

ABSTRACT
MHAA4549A, a human monoclonal antibody targeting the hemagglutinin stalk region of influenza A virus (IAV), is being developed as a therapeutic for patients hospitalized with severe IAV infection. The safety and efficacy of MHAA4549A were assessed in a randomized, double-blind, placebo-controlled, dose-ranging study in a human IAV challenge model. One hundred healthy volunteers were inoculated with A/Wisconsin/67/2005 (H3N2) IAV and, 24 to 36 h later, administered a single intravenous dose of either placebo, MHAA4549A (400, 1,200, or 3,600 mg), or a standard oral dose of oseltamivir. Subjects were assessed for safety, pharmacokinetics (PK), and immunogenicity. The intent-to-treat-infected (ITTI) population was assessed for changes in viral load, influenza symptoms, and inflammatory biomarkers. MHAA4549A was well tolerated in all IAV challenge subjects. The 3,600-mg dose of MHAA4549A significantly reduced the viral burden relative to that of the placebo as determined by the area under the curve (AUC) of nasopharyngeal virus infection, quantified using quantitative PCR (98%) and 50% tissue culture infective dose (TCID50) (100%) assays. Peak viral load, duration of viral shedding, influenza symptom scores, mucus weight, and inflammatory biomarkers were also reduced. Serum PK was linear with a half-life of ∼23 days. No MHAA4549A-treated subjects developed anti-drug antibodies. In conclusion, MHAA4549A was well tolerated and demonstrated statistically significant and substantial antiviral activity in an IAV challenge model. (This study has been registered at ClinicalTrials.gov under identifier NCT01980966.)
KEYWORDS: MHAA4549A, influenza A virus, monoclonal antibodies
INTRODUCTION
The World Health Organization estimates that seasonal influenza affects 5% to 10% of the adult population worldwide, causing 3 to 5 million severe infections and 250,000 to 500,000 deaths annually (1). Reports on clinical outcomes among patients requiring hospitalization have consistently shown high morbidity and mortality (2–5). The standard-of-care therapy for patients with acute uncomplicated influenza consists of administration of neuraminidase (NA) inhibitors that include, but are not limited to, oseltamivir, zanamivir, and peramivir (6–10); however, there are currently no approved treatments for patients hospitalized with severe influenza infection.
To address this unmet medical need, MHAA4549A, a highly specific anti-influenza A virus (anti-IAV) antibody, was cloned from a single human plasmablast cell isolated from an influenza-vaccinated donor (11) and is being developed for the treatment of hospitalized patients with severe influenza A. MHAA4549A (clone 39.29) is a human monoclonal IgG1 antibody containing a VH3-30 heavy chain paired with a Vκ1-15 light chain. MHAA4549A binds to a highly conserved epitope on the stalk of hemagglutinin (HA) and is capable of neutralizing all tested seasonal human IAV strains by two complementary mechanisms of action. First, by binding to HA on viral particles, MHAA4549A neutralizes IAV infectivity by blocking the HA-mediated membrane fusion in the endosome (11–14). Second, by binding to HA on the surface of influenza virus-infected cells, MHAA4549A induces immune cells to lyse the infected cells through antibody-dependent cell-mediated cytotoxicity (ADCC; L. Kamen and E. Kho, unpublished data).
In vivo efficacy studies in mouse and ferret IAV lethal models demonstrated that MHAA4549A provided significant protection over that of control-treated animals, even with administration at 72 h postinoculation (11). This demonstration of proof of activity led to an assessment of the safety, pharmacokinetics, and efficacy in clinical studies.
In two phase 1 studies, MHAA4549A was shown to be well tolerated in healthy volunteers without IAV infection (15). Prior to testing efficacy in the target population, patients hospitalized with severe influenza, proof of activity was assessed in this phase 2, dose-ranging, challenge study to determine the safety, efficacy, and pharmacokinetics (PK) of MHAA4549A in healthy subjects inoculated with IAV. (This study was presented in part as a poster at the 2015 Keystone Symposia Conference on Viral Immunity, Breckenridge, CO, 11 to 16 January 2015 [16].)
RESULTS
Subject demographics.
Key subject demographics for randomized and intent-to-treat infected (ITTI) populations are summarized in Table 1. In the randomized population, gender, ethnicity, and time between challenge virus and administration of study drug were balanced across the groups. The median age of the subjects was 28 years, and 92% were white. The median age and body mass index (BMI) of subjects in the 1,200-mg MHAA4549A group were numerically higher than those of the other groups. To reflect anticipated dosing practice and allow a window to test efficacy, the protocol specified that subjects were to be treated between 24 and 36 h postinoculation (Fig. 1A), which is also typically the exponential phase of viral replication. The median time between inoculation and first dose for all subjects was 27.8 h. Within the ITTI population, similar baseline demographics were observed (Table 1).
TABLE 1.
Subject demographics and baseline characteristics (randomized and ITTI populations)
| Demographic | Value(s) for subjects administered: |
Value for all subjects | ||||
|---|---|---|---|---|---|---|
| Placebo | MHAA4549A |
Oseltamivir | ||||
| 400 mg | 1,200 mg | 3,600 mg | ||||
| Randomized subjects | n = 32 | n = 20 | n = 20 | n = 21 | n = 8 | n = 101 |
| Age (yr), median (range) | 27.5 (20–43) | 26.5 (20–37) | 31.5 (19–45) | 28.0 (18–44) | 24.5 (20–43) | 28.0 (18–45) |
| Male, no. (%) | 21 (65.6) | 10 (50.0) | 14 (70.0) | 11 (52.4) | 6 (75.0) | 42 (61.4) |
| White, no. (%) | 30 (93.8) | 19 (95.0) | 18 (90.0) | 19 (90.5) | 7 (87.5) | 93 (92.1) |
| BMI (kg/m2), median (range) | 23.80 (19.8–30.3) | 22.62 (20.7–28.6) | 24.93 (19.2–31.9) | 23.61 (18.3–29.7) | 24.15 (19.8–28.9) | 23.83 (18.3–31.9) |
| Temp (°C), median (range) | 36.65 (35.5–37.9) | 36.60 (35.7–37.3) | 36.70 (35.6–37.3) | 36.65 (35.7–37.6) | 36.75 (36.3–37.5) | 35.65 (35.5–37.9) |
| Inoculation to first dose, median time (h) (range) | 27.62 (24.4–30.2) | 27.40 (24.5–29.5) | 27.91 (26.4–30.0) | 27.44 (25.5–37.5) | 29.69 (28.9–30.7) | 27.75 (24.4–37.5) |
| ITTI subjects | n = 21 | n = 11 | n = 13 | n = 14 | n = 2 | n = 61 |
| Age (yr), median (range) | 28.0 (20–43) | 25.0 (23–36) | 32.0 (19–44) | 28.0 (18–44) | 27.5 (26–29) | 27.0 (18–44) |
| Male, no. (%) | 14 (66.7) | 4 (36.4) | 11 (84.6) | 8 (57.1) | 2 (100.0) | 39 (63.9) |
| White, no. (%) | 21 (100.0) | 11 (100.0) | 11 (84.6) | 13 (92.9) | 2 (100.0) | 58 (95.1) |
| BMI (kg/m2), median (range) | 23.57 (19.8–28.3) | 22.59 (20.7–27.5) | 25.75 (19.2–31.9) | 23.98 (18.3–28.3) | 27.75 (26.6–28.9) | 24.07 (18.3–31.9) |
| Temp (°C), median (range) | 36.60 (35.9–37.4) | 36.60 (35.7–37.3) | 36.50 (35.7–37.1) | 36.65 (35.7–37.6) | 36.70 (36.6–36.8) | 36.60 (35.7–37.6) |
| Inoculation to first dose, median time (h) (range) | 27.78 (24.7–30.2) | 27.50 (24.5–29.3) | 28.38 (26.9–30.0) | 27.34 (25.9–37.5) | 29.30 (29.1–29.5) | 27.85 (24.5–37.5) |
FIG 1.

Study design (A) and disposition of study subjects (B).
Disposition of study populations.
One hundred randomized subjects were inoculated with challenge virus and received study medications intravenously (i.v.) (Fig. 1B). The ITTI population included a total of 61 subjects: 11 subjects in the 400-mg MHAA4549A group, 13 in the 1,200-mg MHAA4549A group, 14 in the 3,600-mg MHAA4549A group, 2 in the oseltamivir group, and 21 in the placebo group.
Safety.
A total of 207 treatment-emergent adverse events (TEAEs) were reported in 86 of 100 dosed subjects (see Table S1 in the supplemental material). The TEAEs were similar across all groups and primarily reflected active IAV infection. In particular, the most common TEAEs were elevations of alanine aminotransferase (ALT), aspartate aminotransferase (AST), or amylase, which were observed from 4 to 10 days after inoculation with virus in all treatment groups, including the placebo arm, and which have previously been associated with influenza infection (17–19). One or more of these elevations occurred in 9 of 40 (23%) subjects receiving either placebo or oseltamivir and in 17 of 60 (28%) subjects receiving any dose of MHAA4549A. IAV-related adverse events were similar in subjects who received placebo or oseltamivir versus any dose of MHAA4549A. There were no clinically significant changes in vital signs, spirometry, or electrocardiograms and no apparent pattern of study drug-related effects in these parameters. The study had an immunogenicity prevalence rate of 1% overall (1 out of 100 subjects tested positive at baseline). None of the MHAA4549A-treated patients developed an anti-drug antibody (ADA) response.
Efficacy.
In the infected population, compared to the placebo group, the 3,600-mg dose of MHAA4549A almost completely reduced the area under the curve (AUC) of nasopharyngeal (NP) viral load, as measured by quantitative PCR (qPCR) (97.5%; P = 0.005), while the 400-mg group showed a smaller reduction (46%; P = 0.046) (Fig. 2A; Table S2). There was no treatment effect in the 1,200-mg group (3%; P = 0.902).
FIG 2.

NP virus burden determined by qPCR and TCID50 assays. The AUC of NP viral load was measured by qPCR (A) and TCID50 (B) assays. The calculated AUC for each subject is displayed along with the median and interquartile range for each treatment group. Comparisons between the 400-, 1,200-, and 3,600-mg MHAA4549A groups and the placebo group were performed using the nonparametric Wilcoxon rank sum test. Asterisks indicate statistically significant P values (*, P < 0.05). P values were not adjusted for multiple testing. Oseltamivir data were not plotted due to the small number of infected subjects (n = 2). vps, viral particles.
We observed similar reductions in the median viral AUC as measured by 50% tissue culture infective dose (TCID50) assays (Fig. 2B; Table S2), with the greatest reduction in the 3,600-mg group (100%; P = 0.002) and a smaller reduction in the 400-mg group (62.4%; P = 0.009). We observed no treatment effect in the 1,200-mg group (−20.2%; P = 0.874). No differences in clinical variables or demographics between groups, such as BMI or age, were associated with the nonlinear relationship between dose and efficacy. The viral AUCs determined by qPCR and TCID50 assays were reduced for both oseltamivir subjects (n = 2) compared to the placebo group (Table S2). MHAA4549A also significantly reduced the peak viral loads determined by both qPCR and TCID50 assays in the 3,600-mg (77.3% and 100%, respectively) and the 400-mg (20.4% and 58.8%, respectively) treatment groups compared to that of the placebo group, but there was no treatment effect in the 1,200-mg group (Table S2).
By using Kaplan-Meier analysis and censoring subjects not shedding virus, we estimated that the median duration of viral shedding by qPCR (Table 2) was 113.7 h in the placebo group. The duration of viral shedding was reduced to 103.8 h in the 400-mg group and to 71.7 h in the 3,600-mg group (8.7% and 36.9% reductions, respectively). There was no treatment effect in the 1,200-mg group (119.9 h). The duration of viral shedding in the oseltamivir group was also reduced (33.1 h); however, only one of the two oseltamivir-treated subjects in the ITTI group were shedding virus postchallenge, preventing a comparison of the efficacies of MHAA4549A and oseltamivir. We also observed reductions in the duration of viral shedding in both the 400-mg and 3,600-mg MHAA4549A dose groups relative to the placebo group as measured by TCID50 cell culture (40.1% and 20.6%, respectively).
TABLE 2.
Duration of viral shedding in ITTI populationa
| Duration of viral shedding endpointb | Value(s) for subjects given: |
||||
|---|---|---|---|---|---|
| Placebo (n = 21) | MHAA4549A |
Oseltamivir (n = 2) | |||
| 400 mg (n = 11) | 1,200 mg (n = 13) | 3,600 mg (n = 14) | |||
| qPCR assay | |||||
| No. (%) of subjects shedding virus | 15 (71.4) | 8 (72.7) | 11 (84.6) | 7 (50) | 1 (50) |
| Median duration (h) | 113.7 | 103.8 | 119.9 | 71.7 | 33.1 |
| % reduction in duration | 8.7 | 5.5 | 36.9 | 70.9 | |
| TCID50 assay | |||||
| No. (%) of subjects with event | 20 (95.2) | 7 (63.6) | 11 (84.6) | 6 (42.9) | 1 (50) |
| Median duration (h) | 80.5 | 48.2 | 81.2 | 63.9 | 33.1 |
| % reduction in duration | 40.1 | −0.9 | 20.6 | 58.9 | |
Comparison of 400-, 1,200-, and 3,600-mg doses of MHAA4549A and oseltamivir to placebo using Kaplan-Meier methodology.
Percent reduction in duration of shedding was calculated as 100% × [(median value for placebo group − median value for active infection group)/median value for placebo group].
Serum and nasopharyngeal pharmacokinetics.
MHAA4549A demonstrated linear serum pharmacokinetics, which has been characterized more extensively in a previous report (20). Serum MHAA4549A concentrations exhibited a biphasic disposition with a rapid distribution phase followed by a slow elimination phase with a half-life of approximately 23 days (Fig. 3A). The group mean serum AUCs from day 0 to the last time point (AUC0–last) were 1,740, 5,040, and 17,500 day · μg/ml for the 400-, 1,200- and 3,600-mg groups, respectively. However, as previously reported (20), MHAA4549A demonstrated non-dose-proportional NP PK (Fig. 3B), with NP AUC0–last group means of 41.9, 50.4, and 496 day · μg/ml for the 400-, 1,200- and 3,600-mg groups, respectively. In addition, the NP maximum concentration (Cmax) group means were 12.1, 14.9, and 148 μg/ml for the 400-, 1,200- and 3,600-mg groups, respectively. Of note, MHAA4549A demonstrated comparable NP exposures (i.e., AUC0–last and Cmax) between the 400-mg and 1,200-mg groups, while serum PK demonstrated a dose-proportional, 3-fold increase in exposure.
FIG 3.
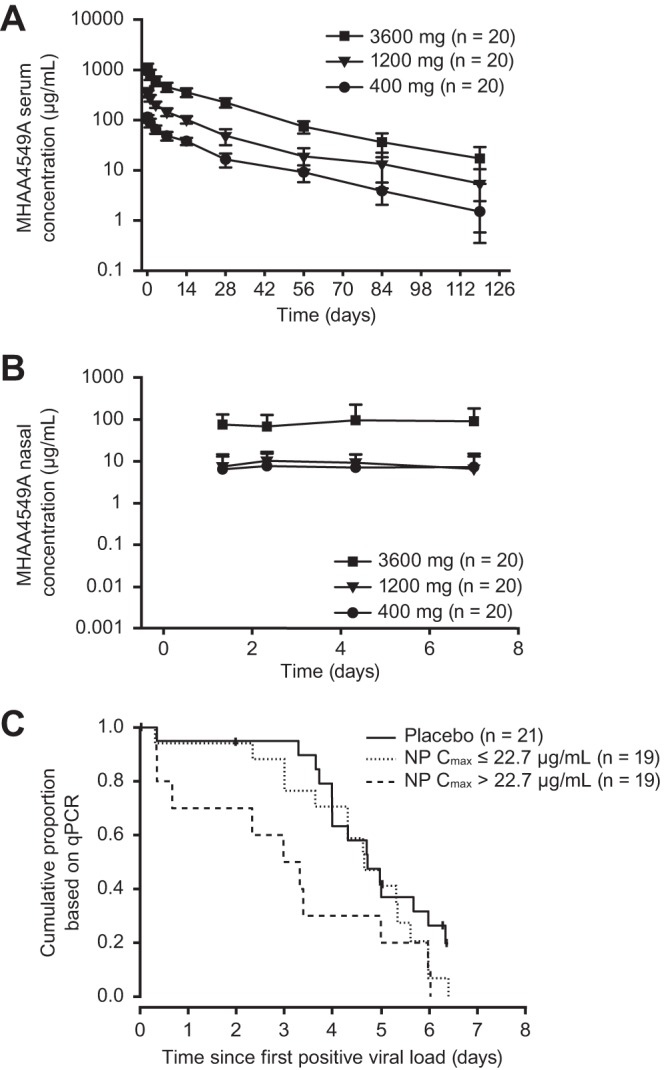
MHAA4549A serum and NP pharmacokinetics for all dosed subjects and MHAA4549A NP exposure-response relationship for all ITTI subjects. (A) Observed group mean (± standard deviation) MHAA4549A serum concentration versus time profile following single i.v. administration (all dosed subjects); (B) observed group mean (±SD) MHAA4549A NP concentration versus time profile following single i.v. administration (all dosed subjects); (C) time to resolution of viral shedding, stratified by the median of the MHAA4549A NP Cmax, 22.7 μg/ml (ITTI subjects). Mean (±SD) MHAA4549A NP concentrations for all treatment groups at nominal time day 0.33 (∼8 h) were not plotted because more than one-third of the values were below the limit of quantification.
Further analyses of all ITTI subjects with an NP Cmax of MHAA4549A that was greater than the median NP Cmax value (22.7 μg/ml), including 2 subjects in the 1,200-mg group, demonstrated a shorter median time to resolution of viral shedding than that of ITTI subjects in the placebo group (75.8 h versus 113.7 h). In addition, subjects with an NP Cmax less than the median Cmax had a median time to resolution of viral shedding that was similar to that of the placebo group (112.1 h versus 113.7 h) (Fig. 3C).
Viral resistance.
Genotypic analyses of primary NP viral isolates from several time points per subject demonstrated no evidence of any majority variant viral populations (>50%) at the HA amino acid residues predicted to be necessary for MHAA4549A binding. We found seven minority and potentially resistant variants (>5% and <50%), each unique and from seven different subjects. All variants were transient and none led to increases in the extent or duration of overall viral titers (our unpublished data). These minority viral populations were selected by plaque purification and are being further characterized for resistance to MHAA4549A, sensitivity to oseltamivir, and additional genotypic analyses (unpublished data).
Clinical symptoms.
In the ITTI population, there were numeric, but not statistically significant, reductions in the median AUC composite symptom score compared to that of the placebo, with the greatest reductions observed in the 3,600-mg group (81.8%; P = 0.289) and 400-mg group (57.9%; P = 0.200) (Table S2). There was no treatment effect in the 1,200-mg group (7.5%; P = 0.874). The AUC composite symptom scores for the two oseltamivir subjects were reduced compared to that of the placebo group (96.1%; P = 0.086).
In addition to the prespecified symptom scores in the ITTI population, fever (temperature of >37.8°C) was observed in few patients postinoculation, i.e., in 3 of 21 placebo recipients (14.3%) and 1 of 13 subjects (7.7%) in the 1,200-mg group on study days 2 and 3 only and in none in the 400-mg or 3,600-mg groups.
There were also reductions in the median total mucus weight (Table S2): both the 3,600-mg and 400-mg MHAA4549A groups had statistically significant reductions compared to the placebo group (96.2%, P = 0.044, and 81.3%, P = 0.038, respectively), but there was no effect in the 1,200-mg group (38.5%, P = 0.272).
Inflammatory biomarkers.
The inflammatory cytokines interleukin 6 (IL-6), tumor necrosis factor alpha (TNF-α), gamma interferon (IFN-γ), IL-10, IL-8, interferon-inducible protein 10 (IP-10), and monocyte chemoattractant protein 1 (MCP-1) were measured in both sera and NP samples of ITTI subjects, and the individual subject AUCs and peak levels were determined. A decline in the median NP AUCs of IFN-γ, IP-10, and MCP-1 was determined relative to the baseline, with significant reductions in IFN-γ (71%) and IP-10 (89%) at the 3,600-mg dose of MHAA4549A (P < 0.05) (Fig. 4). Reductions in the NP AUCs of IL-10, IL-6, IL-8, and TNF-α were also observed but did not reach statistical significance (our unpublished data).
FIG 4.

Effects of MHAA4549A on the AUC of NP and serum cytokines. IFN-γ, IP-10, and MCP-1 cytokine levels were measured in the NP-derived samples (upper panels) and sera (lower panels) of all ITTI subjects. Filled circles represent the AUC of each subject. Box plots show the variance, the median (bold line), and the mean (dashed line) for each group and the 25th/75th percentiles. Treatment comparisons against the placebo group were performed using Dunnett's t test; P values are shown only for groups with results that were statistically significant (*, P < 0.05).
In serum, IL-6, TNF-α, IL-8, and IL-10 cytokine levels were low (<10 pg/ml). For serum cytokines present at higher concentrations, such as IP-10, MCP-1, and IFN-γ, IP-10 was the only inflammatory mediator with statistically significant reductions in the AUC with both the 400-mg (44%) and 3,600-mg (51%) doses of MHAA4549A (Fig. 4). Similar reduction trends were observed in the 3,600-mg group when peak cytokine levels (Cmax) in both NP and serum compartments were compared (Fig. S1).
DISCUSSION
This is the first publication of the safety and efficacy of a broadly neutralizing monoclonal antibody (MHAA4549A) targeted against the stalk region of influenza HA in an IAV challenge model in healthy volunteers. Of the 100 dosed subjects, the numbers and distribution of subjects with confirmed infections (ITTI population) appeared to be balanced across treatment groups, allowing for an adequate assessment of pharmacological activity. The study included a smaller group of subjects dosed with oseltamivir, intended as a positive-control group; however, only two subjects had evidence of infection in this group and thus provided insufficient numbers to assess antiviral activity. Similar challenge models have, however, demonstrated the virologic and clinical efficacy of small molecules such as oseltamivir and peramivir, as well as monoclonal antibodies against other IAV epitopes (21–24).
These results demonstrate that MHAA4549A is well tolerated at the doses tested. Notably, there were no drug-related serious adverse events nor were there study discontinuations due to adverse events. Elevations in ALT, AST, and amylase were the most commonly observed adverse events in both the MHAA4549A group and the placebo group, but no clinically meaningful dose-related pattern in hepatic transaminase elevation was observed. While these events were listed as TEAEs, after researching previous experience with this strain of IAV, reviewing the literature, and comparing treated and control groups, it was clear that these events were not associated with drug treatment but rather with IAV infection (17–19). There was no evidence of immunogenicity or emergence of viral resistance.
The primary objective of this study was to determine the activity of MHAA4549A on viral burden following inoculation with IAV challenge. The 3,600-mg MHAA4549A dose led to substantial and statistically significant reductions in overall viral burden and peak viral load in infected subjects and reduced the duration of viral shedding. Consistent with these findings, additional measures of activity, including self-reported IAV symptoms, total mucus weight, incidence of fever, and inflammatory cytokines, were also notably reduced in the 3,600-mg MHAA4549A-treated group. Reductions in viral burden and other measures were also observed to a lesser degree in the 400-mg dose group; however, neither reductions in viral burden nor any other measure was observed in the 1,200-mg dose group.
The nonmonotonic nature of the results was unexpected; however, as described by Fry et al. (25), the human challenge model has both merits and limitations. The model is helpful in pharmacological assessment of novel antivirals because it allows measurement of antiviral activity while minimizing the clinical severity of infection in inoculated subjects (25). However, one major source of variability is the fact that not all inoculated subjects had a subsequent, productive viral infection. In addition, preexisting CD4+ T cells, titers of antibody capable of ADCC, and preexisting antibodies to neuraminidase can influence the extent of infection (26–28), and viral loads at the time of dosing can be variable. To manage the infection variability in this model, the standard and prespecified approach is to assess efficacy only in the population of subjects showing evidence of a productive infection (ITTI population), which is defined as having either seroconversion or measurable viral replication. Despite this variability, the magnitude of viral reductions in the 3,600-mg group provides evidence of MHAA4549A's significant pharmacological activity.
Serum MHAA4549A levels exhibited linear pharmacokinetics consistent with results from two phase 1 studies (15), and MHAA4549A exhibited a half-life typical of a human IgG1 monoclonal antibody (29). MHAA4549A concentrations in the NP compartment were less than 10% of concentrations in serum and likely due to partitioning from the peripheral blood to the nasal compartment. However, unlike serum, NP pharmacokinetics revealed a different profile of non-dose-proportional exposure (20), which could be due to saturation of neonatal Fc receptors (FcRn) in the nasal mucosa. Ye et al. have hypothesized that FcRn are responsible for transcytosis of monoclonal IgG from the NP compartment to blood (30), which is a nonlinear process. Between the 400-mg and 3,600-mg groups, there was an approximate 10-fold increase in NP exposure, whereas the NP exposure in the 1,200-mg dose group was similar to that of the 400-mg dose group. Given the unexpected lack of efficacy with the 1,200-mg dose, further assessment of the exposure-response relationship across all ITTI subjects confirmed that regardless of dose, higher NP MHAA4549A exposure (NP Cmax of >22.7 μg/ml) was associated with improved efficacy, as defined by a reduction in viral shedding. In particular, 2 subjects out of 13 in the 1,200-mg group had nasal Cmax values that were greater than the median Cmax values; these subjects experienced a shorter time to resolution of viral shedding than the placebo group, indicating that the exposure-response relationship may be more relevant than the dose-response relationship. We did not identify covariates that could explain the unexpected dose response. Of note, this was a human intranasal challenge model in healthy individuals with the infection primarily limited to the upper respiratory tract. It is conceivable that drug exposure could be greater in the lower and upper airways of patients with a more severe and naturally acquired IAV infection due to greater inflammation and vascular leakage. Ongoing phase 2 studies include the 3,600-mg dose, and drug exposure in both compartments in hospitalized patients with natural IAV infection will be assessed.
In conclusion, MHAA4549A was well tolerated and considerably reduced both viral burden and influenza-related symptoms in a human IAV challenge model. Evaluation of both virological and, importantly, clinical activity of a single intravenous dose of MHAA4549A in a phase 2, double-blind, placebo-controlled study in hospitalized patients with severe influenza A and requiring oxygen is ongoing, and those results will inform the dose selection for future phase 3 studies.
MATERIALS AND METHODS
Subjects.
The study enrolled healthy subjects between 18 and 45 years of age who were required to be seronegative in hemagglutination inhibition (HAI) assays specifically toward the A/Wisconsin/67/2005 (H3N2) IAV inoculum. The study subjects had been screened for undetectable or low HAI antibody titers against the challenge virus to decrease the possibility that preexisting neutralizing antibodies would reduce infection. Subjects were not screened for neuraminidase antibody titers. The London City and East Research Ethics Committee approved the study protocol. This trial was registered (ClinicalTrials.gov identifier NCT01980966) and conducted in full conformance with the International Council for Harmonisation E6 guidelines for Good Clinical Practice, the European Union Good Clinical Practice Directive (2005/28/EC), the Declaration of Helsinki, and applicable laws and regulations. All subjects provided written informed consent.
Challenge virus.
The challenge virus, A/Wisconsin/67/2005 (GenBank accession numbers CY114381 to CY114388), was produced by Baxter (Orth an der Donau, Austria) under good manufacturing practices (GMP) and was sterile and mycoplasma-free. The final product had undergone quality testing performed by the manufacturer according to predetermined specifications, including extensive adventitious agent testing. Additional details of the properties of the virus are described by Sobel Leonard et al. (31).
Study design.
This phase 2A randomized, double-blind, placebo-controlled study was conducted from October 2013 to June 2014 and enrolled 101 subjects. The randomization code was computer generated by a biostatistician external to the study team according to a randomization specification using a permuted block algorithm by dose cohort. Subjects were admitted to a quarantine facility (hVIVO, London, England) and received an intranasal inoculation of approximately 5.0 to 5.5 log10 median 50% tissue culture infective doses (TCID50) of influenza A/Wisconsin/67/2005 H3N2 on day 0 (Fig. 1A). One hundred subjects received a single i.v. infusion of placebo or MHAA4549A (Genentech Inc., South San Francisco, CA, USA) at a flat dose of 400, 1,200, or 3,600 mg, which was administered per the standard protocol over 60 min at a rate of 250 ml/h between 24 and 36 h postinoculation (Fig. 1A). Among those who received i.v. placebo for MHAA4549A, 8 patients received a 5-day, twice-daily (BID) course of 75 mg of oral oseltamivir (Roche, Kaiseraugst, Switzerland) 24 to 36 h postinoculation. A double blind was maintained using placebo tablets, such that all subjects received both a single i.v. treatment and a 5-day BID course of tablets, either oseltamivir or placebo. Per the protocol and to prevent subsequent transmission on discharge, all subjects in the study then began a standard course (BID for 5 days) of oseltamivir on day 7 and were discharged after three doses. A follow-up period of 120 days was implemented to evaluate safety, PK, and ADAs.
Safety assessments.
Safety was evaluated in subjects who were randomized, inoculated with influenza virus, and received at least one dose of study treatment. Subjects were allocated to the treatment arm associated with the regimen actually received. Assessments included evaluation of the incidence, severity, seriousness, and relatedness of AEs to either treatment or IAV infection. Vital signs, clinical laboratory results, lung function, physical examinations, and incidence of ADAs were assessed for TEAEs.
Virology sampling.
NP swabs were collected three times per day beginning on day 1 through day 7 and once prior to discharge on day 8. Swabs were stored in universal transport medium (Copan Diagnostics, Inc., Murrieta, CA). Aliquots were frozen at −80°C and subsequently used to measure viral particle concentration (viral particles [vps]/ml) by qPCR and TCID50 (units/ml) in cell culture assays.
Efficacy assessments.
Efficacy was determined in the ITTI population, which was predefined per the standard challenge model protocol as all subjects with a laboratory-confirmed influenza infection: ≥1 positive NP viral result by the TCID50 assays, ≥2 detectable NP qPCR measurements of virus, or evidence of seroconversion (>4-fold over baseline) through study day 29. HAI, qPCR, and TCID50 assays were performed by Viroclinics Biosciences (Rotterdam, Netherlands) using validated assays. Efficacy was determined by evaluating the AUC of virus measured from the NP mucosa by qPCR and TCID50 assays by applying the trapezoid rule to the viral measurements for each subject from day 1 through day 8 (32). Additional virological parameters measured included peak viral load and duration of viral shedding.
Viral resistance.
Viral resistance was assessed in NP samples derived from subjects with a measurable virus load (2 to 4 time points/subject) selected through days 1 to 8 postinoculation. The phenotype of the viral populations was investigated using validated plaque assays (Viroclinics Biosciences, Rotterdam, Netherlands). Major variants were defined as >50% of the total viral populations. HA and NA cDNA amplicons were assessed for genotype by using Illumina MiSEQ (Seqwright, Inc., Houston, TX).
Pharmacokinetics and immunogenicity assessments.
Validated enzyme-linked immunosorbent assays (ELISAs) developed in-house were used to determine serum concentrations of MHAA4549A (15). MHAA4549A was measured in NP samples with a validated ELISA (29). PK parameters were derived from serum and NP concentration-time profiles following administration of MHAA4549A and calculated using noncompartmental methods as described previously (29).
Serum samples were obtained for ADA testing at baseline, before dosing, and at days 29, 85, and 120 after MHAA4549A dosing. The presence of ADAs was detected with a validated MHAA4549A-specific bridging ELISA, using a two-tier testing approach: a screening assay and a confirmatory assay as previously described (15).
Clinical signs and symptoms of influenza infection.
Self-recorded influenza-related symptoms included runny nose, stuffy nose, sneezing, sore throat, earache, malaise, cough, shortness of breath, headache, and muscle/joint ache/stiffness. Severity was graded on a scale of 0 to 3 (0, absence; 1, barely noticeable; 2, bothersome but does not prevent activities; 3, bothersome and interferes with activities). Individual symptom scores at each time point were added to generate a total composite symptom score. Duration, peak, and AUC of the composite symptom scores were determined for individual subjects. Subjects were daily given preweighed clean tissues, and the following day the difference in weight was recorded as the amount of daily mucus.
Serum and nasal inflammatory biomarkers.
Serum (days 0, 1, 2, 4, and 7) and NP samples (days 1 to 8, first daily sample only) from ITTI subjects were assayed for inflammatory biomarkers, including IL-6, TNF-α, IL-8, IL-10, IFN-γ, MCP-1, and IP-10. The cytokines were measured using electrochemiluminescence assays (Genentech, Inc., South San Francisco, CA, USA).
Objectives and statistical analysis.
The primary objective of this study was to evaluate the reduction in the AUC of viral load from the NP mucosa in MHAA4549A-treated subjects compared to placebo group subjects. Secondary objectives included safety, reduction in peak viral load, duration of viral shedding, PK, and duration, peak, and total AUC of clinical symptoms.
The emphasis of this study was the estimation of effect size rather than hypothesis testing. It was assumed for an efficacious dose of MHAA4549A that the true decrease in the mean AUC was 6 log10 TCID50/ml equivalent by days between the placebo- and MHAA4549A-treated groups and that the within-group standard deviations were 7.5 and 5.5 log10 TCID50/ml equivalent by days in these two groups, respectively. Under these assumptions, a 95% confidence interval (CI) for the difference between the placebo and MHAA4549A groups would have a width of ±5.6% for a sample size of 14 infected subjects per group.
Characteristics for all randomized subjects by treatment group were analyzed using descriptive statistics. Continuous data were summarized using descriptive statistics (n, mean, standard deviation, median, minimum, and maximum). Categorical variables were summarized using frequency counts and percentages. For treatment comparisons to placebo, 95% CIs for the mean and median were calculated. The CI for the means was provided under the assumption of a normal distribution with unknown variance. The CI for the median difference was presented using Hodges-Lehman estimates. Viral load data were log10 transformed prior to analysis; other continuous parameters were transformed if considered appropriate. Time-to-event data were computed using Kaplan-Meier analyses and summarized using n, median (when estimable), and 95% CI. Cytokine data were log transformed and analyzed using one-way analysis of variance (ANOVA), and treatment comparisons with placebo were performed using Dunnett's t test. Data analysis was performed using SAS version 9.1 (SAS Institute, Inc., Cary, NC) and R version 3.1.1 (R Foundation for Statistical Computing, Vienna, Austria).
Supplementary Material
ACKNOWLEDGMENTS
We thank the volunteers who participated in the study as well as the clinical and laboratory personnel who performed all the procedures and collections throughout the quarantines.
J.J.L., O.S., and J.M.H. helped design the study. J.M.M. and M.A.D. designed the study and analyzed and interpreted the data. R.L.-W. and H.F. designed the study and acquired the data. P.H. and D.F. acquired the data. T.B., R.D., M.M., D.S., H.C.-H., E.M.N., D.N., C.M.R., M.B.O., and J.A.T. analyzed and interpreted the data. All authors assisted in the preparation of the manuscript.
J.M.M., J.J.L., T.B., R.D., M.M. P.H., D.S., H.C.-H., E.M.N., D.F., D.N., C.M.R., J.M.H., and J.A.T. are employees of Genentech, Inc., and own Roche stock. M.A.D. and M.B.O. were employees of Genentech, Inc., at the time the work was performed and own Roche stock. M.B.O is currently an employee of the University of Texas Southwestern Medical Center (Dallas, TX, USA). O.S. is an employee of Allakos, Inc. (San Carlos, CA, USA). R.L.W. and H.F. are employees of hVIVO (London, United Kingdom). Genentech, Inc., was involved in the study design, data interpretation, and the decision to submit for publication in conjunction with the authors.
This study was supported by Genentech, Inc. Editing and writing support was provided by Deborah Solymar (Genentech, Inc., South San Francisco, CA, USA) and was funded by Genentech, Inc.
hVIVO was formerly Retroscreen Virology Ltd.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.01154-17.
REFERENCES
- 1.World Health Organization. 2016. Influenza (seasonal) fact sheet. http://www.who.int/mediacentre/factsheets/fs211/en/ Accessed 17 August 2015.
- 2.Dao CN, Kamimoto L, Nowell M, Reingold A, Gershman K, Meek J, Arnold KE, Farley M, Ryan P, Lynfield R, Morin C, Baumbach J, Hancock E, Zansky S, Bennett NM, Thomas A, Vandermeer M, Kirschke DL, Schaffner W, Finelli L, Emerging Infections Program Network. 2010. Adult hospitalizations for laboratory-positive influenza during the 2005–2006 through 2007-2008 seasons in the United States. J Infect Dis 202:881–888. doi: 10.1086/655904. [DOI] [PubMed] [Google Scholar]
- 3.Doshi S, Kamimoto L, Finelli L, Perez A, Reingold A, Gershman K, Yousey-Hindes K, Arnold K, Ryan P, Lynfield R, Morin C, Baumbach J, Hancock EB, Bennett NM, Zansky S, Thomas A, Schaffner W, Fry AM. 2011. Description of antiviral treatment among adults hospitalized with influenza before and during the 2009 pandemic: United States, 2005–2009. J Infect Dis 204:1848–1856. doi: 10.1093/infdis/jir648. [DOI] [PubMed] [Google Scholar]
- 4.Lee N, Ison MG. 2012. Diagnosis, management and outcomes of adults hospitalized with influenza. Antivir Ther 17:143–157. doi: 10.3851/IMP2059. [DOI] [PubMed] [Google Scholar]
- 5.Thompson WW, Shay DK, Weintraub E, Brammer L, Bridges CB, Cox NJ, Fukuda K. 2004. Influenza-associated hospitalizations in the United States. JAMA 292:1333–1340. doi: 10.1001/jama.292.11.1333. [DOI] [PubMed] [Google Scholar]
- 6.Kandel R, Hartshorn KL. 2001. Prophylaxis and treatment of influenza virus infection. BioDrugs 15:303–323. doi: 10.2165/00063030-200115050-00003. [DOI] [PubMed] [Google Scholar]
- 7.Kohno S, Kida H, Mizuguchi M, Shimada J, S-021812 Clinical Study Group. 2010. Efficacy and safety of intravenous peramivir for treatment of seasonal influenza virus infection. Antimicrob Agents Chemother 54:4568–4574. doi: 10.1128/AAC.00474-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Monto AS, Moult AB, Sharp SJ. 2000. Effect of zanamivir on duration and resolution of influenza symptoms. Clin Ther 22:1294–1305. doi: 10.1016/S0149-2918(00)83026-X. [DOI] [PubMed] [Google Scholar]
- 9.Monto AS, Fleming DM, Henry D, de Groot R, Makela M, Klein T, Elliott M, Keene ON, Man CY. 1999. Efficacy and safety of the neuraminidase inhibitor zanamivir in the treatment of influenza A and B virus infections. J Infect Dis 180:254–261. doi: 10.1086/314904. [DOI] [PubMed] [Google Scholar]
- 10.Nicholson KG, Aoki FY, Osterhaus AD, Trottier S, Carewicz O, Mercier CH, Rode A, Kinnersley N, Ward P. 2000. Efficacy and safety of oseltamivir in treatment of acute influenza: a randomised controlled trial. Neuraminidase Inhibitor Flu Treatment Investigator Group. Lancet 355:1845–1850. [DOI] [PubMed] [Google Scholar]
- 11.Nakamura G, Chai N, Park S, Chiang N, Lin Z, Chiu H, Fong R, Yan D, Kim J, Zhang J, Lee WP, Estevez A, Coons M, Xu M, Lupardus P, Balazs M, Swem LR. 2013. An in vivo human-plasmablast enrichment technique allows rapid identification of therapeutic influenza A antibodies. Cell Host Microbe 14:93–103. doi: 10.1016/j.chom.2013.06.004. [DOI] [PubMed] [Google Scholar]
- 12.Chai N, Swem LR, Reichelt M, Chen-Harris H, Luis E, Park S, Fouts A, Lupardus P, Wu TD, Li O, McBride J, Lawrence M, Xu M, Tan MW. 2016. Two escape mechanisms of influenza A virus to a broadly neutralizing stalk-binding antibody. PLoS Pathog 12:e1005702. doi: 10.1371/journal.ppat.1005702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bullough PA, Hughson FM, Skehel JJ, Wiley DC. 1994. Structure of influenza haemagglutinin at the pH of membrane fusion. Nature 371:37–43. doi: 10.1038/371037a0. [DOI] [PubMed] [Google Scholar]
- 14.Wiley DC, Skehel JJ. 1987. The structure and function of the hemagglutinin membrane glycoprotein of influenza virus. Annu Rev Biochem 56:365–394. doi: 10.1146/annurev.bi.56.070187.002053. [DOI] [PubMed] [Google Scholar]
- 15.Lim J, Deng R, Derby M, Larouche R, Horn P, Anderson M, Maia M, Carrier S, Pelletier I, Burgess T, Kulkarni P, Newton E, Tavel JA. 2016. Two phase 1, randomized, double-blind, placebo-controlled, single ascending-dose studies to investigate the safety, tolerability, and pharmacokinetics of an anti-influenza A monoclonal antibody, MHAA4549A, in healthy volunteers. Antimicrob Agents Chemother 60:5437–5444. doi: 10.1128/AAC.00607-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McBride J, Rosenberger C, Tavel J, Deng R, Derby M, Burgess T, Chai N, Park S, Xu M, Swem L. 2015. Clinical safety and efficacy of MHAA4549A, a novel monoclonal antibody for the treatment of severe influenza A: results of a randomized, double-blind, placebo-controlled clinical trial, abstr 2061. Abstr 2015 Keystone Symp Conf Viral Immun, Breckenridge, CO. [Google Scholar]
- 17.Monto AS, Ceglarek JP, Hayner NS. 1981. Liver function abnormalities in the course of a type A (H1N1) influenza outbreak: relation to Reye's syndrome. Am J Epidemiol 114:750–759. doi: 10.1093/oxfordjournals.aje.a113246. [DOI] [PubMed] [Google Scholar]
- 18.Polakos NK, Cornejo JC, Murray DA, Wright KO, Treanor JJ, Crispe IN, Topham DJ, Pierce RH. 2006. Kupffer cell-dependent hepatitis occurs during influenza infection. Am J Pathol 168:1169–1178. doi: 10.2353/ajpath.2006.050875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yingying C. 2011. Abnormal liver chemistry in patients with influenza A H1N1. Liver Int 31:902. doi: 10.1111/j.1478-3231.2011.02519.x. [DOI] [PubMed] [Google Scholar]
- 20.Deng R, Lee AP, Maia M, Lim JJ, Burgess T, Horn P, Derby MA, Newton E, Tavel JA, Hanley WD. 21 June 2017. Pharmacokinetics of MHAA4549A, an anti-influenza A monoclonal antibody, in healthy subjects challenged with influenza A virus in a phase IIa randomized trial. Clin Pharmacokinet doi: 10.1007/s40262-017-0564-y. [DOI] [PubMed] [Google Scholar]
- 21.Barroso L, Treanor J, Gubareva L, Hayden FG. 2005. Efficacy and tolerability of the oral neuraminidase inhibitor peramivir in experimental human influenza: randomized, controlled trials for prophylaxis and treatment. Antivir Ther 10:901–910. [PubMed] [Google Scholar]
- 22.Hayden FG, Treanor JJ, Fritz RS, Lobo M, Betts RF, Miller M, Kinnersley N, Mills RG, Ward P, Straus SE. 1999. Use of the oral neuraminidase inhibitor oseltamivir in experimental human influenza: randomized controlled trials for prevention and treatment. JAMA 282:1240–1246. doi: 10.1001/jama.282.13.1240. [DOI] [PubMed] [Google Scholar]
- 23.Ramos EL, Mitcham JL, Koller TD, Bonavia A, Usner DW, Balaratnam G, Fredlund P, Swiderek KM. 2015. Efficacy and safety of treatment with an anti-m2e monoclonal antibody in experimental human influenza. J Infect Dis 211:1038–1044. doi: 10.1093/infdis/jiu539. [DOI] [PubMed] [Google Scholar]
- 24.Patel K, Smith PF, Dall G, Lovern M, Sloan S, Trevejo J, Hershberger E. 2017. Population pharmacokinetic and viral dynamic modeling of VIS410, a monoclonal antibody against influenza A virus in a human challenge model, abstr 3648. Abstr 2nd Annu Meet ASM Microbe, New Orleans, LA. [Google Scholar]
- 25.Fry AM, Zhong W, Gubareva LV. 2015. Advancing treatment options for influenza: challenges with the human influenza challenge. J Infect Dis 211:1033–1035. doi: 10.1093/infdis/jiu543. [DOI] [PubMed] [Google Scholar]
- 26.Wilkinson TM, Li CK, Chui CS, Huang AK, Perkins M, Liebner JC, Lambkin-Williams R, Gilbert A, Oxford J, Nicholas B, Staples KJ, Dong T, Douek DC, McMichael AJ, Xu XN. 2012. Preexisting influenza-specific CD4+ T cells correlate with disease protection against influenza challenge in humans. Nat Med 18:274–280. doi: 10.1038/nm.2612. [DOI] [PubMed] [Google Scholar]
- 27.Jegaskanda S, Luke C, Hickman HD, Sangster MY, Wieland-Alter WF, McBride JM, Yewdell JW, Wright PF, Treanor J, Rosenberger CM, Subbarao K. 2016. The generation and protective ability of influenza-specific antibody-dependent cellular cytotoxicity in humans elicited by vaccination, natural infection or experimental challenge. J Infect Dis 214:945–952. doi: 10.1093/infdis/jiw262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Monto AS, Petrie JG, Cross RT, Johnson E, Liu M, Zhong W, Levine M, Katz JM, Ohmit SE. 2015. Antibody to influenza virus neuraminidase: an independent correlate of protection. J Infect Dis 212:1191–1199. doi: 10.1093/infdis/jiv195. [DOI] [PubMed] [Google Scholar]
- 29.Deng R, Jin F, Prabhu S, Iyer S. 2012. Monoclonal antibodies: what are the pharmacokinetic and pharmacodynamic considerations for drug development? Expert Opin Drug Metab Toxicol 8:141–160. doi: 10.1517/17425255.2012.643868. [DOI] [PubMed] [Google Scholar]
- 30.Ye L, Zeng R, Bai Y, Roopenian DC, Zhu X. 2011. Efficient mucosal vaccination mediated by the neonatal Fc receptor. Nat Biotechnol 29:158–163. doi: 10.1038/nbt.1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sobel Leonard A, McClain MT, Smith GJ, Wentworth DE, Halpin RA, Lin X, Ransier A, Stockwell TB, Das SR, Gilbert AS, Lambkin-Williams R, Ginsburg GS, Woods CW, Koelle K. 2016. Deep sequencing of influenza A virus from a human challenge study reveals a selective bottleneck and only limited intrahost genetic diversification. J Virol 90:11247–11258. doi: 10.1128/JVI.01657-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dey S, Gupta S. 2013. Numerical methods. McGraw Hill Education (India) Private Limited, New Delhi, India. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.