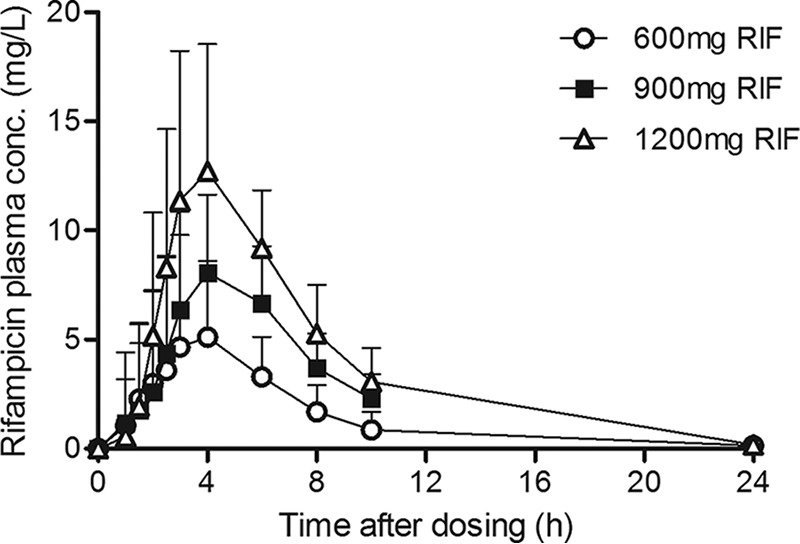
Plasma concentration-time profiles of rifampin in TB patients who received 600, 900, or 1,200 mg rifampin daily (means and standard deviations, recorded at 6 weeks of treatment).
The pharmacokinetics of isoniazid and pyrazinamide did not differ between the study arms, indicating that the higher doses of rifampin did not affect the levels of exposure to these drugs (Table 2). In univariable analyses, gender was not tested in view of the limited numbers of female patients, and age and weight were not associated with the AUC0–24 or the Cmax of each of the anti-TB drugs in each of the dose groups. The relationship between pyrazinamide and weight in the 900-mg rifampin dose group was the exception to this observation (Spearman's rho = −0.442, P = 0.045). When the data for the dosing groups were combined, neither gender, age, nor weight was found to be a significant predictor of any of the main drug exposure parameters (AUC0–24 and Cmax).
Safety and tolerability.
A total of 1,004 adverse events were reported in the study, among which there were 821 grade 1 events, 160 grade 2 events, 20 grade 3 events, and 3 grade 5 events (deaths), and these adverse events were equally distributed over the study arms (Table 3). Of the 20 grade 3 (severe) adverse events in total, 6 adverse events occurred in 2 patients (with, in total, 5 out of the 6 adverse events at least possibly related to the study drug in both patients) in the 600-mg arm, 5 adverse events (with 1 out of the 5 adverse events possibly related to the study drug) occurred in 4 patients in the 900-mg arm, and finally, 9 adverse events occurred in 5 patients (with 5 out of the 9 adverse events at least possibly related to the study drug in 3 patients) in the 1,200-mg arm (Tables 3 and 4). Grade 3 increases in alanine aminotransferase (AST) and/or aspartate aminotransferase (ALT) levels occurred in 3 patients, 1 in the 600-mg arm and 2 in the 1,200-mg arm. Table 4 shows the number of patients experiencing a grade 3 (severe) adverse event per system organ class (SOC). Overall, grade 3 events occurred in 2 patients in the 600-mg arm, 4 patients in the 900-mg arm, and 5 patients in the 1,200-mg arm. The three patients who died, one in each arm, had advanced TB and died of worsening cough with respiratory failure and choking due to hemoptysis. All were deemed not related or doubtfully related to rifampin.
TABLE 3.
Summary of frequency of adverse events according to CTCAE criteriaa
| AE grade | No. of AEs for subjects receiving: |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| All subjects (n = 150) | 600 mg rifampin (n = 50) |
900 mg rifampin (n = 50) |
1,200 mg rifampin (n = 50) |
|||||||
| All | Related | Unrelated | All | Related | Unrelated | All | Related | Unrelated | ||
| Grade 1 (mild AEs) | 821 | 273 | 120 | 153 | 239 | 110 | 129 | 309 | 105 | 204 |
| Grade 2 (moderate AEs) | 160 | 48 | 16 | 32 | 48 | 10 | 38 | 64 | 9 | 55 |
| Grade 3 (severe AEs) | 20 | 6 | 5 | 1 | 5 | 1 | 4 | 9 | 5 | 4 |
| Grade 4 (life-threatening AEs) | 0 | 0 | 0 | 0 | ||||||
| Grade 5 (death related to AE) | 3 | 1 | 1 | 1 | 1 | 1 | 1 | |||
The CTCAE criteria are described elsewhere (24). AE, adverse event; related, the AE is considered associated with the use of the investigational product if the attribution is possible, probable, or very likely.
TABLE 4.
Summary of patients with grade 3 adverse events classified according to CTCAE criteriaa
| Characteristic | Value(s) for subjects receiving: |
||
|---|---|---|---|
| 600 mg rifampin | 900 mg rifampin | 1,200 mg rifampin | |
| Total no. of patients randomized | 50 | 50 | 50 |
| Total no. of patients with grade 3 events | 2 | 4 | 5 |
| AEs by SOC | |||
| Gastrointestinal disorders | |||
| Total no. (%) of patients | 1 (2) | 2 (4) | |
| No. of AEs | |||
| Oral pain | 1 | ||
| Dental caries | 1 | 1 | |
| General disorders and administration site conditions | |||
| Total no. (%) of patients | 1 (2) | ||
| No. of AEs of fever | 1 | ||
| Laboratory investigations | |||
| Total no. (%) of patients | 2 (4) | 1 (2) | 3 (6) |
| No. of AEs | |||
| Increased ALT levels | 1 | 2 | |
| Increased AST levels | 1 | 2 | |
| Increased GGT levels | 1 | ||
| Decreased lymphocyte count | 2 | ||
| Decreased neutrophil count | 1 | ||
| Decreased platelet count | 2 | ||
| Respiratory, thoracic, and mediastinal disorders | |||
| Total no. (%) of patients | 2 (4) | 2 (4) | |
| No. of AEs | |||
| Cough | 1 | 1 | |
| Dyspnea | 1 | ||
| Hiccups | 1 | ||
The CTCAE criteria are described elsewhere (25). No grade 4 adverse events were reported. SOC, system organ class; ALT, alanine aminotransferase; AST, aspartate aminotransferase; GGT, γ-glutamyl transpeptidase.
Bacteriological response.
Five patients (2 patients each in the 1,200-mg and 900-mg arms and 1 patient in the 600-mg arm) were excluded from the bacteriological analysis due to the absence of a positive culture on Lowenstein-Jensen medium (LJ) or in mycobacterial growth indicator tubes (MGIT) (4 patients) within 2 weeks of randomization or isoniazid resistance at the baseline (1 patient, 900-mg arm). Although estimates of the proportions of patients achieving culture conversion by 8 weeks in the 1,200-mg arm (60% in MGIT and 82% on LJ) were numerically higher than those for patients in the 600-mg arm (49% in MGIT and 74% on LJ), there was no evidence for a difference between arms in the time to culture conversion in MGIT (P = 0.237) or on LJ (P = 0.345) (Table 5; Fig. 2). There was no significant difference between arms in the change in the bacillary load over time (P = 0.057 for log time to positivity [TTP; in days] in MGIT and P = 0.628 for number of log CFU on Middlebrook 7H11 plates) (Fig. 3).
TABLE 5.
Summary of analyses of time to culture conversion in MGIT culture and on solid LJa
| Medium and parameter | Value(s) for subjects receiving rifampin at: |
||
|---|---|---|---|
| 600 mg | 900 mg | 1,200 mg | |
| MGIT | |||
| Total no. (%) of patients with culture conversion/total no. in analysis | 33 (67)/49 | 26 (54)/48 | 32 (67)/48 |
| Hazard ratio (95% CI) compared to 600-mg dose | 0.91 (0.55, 1.52) | 1.34 (0.82, 2.18) | |
| P value for hazard ratio | 0.723 | 0.237 | |
| % of patients with culture conversion at: | |||
| 28 days | 6.2 | 8.7 | 32.4 |
| 42 days | 29.7 | 36.5 | 51 |
| 56 days | 48.8 | 51 | 60.4 |
| 70 days | 66.6 | 56.1 | 65.7 |
| 84 days | 72.2 | 62.9 | 73.8 |
| LJ | |||
| Total no. (%) of patients with culture conversion/total no. in analysis | 42 (86)/49 | 37 (77)/48 | 42 (88)/48 |
| Hazard ratio (95% CI) compared to 600-mg dose | 1.01 (0.65, 1.57) | 1.23 (0.80, 1.89) | |
| P value for hazard ratio | 0.971 | 0.345 | |
| % of patients with culture conversion at: | |||
| 28 days | 41.2 | 52.4 | 47.4 |
| 42 days | 60.8 | 64.2 | 72.5 |
| 56 days | 73.9 | 76.2 | 81.7 |
| 70 days | 85.8 | 78.8 | 86.9 |
| 84 days | 90.5 | 85.5 | 92.2 |
LJ, Lowenstein-Jensen medium; MGIT, mycobacterial growth indicator tube; CI, confidence interval.
FIG 2.
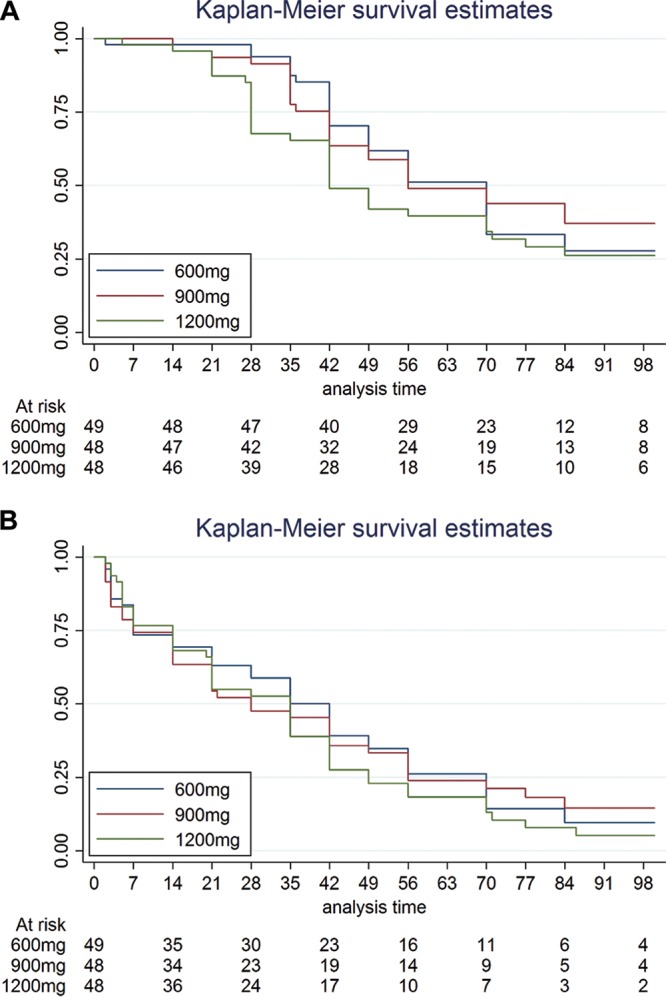
Kaplan-Meier survival estimates by dose. (A) Time to culture conversion in mycobacterial growth indicator tube; (B) time to culture conversion on Lowenstein-Jensen medium.
FIG 3.

Slope of log time to positivity (TTP) (left) and number of log CFU (right) over time.
Pharmacokinetic-pharmacodynamic relationships.
Dichotomization of the parameters actual dose received, rifampin AUC0–24, and rifampin Cmax and comparison of the groups with high values (≥16 mg/kg, ≥48 mg · h/liter, and ≥9.12 mg/liter, respectively) and groups with low values (<16 mg/kg, <48 mg · h/liter, and <9.12 mg/liter, respectively) showed that there was no evidence that the time to culture conversion in MGIT or on LJ differed for these groups (Table 6). Also, AUC0–24 (n = 60) and Cmax (n = 61) had no significant impact on the time to culture conversion in MGIT or on LJ (up to 12 weeks), when the exposure parameters were entered as a continuous variable in the Cox regression analysis (Table 6). However, in these analyses, data for the last culture for 12 out of 61 patients (20%) were censored because they had reached the time of collection of their last sample for culture prior to 12 weeks of treatment but they had not yet achieved culture conversion.
TABLE 6.
Analysis of time to culture conversion by actual dose of rifampin received and by exposure to rifampina
| Parameter | MGIT |
LJ |
||
|---|---|---|---|---|
| Hazard ratio (95% CI) | P value | Hazard ratio (95% CI) | P value | |
| Actual wt-adjusted dose of rifampin received (≥16 mg/kg vs <16 mg/kg) | 1.20 (0.80, 1.82) | 0.376 | 1.22 (0.85, 1.75) | 0.271 |
| AUC0–24 of rifampin (≥48 mg · h/liter vs <48 mg · h/liter) | 0.50 (0.22, 1.13) | 0.096 | 1.39 (0.77, 2.51) | 0.276 |
| Cmax of rifampin (≥9.12 mg/liter vs <9.12 mg/liter) | 0.73 (0.34, 1.60) | 0.436 | 1.42 (0.79, 2.56) | 0.240 |
| AUC0–24 of rifampin (per 1 unit) | 0.99 (0.98–1.01) | 0.432 | 1.01 (1.00–1.02) | 0.140 |
| Cmax of rifampin (per 1 unit) | 0.99 (0.91–1.07) | 0.703 | 1.03 (0.98–1.09) | 0.211 |
AUC0–24, area under the concentration-versus-time curve up to 24 h after the dose; Cmax, maximum plasma concentration; LJ, Lowenstein-Jensen medium; MGIT, mycobacterial growth indicator tube; CI, confidence interval.
Pharmacokinetic data were available for seven of the patients with at least one grade 3 adverse event. For five out of seven of these patients, the events were not considered related to rifampin. The geometric mean rifampin AUC0–24 for those with and without at least a grade 3 adverse event were 65.0 and 41.5 mg · h/liter, respectively (P = 0.111). Importantly, there was no significant difference in the geometric mean AUC0–24 or Cmax of rifampin, isoniazid, or pyrazinamide between those with (n = 7) and those without (n = 56) grade 3 to 5 adverse events.
DISCUSSION
This study represents a comprehensive account of the pharmacokinetics of rifampin administered beyond the standard dose of 10 mg/kg. The results demonstrate a more than proportional increase in the level of exposure to rifampin in plasma upon an increase in the rifampin dose from 600 to 900 and 1,200 mg daily. Doses of 900 and 1,200 mg of rifampin in combination with other first-line anti-TB drugs were safe and as well tolerated as the standard dose when administered for a period of 2 months.
The concept of the use of a higher dose of rifampin is based on available data on the concentration-effect relationship of this drug. The efficacy of rifampin is exposure or concentration dependent, which means that the effect correlates with total exposure to the drug (AUC0–24) and/or its peak concentration (Cmax) (4). An increase in the rifampin dose is expected to increase the total exposure and peak concentration in plasma and, in turn, to increase the levels of those parameters at other sites of action. The dose increase in the current study resulted in a more than dose-proportional (superproportional) increase in geometric mean rifampin AUC0–24 and Cmax values, showing that the intervention was effective from a pharmacokinetic point of view and reflecting the nonlinear pharmacokinetics of rifampin, as observed before (11, 14).
The geometric mean rifampin AUC0–24 and Cmax values after the administration of 600 mg to Tanzanian pulmonary TB patients in this study (23.9 mg · h/liter and 5.3 mg/liter, respectively) (Table 2) appeared to be lower than the values that we have previously reported for Tanzanian patients (39.9 mg · h/liter and 8.9 mg/liter, respectively [15]). This could not be explained by the different percentages of HIV-infected patients in each of the studies (possibly resulting in lower exposures) but may at least partly be related to the time of sampling. The pharmacokinetics of rifampin in the current trial were studied after 6 weeks of anti-TB treatment, whereas patients in the previous study were evaluated after a median of 19 days. The autoinduction of rifampin has been thought to be largely completed after 2 weeks (14), but more recent data suggest that autoinduction is completed and steady state is achieved after 23 days (16) and up to 40 days (17), and this may possibly explain some of the between-study differences in the levels of exposure to rifampin.
The levels of exposures achieved with the higher 1,200-mg rifampin dose in the current study were also lower than those assessed in our previous multiple-dose-ranging trial in South African patients (13), which, again, may be due to the later time of pharmacokinetic sampling in the current trial compared to the time of pharmacokinetic sampling used in our previous study with the short-term use of rifampin, or it may be due to interethnic differences in genetic polymorphisms for drug transporters or enzymes involved in the metabolism of rifampin.
This study also showed a large interindividual variability in the levels of exposure to rifampin at each of the dose levels. The lowest observed AUC0–24 and Cmax values also increased with an increase in the dose. This is actually what we aimed for, as we considered that these lowest exposures may increase the chance of treatment failures, relapses, and the emergence of resistance.
As to the pharmacokinetics of the first-line anti-TB drugs isoniazid and pyrazinamide, these were comparable to those seen in previous data for Tanzanian subjects (15). The higher doses of rifampin did not affect the average AUC0–24 or Cmax of these drugs. Such a pharmacokinetic interaction was not anticipated, considering that the extent of induction of metabolic enzymes by rifampin is nearly maximal at a dose of 300 mg once daily (18).
Importantly, the higher doses of rifampin (900 and 1,200 mg) were safe and well tolerated during 2 months of treatment in combination with other first-line anti-TB drugs. Adverse events of all severities considered related or not related to rifampin were equally distributed over the three study arms. One patient in each of the study arms died, and the death could be explained by advanced disease. Exposure to rifampin or other anti-TB drugs was not related to the occurrence of grade 3 or grade 5 adverse events. As to the type of adverse event, the possibility of hepatotoxicity was feared, but it was rarely observed. In the past, attempts to use high-dose rifampin (900 mg, or 15 mg/kg, and more) were intermittently met with a high incidence of a flu-like syndrome, an allergic reaction (10, 19). Such reactions also were not observed, and they were previously ascribed more to the intermittency of dosing than the higher rifampin dose used (10, 19). The good tolerability of 900 and 1,200 mg of rifampin is in agreement with that when such doses are used to treat other diseases, such as brucellosis, leishmaniasis, and bone and joint infections (20–22), although treatment of these diseases requires less toxic drugs to be coadministered with rifampin.
This study showed no differences between the three doses of rifampin in the time to culture conversion in MGIT or on LJ. More specifically, there was no evidence (the confidence intervals of the hazard ratios still included 1) that culture conversion in the 1,200-mg arm was faster than that in the 600-mg arm. The finding that hazard ratios were numerically greater than 1 for both MGIT and LJ might indicate there was a small reduction in the time to culture conversion that our study was too small to show. So, the lack of an additional bacteriological response may be explained by the small number of participants in this study, which was not powered for bacteriological endpoints. Another explanation may be that there was large interindividual variability in the rifampin exposures achieved, with the rifampin exposures in the three study arms overlapping. However, the levels of exposure to rifampin were also not related to the bacteriological response, even though a possible effect on the time to culture conversion may have been obscured by patients (20%) for whom the last sample for culture was collected prior to 12 weeks of treatment and before they achieved culture conversion. Nonetheless, the most likely explanation for the absence of an effect in this trial is that the doses of rifampin used and the levels of exposure achieved were not high enough to expect an improvement in bacteriological response. In line with this, a study by Jayaram et al. (2003) with a murine model of TB is most illustrative in showing a classical sigmoidal curve which describes the relationship between exposure to rifampin (AUC/MIC) and the response in terms of the reduction in the number of CFU in lung tissue (5). Another study in mice showed that a reduction in the number of CFU in lung tissue occurs only beyond a certain high dose (6). In other words, the exposures achieved with 900 and 1,200 mg of rifampin may not be on the steep part of the concentration-response curve for rifampin in humans. A further similar 2-month study is currently evaluating 15 and 20 mg/kg of rifampin in Peruvian pulmonary TB patients (ClinicalTrials.gov identifier NCT01408914). This study will shed more light on the pharmacokinetics, safety, and bacteriological response of these rifampin doses in South American patients.
Within our series of trials on high-dose rifampin, we have recently reported the findings of our multiple-dose-ranging (or maximum-dose-tolerability) study of rifampin, showing that doses of up to 35 mg/kg rifampin were tolerated by South African TB patients for a short (2-week) period (HIGHRIF study 1) (13). On the basis of the results of HIGHRIF study 1 and the findings of the current study on the long-term use of rifampin at 900-mg (15-mg/kg) and 1,200-mg (20-mg/kg) doses, we have performed a third clinical trial comparing an arm with 35 mg/kg rifampin and other first-line anti-TB drugs to other investigational drug combinations during 3 months of administration in both Tanzanian and South Africa patients (23). This trial showed that rifampin at the higher 35-mg/kg dose was well tolerated and significantly decreased the time to culture conversion in MGIT.
In conclusion, higher daily doses of rifampin (900 and 1,200 mg, corresponding to 15 and 20 mg/kg, respectively) resulted in a more than proportional increase in the level of rifampin exposure in plasma compared to that achieved with the 600-mg (10-mg/kg) dose and were safe and well tolerated when combined with other first-line anti-TB drugs for 2 months, but they did not result in an improved bacteriological response in patients with pulmonary TB. These findings from HIGHRIF study 2 warrant the further evaluation of higher doses of rifampin in a larger study population and led to the evaluation of higher doses of rifampin in a follow-up trial (PanACEA-MAMS-TB-01).
MATERIALS AND METHODS
Research sites and ethics.
This study (ClinicalTrials.gov identifier NCT00760149) was conducted at the Kilimanjaro Clinical Research Institute (KCRI), Kilimanjaro Christian Medical Centre (KCMC), and affiliated study sites (Kibong'oto National Tuberculosis Hospital and Mawenzi Regional Hospital, Kilimanjaro region, Tanzania) and at the Ifakara Health Institute (IHI)/Bagamoyo Research and Training Centre (BRTC) and affiliated study sites (Bagamoyo and Dar es Salam, Tanzania).
Approval for this study was obtained from the Kilimanjaro Christian Medical College Research Ethics and Review Committee (CRERC), the Ifakara Health Institute Institutional Review Board (IHI-IRB), and the Tanzanian National Health Research Ethics Subcommittee (NatHREC).
Patients.
Patients were included in the trial if they were between the ages of 18 and 65 years, had newly diagnosed pulmonary TB (confirmed by a positive smear of at least two spontaneously produced specimens using Ziehl-Neelsen [ZN] staining), and provided informed consent. Female patients agreed to take measures to prevent pregnancy during the initial phase of treatment. Exclusion criteria included treatment with anti-TB drugs in the past 3 years; a body weight of less than 50 kg; ALT or AST levels >3 times the upper limit of normal; clinical liver disease (presenting with nausea, jaundice, and tender hepatomegaly); a serum creatinine level higher than the upper limit of normal; a relevant history or current condition that might interfere with drug absorption, distribution, metabolism, or excretion; the use of antiretroviral treatment or the expectation of the use of antiretroviral treatment within 2 months; a Karnofsky score of <40; pregnancy or breast-feeding; and rifampin-resistant or multidrug-resistant (MDR) TB.
Study design.
The study was a double-blind, randomized, placebo-controlled, phase II clinical trial. After stratification for gender and HIV status, eligible patients were randomized to 600 mg, 900 mg, or 1,200 mg rifampin as an oral daily dose during the 2-month intensive phase.
Randomization occurred in blocks of 6 patients containing 2 subjects in each arm. A randomization list was created using the ASELECT formula in Microsoft Excel 2007 software. In accordance with the guidelines of the Tanzanian National TB Program, additional treatment of TB during the intensive phase further consisted of isoniazid (300 mg), pyrazinamide (1,600 mg), and ethambutol (1,100 mg) once daily for 2 months.
Dependent on the study site, subjects were hospitalized for the intensive phase of treatment or were treated at home. During hospitalization, medication intake was directly observed by study staff. If the subjects were not hospitalized, treatment adherence was verified by home-based or facility-based directly observed therapy (DOT), meaning that patients were instructed to take their medication after breakfast in the presence of a counselor or a nurse. Furthermore, administration of study drug was documented with dispensing and drug accountability forms.
Drug treatment.
TB treatment during the intensive phase was administered as fixed-dose combination (FDC) tablets containing rifampin, isoniazid, pyrazinamide, and ethambutol (the tablets contained these four drugs at 150, 75, 400, and 275 mg, respectively) manufactured by Sandoz, Mumbai, India, and donated to the Tanzanian TB program by Novartis through the WHO Global Drug Facility, which monitors the quality of anti-TB drugs according to WHO standards. The study medication consisted of four of these FDC tablets together with either two capsules of placebo (600-mg arm), one capsule with 300 mg rifampin (Rifadin; Sanofi-Aventis, France) plus one capsule of placebo (900 mg arm), or two capsules with 300 mg rifampin (1,200-mg arm).
The study medications were taken together once daily in the morning 7 days per week with a glass of water. In the case of gastrointestinal adverse effects, patients were allowed to take the pills with a light meal.
After the intensive phase, all patients were treated according to Tanzanian guidelines; i.e., they received 300 mg isoniazid and 600 mg rifampin daily 7 days per week for 4 months.
Pharmacokinetics.
A full pharmacokinetic curve was recorded at steady state after 6 weeks of treatment in hospitalized patients (day 39 ± 3), more specifically, in 23 patients in the 600-mg arm and 20 patients each in the 900- and 1,200-mg arms. After an overnight fast, patients took all anti-TB drugs with a standardized meal. Serial venous blood samples were collected predosing and at 1, 1.5, 2, 2.5, 3, 4, 6, 8, 10, and 24 h after witnessed drug intake (the sample collected at 24 h postdosing was only from hospitalized patients). Plasma was separated within 1 h of collection of blood and was frozen at −20°C, transferred to −80°C within 72 h, and transported on dry ice to the Radboud University Medical Center, Nijmegen, the Netherlands, for bioanalysis.
Total (protein bound plus unbound) plasma concentrations of rifampin, isoniazid, and pyrazinamide were assessed by validated ultraperformance liquid chromatographic (UPLC) methods at the laboratory of the Department of Pharmacy, Radboud University Medical Center, Nijmegen, the Netherlands. The bioanalysis of ethambutol was not prioritized, as ongoing efforts to shorten TB treatment have mostly focused on replacement of the relatively weak anti-TB drug ethambutol with more promising anti-TB drugs (24). The accuracy for standard rifampin concentrations was between 99.5 and 101.2%, depending on the concentration. The intraday and interday coefficients of variation were 0.7 to 3.0% and 0.2 to 0.9%, respectively, and the range of detection of the method was 0.13 to 31.0 mg/liter. For isoniazid, accuracy was between 99.4 and 108.8%, the intra- and interday coefficients of variation were 1.8 to 12.6% and 0 to 2.7%, respectively, and the range of detection of the method was 0.025 to 15.1 mg/liter. Finally, for pyrazinamide, accuracy was between 100.0 and 102.1%, intra- and interday coefficients of variation were 1.8 to 4.0% and 0.0 to 2.0%, respectively, and the range of detection of the method was 0.2 to 60.1 mg/liter.
Subsequently, a noncompartmental analysis with WinNonLin (version 6.3) software (Pharsight Corp., Mountain View, CA, USA) was performed to compute the values of the pharmacokinetic parameters for rifampin, as described previously (11).
Safety and tolerability.
Physical examination, testing of vital signs (including systolic and diastolic blood pressure and heart rate), assessment of weight, biochemistry testing (AST, ALT, γ-glutamyl transpeptidase, alkaline phosphatase, and creatinine), and hematology testing (hemoglobin, hematocrit, red cell count, platelet count, total white cell count with differential) took place at screening and after 1, 2, 3, 4, 6, 8, 10, and 12 weeks of treatment. In addition, at these visits the safety history was assessed and concomitant medication was registered. Determination of fasting blood glucose levels was performed only at screening.
Investigators reviewed each adverse event and assessed its relationship to drug treatment as unrelated, possibly related, or related according to predefined definitions.
The severity of adverse events, including laboratory abnormalities, was classified following the U.S. National Institutes of Health Common Terminology Criteria for Adverse Events (CTCAE; version 4.0) (25) and reviewed by a data safety monitoring board.
Microbiological assessments.
Spot sputum samples were collected for Ziehl-Neelsen (ZN) staining and direct microscopy before enrollment. For confirmation of the diagnosis, isolates from screening cultures were identified as members of the M. tuberculosis complex using an AccuProbe assay (Gen-Probe, San Diego, CA, USA) or a BD MGIT TBc identification test (BD Diagnostics, Sparks, MD, USA). These screening isolates were also tested for susceptibility to streptomycin, isoniazid, rifampin, and ethambutol by a commercial broth macrodilution test for the mycobacterial growth indicator tube (MGIT) platform (SIRE; BD Bioscience, Erembodegem, Belgium) or a GenoType MTBDRplus assay (Hain Lifescience GmbH, Nehren, Germany).
Pooled overnight (16 h) sputum samples were collected at the baseline and at days 2, 4, 7, 14, 21, 28, 35, 42, 49, and 56. All samples were processed for culture in the MGIT automated liquid culture system as well as for culture on solid Lowenstein-Jensen medium (LJ); the latter was incubated for 8 weeks at 37°C. Pooled overnight sputum samples were also processed for counting of the colonies in serial sputum samples; samples were diluted and plated in quadruplicate on selective Middlebrook 7H11 plates. The numbers of CFU per milliliter of sputum were calculated after a minimum of 21 days of incubation at 37°C. In addition, the time to positivity (TTP) of the MGIT liquid culture was determined for these samples to assess the change in TTP over time.
The study endpoints were defined as the time to culture conversion on LJ and in MGIT and the proportion of patients that achieved culture conversion after 4, 6, 8, 10, and 12 weeks, in addition to the change in TTP in MGIT and the number of log CFU on Middlebrook 7H11 plates over time.
All sputum samples for microbiology were processed in the research microbiological laboratory of KCMC. Procedures were performed according to the REMoxTB laboratory and quality manuals (26).
Statistical analysis.
On the basis of experience obtained during a pharmacokinetic study of anti-TB drugs in Botswana (27), it was estimated that 20 patients per arm would be needed to detect an increase in total rifampin exposure in plasma of at least 35%, defined from the area under the concentration-versus-time curve up to 24 h after the dose (AUC0–24), after an increase in the rifampin dose from 600 to 900 mg and from 900 to 1,200 mg (one-sided type I error, 0.05; 80% power; between-arm t test). To obtain a more thorough insight into the safety and tolerability of higher than standard doses of rifampin, the total number of patients per study arm was targeted at 50.
Pharmacokinetic parameters were presented descriptively and were log transformed before further statistical analysis. Between-group differences in pharmacokinetic parameters were tested by one-way analysis of variance (ANOVA) or the Kruskal-Wallis test for the time to the maximum plasma concentration (Tmax). Univariable and multivariable linear regression analyses were performed for each of the arms separately to assess the effects of gender, age, and body weight on the AUC0–24 and the maximum plasma concentration (Cmax) of rifampin.
Safety was expressed as the incidence of adverse events by severity and relatedness, according to the CTCAE criteria (25). The number of patients experiencing each event classified by system organ class (SOC) was further specified for grade 3 adverse events.
The time to culture conversion between arms was compared using Cox proportional hazards regression and by plotting Kaplan-Meier survival estimates. Cox proportional hazards regression was also used to evaluate the effect of the actual rifampin dose received (in milligrams per kilogram) and the rifampin exposures achieved (AUC0–24 and Cmax). The effect of exposure was evaluated by dichotomizing each continuous parameter and by entering total and peak exposures as continuous variables. The AUC0–24 or Cmax values of rifampin, isoniazid, or pyrazinamide were compared between those with and those without grade 3 to 5 adverse events using an independent-groups t test.
The distribution of TTP in MGIT was positively skewed, with the log-transformed TTP more closely following a symmetric normal distribution. Mixed-effects models, including the rifampin dose and treatment day interaction terms, were used to evaluate the effect of dose on the change in the number of log CFU and the log TTP over time. A number of log CFU of 0.5 or a TTP of 30 days was used for negative cultures in this analysis up to and including the first negative culture after the last positive culture. Subsequent negative cultures were excluded from the mixed-effects analysis.
Statistical evaluations were performed with IBM SPSS Statistics for Windows software, version 22, and Stata software, version 14.0.
ACKNOWLEDGMENTS
Capsules with 300 mg rifampin (Rifadin) and placebo were donated by Sanofi-Aventis, France. The capsules were repackaged and relabeled by the Department of Pharmacy at the Radboud University Medical Center. This study was funded by NACCAP, a Dutch contribution to the European and Developing Countries Clinical Trials Partnership (EDCTP) that enabled the African Poverty Related Infection Oriented Research Initiative (APRIORI), and by the European and Developing Countries Clinical Trials Partnership (EDCTP) that funded the Pan-African Consortium for the Evaluation of Antituberculosis Antibiotics (PanACEA Consortium; project code IP.2007.32011.012 [HIGHRIF]) and Rapid Evaluation of Moxifloxacin in Tuberculosis (REMoxTB for microbiology support; project code IP.2007.32011.011).
We thank the study team, including the physicians, nurses, pharmacists, laboratory technicians, drivers, and administrative units, who participated in this study.
The Pan African Consortium for the Evaluation of Antituberculosis Antibiotics (PanACEA) comprises the following institutions (and the indicated individuals at those institutions): Medical Centre of the University of Munich, Munich, Germany (Anna Maria Mekota, Norbert Heinrich, Andrea Rachow, Elmar Saathoff, Michael Hoelscher); University of St. Andrews, St. Andrews, United Kingdom (Stephen Gillespie); Radboud University Medical Center, Nijmegen, the Netherlands (Georgette Plemper van Balen, Marloes Weijers, Angela Colbers, Rob Aarnoutse, Martin Boeree); University College of London, London, United Kingdom (Anna Bateson, Timothy McHugh, Kasha Singh, Robert Hunt, Alimuddin Zumla); MRC Clinical Trials Unit at UCL, London, United Kingdom (Andrew J. Nunn, Patrick P. J. Phillips, Sunita Rehal); University of Cape Town, Cape Town, South Africa (Rodney Dawson, Kim Narunsky); University of Stellenbosch, Cape Town, South Africa (Andreas Diacon, Jeannine du Bois, Amour Venter, Sven Friedrich); University of the Witwatersrand, Johannesburg, South Africa (Ian Sanne, Karla Mellet, Eefje de Jong); The Aurum Institute, Johannesburg, South Africa (Gavin Churchyard, Salome Charalambous, Nomagugu Ndlovu, Vinodh Edward, Madulagotla Sebe, Lungile Mbata, Robert Wallis); University of Zambia, Lusaka, Zambia (Peter Mwaba); NIMR-Mbeya Medical Research Centre, Mbeya, Tanzania (Nyanda Elias Ntinginya, Chacha Mangu, Christina Manyama, Gabriel Rojas-Ponce, Bariki Mtafya, Leonard Maboko); Ifakara Health Institute, Bagamoyo, Tanzania (Frederick Haraka, Alphonce Kelemani, Khadija Said, Mohamed Sasamalo); Swiss Tropical and Public Health Institute, Basel, Switzerland, and University of Basel, Basel, Switzerland (Klaus Reither, Levan Jugheli); Kilimanjaro Clinical Research Institute, Moshi, Tanzania (Noel Sam, Gibson Kibiki, Hadija Semvua, Stellah Mpagama); Medical Research Unit, Albert Schweitzer Hospital, Lambarene, Gabon (Abraham Alabi, Ayola Akim Adegnika); Kenya Medical Research Institute, Nairobi, Kenya (Evans Amukoye); and Makerere University, Kampala, Uganda (Alphonse Okwera).
REFERENCES
- 1.WHO. 2016. Global tuberculosis report 2016. WHO, Geneva, Switzerland: http://www.who.int/tb/publications/global_report/en/. [Google Scholar]
- 2.Fox W, Ellard GA, Mitchison DA. 1999. Studies on the treatment of tuberculosis undertaken by the British Medical Research Council tuberculosis units, 1946–1986, with relevant subsequent publications. Int J Tuberc Lung Dis 3(Suppl 2):S231–S279. [PubMed] [Google Scholar]
- 3.van Ingen J, Aarnoutse RE, Donald PR, Diacon AH, Dawson R, Plemper van Balen G, Gillespie SH, Boeree MJ. 2011. Why do we use 600 mg of rifampicin in tuberculosis treatment? Clin Infect Dis 52:e194–e199. doi: 10.1093/cid/cir184. [DOI] [PubMed] [Google Scholar]
- 4.Gumbo T, Louie A, Deziel MR, Liu W, Parsons LM, Salfinger M, Drusano GL. 2007. Concentration-dependent Mycobacterium tuberculosis killing and prevention of resistance by rifampin. Antimicrob Agents Chemother 51:3781–3788. doi: 10.1128/AAC.01533-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jayaram R, Gaonkar S, Kaur P, Suresh BL, Mahesh BN, Jayashree R, Nandi V, Bharat S, Shandil RK, Kantharaj E, Balasubramanian V. 2003. Pharmacokinetics-pharmacodynamics of rifampin in an aerosol infection model of tuberculosis. Antimicrob Agents Chemother 47:2118–2124. doi: 10.1128/AAC.47.7.2118-2124.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Steenwinkel JE, Aarnoutse RE, de Knegt GJ, ten Kate MT, Teulen M, Verbrugh HA, Boeree MJ, van Soolingen D, Bakker-Woudenberg IA. 2013. Optimization of the rifampin dosage to improve the therapeutic efficacy in tuberculosis treatment using a murine model. Am J Respir Crit Care Med 187:1127–1134. doi: 10.1164/rccm.201207-1210OC. [DOI] [PubMed] [Google Scholar]
- 7.Rosenthal IM, Tasneen R, Peloquin CA, Zhang M, Almeida D, Mdluli KE, Karakousis PC, Grosset JH, Nuermberger EL. 2012. Dose-ranging comparison of rifampin and rifapentine in two pathologically distinct murine models of tuberculosis. Antimicrob Agents Chemother 56:4331–4340. doi: 10.1128/AAC.00912-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hu Y, Liu A, Ortega-Muro F, Alameda-Martin L, Mitchison D, Coates A. 2015. High-dose rifampicin kills persisters, shortens treatment duration, and reduces relapse rate in vitro and in vivo. Front Microbiol 6:641. doi: 10.3389/fmicb.2015.00641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Steingart KR, Jotblad S, Robsky K, Deck D, Hopewell PC, Huang D, Nahid P. 2011. Higher-dose rifampin for the treatment of pulmonary tuberculosis: a systematic review. Int J Tuberc Lung Dis 15:305–316. [PubMed] [Google Scholar]
- 10.Peloquin C. 2003. What is the ‘right’ dose of rifampin? Int J Tuberc Lung Dis 7:3–5. [PubMed] [Google Scholar]
- 11.Ruslami R, Nijland HM, Alisjahbana B, Parwati I, van Crevel R, Aarnoutse RE. 2007. Pharmacokinetics and tolerability of a higher rifampin dose versus the standard dose in pulmonary tuberculosis patients. Antimicrob Agents Chemother 51:2546–2551. doi: 10.1128/AAC.01550-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Diacon AH, Patientia RF, Venter A, van Helden PD, Smith PJ, McIlleron H, Maritz JS, Donald PR. 2007. Early bactericidal activity of high-dose rifampin in patients with pulmonary tuberculosis evidenced by positive sputum smears. Antimicrob Agents Chemother 51:2994–2996. doi: 10.1128/AAC.01474-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boeree MJ, Diacon AH, Dawson R, Narunsky K, du Bois J, Venter A, Phillips PP, Gillespie SH, McHugh TD, Hoelscher M, Heinrich N, Rehal S, van Soolingen D, van Ingen J, Magis-Escurra C, Burger D, Plemper van Balen G, Aarnoutse RE. 2015. A dose-ranging trial to optimize the dose of rifampin in the treatment of tuberculosis. Am J Respir Crit Care Med 191:1058–1065. doi: 10.1164/rccm.201407-1264OC. [DOI] [PubMed] [Google Scholar]
- 14.Acocella G. 1978. Clinical pharmacokinetics of rifampicin. Clin Pharmacokinet 3:108–127. doi: 10.2165/00003088-197803020-00002. [DOI] [PubMed] [Google Scholar]
- 15.Tostmann A, Mtabho CM, Semvua HH, van den Boogaard J, Kibiki GS, Boeree MJ, Aarnoutse RE. 2013. Pharmacokinetics of first-line tuberculosis drugs in Tanzanian patients. Antimicrob Agents Chemother 57:3208–3213. doi: 10.1128/AAC.02599-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chirehwa MT, Rustomjee R, Mthiyane T, Onyebujoh P, Smith P, McIlleron H, Denti P. 2015. Model-based evaluation of higher doses of rifampin using a semimechanistic model incorporating autoinduction and saturation of hepatic extraction. Antimicrob Agents Chemother 60:487–494. doi: 10.1128/AAC.01830-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smythe W, Khandelwal A, Merle C, Rustomjee R, Gninafon M, Bocar Lo M, Sow OB, Olliaro PL, Lienhardt C, Horton J, Smith P, McIlleron H, Simonsson US. 2012. A semimechanistic pharmacokinetic-enzyme turnover model for rifampin autoinduction in adult tuberculosis patients. Antimicrob Agents Chemother 56:2091–2098. doi: 10.1128/AAC.05792-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burman WJ, Gallicano K, Peloquin C. 2001. Comparative pharmacokinetics and pharmacodynamics of the rifamycin antibacterials. Clin Pharmacokinet 40:327–341. doi: 10.2165/00003088-200140050-00002. [DOI] [PubMed] [Google Scholar]
- 19.Niemi M, Backman JT, Fromm MF, Neuvonen PJ, Kivisto KT. 2003. Pharmacokinetic interactions with rifampicin: clinical relevance. Clin Pharmacokinet 42:819–850. doi: 10.2165/00003088-200342090-00003. [DOI] [PubMed] [Google Scholar]
- 20.Solera J, Rodriguez-Zapata M, Geijo P, Largo J, Paulino J, Saez L, Martinez-Alfaro E, Sanchez L, Sepulveda MA, Ruiz-Ribo MD, the GECMEI Group. 1995. Doxycycline-rifampin versus doxycycline-streptomycin in treatment of human brucellosis due to Brucella melitensis. Antimicrob Agents Chemother 39:2061–2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kochar DK, Aseri S, Sharma BV, Bumb RA, Mehta RD, Purohit SK. 2000. The role of rifampicin in the management of cutaneous leishmaniasis. QJM 93:733–737. doi: 10.1093/qjmed/93.11.733. [DOI] [PubMed] [Google Scholar]
- 22.Kissling M, Bergamini N. 1981. Rifampicin in free combination with other antimicrobial drugs in non-Tb infections. Clinical data on 650 patients (a review). Chemotherapy 27:368–402. [DOI] [PubMed] [Google Scholar]
- 23.Boeree MJ, Heinrich N, Aarnoutse R, Diacon AH, Dawson R, Rehal S, Kibiki GS, Churchyard G, Sanne I, Ntinginya NE, Minja LT, Hunt RD, Charalambous S, Hanekom M, Semvua HH, Mpagama SG, Manyama C, Mtafya B, Reither K, Wallis RS, Venter A, Narunsky K, Mekota A, Henne S, Colbers A, van Balen GP, Gillespie SH, Phillips PP, Hoelscher M. 2017. High-dose rifampicin, moxifloxacin, and SQ109 for treating tuberculosis: a multi-arm, multi-stage randomised controlled trial. Lancet Infect Dis 17:39–49. doi: 10.1016/S1473-3099(16)30274-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gillespie SH. 2016. The role of moxifloxacin in tuberculosis therapy. Eur Respir Rev 25:19–28. doi: 10.1183/16000617.0085-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.National Cancer Institute. Common terminology criteria for adverse events 4.0 (CTCAE). Division of Cancer Treatment and Diagnosis, National Cancer Institute, Rockville, MD: http://ctep.cancer.gov/protocolDevelopment/electronic_applications/ctc.htm. [Google Scholar]
- 26.Global Alliance for TB Drug Development. 2011. REMox clinical TB trial laboratory manual. Global Alliance for TB Drug Development, New York, NY: https://www.tballiance.org/portfolio/trial/5093. [Google Scholar]
- 27.Tappero JW, Bradford WZ, Agerton TB, Hopewell P, Reingold AL, Lockman S, Oyewo A, Talbot EA, Kenyon TA, Moeti TL, Moffat HJ, Peloquin CA. 2005. Serum concentrations of antimycobacterial drugs in patients with pulmonary tuberculosis in Botswana. Clin Infect Dis 41:461–469. doi: 10.1086/431984. [DOI] [PubMed] [Google Scholar]