

ABSTRACT
Favipiravir is an antiviral agent effective against several RNA viruses. The drug has been shown to protect mice against experimental infection with a lethal dose of West Nile virus (WNV), a mosquito-borne flavivirus responsible for outbreaks of meningitis and encephalitis for which no antiviral therapy has been licensed; however, the mechanism of action of the drug is still not well understood. Here, we describe the potent in vitro antiviral activity of favipiravir against WNV, showing that it decreases virus-specific infectivity and drives the virus to extinction. Two passages of WNV in the presence of 1 mM favipiravir—a concentration that is more than 10-fold lower than its 50% cytotoxic concentration (CC50)—resulted in a significant increase in mutation frequency in the mutant spectrum and in a bias toward A→G and G→A transitions relative to the population passaged in the absence of the drug. These data, together with the fact that the drug is already licensed in Japan against influenza virus and in a clinical trial against Ebola virus, point to favipiravir as a promising antiviral agent to fight medically relevant flaviviral infections, such as that caused by WNV.
KEYWORDS: favipiravir, lethal mutagenesis, West Nile virus, antiviral agents, flavivirus
INTRODUCTION
West Nile virus (WNV) is an enveloped plus-strand RNA neurotropic flavivirus transmitted by mosquitoes that constitutes a worrisome threat to global human and animal health (1). About 80% of human infections are asymptomatic, and of the 20% that display disease symptoms, 1% progress to severe neurological disease, including meningitis, encephalitis, and flaccid paralysis, which can be fatal (2). Nowadays, no specific drug is licensed for the control of any flavivirus, including WNV, and current treatment is only palliative (3).
RNA viruses encode an RNA-dependent RNA polymerase (RdRp) that lacks proofreading activity (4). The intrinsic error rates of flaviviral RNA polymerases have been established as ∼1 mutation per genome and round of copying (5), which is the range estimated for other RNA viruses and seems adequate to maintain viral fitness. Variation of template-copying fidelity often entails a fitness cost for RNA viruses (6). An increase in replication fidelity (i.e., decreasing mutation rates of RdRp) restricts genetic diversity and limits phenotypic plasticity, which is particularly disadvantageous for arthropod-borne viruses, as they require successful infection, replication, and transmission in taxonomically divergent vertebrate and invertebrate hosts. Conversely, an induced decrease in replication fidelity lowers population fitness through the irreversible accumulation of deleterious and lethal mutations (7). Virus extinction through increased mutation rates is known as lethal mutagenesis. This antiviral strategy has been studied recently in the search of antiviral compounds that exploit the high error rates of RNA viruses, pushing them above an error threshold incompatible with viability (8). The purine analogue ribavirin reduced WNV infectivity and viral RNA synthesis, with a significant increase of transition mutations, suggesting error catastrophe as one of the modes of action of ribavirin against WNV (9).
Favipiravir (6-fluoro-3-hydroxy-2-pyrazinecarboxamide) is a broad-spectrum nucleoside analogue (10) that shows potent antiviral activity against several RNA viruses (11–28). It is known that intracellular host enzymes act upon favipiravir, converting it to its active form (favipiravir-4-ribofuranosyl-5′-triphosphate), which functions as a purine nucleotide analog (29). However, its antiviral mechanisms of action are still controversial, with some reports supporting that it directly inhibits the RdRp (11, 15, 29–32) and others favoring that it acts through a lethal mutagenesis mechanism (33–37). Here we quantify the antiviral activity of favipiravir on WNV infection in cell culture and provide for the first time evidence supporting lethal mutagenesis as at least part of its mode of action. The results open the possibility to apply lethal mutagenesis to combat WNV infections.
RESULTS
Antiviral activity and cellular toxicity of favipiravir in vitro.
To test the effect of favipiravir on WNV infectious progeny production, Vero cells were infected in the presence of different concentrations of the drug (50 to 250 μM) and the virus titer was determined by plaque assay at 48 h postinfection (p.i.). Favipiravir significantly reduced the virus yield in a dose-dependent manner (Fig. 1A), with a 50% inhibitory concentration (IC50) of 51.2 ± 6.5 μM.
FIG 1.

Reduction of WNV yield and cytotoxicity of favipiravir. (A) Percentage of WNV viral titer upon favipiravir treatment. Vero cells were pretreated for 16 h with the indicated favipiravir concentration and then infected with WNV (MOI, 0.1 PFU/cell) in the presence of the same drug concentration; at 48 h p.i., the cell culture supernatants were titrated by plaque assay, and the 50% inhibitory concentration (IC50) was determined. (B) Percentage of viable Vero cells after favipiravir treatment. The 50% cytotoxic concentration was determined by measuring the cellular ATP content at 48 h posttreatment. Data are presented as the mean ± standard deviation (SD) from three independent determinations of both IC50 and CC50.
To exclude the possibility that the observed antiviral activity of favipiravir could be due to an effect on the cellular polymerase or a metabolic inhibition of Vero cells, the drug-induced cytotoxicity was assayed. No apparent effect was observed by light microscopy in infected cells treated with the highest drug concentration after 48 h (data not shown), and the 50% cytotoxic concentration (CC50) was >10,000 μM (Fig. 1B), indicating that favipiravir has low toxicity under the tested conditions. These data resulted in a selective index (SI [CC50/IC50]), of over 195.
Favipiravir reduces virus-specific infectivity in vitro.
Based on the antiviral activity and cytotoxicity of favipiravir, serial passages of WNV in Vero cells were carried out in the presence of different drug concentrations (0.5 to 2 mM). All tested concentrations decreased viral infectivity, measured by plaque assay, although 0.5 mM favipiravir was unable to completely inhibit the infection, even when the drug was maintained for 5 consecutive passages (Fig. 2A). Then viral RNA present in the supernatants of cells infected in the absence or presence of 1 mM favipiravir was quantified by quantitative reverse transcription-PCR (qRT-PCR). Whereas the viral RNA load in the supernatant of cells infected in the absence of favipiravir remained almost invariable after 5 passages, viral RNA when favipiravir was present drastically decreased after just one passage and was undetectable beginning on the third passage (Fig. 2B). The specific infectivity, determined as the ratio between the number of infectious viruses and the total amount of viral RNA, was significantly reduced (Fig. 2C). No infectivity or viral RNA was rescued when the cell culture supernatant from the fifth passage in the presence of 1 mM favipiravir was subjected to five additional passages in the absence of the drug.
FIG 2.

Antiviral activity of favipiravir against WNV in Vero cells. (A) Viral titers of WNV after each passage (n = 5) on Vero cells in the presence or absence of favipiravir. Virus yield in the supernatant of cells was titrated by plaque assay. (B) WNV RNA was extracted from the supernatants of untreated or 1 mM favipiravir-treated Vero cells after each passage (n = 5) and quantified in triplicate by qRT-PCR. (C) Specific infectivity of WNV in the presence of 1 mM favipiravir, calculated as the ratio between the number of infectious viruses and the total amount of viral RNA. Data are presented as the mean ± SD. Statistically significant differences are indicated: **, P < 0.005, and ***, P < 0.0001, by two-way ANOVA with Bonferroni's correction.
Mutagenic effect of favipiravir on WNV.
Since a reduction in specific infectivity is a hallmark of lethal mutagenesis, the mechanism behind the observed viral extinction induced by favipiravir was investigated at the level of nucleotide sequences of viral RNA. For this purpose, a fragment (909 bp) from the relatively conserved NS5 viral genomic region of WNV (38), which encodes the viral RdRp, was amplified by RT-PCR from the supernatant of infected cells at the second viral passage in absence or presence of 1 mM favipiravir, cloned, and bidirectionally sequenced. A total of 43 and 48 clones were sequenced from the populations passaged in presence and absence of favipiravir, respectively, representing a total of 39,087 and 43,632 nucleotides sequenced. Favipiravir induced a statistically significant 5.6-fold increase of mutation frequency in the mutant spectrum (Fig. 3). A significant difference was also found in the proportion of mutated clones: 0.74 (32/43) with favipiravir and 0.20 (10/48) without it. No modification of the consensus sequence of the populations passaged in the presence or absence of favipiravir was noted.
FIG 3.
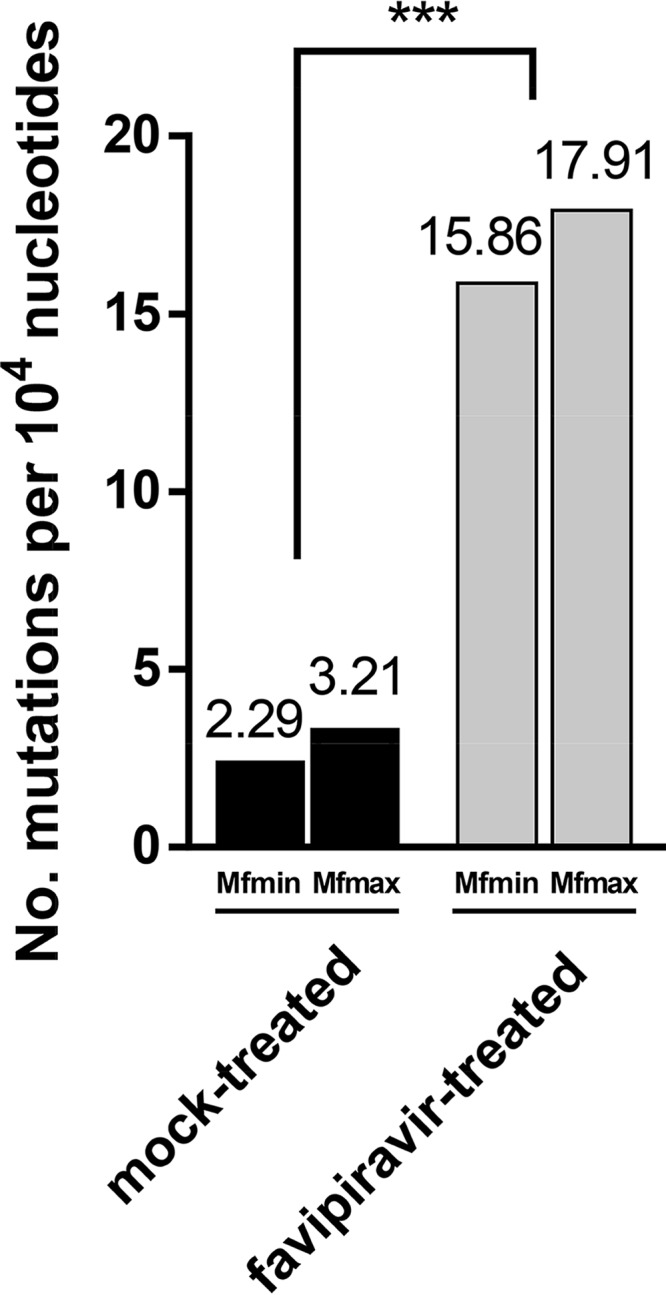
Increased WNV mutation frequency induced by favipiravir. The minimum (Mfmin) and maximum (Mfmax) mutation frequencies of a 909-bp fragment of the NS5 gene amplified by RT-PCR from WNV were determined from supernatants of infected Vero cells after two passages in the absence or presence of 1 mM favipiravir. The mean number of mutations per 104 nucleotides sequenced is displayed. The minimum mutation frequency is the number of different mutations found in the mutant spectrum relative to the consensus sequence of the corresponding population divided by the total number of nucleotides sequenced and normalized to 104 nucleotides. The maximum mutation frequency is the number of total mutations found in the mutant spectrum relative to the consensus sequence of the corresponding population divided by the total number of nucleotides sequenced and normalized to 104 nucleotides. Statistically significant difference is indicated: ***, P < 0.0001, by χ2 test with Yates' correction.
In addition to the difference in point mutations, one deletion (G at position 8294) was detected in the genomic region analyzed of the population passaged in the presence of favipiravir. In line with the ambiguous purine-pseudobase feature of favipiravir (39), a significant increase of G→A, A→G, and, to a lesser extent, U→C mutations was observed (Table 1). No differences in mutation hot spots, ratios of synonymous to nonsynonymous mutations, or nucleotide types present at the 3′ or 5′ side of the mutated sites were noted (Fig. 4), suggesting that the observed mutagenesis was basically random. The results demonstrate a mutagenic activity of favipiravir for WNV and suggest that the observed viral extinction was associated with an accumulation of mutations that led to an unviable viral progeny. We conclude that the broad-spectrum antiviral agent favipiravir can act as a lethal mutagen for WNV.
TABLE 1.
Type of mutations found in WNV populations untreated or treated with 1 mM favipiravira
| WNV population | No. of mutated clones/total | No. of deletions | No. of mutationsb |
Total nucleotides sequenced | |||
|---|---|---|---|---|---|---|---|
| A→G | U→C | G→A | C→U | ||||
| Untreated | 10/48 | 0 | 4 (0.01) | 2 (0.01) | 0 (0.00) | 8 (0.04) | 43,632 |
| Treated | 32/43** | 1 | 26** (0.1)* | 9* (0.05) | 21** (0.08)* | 13 (0.06) | 39,087 |
Shown are the number of mutated clones and type of mutated nucleotides observed in the untreated or favipiravir-treated WNV population relative to the consensus sequence. The proportion of each type of mutation normalized to the number of each nucleotide type in the 909-base fragment sequenced is given in parentheses.
Statistically significant differences compared to untreated samples are indicated: *, P = 0.05, and **, P < 0.0001, by Fisher's exact test and χ2 test with Yates' correction, respectively.
FIG 4.

Distribution of mutations induced by favipiravir. Schematic representation of the number of clones with point mutations found in the mutant spectrum of the WNV NS5 gene fragment analyzed in the absence (A and B) or presence (C and D) of 1 mM favipiravir. Nucleotide positions of synonymous (open triangles in panels A and C) and nonsynonymous (solid circles in panels B and D) mutations are displayed. Pie charts representing the percentages of synonymous (black) and nonsynonymous (gray) mutations are included.
DISCUSSION
The mechanism of favipiravir activity as a broad-spectrum antiviral nucleoside analogue is still controversial, as both direct inhibition of the viral RdRp (11, 15, 29–32) and a lethal mutagenesis activity that increases the virus mutation frequency (33–37) have been proposed. It is difficult to discern whether an inhibitory or mutagenic activity prevails in the antiviral activity of a nucleoside analogue: there are several reasons for the difficulty, including that a mutagenic activity per se often entails inhibition of viral progeny production. It is possible that, depending on the drug concentration and on the RdRp targeted by favipiravir-4-ribofuranosyl-5′-triphosphate, an inhibitory or mutagenic activity may predominate. In fact, even with picornaviral RdRps that show remarkable amino acid sequence identity, the same amino acid substitution can have very different effects on the recognition of mutagenic nucleotide analogues (compare references 40 and 41). Studies of favipiravir-induced mutagenesis with purified RdRps may help in settling this issue.
Favipiravir administered orally (200 mg/kg of body weight) twice a day protected mice against experimental infection with WNV and reduced WNV RNA loads in animal tissues (42). Similar results were observed in a hamster treated with the same doses of favipiravir and infected with yellow fever virus (YFV) (43), a flavivirus also transmitted by mosquitoes, but the underlying mechanism was not addressed in any of these reports. Here, the mechanism of favipiravir inhibition of WNV multiplication in vitro has been analyzed. First, once it was established that favipiravir presented a low toxicity in Vero cells, its anti-WNV capacity was assayed using a wide range of drug concentrations. The CC50, IC50, and SI values suggest that favipiravir can be a good anti-WNV candidate, in agreement with its in vivo effect (42). These results are in line with those first reported for influenza virus (10, 29) and then for other RNA viruses, such as filoviruses (44), bunyaviruses (45), and hantaviruses (46).
To assess whether the anti-WNV activity of favipiravir could be associated with a mutagenic activity, the virus was serially propagated in Vero cells in the presence of different concentrations of the drug. At 2 mM, the virus was no longer detected after just one passage, whereas at 1 mM, it was undetectable by the third passage—results that are similar to those reported for influenza virus (34). Likewise, no virus was recovered after 10 serial passages in which the drug was present only during the first five passages. In addition, the specific infectivity, measured as the ratio of viral yield to genomic RNA, was reduced 26- and 95-fold after the first and second passages, respectively, in the presence of 1 mM favipiravir. The consensus sequence remained invariant. All these data, together with the low toxicity observed, point toward WNV extinction being associated with lethal mutagenesis.
Viral RNA from cell culture supernatants after two passages in the absence or presence of 1 mM favipiravir was amplified by RT-PCR, cloned, and sequenced to analyze the mutant spectra of the viral populations. Sequence analysis showed a 5.6-fold increase in the mutation frequency of the mutant spectrum associated with replication in the presence of favipiravir. Consistent with the structure of favipiravir, which renders it a substrate alternative to GTP and ATP (31, 39), a significant increase in G→A (21-fold) and A→G (6.5-fold) frequencies was associated with the presence of favipiravir. Different mutation patterns upon replication in the presence of favipiravir have been reported for other viruses: an enrichment in G→A and C→U mutations was observed in the case of influenza virus (32, 34) and hepatitis C virus (35), a slight increase of A→G and U→C mutations was found for murine norovirus (33), and no obvious outstanding mutation type was found in the case of foot-and-mouth disease virus (37). These differences suggest that the RdRp recognition of favipiravir varies among RNA viruses, again rendering plausible that depending on the virus-host system, an inhibitory or mutagenic activity by favipiravir may prevail. To have elucidated that mutagenesis is at least part of the mechanism of favipiravir against WNV is relevant to the design of effective antiviral strategies against this important pathogen. It is essential to use two or more antiviral agents to delay or prevent selection of RNA virus mutants that escape antiviral interventions. In fact, when one of the agents has a mutagenic activity, maximum efficacy is predicted with the sequential administration of a nonmutagenic inhibitor first, followed by the mutagen, but not the converse (47, 48). Therefore, our study positions favipiravir as an adequate antiviral agent to reach its maximum efficacy not in combination with nonmutagenic inhibitors but as the second drug in a sequential administration protocol.
Overall, our results demonstrate, for the first time, a mutagenic activity as part of the antiviral action of favipiravir on WNV infection in cell culture. This, together with the clinical safety record of favipiravir, which is already licensed in Japan against influenza virus (Avigan; Toyama Chemical Co., Ltd.) and in a clinical trial against Ebola virus (U.S. National Institutes of Health identifier NCT02662855; https://clinicaltrials.gov/ct2/show/NCT02662855) point to favipiravir as an interesting antiviral candidate against WNV and other medically relevant flaviviruses.
MATERIALS AND METHODS
Cells, virus infections, and virus titrations.
Vero CCL-81 cells (ATCC, Manassas, VA) were grown in Eagle's minimal essential medium (EMEM [Lonza, Verviers, Belgium]) supplemented with 5% fetal bovine serum (HyClone; GE Healthcare, United Kingdom), 2 mM l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin (Lonza). Cells were maintained at 37°C and 5% CO2 in a humidified incubator and used at low passage. The origin and passage history of the WNV strain NY99 (GenBank accession no. KC407666) have been described previously (49). For infections in liquid medium, WNV was incubated with Vero CCL-81 cell monolayers for 1 h at 37°C, and then the inoculum was removed and fresh medium containing 2.5% fetal bovine serum was added. The viral titer was determined 48 h postinfection by plaque assay in semisolid agarose medium (50). All infectious virus manipulations were performed in biosafety level 3 (BSL3) facilities.
Favipiravir treatment.
Favipiravir (Atomax Chemicals Co., Ltd., Shenzhen, China) was diluted in distilled H2O at a concentration of 20 mM, sterilized by filtration, and stored at −80°C until use. Vero cells were infected with WNV NY99 as described above in the presence of different concentrations of favipiravir. The 50% inhibitory concentration (IC50) was determined as the concentration required to reduce the number of plaques to 50% relative to the untreated controls. WNV was subjected to serial passages in the presence of different concentrations of favipiravir in Vero cells seeded in 6-well plates (2 × 105 cells per well). In the first passage, cell monolayers were pretreated with the drug (or EMEM in untreated wells) for 16 h prior to infection. Viral adsorption (multiplicity of infection [MOI], 0.01 PFU/cell) was carried out in fresh medium containing the drug for 1 h at 37°C. Particles not attached to the cell were discarded by washing three times with EMEM, and infected cells were left in fresh medium in the presence or absence of favipiravir for 48 h, a time point at which 1/4 of the supernatant was used to infect fresh new monolayers of Vero cells in the presence of the same concentration of the drug. For serial blind passages (n = 10), the same protocol was used, except that no drug was present during the last five passages. Plaque assays were performed as previously described (50).
Selective index determination.
Drug toxicity was examined by measuring the cellular ATP content with the CellTiter-Glo luminescent cell viability assay (Promega, Madison, WI). The 50% cytotoxic concentration (CC50 [i.e., the concentration required to kill 50% of the cell population]) was determined after culturing confluent Vero cell monolayers with a wide range of drug concentrations (from 0 to 10 mM) in triplicate for 48 h. Then the selective index (SI) was calculated as the CC50/IC50 ratio.
RNA extraction and qRT-PCR.
Viral RNA was extracted from clarified supernatants of infected cells using a commercial kit (Speedtools RNA virus extraction kit, Biotools B&M Labs S.A, Madrid, Spain). Quantitative reverse transcription-PCR (qRT-PCR) was performed on a 7500 Fast real-time PCR system (Applied Biosystems, Foster City, CA) using a High Scriptools-Quantimix Easy Probes kit (Biotools) and previously described (51) primers and probe matching the 3′ noncoding (NC) region to ensure detection of full-length RNA genomes. Viral RNA was quantified as genomic equivalents (GE) to PFU by comparison with RNA extracted from previously titrated samples (52, 53).
Viral mutation analysis.
RNA extracted from supernatants of infected cells was amplified by RT-PCR (SuperScript One Step RT-PCR system, Invitrogen, Carlsbad, CA, USA), using specific primers (forward, 5′-CGAACCGGTCGGAAAAGTGA-3′; reverse, 5′-CAAAAACGCCCACAACCAGT-3′; enclosing nucleotides 7893 to 8841 of the WNV strain [GenBank accession no. KC407666]) (49), to yield a 949-bp cDNA fragment of the virus NS5 gene. Amplified fragments were cloned into pGEM-T Easy vector (Promega, Madison, WI), and over 40 positive colonies from each sample were randomly chosen for bidirectional sequencing (Macrogen, The Netherlands). To avoid complexity biases due to redundant amplifications of the same initial RNA templates (54), amplifications were carried out with the undiluted samples only if a 1:1,000 dilution of the sample yielded an amplification band that was visible in stained agarose gels. The sequences were aligned and compared (SeqMan II software in DNAStar Package version 5.01) with the parental virus sequence as a reference, and each point mutation was recorded.
Statistical analyses.
Statistical analysis was performed using Graph Pad Prism for Windows, version 6 (Graph Pad Software, Inc., San Diego, CA, 2005). Two-way analysis of variance (ANOVA) with Bonferroni's correction for multiple comparisons and the chi-square test were used. Statistically significant differences are indicated by asterisks.
ACKNOWLEDGMENTS
We thank M. Calvo for technical assistance.
This work was supported by grants RTA2013-00013-C04-2014 and RTA2015-00009 from INIA to J.-C.S., SAF2014-52400-R from the Spanish Ministry of Economy and Competitiveness (MINECO) to E.D., and S2013/ABI-2906 (PLATESA) from Comunidad de Madrid/FEDER to J.C.-S. and E.D. Work at Centro de Biología Molecular “Severo Ochoa” was also supported by Fundación R. Areces. CIBERehd is funded by Instituto de Salud Carlos III.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
REFERENCES
- 1.Martin-Acebes MA, Saiz JC. 2012. West Nile virus: a re-emerging pathogen revisited. World J Virol 1:51–70. doi: 10.5501/wjv.v1.i2.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sejvar JJ. 2014. Clinical manifestations and outcomes of West Nile virus infection. Viruses 6:606–623. doi: 10.3390/v6020606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kok WM. 2016. New developments in flavivirus drug discovery. Expert Opin Drug Discov 11:433–445. doi: 10.1517/17460441.2016.1160887. [DOI] [PubMed] [Google Scholar]
- 4.Hamer GL, Kitron UD, Goldberg TL, Brawn JD, Loss SR, Ruiz MO, Hayes DB, Walker ED. 2009. Host selection by Culex pipiens mosquitoes and West Nile virus amplification. Am J Trop Med Hyg 80:268–278. [PubMed] [Google Scholar]
- 5.Zhang S, Vogt MR, Oliphant T, Engle M, Bovshik EI, Diamond MS, Beasley DW. 2009. Development of resistance to passive therapy with a potently neutralizing humanized monoclonal antibody against West Nile virus. J Infect Dis 200:202–205. doi: 10.1086/599794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Borderia AV, Rozen-Gagnon K, Vignuzzi M. 2016. Fidelity variants and RNA quasispecies. Curr Top Microbiol Immunol 392:303–322. doi: 10.1007/82_2015_483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van Slyke GA, Arnold JJ, Lugo AJ, Griesemer SB, Moustafa IM, Kramer LD, Cameron CE, Ciota AT. 2015. Sequence-specific fidelity alterations associated with West Nile virus attenuation in mosquitoes. PLoS Pathog 11:e1005009. doi: 10.1371/journal.ppat.1005009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perales C, Domingo E. 2016. Antiviral strategies based on lethal mutagenesis and error threshold. Curr Top Microbiol Immunol 392:323–339. doi: 10.1007/82_2015_459. [DOI] [PubMed] [Google Scholar]
- 9.Day CW, Smee DF, Julander JG, Yamshchikov VF, Sidwell RW, Morrey JD. 2005. Error-prone replication of West Nile virus caused by ribavirin. Antiviral Res 67:38–45. doi: 10.1016/j.antiviral.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 10.Furuta Y, Takahashi K, Fukuda Y, Kuno M, Kamiyama T, Kozaki K, Nomura N, Egawa H, Minami S, Watanabe Y, Narita H, Shiraki K. 2002. In vitro and in vivo activities of anti-influenza virus compound T-705. Antimicrob Agents Chemother 46:977–981. doi: 10.1128/AAC.46.4.977-981.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Furuta Y, Gowen BB, Takahashi K, Shiraki K, Smee DF, Barnard DL. 2013. Favipiravir (T-705), a novel viral RNA polymerase inhibitor. Antiviral Res 100:446–454. doi: 10.1016/j.antiviral.2013.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scharton D, Bailey KW, Vest Z, Westover JB, Kumaki Y, Van Wettere A, Furuta Y, Gowen BB. 2014. Favipiravir (T-705) protects against peracute Rift Valley fever virus infection and reduces delayed-onset neurologic disease observed with ribavirin treatment. Antiviral Res 104:84–92. doi: 10.1016/j.antiviral.2014.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tani H, Fukuma A, Fukushi S, Taniguchi S, Yoshikawa T, Iwata-Yoshikawa N, Sato Y, Suzuki T, Nagata N, Hasegawa H, Kawai Y, Uda A, Morikawa S, Shimojima M, Watanabe H, Saijo M. 2016. Efficacy of T-705 (Favipiravir) in the treatment of infections with lethal severe fever with thrombocytopenia syndrome virus. mSphere 1:e00061-15. doi: 10.1128/mSphere.00061-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yamada K, Noguchi K, Komeno T, Furuta Y, Nishizono A. 2016. Efficacy of favipiravir (T-705) in rabies postexposure prophylaxis. J Infect Dis 213:1253–1261. doi: 10.1093/infdis/jiv586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jochmans D, van Nieuwkoop S, Smits SL, Neyts J, Fouchier RA, van den Hoogen BG. 2016. Antiviral activity of favipiravir (T-705) against a broad range of paramyxoviruses in vitro and against human metapneumovirus in hamsters. Antimicrob Agents Chemother 60:4620–4629. doi: 10.1128/AAC.00709-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gowen BB, Juelich TL, Sefing EJ, Brasel T, Smith JK, Zhang L, Tigabu B, Hill TE, Yun T, Pietzsch C, Furuta Y, Freiberg AN. 2013. Favipiravir (T-705) inhibits Junin virus infection and reduces mortality in a guinea pig model of Argentine hemorrhagic fever. PLoS Negl Trop Dis 7:e2614. doi: 10.1371/journal.pntd.0002614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gowen BB, Westover JB, Sefing EJ, Van Wettere AJ, Bailey KW, Wandersee L, Komeno T, Furuta Y. 2017. Enhanced protection against experimental Junin virus infection through the use of a modified favipiravir loading dose strategy. Antiviral Res 145:131–135. doi: 10.1016/j.antiviral.2017.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gowen BB, Wong MH, Jung KH, Sanders AB, Mendenhall M, Bailey KW, Furuta Y, Sidwell RW. 2007. In vitro and in vivo activities of T-705 against arenavirus and bunyavirus infections. Antimicrob Agents Chemother 51:3168–3176. doi: 10.1128/AAC.00356-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gowen BB, Wong MH, Jung KH, Smee DF, Morrey JD, Furuta Y. 2010. Efficacy of favipiravir (T-705) and T-1106 pyrazine derivatives in phlebovirus disease models. Antiviral Res 86:121–127. doi: 10.1016/j.antiviral.2009.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mendenhall M, Russell A, Juelich T, Messina EL, Smee DF, Freiberg AN, Holbrook MR, Furuta Y, de la Torre JC, Nunberg JH, Gowen BB. 2011. T-705 (favipiravir) inhibition of arenavirus replication in cell culture. Antimicrob Agents Chemother 55:782–787. doi: 10.1128/AAC.01219-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mendenhall M, Russell A, Smee DF, Hall JO, Skirpstunas R, Furuta Y, Gowen BB. 2011. Effective oral favipiravir (T-705) therapy initiated after the onset of clinical disease in a model of arenavirus hemorrhagic fever. PLoS Negl Trop Dis 5:e1342. doi: 10.1371/journal.pntd.0001342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oestereich L, Rieger T, Ludtke A, Ruibal P, Wurr S, Pallasch E, Bockholt S, Krasemann S, Munoz-Fontela C, Gunther S. 2016. Efficacy of favipiravir alone and in combination with ribavirin in a lethal, immunocompetent mouse model of Lassa fever. J Infect Dis 213:934–938. doi: 10.1093/infdis/jiv522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Safronetz D, Falzarano D, Scott DP, Furuta Y, Feldmann H, Gowen BB. 2013. Antiviral efficacy of favipiravir against two prominent etiological agents of hantavirus pulmonary syndrome. Antimicrob Agents Chemother 57:4673–4680. doi: 10.1128/AAC.00886-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Safronetz D, Rosenke K, Westover JB, Martellaro C, Okumura A, Furuta Y, Geisbert J, Saturday G, Komeno T, Geisbert TW, Feldmann H, Gowen BB. 2015. The broad-spectrum antiviral favipiravir protects guinea pigs from lethal Lassa virus infection post-disease onset. Sci Rep 5:14775. doi: 10.1038/srep14775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Westover JB, Sefing EJ, Bailey KW, Van Wettere AJ, Jung KH, Dagley A, Wandersee L, Downs B, Smee DF, Furuta Y, Bray M, Gowen BB. 2016. Low-dose ribavirin potentiates the antiviral activity of favipiravir against hemorrhagic fever viruses. Antiviral Res 126:62–68. doi: 10.1016/j.antiviral.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Furuta Y, Takahashi K, Shiraki K, Sakamoto K, Smee DF, Barnard DL, Gowen BB, Julander JG, Morrey JD. 2009. T-705 (favipiravir) and related compounds: novel broad-spectrum inhibitors of RNA viral infections. Antiviral Res 82:95–102. doi: 10.1016/j.antiviral.2009.02.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zmurko J, Marques RE, Schols D, Verbeken E, Kaptein SJ, Neyts J. 2016. The viral polymerase inhibitor 7-deaza-2′-C-methyladenosine is a potent inhibitor of in vitro Zika virus replication and delays disease progression in a robust mouse infection model. PLoS Negl Trop Dis 10:e0004695. doi: 10.1371/journal.pntd.0004695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tokunaga T, Yamamoto Y, Sakai M, Tomonaga K, Honda T. 2017. Antiviral activity of favipiravir (T-705) against mammalian and avian bornaviruses. Antiviral Res 143:237–245. doi: 10.1016/j.antiviral.2017.04.018. [DOI] [PubMed] [Google Scholar]
- 29.Furuta Y, Takahashi K, Kuno-Maekawa M, Sangawa H, Uehara S, Kozaki K, Nomura N, Egawa H, Shiraki K. 2005. Mechanism of action of T-705 against influenza virus. Antimicrob Agents Chemother 49:981–986. doi: 10.1128/AAC.49.3.981-986.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Delang L, Segura Guerrero N, Tas A, Querat G, Pastorino B, Froeyen M, Dallmeier K, Jochmans D, Herdewijn P, Bello F, Snijder EJ, de Lamballerie X, Martina B, Neyts J, van Hemert MJ, Leyssen P. 2014. Mutations in the chikungunya virus non-structural proteins cause resistance to favipiravir (T-705), a broad-spectrum antiviral. J Antimicrob Chemother 69:2770–2784. doi: 10.1093/jac/dku209. [DOI] [PubMed] [Google Scholar]
- 31.Sangawa H, Komeno T, Nishikawa H, Yoshida A, Takahashi K, Nomura N, Furuta Y. 2013. Mechanism of action of T-705 ribosyl triphosphate against influenza virus RNA polymerase. Antimicrob Agents Chemother 57:5202–5208. doi: 10.1128/AAC.00649-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vanderlinden E, Vrancken B, Van Houdt J, Rajwanshi VK, Gillemot S, Andrei G, Lemey P, Naesens L. 2016. Distinct effects of T-705 (favipiravir) and ribavirin on influenza virus replication and viral RNA synthesis. Antimicrob Agents Chemother 60:6679–6691. doi: 10.1128/AAC.01156-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arias A, Thorne L, Goodfellow I. 2014. Favipiravir elicits antiviral mutagenesis during virus replication in vivo. eLife 3:e03679. doi: 10.7554/eLife.03679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baranovich T, Wong SS, Armstrong J, Marjuki H, Webby RJ, Webster RG, Govorkova EA. 2013. T-705 (favipiravir) induces lethal mutagenesis in influenza A H1N1 viruses in vitro. J Virol 87:3741–3751. doi: 10.1128/JVI.02346-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.de Avila AI, Gallego I, Soria ME, Gregori J, Quer J, Esteban JI, Rice CM, Domingo E, Perales C. 2016. Lethal mutagenesis of hepatitis C virus induced by favipiravir. PLoS One 11:e0164691. doi: 10.1371/journal.pone.0164691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thorne L, Arias A, Goodfellow I. 2016. Advances toward a norovirus antiviral: from classical inhibitors to lethal mutagenesis. J Infect Dis 213(Suppl 1):S27–S31. doi: 10.1093/infdis/jiv280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.de Avila AI, Moreno E, Perales C, Domingo E. 2017. Favipiravir can evoke lethal mutagenesis and extinction of foot-and-mouth disease virus. Virus Res 233:105–112. doi: 10.1016/j.virusres.2017.03.014. [DOI] [PubMed] [Google Scholar]
- 38.Lanciotti RS, Ebel GD, Deubel V, Kerst AJ, Murri S, Meyer R, Bowen M, McKinney N, Morrill WE, Crabtree MB, Kramer LD, Roehrig JT. 2002. Complete genome sequences and phylogenetic analysis of West Nile virus strains isolated from the United States, Europe, and the Middle East. Virology 298:96–105. doi: 10.1006/viro.2002.1449. [DOI] [PubMed] [Google Scholar]
- 39.Jin Z, Smith LK, Rajwanshi VK, Kim B, Deval J. 2013. The ambiguous base-pairing and high substrate efficiency of T-705 (favipiravir) ribofuranosyl 5′-triphosphate towards influenza A virus polymerase. PLoS One 8:e68347. doi: 10.1371/journal.pone.0068347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pfeiffer JK, Kirkegaard K. 2003. A single mutation in poliovirus RNA-dependent RNA polymerase confers resistance to mutagenic nucleotide analogs via increased fidelity. Proc Natl Acad Sci U S A 100:7289–7294. doi: 10.1073/pnas.1232294100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ferrer-Orta C, Sierra M, Agudo R, de la Higuera I, Arias A, Perez-Luque R, Escarmis C, Domingo E, Verdaguer N. 2010. Structure of foot-and-mouth disease virus mutant polymerases with reduced sensitivity to ribavirin. J Virol 84:6188–6199. doi: 10.1128/JVI.02420-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morrey JD, Taro BS, Siddharthan V, Wang H, Smee DF, Christensen AJ, Furuta Y. 2008. Efficacy of orally administered T-705 pyrazine analog on lethal West Nile virus infection in rodents. Antiviral Res 80:377–379. doi: 10.1016/j.antiviral.2008.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Julander JG, Shafer K, Smee DF, Morrey JD, Furuta Y. 2009. Activity of T-705 in a hamster model of yellow fever virus infection in comparison with that of a chemically related compound, T-1106 Antimicrob Agents Chemother 53:202–209. doi: 10.1128/AAC.01074-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Oestereich L, Ludtke A, Wurr S, Rieger T, Munoz-Fontela C, Gunther S. 2014. Successful treatment of advanced Ebola virus infection with T-705 (favipiravir) in a small animal model. Antiviral Res 105:17–21. doi: 10.1016/j.antiviral.2014.02.014. [DOI] [PubMed] [Google Scholar]
- 45.Caroline AL, Powell DS, Bethel LM, Oury TD, Reed DS, Hartman AL. 2014. Broad spectrum antiviral activity of favipiravir (T-705): protection from highly lethal inhalational Rift Valley fever. PLoS Negl Trop Dis 8:e2790. doi: 10.1371/journal.pntd.0002790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Buys KK, Jung KH, Smee DF, Furuta Y, Gowen BB. 2011. Maporal virus as a surrogate for pathogenic New World hantaviruses and its inhibition by favipiravir. Antivir Chem Chemother 21:193–200. doi: 10.3851/IMP1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Iranzo J, Perales C, Domingo E, Manrubia SC. 2011. Tempo and mode of inhibitor-mutagen antiviral therapies: a multidisciplinary approach. Proc Natl Acad Sci U S A 108:16008–16013. doi: 10.1073/pnas.1110489108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Perales C, Iranzo J, Manrubia SC, Domingo E. 2012. The impact of quasispecies dynamics on the use of therapeutics. Trends Microbiol 20:595–603. doi: 10.1016/j.tim.2012.08.010. [DOI] [PubMed] [Google Scholar]
- 49.Merino-Ramos T, Blazquez AB, Escribano-Romero E, Canas-Arranz R, Sobrino F, Saiz JC, Martin-Acebes MA. 2014. Protection of a single dose West Nile virus recombinant subviral particle vaccine against lineage 1 or 2 strains and analysis of the cross-reactivity with Usutu virus. PLoS One 9:e108056. doi: 10.1371/journal.pone.0108056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Martin-Acebes MA, Saiz JC. 2011. A West Nile virus mutant with increased resistance to acid-induced inactivation. J Gen Virol 92:831–840. doi: 10.1099/vir.0.027185-0. [DOI] [PubMed] [Google Scholar]
- 51.Lanciotti RS, Kerst AJ, Nasci RS, Godsey MS, Mitchell CJ, Savage HM, Komar N, Panella NA, Allen BC, Volpe KE, Davis BS, Roehrig JT. 2000. Rapid detection of West Nile virus from human clinical specimens, field-collected mosquitoes, and avian samples by a TaqMan reverse transcriptase-PCR assay. J Clin Microbiol 38:4066–4071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Blazquez AB, Saiz JC. 2010. West Nile virus (WNV) transmission routes in the murine model: intrauterine, by breastfeeding and after cannibal ingestion. Virus Res 151:240–243. doi: 10.1016/j.virusres.2010.04.009. [DOI] [PubMed] [Google Scholar]
- 53.Escribano-Romero E, Gamino V, Merino-Ramos T, Blazquez AB, Martin-Acebes MA, de Oya NJ, Gutierrez-Guzman AV, Escribano JM, Hofle U, Saiz JC. 2013. Protection of red-legged partridges (Alectoris rufa) against West Nile virus (WNV) infection after immunization with WNV recombinant envelope protein E (rE). Vaccine 31:4523–4527. doi: 10.1016/j.vaccine.2013.07.071. [DOI] [PubMed] [Google Scholar]
- 54.Airaksinen A, Pariente N, Menendez-Arias L, Domingo E. 2003. Curing of foot-and-mouth disease virus from persistently infected cells by ribavirin involves enhanced mutagenesis. Virology 311:339–349. doi: 10.1016/S0042-6822(03)00144-2. [DOI] [PubMed] [Google Scholar]