

ABSTRACT
Fosfomycin exhibits broad-spectrum antibacterial activity and is being reevaluated for the treatment of extensively drug-resistant pathogens. Its activity in Gram-negative organisms, however, can be compromised by expression of FosA, a metal-dependent transferase that catalyzes the conjugation of glutathione to fosfomycin, rendering the antibiotic inactive. In this study, we solved the crystal structures of two of the most clinically relevant FosA enzymes: plasmid-encoded FosA3 from Escherichia coli and chromosomally encoded FosA from Klebsiella pneumoniae (FosAKP). The structure, molecular dynamics, catalytic activity, and fosfomycin resistance of FosA3 and FosAKP were also compared to those of FosA from Pseudomonas aeruginosa (FosAPA), for which prior crystal structures exist. E. coli TOP10 transformants expressing FosA3 and FosAKP conferred significantly greater fosfomycin resistance (MIC, >1,024 μg/ml) than those expressing FosAPA (MIC, 16 μg/ml), which could be explained in part by the higher catalytic efficiencies of the FosA3 and FosAKP enzymes. Interestingly, these differences in enzyme activity could not be attributed to structural differences at their active sites. Instead, molecular dynamics simulations and hydrogen-deuterium exchange experiments with FosAKP revealed dynamic interconnectivity between its active sites and a loop structure that extends from the active site of each monomer and traverses the dimer interface. This dimer interface loop is longer and more extended in FosAKP and FosA3 than in FosAPA, and kinetic analyses of FosAKP and FosAPA loop-swapped chimeric enzymes highlighted its importance in FosA activity. Collectively, these data yield novel insights into fosfomycin resistance that could be leveraged to develop new strategies to inhibit FosA and potentiate fosfomycin activity.
KEYWORDS: Escherichia coli, FosA, FosA3, FosAKP, FosAPA, Klebsiella, X-ray crystallography, fosfomycin
INTRODUCTION
Antimicrobial resistance is a global health crisis responsible for upwards of 700,000 deaths per year (1). As therapeutic options for the treatment of antibiotic-resistant bacteria become limited, clinicians have begun to revive older antibiotics. One such antibiotic, fosfomycin, works by inhibiting UDP-N-acetylglucosamine enolpyruvyl transferase, the enzyme that catalyzes the first step in peptidoglycan biosynthesis. Fosfomycin has regained popularity in the 21st century, with the Infectious Diseases Society of America and the European Society of Clinical Microbiology and Infectious Diseases jointly recommending it as a first-line agent for the treatment of uncomplicated urinary tract infections (2). In the United States, fosfomycin is approved only as an oral formulation for the treatment of urinary tract infections. However, due to its favorable safety profile and broad-spectrum activity against both Gram-positive and -negative bacteria, intravenous fosfomycin is approved in Europe and Asia for a broader range of indications (3). In this regard, a phase III clinical trial evaluating an intravenous formulation of fosfomycin for complicated urinary tract infections and acute pyelonephritis has recently been completed (ClinicalTrials.gov registration no. NCT02753946). However, as with all antibiotics, emerging resistance threatens to limit the clinical utility of fosfomycin.
In Gram-negative bacteria, the most clinically relevant mechanism of fosfomycin resistance involves expression of FosA, an enzyme that catalyzes the Mn2+- and K+-dependent glutathione-mediated degradation of fosfomycin (Fig. 1) (4, 5). FosA functions as a homodimer, with each of the two active sites per dimer coordinating an Mn2+ ion that binds fosfomycin and acts as a Lewis acid during the nucleophilic attack by glutathione (6). A K+ ion also binds close to the active site, which balances the negatively charged active site and increases the rate of reaction ∼100-fold (5).
FIG 1.
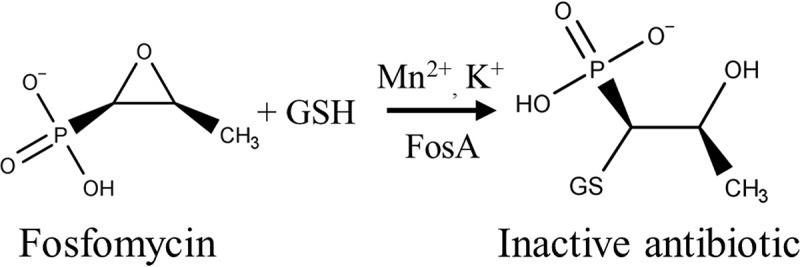
FosA-catalyzed degradation of fosfomycin.
The fosA gene is frequently present in the chromosomes of many Gram-negative bacteria, including Klebsiella pneumoniae, Enterobacter cloacae, and Serratia marcescens, and confers inherent resistance to fosfomycin (7). Furthermore, plasmid-mediated fosA genes can be transferred between species in the Enterobacteriaceae family. By far the most widespread plasmid-borne fosA is fosA3, which has been reported to be present in up to 90% of fosfomycin-resistant extended-spectrum-β-lactamase (ESBL)-producing E. coli isolates in China (8). Chromosomally encoded fosA from K. pneumoniae (fosAKP) also represents a substantial risk to human health because it confers intrinsic reduced fosfomycin susceptibility in K. pneumoniae and can also serve as a reservoir for the nearly identical plasmid-borne genes fosA5 and fosA6 (9, 10).
FosA enzymes from different bacterial pathogens are highly divergent, and the extents to which they confer fosfomycin resistance differ. In this regard, there is a significant knowledge gap regarding the structures and activities of different FosA enzymes and the molecular basis for fosfomycin resistance. Crystal structures of the chromosomally encoded FosA from P. aeruginosa (FosAPA) in the absence and presence of fosfomycin have been reported (11). A crystal structure of FosATn2921, which was described as a component of Tn2921 on plasmid pSU912 in S. marcescens (12), has also been solved (13). However, the clinical significance of FosATn2921 is unclear given that it has only ever been reported once and was found in S. marcescens, which typically harbors its own chromosomal copy of fosA (7). Therefore, in this study, we carried out a comprehensive structural and functional analysis of FosA3 and FosAKP, which share only 60 to 70% amino acid sequence identity with FosAPA. Our data highlight the importance of a loop structure that extends from the active site of each monomer and traverses the dimer interface in FosA activity and resistance. Importantly, the information derived from this work could be leveraged to develop new strategies to inhibit FosA activity and potentiate fosfomycin activity.
RESULTS
FosA3 and FosAKP confer greater fosfomycin resistance and are more active than FosAPA.
To compare the degrees of resistance conferred by different FosA enzymes, we cloned the wild-type genes fosAPA, fosA3, and fosAKP into pUC57-transformed Escherichia coli TOP10 and measured MICs of fosfomycin. The MICs for the fosA3 and fosAKP transformants were >1,024 μg/ml, while the MIC for the fosAPA transformant was only 16 μg/ml (Table 1), as previously reported (7). Notably, the Clinical and Laboratory Standards Institute (CLSI) breakpoint for fosfomycin susceptibility is 64 μg/ml for uncomplicated urinary tract infections (14). Although the fosAPA transformant is considered susceptible to fosfomycin, this transformation still produces a 16-fold increase from the baseline MIC of 1 μg/ml (data not shown). Because MIC values depend on both the inherent activity of the enzyme and its stable expression within the bacterium, we determined the relative expression levels of FosAPA and FosAKP in E. coli. We found that FosAKP expression was substantially greater than that of FosAPA, which may explain in part the observed differences in the MICs (Fig. 2A).
TABLE 1.
Fosfomycin resistance conferred by, and in vitro activities of, FosA enzymesa
| Enzyme | MIC (μg/ml)b | kcat (s−1) | KmFos (mM) | kcat/Km (M−1 s−1) |
|---|---|---|---|---|
| Wild type | ||||
| FosAPA | 16 | 42.1 ± 4.5 | 12 ± 4 | (3.7 ± 1.0) × 103 |
| FosA3 | >1,024 | 99.4 ± 3.3 | 13 ± 3 | (8.0 ± 1.9) × 103 |
| FosAKP | >1,024 | 140 ± 15 | 13 ± 3 | (1.0 ± 1.3) × 104 |
| Chimeric | ||||
| FosAPAc | 52.3 ± 0.8* | 17 ± 3 | (3.2 ± 0.7) × 103 | |
| FosAKPc | 26.2 ± 3.2** | 14 ± 4 | (2.0 ± 0.6) × 103** |
MICs were determined using the agar dilution method. The steady-state kinetic parameters for fosfomycin were determined in the presence of 30 mM glutathione. Kinetic parameters are reported as the means ± standard deviations from three or four independent biological replicates. *, P < 0.05 compared to value for the wild type by 2-tailed Student's t test; **, P < 0.001 compared to value for the wild type by 2-tailed Student's t test.
Reported in reference 7.
FIG 2.

(A) Relative expression of His6-tagged FosAKP versus FosAPA when transformed into E. coli TOP10 cells. Shown is a Coomassie gel with 25 μg of lysate protein loaded (left) and anti-His6 Western blot (right). (B) Steady-state kinetics of wild-type enzymes, determined in the presence of 30 mM glutathione with various fosfomycin concentrations. Values are reported as the means ± standard deviations from three or four independent biological replicates.
In order to deconvolute the contributions to resistance by FosA expression and activity, we determined steady-state kinetic parameters for FosA3, FosAKP, and FosAPA using recombinantly purified enzymes (Fig. 2B). We found that FosA3 and FosAKP were more active than FosAPA. This increase in activity for FosA3 and FosAKP was driven by a 2- to 3-fold increase in kcat, with no difference in Km for fosfomycin (KmFos) (Table 1). Collectively, these data indicate that FosA3 and FosAKP confer greater fosfomycin resistance than does FosAPA due to increases in both protein expression in E. coli and enzymatic activity.
Diverse FosA enzymes construct structurally conserved active sites.
To gain insight to the molecular basis for the different activities of these enzymes, we solved the atomic resolution crystal structures of unliganded (or apo) FosA3, apo FosAKP, and FosAKP in complex with fosfomycin (holo FosAKP) (Table 2). Although FosA3 and FosAKP are only 60 to 70% identical in amino acid sequence to FosAPA, superimposition of all three apo enzymes reveals that the overall structure is largely conserved, with a Cα root mean square deviation (RMSD) between any two of these structures less than 0.7 Å (Fig. 3A). Of note, there is conservation in both sequence and structure of the residues that coordinate the divalent cation (H7, H67, and E113) and other residues known to be involved in fosfomycin binding (e.g., T9, K93, S97, S101, Y103, and R122) (15) (Fig. 3B). Tyrosine 65, which bridges the active site and the dimer interface loop, is also conserved (Fig. 3A and B). Although the only added metal was Mg2+ (present as 250 mM MgCl2 in the mother liquor), we identified Zn2+ bound in the active site, as supported by electron density and X-ray fluorescence scanning (Fig. 3C). Although this is the first reported crystal structure of a FosA protein with a bound metal ion other than Mn2+, previous biochemical studies have shown that FosA enzymes can bind a variety of divalent cations (4).
TABLE 2.
Data collection and refinement statisticsa
| Parameter | Value for: |
||
|---|---|---|---|
| Apo FosA3 | Holo FosAKP | Apo FosAKP | |
| Data collection statistics | |||
| Resolution range | 29.44–2.689 (2.785–2.689) | 28.27–1.539 (1.594–1.539) | 28.69–1.3 (1.346–1.3) |
| Space group | P 41 2 2 | P 21 21 21 | P 2 21 21 |
| Unit cell (a, b, c, α, β, γ) | 87.608, 87.608, 357.038, 90, 90, 90 | 40.071, 47.155, 149.517, 90, 90, 90 | 44.875, 67.54, 89.508, 90, 90, 90 |
| No. of: | |||
| Total reflections | 376,188 (37,524) | 183,460 (17,829) | 349,328 (7,547) |
| Unique reflections | 39,913 (3,873) | 41,623 (4,170) | 60,254 (3,033) |
| Multiplicity | 9.4 (9.7) | 4.4 (4.3) | 5.8 (2.5) |
| Completeness (%) | 1.00 (1.00) | 0.97 (0.99) | 0.89 (0.46) |
| Mean I/sigma I | 20.50 (3.15) | 14.74 (2.22) | 19.77 (2.32) |
| Wilson B-factor | 57.2 | 17.87 | 12.51 |
| Rmerge | 0.0800 (0.6252) | 0.0509 (0.6815) | 0.04812 (0.3153) |
| Rmeas | 0.0847 (0.6605) | 0.0577 (0.7737) | 0.0525 (0.4066) |
| CC1/2 | 0.999 (0.951) | 0.999 (0.924) | 0.999 (0.837) |
| CC* | 1 (0.987) | 1 (0.98) | 1 (0.955) |
| Refinement | |||
| Reflections used in refinement | 39,869 (3,864) | 41,577 (4,164) | 60,246 (3,031) |
| No. of reflections used for Rfree | 1,918 (185) | 2,065 (214) | 3,094 (162) |
| Rwork | 0.2051 (0.2963) | 0.1682 (0.2709) | 0.1254 (0.1772) |
| Rfree | 0.2493 (0.3524) | 0.2059 (0.3171) | 0.1491 (0.2142) |
| CC(work) | 0.947 (0.872) | 0.967 (0.947) | 0.977 (0.941) |
| CC(free) | 0.913 (0.732) | 0.952 (0.906) | 0.971 (0.921) |
| No. of: | |||
| Nonhydrogen atoms | 8,600 | 2,480 | 2,624 |
| Macromolecules | 8,586 | 2,172 | 2,220 |
| Ligands | 14 | 31 | 2 |
| Protein residues | 1,091 | 275 | 276 |
| Water molecules | 0 | 277 | 402 |
| RMSD | |||
| Bond length (Å) | 0.006 | 0.006 | 0.007 |
| Angles (°) | 0.97 | 0.78 | 0.91 |
| Average B-factor | 65.61 | 29.5 | 17.27 |
Overall values are reported, with highest-resolution shell values in parentheses.
FIG 3.

Three-dimensional structures of the apo and holo forms of FosAKP and FosA3. (A) Overlay of the apo forms of FosAPA (red), FosA3 (blue), and FosAKP (green). (B) Active-site residues in the apo forms of FosA with residues labeled using FosAKP numbering. (C) Excitation scan of Zn-K edge in apo FosAKP crystal. (D) E98, which sits on the K+-binding loop, exhibits two conformations in apo FosAKP. (E) Superimposition of holo forms of FosAPA (cyan) and FosAKP (green) with a second nonphysiological fosfomycin (gray) bound in the FosAPA structure. (F) The holo active site is highly conserved with the exception of Y65, which adopts a different rotamer in FosAPA, likely due to a contact with a second fosfomycin molecule (not shown).
Glutamate 98, which resides in the mobile K+-binding loop, adopts two distinct conformations in the apo FosAKP structure (Fig. 3D). In one conformation (E98a), it is located in the active site, where it coordinates the Zn2+ metal, whereas in a second conformation (E98b), the carboxylate Oε atoms are located ∼11 Å from the Zn2+ ion. Although this may be due to asymmetric crystal packing, it highlights the flexibility of the K+-binding loop in the absence of K+ and fosfomycin, consistent with previous reports (13).
The crystal structure of the holo FosAKP closely resembles that of the holo FosAPA (PDB code 1LQP), with an overall Cα RMSD of 0.5 Å (Fig. 3E). While the active sites in both enzymes are conserved, one notable difference relates to Y65, which forms a hydrogen bond with a fosfomycin phosphonate oxygen in FosAKP but adopts a different rotamer conformation in FosAPA (Fig. 3F). In the FosAPA structure, it contacts a second fosfomycin molecule that has not been shown to be physiologically relevant and may have resulted from soaking these crystals with very high concentrations of fosfomycin (11).
The dimer interface loop exhibits notable divergence among FosA structures.
The differences in enzymatic activity observed between FosA3 or FosAKP and FosAPA (Table 1) are not readily explained by differences in their active sites or K+-binding loops. Therefore, we searched for structural divergence in other regions of the protein that could account for the kinetic differences. We found that a loop that extends from each active site and traverses the dimer interface exhibits notable structural variability between FosA enzymes. In FosAPA, this dimer interface loop is three residues shorter than in the loops found in either FosA3 or FosAKP. Consequently, the loop crosses the dimer interface relatively directly in FosAPA, whereas it forms a short α-helix and takes a more circuitous route in FosA3 and FosAKP (Fig. 4A and B). These distinct conformations alter accessibility to a deep cavity that is found in the dimer interface of all FosA enzymes (Fig. 4C and D). This pocket is contiguous with both active sites, and residues that form the floor of the pocket, such as Y68, are immediately adjacent to residues that coordinate the divalent cation (e.g., H67) required for catalysis (Fig. 4E).
FIG 4.

(A and B) Cartoon representations of apo FosAPA (A) and FosAKP (B) with dimer interface loops highlighted in yellow. Y65, which bridges the active site and the dimer interface loop, is also marked. (C and D) Apo FosAPA (C) and FosAKP (D) color coded by distance (Å) from the center of mass, with active sites marked by yellow arrows. The central dimer interface cavity can be seen in blue. (E) Apo FosAKP exhibiting close proximity between Y68, which forms the floor of the interface cavity, and H67, which coordinates the active-site metal.
The active sites and dimer interface loops are interconnected in FosAKP.
To determine whether the differences in the dimer interface loop influence FosA activity, we ran molecular dynamics (MD) simulations of Mn2+-bound FosAKP in the presence and absence of fosfomycin and K+. We first calculated covariance matrices for all backbone atoms in the apo and holo FosAKP dimers, which were then postprocessed with the DIRECT-ID algorithm (16), which computes the norm difference between these matrices to identify the residues with the greatest changes in their dynamics as a function of fosfomycin and K+ binding. The residues identified in this analysis included the dimer interface loop residues (P53 to S63, except P60), the K+-binding loop residues (R96 to G99), and several residues that may contribute to the putative glutathione channel (e.g., A120 and E128 to G133) (15) (Fig. 5A). This finding suggests that fosfomycin binding modulates cooperative motions in the K+-binding loop, glutathione channel, and dimer interface loop. The K+-binding loop itself exhibited distinct ligand-dependent behavior. Using the distance between carboxylate Oε atoms of E98 and the active-site Mn2+ as a metric to define the loop conformational ensembles, the apo and holo forms of FosAKP exhibited different behaviors (Fig. 3D and 5B). Whereas the apo form sampled three different conformational states, including closed (∼8 Å), intermediate (∼15 Å), and open (∼20 Å), the holo form exhibited a dominant peak in the closed state (∼8 Å) and a much smaller peak at ∼16 Å. These results indicate that in the presence of fosfomycin, the K+-binding loops remained more proximal to the active sites and sampled a more restricted conformational ensemble. To further quantify differences in the dynamic motions of the apo and holo forms of FosAKP, we performed quasiharmonic analysis of the backbone atoms and computed the entropy along the vibrational modes (17). The per-residue entropy differences between apo and holo FosAKP are provided in Fig. 5C. These differences indicate that in general, the amino acid residues in the apo form of the enzyme exhibit more freedom of motion than in the holo form, particularly in the regions identified by DIRECT-ID, providing further evidence that fosfomycin binding restricts the motions of the K+-binding loop, glutathione channel, and dimer interface. The dynamic connectivity between the active site and dimer interface loop may be mediated via a hydrogen bond between Y65 and fosfomycin that was observed in both the crystal structure (Fig. 5F) and MD simulations.
FIG 5.

Dynamic analysis of FosAKP from MD (A to C) and HDX-MS (D and E). (A) Regions of FosAKP that showed the greatest differences in dynamics between the apo and holo forms. The protein dimer is rendered in gray, with residues identified by DIRECT-ID to undergo changes in their dynamics in blue. The bound Mn2+ ion (gold), K+ ion (purple), and fosfomycin (stick) are shown for perspective. (B) Distance distributions between E98 Oε atoms and Mn2+ for apo and holo forms. (C) Per-residue entropy differences between the apo and holo forms of FosAKP with glutathione (GSH) channel, dimer interface, and K+-binding loop residues highlighted. (D) Fosfomycin-induced changes in hydrogen-deuterium exchange of FosAKP, with peptides that contain residues from the GSH channel, dimer interface, and K+-binding loop highlighted. Differences in deuteration between peptides from apo and holo FosAKP at individual time points are plotted as colored lines, with the sum of the differences over all time points plotted as gray bars. Confidence intervals (98%) for individual time points and sums are plotted as thin and thick black lines, respectively. (E) Cartoon representation of fosfomycin-induced changes in hydrogen-deuterium exchange of FosAKP. Regions with statistically significant decreases in deuteration at the earliest time point (10 s) are colored dark blue. Regions with decreases in deuteration observed only at later time points are colored light blue. Regions with increased deuteration are colored red.
Delineating the effects of fosfomycin and K+ binding on FosAKP dynamics.
To determine whether the observed changes in the dynamics of the active site and dimer interface loop were due to fosfomycin or K+ binding, we initiated apo simulations following removal of both fosfomycin and K+ ions from the holo crystal structure. A bulk concentration of ∼150 mM KCl, typical of intracellular levels, was included in the simulation system. We observed that the mobile K+ ions bound to residues D95, S97, E98, and G99 such that in over 99% of the simulation frames a K+ ion was within 3 Å of the backbone carbonyl O atom of at least one of these residues in each subunit of the dimer. We also observed reversible dissociation of K+ over the course of both the apo and holo simulations, suggesting that the applied NBFIX correction (see Materials and Methods and Fig. S2 and Table S2 in the supplemental material) was an appropriate physical model of the carbonyl O-K+ interaction. Given the high occupancy of the K+-binding loop in both the fosfomycin-bound and apo forms of FosAKP, the observed differences in dynamics are likely due to fosfomycin binding only.
HDX-MS supports interconnectivity between the FosA active sites and dimer interface loops.
To validate our in silico analysis, we performed hydrogen-deuterium exchange-mass spectrometry (HDX-MS) of FosAKP in the presence and absence of fosfomycin. HDX-MS relies on the exchange of hydrogen with deuterium on peptide backbone amides and provides information on backbone solvent accessibility and dynamics, because residues that are more frequently solvent exposed will undergo faster deuteration. By subtracting the percent deuteration for holo FosAKP from apo FosAKP peptides, we determined which regions of the protein displayed statistically significant protection as a result of fosfomycin binding (Fig. 5D). These regions were mapped onto the structure of holo FosAKP (Fig. 5E). The results illustrate that fosfomycin binding decreases deuteration in multiple places throughout the enzyme, although this was particularly prominent in the K+-binding loop, glutathione channel, and dimer interface loop (Fig. 5E), in close agreement with the MD simulations (Fig. 5A and C). In some cases, we observed protection at the earliest time point probed (10 s). Early protection is likely due to solvent exclusion upon fosfomycin binding of otherwise solvent-accessible amide hydrogens. Notably, early protection occurs in the K+-binding loop (see Fig. S3A) and other residues close to the active site (Fig. S3B). In other cases, protection occurs only at later time points, which reflects changes in conformational dynamics in peripheral secondary structure elements. For instance, such protection is observed in α4, which forms the floor of the glutathione channel (Fig. S3C). Other regions that contain residues that contribute to the glutathione channel (e.g., W34, S36, Y39, R122, and Y131) also displayed decreased deuteration in the fosfomycin-bound state (Fig. S3D to F). Together, these results indicate that fosfomycin binding reduces flexibility of the structural elements that form the active site.
Fosfomycin binding also causes a decrease in deuteration in the dimer interface loop. The peptide from residues 48 to 64 (peptide 48-64), which encompasses the majority of the loop, exhibited an ∼15% reduction in deuteration at 10 s (Fig. S4A), which amounts to the protection of ∼2 backbone amides upon fosfomycin binding. Residue Y65, which also resides on the loop and anchors it to the active site, likewise displayed a fosfomycin-induced decrease in deuteration. A comparison of overlapping peptides 64-70 and 65-70 isolates the exchange contribution of Y65's amide hydrogen and points to mild fosfomycin-induced protection (Fig. S4C and D). In only one instance did we observe a fosfomycin-induced increase in deuteration. A comparison of overlapping peptic fragments suggests that the observed deuterium increase is attributable to the amide hydrogen of W46 (Fig. S5). This residue's side chain on one monomer inserts into the active site of the second monomer, forming the floor of the fosfomycin-binding pocket. This outcome suggests that fosfomycin binding conformationally constrains the dimer interface at the top while loosening it at the bottom. Together, these data support the concept that fosfomycin binding induces conformational changes in the active sites that propagate through the dimer interface loops and other nearby regions.
The dimer interface loops modulate FosA enzyme activity.
As described above, the MD simulations and HDX-MS analysis suggest dynamic interconnectivity between the FosA active sites and dimer interface loops. To ascertain whether the structure of the dimer interface loop impacts FosA enzyme activity, we created dimer interface loop-swapped chimeric enzymes of FosAPA and FosAKP and evaluated enzyme activity. Briefly, the FosAPA chimera (FosAPAc) harbored the FosAKP dimer interface loop, whereas the FosAKP chimera (FosAKPc) harbored the FosAPA loop (Fig. S1). As expected, the kcat for FosAKPc was significantly decreased compared to that for FosAKP (P < 0.001) and more closely resembled the kinetic parameters determined for FosAPA (Fig. 6; Table 1). Also as expected, introduction of the FosAKP dimer interface loop into FosAPA significantly increased the kcat value (P < 0.05), but FosAPAc activity was still less than that of FosAKP. Taken together, these results support the notion that the length of the dimer interface loop impacts FosA activity.
FIG 6.
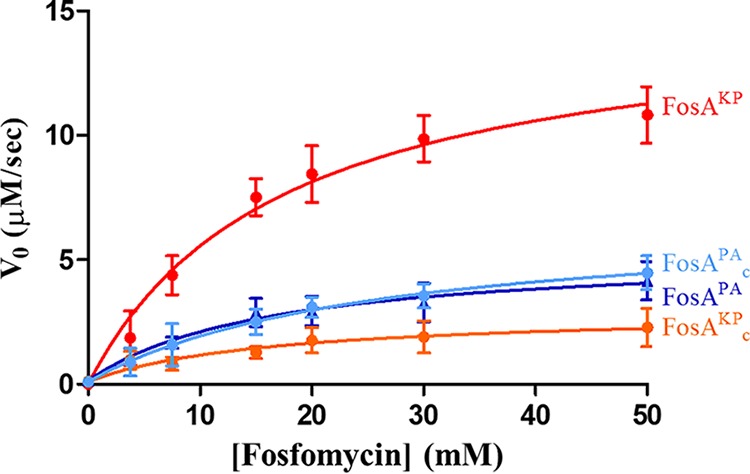
Steady-state kinetics of FosAKPc and FosAPAc compared to wild-type enzymes (Fig. 2B), determined in the presence of 30 mM glutathione with various fosfomycin concentrations. Values are reported as the means ± standard deviations from three or four independent biological replicates.
DISCUSSION
Fosfomycin use is becoming more popular due to its favorable safety profile and broad-spectrum activity against Gram-negative bacteria. However, its activity is frequently mitigated by the fosA gene, which can be carried either on the chromosome in Gram-negative bacteria (e.g., in P. aeuroginosa and K. pneumoniae) or on a plasmid (e.g., in E. coli). In this study, we demonstrated that FosA3 and FosAKP, key enzymes that contribute to fosfomycin resistance in Gram-negative bacteria, exhibit greater enzymatic activity and fosfomycin resistance than FosAPA. These differences appear to be driven in part by the presence of extended dimer interface loops compared to those in FosAPA. The dimer interface loops exhibit decreased flexibility upon fosfomycin binding, potentially mediated by a hydrogen bond between fosfomycin and Y65. Furthermore, perturbing the dimer interface loops in either FosAKP or FosAPA significantly impacts enzymatic activity. This effect was dramatic in the loop-swapped chimeric enzyme FosAKPc, whose activity was completely reverted to that of FosAPA. The effect was more subtle in FosAPAc, whose activity only partially reverted to that of FosAKP, suggesting that FosAKP may have evolved differences elsewhere to allow it to benefit from a longer loop. Collectively, these studies show significant differences between enzymes in the FosA superfamily and provide novel and unique insights into the underlying mechanisms of fosfomycin resistance.
The discovery and development of specific FosA inhibitors could significantly expand fosfomycin use to Gram-negative pathogens that carry the fosA gene. Along these lines, we demonstrated that diverse FosA enzymes construct highly conserved active sites, providing justification for the search for broad-spectrum FosA inhibitors that bind in the active site of the enzyme. However, the current study also suggests that the dimer interface could act as an allosteric site in FosA and could also be targeted for inhibitor discovery. The dimer interface loops mediate access to a large central cavity, which lies directly between both active sites (Fig. 4E). Molecules that bind to this pocket could potentially inhibit FosA activity in two ways. First, inhibitor binding may alter dimer interface loop stability, prolonging contacts between Y65 and the glutathione-fosfomycin product, favoring a lower turnover rate. Second, inhibitor binding at the dimer interface could potentially disrupt coordination of the divalent metal ion in the FosA active site. The Mn2+ ion plays a central role in activating the oxirane ring of fosfomycin for nucleophilic attack, and the residues that coordinate Mn2+ are highly conserved across FosA enzymes and sensitive to mutation (4, 15). Because the dimer interface pocket is contiguous with residues that coordinate the divalent cation in both active sites, an inhibitor that binds at the dimer interface could potentially distort both active sites such that divalent cation coordination and catalysis are disrupted. Although FosA shares very little structural homology with human glutathione S-transferases (GSTs), both are composed of homodimers with two active sites and a central cavity. Furthermore, human GSTs are known to exhibit allosteric behavior with respect to the central cavity (18–20), and compounds that allosterically inhibit human GSTs have been identified (21), giving credence to the possibility that this may also be a viable strategy for FosA inhibition.
MATERIALS AND METHODS
MIC determination.
The wild-type genes fosAPA (GenBank accession number CP017149), fosAKP (GenBank accession number CP006923), and fosA3 (GenBank accession number KR078259) were synthesized by GenScript (Piscataway, NJ) and cloned into pUC57 under the control of the lacZ promoter. The recombinant plasmids were introduced into E. coli TOP10 by electroporation. The fosfomycin MICs for these transformant strains expressing FosA were obtained using the agar dilution method according to CLSI guidelines, with Mueller-Hinton agar supplemented with 25 mg/ml of glucose-6-phosphate (14).
FosA expression determination.
fosAKP and fosAPA genes followed by a C-terminal His6 tag in pUC57 were purchased from Genescript (Piscataway, NJ). The plasmids were transformed into E. coli TOP10 by electroporation. Recombinant strains were grown in LB supplemented with 50 μg/ml of ampicillin to an optical density at 600 nm (OD600) of 0.5. Cultures were pelleted, suspended in 1× phosphate-buffered saline (PBS) containing 1× complete EDTA-free protease inhibitor (Roche Diagnostics, Indianapolis, IN), and lysed at 4°C by sonication. Protein quantities were determined using a modified Lowry protein assay with bovine serum albumin as a standard (22). Samples resuspended in Laemmli buffer were heated to 100°C for 5 min, and 25 μg of total protein per well was separated by 12.5% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Separated proteins were either stained with Coomassie stain or transferred to a 0.45-μm Transblot nitrocellulose membrane (Bio-Rad) by the method of Towbin et al. (23). Membranes were stained with 0.1% naphthol blue black (Sigma) in 1% acetic acid, and standards were marked. Membranes were blocked in 5% nonfat dry milk in Tris-buffered saline-Tween 20 (TBS-T20; 10 mM Tris-HCl [pH 8], 150 mM NaCl, 0.1% Tween 20 [Fisher]) and probed with 1:1,000 anti-His6 monoclonal antibody (Thermo) for 1 h at 25°C. After a washing, the membrane was probed with 1:2,000 horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (Thermo) for 1 h at 25°C. Membrane bands were visualized with Clarity Western ECL reagent (Bio-Rad) in accordance with the manufacturer's specification.
Synthesis, cloning, expression, and purification of FosA proteins for kinetic studies.
The fosAKP, fosA3, and fosAPA genes were synthesized by Genscript (Piscataway, NJ) and cloned into pET-22(b+) or pE-SUMOstar (LifeSensors, Malvern, PA) for protein expression and purification. fosA was cloned between the NdeI and BamHI restriction sites in the pET-22(b+) vector, and a hexahistidine tag was added to the 3′ terminus of the gene. Cloning into the pE-SUMOstar vector occurred between the SUMOstar fusion protein sequence and BsaI restriction site, according to the manufacturer's instructions. The FosAKP chimera (FosAKPc), which included the FosAPA interface loop (resides 51 to 60 of FosAPA), and FosAPA chimera (FosAPAc), which included the FosAKP interface loop (residues 51 to 63 of FosAKP), were also synthesized by Genscript and were cloned into pET-22(b+), as described above (see Fig. S1 for amino acid alignments of proteins included in this study). E. coli BL21(DE3)/pLysS was then transformed with fosA-containing pET-22b(+) or pE-SUMOstar. Protein expression and purification were performed as described previously (9). Although the histidine tag was left on all enzymes in our studies, it had no effect on their structure or function, as the C termini of the tagged enzymes were identical to those of previously solved untagged enzymes (Fig. 3) and enzymes purified from either pET-22b(+) or pE-SUMOstar showed identical activities (data not shown).
Steady-state kinetic analyses of FosA.
Fosfomycin-dependent glutathione conjugation was detected spectrophotometrically using monochlorobimane (Sigma-Aldrich) to detect unreacted free glutathione. Assays were carried out in a volume of 50 μl at 25°C in 0.1 M sodium phosphate buffer (pH 8.0) containing 50 mM KCl, 25 μM MnCl2, 30 mM glutathione, and various concentrations of fosfomycin (0 to 50 mM). A 100 nM concentration of FosA was used to initiate the reaction, which was quenched after 20 min for FosAKP and FosA3 and after 40 min for FosAPA, FosAPAc, and FosAKPc. A no-enzyme control was also performed. Reactions were quenched by the addition of 150 μl of methanol. Quenched reactions were diluted in 100 μl of 0.1 M sodium phosphate buffer (pH 8.0) containing 1 mM EDTA. Following the addition of 500 μM monochlorobimane, the concentration of glutathione was established by fluorescence spectroscopy using a SpectraMax M2 plate reader (Molecular Devices). A standard curve was prepared using 0 to 750 μM glutathione. Data were fitted to Michaelis-Menten equations using SigmaPlot (Systat Software Inc., San Jose, CA).
Protein expression and purification for crystallization.
FosA3 and FosAKP were cloned into the pET-22(b+) vector (Novagen) with the inclusion of a C-terminal His6 tag, as described above. Both constructs were expressed in 6 liters of LB medium overnight at 18°C in E. coli BL21(DE3)/pLysS cells after induction with 0.5 mM isopropyl-β-d-thiogalactopyranoside (IPTG) at an OD600 of 0.6. Cells were harvested by centrifugation (5,000 × g for 15 min) and lysed in PBS by sonication. The soluble fraction was purified using HisPur nickel-nitrilotriacetic acid (Ni-NTA) resin (Thermo Scientific) using a gradient of 10 to 500 mM imidazole. The protein was then dialyzed into 150 mM NaCl–50 mM Tris (pH 7.5) and then further purified by size exclusion chromatography (Superdex 200 10/300 GL; GE Healthcare). It was then buffer exchanged into 75 mM NaCl–10 mM Tris, (pH 7.5) and concentrated.
Protein crystallization.
For FosA3, protein was concentrated to 9 mg/ml and combined with 6 mM fosfomycin and 6 mM MnCl2. The solution was centrifuged (19,150 × g for 5 min), and 250 nl of the supernatant was combined with 250 nl of mother liquor (7% [vol/vol] ethylene glycol, 7% [wt/vol] polyethylene glycol 6000 [PEG 6000], 0.1 M HEPES [pH 6.95]) in sitting drops. For fosfomycin-bound FosAKP, protein was concentrated to 9 mg/ml, combined with 6 mM fosfomycin disodium salt (Sigma-Aldrich; purity as determined by thin-layer chromatography [TLC], >98%) and 6 mM MnCl2, and centrifuged (19,150 × g for 5 min). One microliter of supernatant was combined in hanging drops with 1 μl of mother liquor (0.22 M KBr, 20% [wt/vol] PEG 2000 monomethyl ether). For fosfomycin-unbound FosAKP, protein was concentrated to 13 mg/ml and 1 μl of protein was combined in hanging drops with 1 μl of mother liquor (0.25 M MgCl2, 20% [wt/vol] PEG 3350, 0.1 M bis-Tris [pH 5.5]). Resulting crystals were improved by streak seeding. Crystals were harvested and flash-cooled with liquid nitrogen in mother liquor supplemented with 20% (vol/vol) glycerol as a cryoprotectant.
X-ray diffraction, data processing, structure determination, and refinement.
X-ray diffraction data for FosA3 and fosfomycin-bound FosAKP were collected using a Dectris 6M PILATUS detector on beamline 23-ID-D at the Advanced Photon Source (APS), processed using XDS (24), and scaled in AIMLESS (25, 26). Both data sets were phased by molecular replacement in PHENIX using Phaser-MR (27) with PDB accession code 1LQP as a search model and further built and refined using Coot (28) and PHENIX (29), respectively. Data for fosfomycin-unbound FosAKP was collected on beamline 23-ID-B at APS and processed as described above, except for fosfomycin-bound FosAKP as the search model for molecular replacement.
MD system preparation and parametrization.
Parameters for the protein and K+ ion were taken from the CHARMM36 force field (30, 31). The water model was the CHARMM-modified TIP3P (32–34), and Mn2+ parameters were taken from work by Won (35). Parameters for fosfomycin were generated using CGenFF (36). Coordination of Mn2+ by histidine residues was modeled via covalent bonds between ligating N atoms and the Mn2+ ion. The equilibrium bond lengths, angles, and force constants for these bonds were determined by performing a quantum mechanical (QM) optimization of a system containing imidazole-Mn2+-imidazole, optimized using the MP2/6-31+G* model chemistry using Gaussian09 (revision D.01) (37). Force field parameters (see Table S2) were assigned to achieve a balance between the optimized QM geometry and the crystallographically assigned distances, which were slightly longer than the in vacuo QM geometry. The interaction between K+ and carbonyl O atoms was evaluated by calculating the interaction energy between K+ and N-methylacetamide (NMA; held in a rigid gas-phase optimized geometry) in intervals of 0.1 Å from 1.0 to 4.0 Å and in intervals of 1.0 Å from 4.0 to 10.0 Å. Energies were evaluated in Gaussian09 using the MP2/6-311++G(2df,2pd) model chemistry with counterpoise correction (38) for basis set superposition error (39). The interaction energy profile using the standard CHARMM force field parameters was too weak compared to the QM interaction energy, and therefore, an off-diagonal Lennard-Jones term (NBFIX in CHARMM) was applied between the O and K+ atom types to better model this interaction (see Fig. S2).
MD simulations.
All simulation systems were built using the CHARMM program (40). The apo form of FosAKP was generated by deleting the ligated K+ ions and bound fosfomycin from the holo crystal structure. Both the apo and holo forms were solvated in TIP3P water boxes with a minimum box-protein distance of 10 Å; simulation boxes also contained ∼150 mM KCl. Energy minimization was performed in CHARMM, and position-restrained equilibration was performed in NAMD (41) for 1 ns by restraining all nonhydrogen atoms (k = 5.0 kcal mol−1 Å−2). Following equilibration, restraints were removed and simulations were carried out in OpenMM (42) for 1 μs under an NPT ensemble, with snapshots saved every 20 ps for analysis. Temperature was regulated at 298 K using the Andersen method (43) with a collision frequency of 1 ps−1, and pressure was maintained at 1 atm using a Monte Carlo barostat with exchanges attempted every 25 integration steps. The time step for the simulations was 2 fs, and bonds involving H atoms were constrained using the SHAKE algorithm (44). Periodic boundary conditions were applied in all three dimensions. Electrostatic interactions were evaluated with the particle mesh Ewald method (45), and Lennard-Jones forces were switched to zero from 10 to 12 Å. All analysis was performed using the CHARMM program. Covariance matrices were postprocessed with DIRECT-ID (16).
HDX-MS.
The coverage maps for all proteins were obtained from undeuterated controls as follows: 3 μl of ∼50 μM FosAKP in 20 mM Tris (pH 7.8), 150 mM KCl, and 50 μM MnCl2 was diluted with 27 μl of the same buffer at room temperature, followed by the addition of 30 μl of ice-cold quencher [100 mM glycine, 1.5 M guanidine hydrochloride, 15 mM tris(2-carboxyethyl)phosphine (TCEP; pH 2.4)]. The quenched samples were injected into a Waters HDX nanoAcquity ultraperformance liquid chromatograph (UPLC; Waters, Milford, MA) with in-line pepsin digestion (Waters Enzymate BEH pepsin column). Peptic fragments were trapped on an Acquity UPLC BEH C18 peptide trap and separated on an Acquity UPLC BEH C18 column. A 7-min, 5% to 35% acetonitrile (0.1% formic acid) gradient was used to elute peptides directly into a Waters Synapt G2 mass spectrometer. MSe fragmentation data were acquired with a 20- to 30-V ramp trap CE for high-energy acquisition of product ions as well as continuous lock mass (Leu-Enk) for mass accuracy correction. Peptides were identified using ProteinLynx Global Server 2.5.1 (PLGS) from Waters. Further filtering of 0.3 fragment per residue was applied in DynamX 3.0.
HD exchange reactions, quenching, and injection were performed using a LEAP autosampler controlled with HDxDirector. Briefly, 3 μl of ∼50 μM FosAKP in 20 mM Tris (pH 7.8), 150 mM KCl, and 50 μM MnCl2 was incubated in 27 μl of 20 mM Tris, 99.99% D2O (pH in D2O [pD] 7.8), 150 mM KCl, 50 μM MnCl2. All reactions were performed at 25°C. Prior to injection, deuteration reactions were quenched at various times (10 s, 1 min, 10 min, 1 h, and 2 h) with 30 μl of ice-cold 100 mM glycine buffer, 1.5 M guanidine hydrochloride, and 15 mM TCEP (pH 2.4). Back exchange correction was performed against fully deuterated controls acquired by incubating 3 μl of 50 μM FosAKP in 17 μl of 20 mM Tris, 99.99% D2O (pD 7.8), 150 mM KCl, and 50 μM MnCl2 containing 6.4 M deuterated guanidine deuteriochloride and 10 mM TCEP for 1 h at 25°C, followed by the addition of 10 μl of 20 mM Tris, 99.99% D2O (pD 7.8), 150 mM KCl, and 50 μM MnCl2 for 1 h prior to quenching (without guanidine HCl). All deuteration time points and controls were acquired in triplicates.
The deuterium uptake by the identified peptides through increasing deuteration time and for the fully deuterated control was determined using Waters DynamX 3.0 software. The normalized percentage of deuterium uptake (%D) at an incubation time t for a given peptide was calculated as follows: %D = 100(mt − m0)/(mf − m0), where mt is the centroid mass at incubation time t, m0 is the centroid mass of the undeuterated control, and mf is the centroid mass of the fully deuterated control. Percent deuteration difference plots (Δ%D) were generated using the percent deuteration calculated. Confidence intervals for the Δ%D plots were determined using the method outlined in reference 46, adjusted to percent deuteration using the fully deuterated controls. Confidence intervals (98%) were plotted on the Δ%D plots as horizontal dashed lines.
Accession number(s).
The atomic coordinates have been deposited in the Protein Data Bank with codes 5V91, 5V3D, and 5VB0.
Supplementary Material
ACKNOWLEDGMENTS
This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under contract no. DE-AC02-06CH11357. Computational resources were provided by the University of Maryland, Baltimore, Computer-Aided Drug Design Center.
This work is also supported in part by the University of Maryland, Baltimore, School of Pharmacy Mass Spectrometry Center (SOP1841-IQB2014), as well as NIH grant F32GM109632 (J.A.L.), NIH grants GM072558 and GM051501 (A.D.M.), and NIH grant R21AI123747 (Y.D.).
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
A.D.M. is the cofounder and CSO of SilcsBio LLC.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.01572-17.
REFERENCES
- 1.O'Neill J, Review on Antimicrobial Resistance. 2014. Antimicrobial resistance: tackling a crisis for the health and wealth of nations. HM Government, London, United Kingdom. [Google Scholar]
- 2.Gupta K, Hooton TM, Naber KG, Wullt B, Colgan R, Miller LG, Moran GJ, Nicolle LE, Raz R, Schaeffer AJ, Soper DE, Infectious Diseases Society of America, European Society for Microbiology and Infectious Diseases. 2011. International clinical practice guidelines for the treatment of acute uncomplicated cystitis and pyelonephritis in women: a 2010 update by the Infectious Diseases Society of America and the European Society for Microbiology and Infectious Diseases. Clin Infect Dis 52:e103–e120. doi: 10.1093/cid/ciq257. [DOI] [PubMed] [Google Scholar]
- 3.Grabein B, Graninger W, Rodriguez Bano J, Dinh A, Liesenfeld DB. 9 December 2016. Intravenous fosfomycin—back to the future. Systematic review and meta-analysis of the clinical literature. Clin Microbiol Infect doi: 10.1016/j.cmi.2016.12.005. [DOI] [PubMed] [Google Scholar]
- 4.Bernat BA, Laughlin LT, Armstrong RN. 1997. Fosfomycin resistance protein (FosA) is a manganese metalloglutathione transferase related to glyoxalase I and the extradiol dioxygenases. Biochemistry 36:3050–3055. doi: 10.1021/bi963172a. [DOI] [PubMed] [Google Scholar]
- 5.Bernat BA, Laughlin LT, Armstrong RN. 1999. Elucidation of a monovalent cation dependence and characterization of the divalent cation binding site of the fosfomycin resistance protein (FosA). Biochemistry 38:7462–7469. doi: 10.1021/bi990391y. [DOI] [PubMed] [Google Scholar]
- 6.Liao RZ, Thiel W. 2013. Determinants of regioselectivity and chemoselectivity in fosfomycin resistance protein FosA from QM/MM calculations. J Phys Chem B 117:1326–1336. doi: 10.1021/jp4002719. [DOI] [PubMed] [Google Scholar]
- 7.Ito R, Mustapha M, Tomich AD, Callaghan JD, McElheny CL, Mettus RT, Shanks RMQ, Sluis-Cremer N, Doi Y. 2017. Widespread fosfomycin resistance in Gram-negative bacteria attributable to the chromosomal fosA gene. mBio 8(4):e00749-17. doi: 10.1128/mBio.00749-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cao XL, Shen H, Xu YY, Xu XJ, Zhang ZF, Cheng L, Chen JH, Arakawa Y. 2016. High prevalence of fosfomycin resistance gene fosA3 in bla CTX-M-harbouring Escherichia coli from urine in a Chinese tertiary hospital during 2010–2014. Epidemiol Infect 145:818–824. doi: 10.1017/S0950268816002879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guo QL, Tomich AD, McElheny CL, Cooper VS, Stoesser N, Wang MG, Sluis-Cremer N, Doi YH. 2016. Glutathione-S-transferase FosA6 of Klebsiella pneumoniae origin conferring fosfomycin resistance in ESBL-producing Escherichia coli. J Antimicrob Chemother 71:2460–2465. doi: 10.1093/jac/dkw177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma Y, Xu X, Guo Q, Wang P, Wang W, Wang M. 2015. Characterization of fosA5, a new plasmid-mediated fosfomycin resistance gene in Escherichia coli. Lett Appl Microbiol 60:259–264. doi: 10.1111/lam.12366. [DOI] [PubMed] [Google Scholar]
- 11.Rife CL, Pharris RE, Newcomer ME, Armstrong RN. 2002. Crystal structure of a genomically encoded fosfomycin resistance protein (FosA) at 1.19 A resolution by MAD phasing off the L-III edge of Tl(+). J Am Chem Soc 124:11001–11003. doi: 10.1021/ja026879v. [DOI] [PubMed] [Google Scholar]
- 12.García-Lobo JM, Ortiz JM. 1982. Tn292l, a transposon encoding fosfomycin resistance. J Bacteriol 151:477–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pakhomova S, Rife CL, Armstrong RN, Newcomer ME. 2004. Structure of fosfomycin resistance protein FosA from transposon Tn2921. Protein Sci 13:1260–1265. doi: 10.1110/ps.03585004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clinical and Laboratory Standards Institute. 2017. Performance standards for antimicrobial susceptibility testing, 27th ed (M100-S27) Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 15.Beharry Z, Palzkill T. 2005. Functional analysis of active site residues of the fosfomycin resistance enzyme FosA from Pseudomonas aeruginosa. J Biol Chem 280:17786–17791. doi: 10.1074/jbc.M501052200. [DOI] [PubMed] [Google Scholar]
- 16.Lakkaraju SK, Lemkul JA, MacKerell AD Jr. 2016. DIRECT-ID: an automated method to identify and quantify conformational variations—application to β2-adrenergic GPCR. J Comput Chem 37:416–425. doi: 10.1002/jcc.24231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Andricioaei I, Karplus M. 2001. On the calculation of entropy from covariance matrices of the atomic fluctuations. J Chem Phys 115:6289–6292. doi: 10.1063/1.1401821. [DOI] [Google Scholar]
- 18.Asakura T, Sasagawa A, Takeuchi H, Shibata S, Marushima H, Mamori S, Ohkawa K. 2007. Conformational change in the active center region of GST P1-1, due to binding of a synthetic conjugate of DXR with GSH, enhanced JNK-mediated apoptosis. Apoptosis 12:1269–1280. doi: 10.1007/s10495-007-0053-0. [DOI] [PubMed] [Google Scholar]
- 19.Axarli I, Muleta AW, Vlachakis D, Kossida S, Kotzia G, Maltezos A, Dhavala P, Papageorgiou AC, Labrou NE. 2016. Directed evolution of Tau class glutathione transferases reveals a site that regulates catalytic efficiency and masks co-operativity. Biochemical J 473:559–570. doi: 10.1042/BJ20150930. [DOI] [PubMed] [Google Scholar]
- 20.Hegazy UM, Musdal Y, Mannervik B. 2013. Hidden allostery in human glutathione transferase P1-1 unveiled by unnatural amino acid substitutions and inhibition studies. J Mol Biol 425:1509–1514. doi: 10.1016/j.jmb.2013.01.038. [DOI] [PubMed] [Google Scholar]
- 21.Musdal Y, Hegazy UM, Aksoy Y, Mannervik B. 2013. FDA-approved drugs and other compounds tested as inhibitors of human glutathione transferase P1-1. Chem Biol Interact 205:53–62. doi: 10.1016/j.cbi.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 22.Markwell MA, Haas SM, Bieber LL, Tolbert NE. 1978. A modification of the Lowry procedure to simplify protein determination in membrane and lipoprotein samples. Anal Biochem 87:206–210. doi: 10.1016/0003-2697(78)90586-9. [DOI] [PubMed] [Google Scholar]
- 23.Towbin H, Staehelin T, Gordon J. 1979. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci U S A 76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kabsch W. 2010. XDS. Acta Crystallogr D Biol Crystallogr 66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Evans PR, Murshudov GN. 2013. How good are my data and what is the resolution? Acta Crystallogr D Biol Crystallogr 69:1204–1214. doi: 10.1107/S0907444913000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS. 2011. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr 67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. 2007. Phaser crystallographic software. J Appl Crystallogr 40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 29.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Best RB, Zhu X, Shim J, Lopes PEM, Mittal J, Feig M, MacKerell AD Jr. 2012. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ,ψ and side-chain χ1 and χ2 dihedral angles. J Chem Theory Comput 8:3257–3273. doi: 10.1021/ct300400x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beglov D, Roux B. 1994. Finite representation of an infinite bulk system: solvent boundary potential for computer simulations. J Chem Phys 100:9050–9063. [Google Scholar]
- 32.Durell SR, Brooks BR, Ben-Naim A. 1994. Solvent-induced forces between two hydrophilic groups. J Phys Chem 98:2198–2202. [Google Scholar]
- 33.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. 1983. Comparison of simple potential functions for simulating liquid water. J Chem Phys 79:926–935. [Google Scholar]
- 34.Neria E, Fischer S, Karplus M. 1996. Simulation of activation free energies in molecular systems. J Chem Phys 105:1902. [Google Scholar]
- 35.Won Y. 2012. Force field for monovalent, divalent, and trivalent cations developed under the solvent boundary potential. J Phys Chem A 116:11763–11767. doi: 10.1021/jp309150r. [DOI] [PubMed] [Google Scholar]
- 36.Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, Darian E, Guvench O, Lopes P, Vorobyov I, MacKerell AD Jr. 2010. CHARMM general force field: a force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J Comput Chem 31:671–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas ¨O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. 2009. Gaussian 09, revision D.01. Gaussian, Inc, Wallingford, CT. [Google Scholar]
- 38.Boys SF, Bernardi F. 1970. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol Phys 19:553–566. [Google Scholar]
- 39.Ransil BJ. 1961. Studies in molecular structure. IV. Potential curve for the interaction of two helium atoms in single-configuration LCAO MO SCF approximation. J Chem Phys 34:2109–2118. [Google Scholar]
- 40.Brooks BR, Brooks CL III, MacKerell AD Jr, Nilsson L, Petrella RJ, Roux B, Wom Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, Kuczera K, Lazaridis T, Ma J, Ovchinnikov V, Paci E, Pastor RW, Post CB, Pu JZ, Schaefer M, Tidor B, Venable RM, Woodcock HL, Wu X, Yan W, York DM, Karplus M. 2009. CHARMM: the biomolecular simulation program. J Comput Chem 30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kalé L, Schulten K. 2005. Scalable molecular dynamics with NAMD. J Comput Chem 26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eastman P, Friedrichs MS, Chodera JD, Radmer RJ, Bruns CM, Ku JP, Beauchamp KA, Lane TJ, Wang L-P, Shukla D, Tye T, Houston M, Stich T, Klein C, Shirts MR, Pande VS. 2013. OpenMM 4: a reusable, extensible, hardware independent library for high performance molecular simulation. J Chem Theory Comput 9:461–469. doi: 10.1021/ct300857j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Andersen HC. 1980. Molecular dynamics simulations at constant pressure and/or temperature. J Chem Phys 72:2384–2393. [Google Scholar]
- 44.Ryckaert J-P, Ciccotti G, Berendsen HJC. 1977. Numerical integration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys 23:327–341. [Google Scholar]
- 45.Darden T, York D, Pedersen L. 1993. Particle mesh Ewald: an N · log(N) method for Ewald sums in large systems. J Chem Phys 98:10089–10092. [Google Scholar]
- 46.Houde D, Berkowitz SA, Engen JR. 2011. The utility of hydrogen/deuterium exchange mass spectrometry in biopharmaceutical comparability studies. J Pharm Sci 100:2071–2086. doi: 10.1002/jps.22432. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.