

ABSTRACT
The mycobacterial phosphoglycosyltransferase WecA, which initiates arabinogalactan biosynthesis in Mycobacterium tuberculosis, has been proposed as a target of the caprazamycin derivative CPZEN-45, a preclinical drug candidate for the treatment of tuberculosis. In this report, we describe the functional characterization of mycobacterial WecA and confirm the essentiality of its encoding gene in M. tuberculosis by demonstrating that the transcriptional silencing of wecA is bactericidal in vitro and in macrophages. Silencing wecA also conferred hypersensitivity of M. tuberculosis to the drug tunicamycin, confirming its target selectivity for WecA in whole cells. Simple radiometric assays performed with mycobacterial membranes and commercially available substrates allowed chemical validation of other putative WecA inhibitors and resolved their selectivity toward WecA versus another attractive cell wall target, translocase I, which catalyzes the first membrane step in the biosynthesis of peptidoglycan. These assays and the mutant strain described herein will be useful for identifying potential antitubercular leads by screening chemical libraries for novel WecA inhibitors.
KEYWORDS: cell wall, drug targets, tuberculosis
INTRODUCTION
According to the latest WHO report, 10.4 million people developed tuberculosis (TB) and 1.8 million lives were lost to this devastating disease in 2015 (1), making TB the leading cause of mortality from infection. The global health threat of TB has been heightened significantly by the alarming rise in drug-resistant disease, with 480,000 people developing multidrug-resistant (TB MDR-TB) in 2015, 9.5% of whom had extensively drug-resistant (XDR-TB). To reduce the global burden of TB, there is a clear and urgent need to develop new drugs for the treatment of this disease. Among other requirements, new TB drugs should (i) be more effective than the current drugs in order to shorten the duration of treatment, (ii) have novel mechanisms of action to combat resistant forms of TB, (iii) be compatible with antiretroviral therapy to enable the treatment of patients coinfected with HIV, and (iv) not antagonize existing TB drugs used in first- and second-line regimens (2).
Fundamental to the discovery of antimycobacterial agents with novel mechanisms of action are the identification, characterization, and validation of new TB drug targets. The prominence of the cell envelope of Mycobacterium tuberculosis as a target for TB drugs is underlined by the fact that two of the first-line drugs, isoniazid and ethambutol, act on cell envelope biogenesis. Importantly, several new TB drug candidates in preclinical or clinical development, including the benzothiazinone PBTZ169 (3), also inhibit components of M. tuberculosis cell envelope metabolism (http://www.newtbdrugs.org/pipeline/clinical), underscoring the richness of biosynthesis of the cell envelope as a source of targets for the development of new drugs (4). Among these compounds, the caprazamycin derivative CPZEN-45 was shown by Ishizaki et al. to inhibit the activity of the enzyme WecA, based on activity assays performed with membranes from a derivative of Mycobacterium smegmatis mc2155 that lacks its own wecA homologue (MSMEG_4947) and overexpresses wecA (Rv1302) from M. tuberculosis H37Rv (5). Catalytic activity attributable to WecA in M. smegmatis and its sensitivity to the uridine-nucleoside antibiotic tunicamycin were originally described more than 20 years ago (6) while defining the first steps in the biosynthesis of the mycobacterial arabinogalactan. These steps involve the production of glycolipid 1 (GL1, decaprenyl-P-P-GlcNAc) from UDP-GlcNAc and decaprenyl-P, which is then extended by rhamnosyl transferase WbbL (7) to form glycolipid 2 (GL2, decaprenyl-P-P-GlcNAc-Rha) that serves as a basis for polymerization of arabinogalactan (8) (Fig. 1). In a later study, Jin et al. (9) showed that Rv1302 and MSMEG_4947 can functionally complement a wecA mutant of Escherichia coli, and they confirmed the essentiality of MSMEG_4947 in M. smegmatis.
FIG 1.

Biosynthesis of lipid-linked intermediates involved in mycobacterial cell wall biogenesis catalyzed by membrane proteins WecA and translocase I (MurX) in M. tuberculosis. WecA transfers GlcNAc-1-P on decaprenyl-P to form glycolipid 1 (GL1) in arabinogalactan (AG) biosynthesis, while translocase I (MurX) transfers phospho-MurNAc-pentapeptide to form lipid I intermediate involved in peptidoglycan (PG) biosynthesis.
In Bacillus subtilis, both genetic and biochemical evidence revealed that the WecA orthologue TagO, which is involved in the synthesis of teichoic acids, is a target of CPZEN-45 (5). Remarkably, however, in the same organism, caprazamycin B, one of the most active antitubercular agents from the group of naturally occurring caprazamycins (10, 11), was shown to target translocase I, another enzyme from the same family of polyprenyl phosphate–N-acetyl-hexosamine-1-phosphate transferases (5). Bacterial translocase I (also known as MraY in various bacteria, or MurX in M. tuberculosis H37Rv) catalyzes the first membrane step in peptidoglycan biosynthesis, i.e., the transfer of MurNAc-pentapeptide-1-P from its activated donor UDP-MurNAc-pentapeptide to polyprenyl-P (12), resulting in the production of lipid I (Fig. 1).
In the present study, we investigated WecA as a novel pharmacological target for TB through a series of biochemical and chemical-genetic experiments. First, we biochemically characterized the activity of mycobacterial WecA. We then analyzed the impact of transcriptional silencing of wecA on the viability of M. tuberculosis in vitro and ex vivo and on its susceptibility to putative WecA inhibitors. Finally, we developed simple radiometric assays for WecA and translocase I for an evaluation of potential dual activity or a switch in the activities of selected inhibitors.
RESULTS
Rv1302 from M. tuberculosis H37Rv and MSMEG_4947 from M. smegmatis mc2155 have UDP-GlcNAc–decaprenyl-phosphate GlcNAc-1-phosphate transferase activity.
To investigate the function of WecA proteins from M. tuberculosis and M. smegmatis, we employed a prior successful approach for the functional characterization of several other enzymes involved in biosynthesis of the mycobacterial cell envelope (13–16). The technique relied on constitutive production of recombinant N-terminally or C-terminally His-tagged proteins in M. smegmatis using pVV2 (17) and pVV16 (18) expression vectors, followed by comparison of the target enzyme activities in cell-free assays using membrane/cell wall fractions of the control cells harboring an empty vector versus the overproducers. In a pilot experiment, M. smegmatis transformed with pVV16-rv1302 did not grow. We therefore switched to using the acetamide-inducible pJAM2 system (19) to avoid possible toxicity issues due to the overproduction of a protein with 11 predicted transmembrane segments, as predicted using hidden Markov models (http://tuberculist.epfl.ch/tmhmm/Rv1302.html). Analysis of proteins from the induced fractionated cells by SDS-PAGE and Western blotting confirmed the presence of recombinant MSMEG_4947 in mycobacterial membrane and cell wall (P60) fractions (Fig. 2A), while the production of Rv1302 was much lower and minimally detectable only in the membranes (data not shown). The apparent molecular masses of these recombinant proteins (ca. 30 kDa) did not correspond to the expected values, which were approximately 10 kDa higher. However, a similar behavior was also described for WecA from E. coli (20, 21), suggesting that the anomalous migration on SDS-PAGE can very likely be attributed to detergent binding, as described for membrane proteins (22).
FIG 2.

Localization of recombinant MSMEG_4947 and examination of its activity in membranes. (A) M. smegmatis mc2155/pJAM2-MSMEG_4947 was disrupted by sonication, and cell fractions were obtained by differential centrifugation. The presence of His-tagged MSMEG_4947 in cytosol, membrane fraction, and cell wall (P60) fractions was analyzed by SDS-PAGE (left) and Western blotting (right). (B) The activity of recombinant MSMEG_4947 was analyzed by enzymatic reaction mixtures containing membrane fractions from M. smegmatis mc2155/pJAM2 (control) and M. smegmatis mc2155 pJAM2-MSMEG_4947 (overproducer) and UDP-[14C]-GlcNAc. Reaction products [14C]-glycolipid 1 (GL1) and [14C]-glycolipid 2 (GL2) were extracted by organic solvents. Twenty percent of the lipid extract was loaded on silica-gel TLC plate, developed in CHCl3-CH3OH-NH4OH-H2O (65:25:0.5:3.6), and then exposed to autoradiography film for 3 days. (C) The amount of 14C-label incorporated into organic phase was quantified by scintillation counting. Data represent the means ± standard deviation (SD) of the results from two independent experiments (from two batches of cells) performed in triplicates.
Membrane fractions prepared from M. smegmatis pJAM2 and M. smegmatis pJAM2-MSMEG_4947 served as enzyme sources for an evaluation of recombinant WecA activity. After incubation of UDP-[14C]-GlcNAc with mycobacterial membrane fractions, which also provided decaprenyl-phosphate for the WecA-catalyzed reaction, the 14C-labeled glycolipid products were extracted with CHCl3-CH3OH (2:1) and separated from the water-soluble radioactive substrate UDP-[14C]-GlcNAc by a biphasic Folch wash. The fractions were quantified by scintillation spectrometry and then analyzed by thin-layer chromatography (TLC), followed by autoradiography. The TLC profile (Fig. 2B) revealed two bands corresponding to [14C]-GL1 and [14C]-GL2. Biosynthesis of [14C]-GL2 in the reaction mixture occurs due to the presence of endogenous TDP-Rha and rhamnosyltransferase activity in the membrane preparations, but because the entire radiolabeled [14C]-GL2 is formed from its de novo-synthetized [14C]-GL1 precursor, both of these products were included for overall quantification of the WecA-catalyzed reaction. A comparison of the incorporation of [14C]-GlcNAc to organic fractions containing [14C]-GL1/[14C]-GL2 catalyzed by membranes prepared from the control strains and overproducers revealed an approximately 2-fold increase in the products in the reaction mixtures comprising the recombinant proteins (Fig. 2C). Given that MSMEG_4947 is an experimentally confirmed orthologue of Rv1302 (5), we concluded that these proteins exhibit WecA activity in M. smegmatis and M. tuberculosis, respectively. As reported for WecA from Thermotoga maritima (23), M. smegmatis WecA strictly requires Mg2+ ions for activity (see Fig. S1 in the supplemental material).
WecA from Mycobacterium thermoresistibile can be partially purified in an active form.
We next attempted to produce recombinant forms of the WecA orthologues Rv1302 and MSMEG_4947 in E. coli for purification and further biochemical characterization. Inspired by the success in expressing WecA from T. maritima using the pET3210 expression vector in E. coli C43(DE3), as reported by Al-Dabbagh et al. (23), we cloned Rv1302 and MSMEG_4947 into pET28a, pET29a, and pET-SUMO vectors and analyzed the expression of the recombinant proteins in E. coli BL21(DE3), E. coli BL21(DE3)/pLysS, E. coli C41(DE3), and E. coli C43(DE3) under various induction conditions. Optimization of the conditions for overexpression was followed by solubilization and purification trials. However, the results were unsatisfactory. Based on our successful purification of mycobacterial acyltransferase PatA (24, 25) using the mycobacterial expression system pJAM2, we then attempted solubilization and isolation of recombinant WecA from induced M. smegmatis pJAM2-MSMEG_4947 cells. However, we were again unable to obtain purified WecA protein in sufficient amounts using this approach.
We therefore decided to produce recombinant protein from M. thermoresistibile, which is an attractive surrogate for the production of problematic mycobacterial proteins (26). Importantly, WecA is highly conserved in mycobacteria (Fig. S2), with the WecA from M. thermoresistibile ATCC 19527 sharing 84.5% amino acid identity to that of M. tuberculosis H37Rv. The WecA M. thermoresistibile (WecAMth) coding sequence was cloned into pET28a, pET29a, and pET-SUMO vectors. Based on expression trials in various host strains, E. coli C43(DE3)/pET28a-wecAMth was selected as the expression system of choice. The isolation procedure, which included solubilization of membrane proteins with 8% 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate (CHAPS), followed by metal-affinity chromatography, yielded a protein fraction with WecAMth as the major component (Fig. S3), as confirmed by mass spectrometry analysis. Importantly, WecA from the E. coli host was not detected in the final fraction (Data Set S1), which confirms that the WecA activity of this sample could be attributed specifically to recombinant WecAMth (Fig. S3).
WecA is essential in M. tuberculosis.
Saturating transposon mutagenesis studies have suggested that the wecA gene, Rv1302, is essential in M. tuberculosis (27, 28). To confirm this, we constructed an anhydrotetracycline (ATc)-regulated conditional knockdown mutant (cKD) in wecA by promoter replacement using the same approach as applied for other genes in M. tuberculosis (29–32). The wecA Tet-Off mutant showed ATc-dependent growth both in liquid culture and on agar (Fig. 3A and B). Quantitative analysis of the effect of ATc treatment on wecA gene expression in the wild-type and mutant strains was performed by droplet digital PCR (ddPCR) (Fig. 3C). In the absence of ATc, the level of wecA transcript in the wecA Tet-Off mutant was comparable to that in H37Rv. However, ATc treatment resulted in time-dependent reduction in the level of wecA transcript to ∼25% of that in the untreated control after 24 h and ∼2.8% after 48 h. Thus, the inhibitory effect of ATc on the growth of the wecA Tet-Off mutant correlated with transcriptional silencing of wecA.
FIG 3.
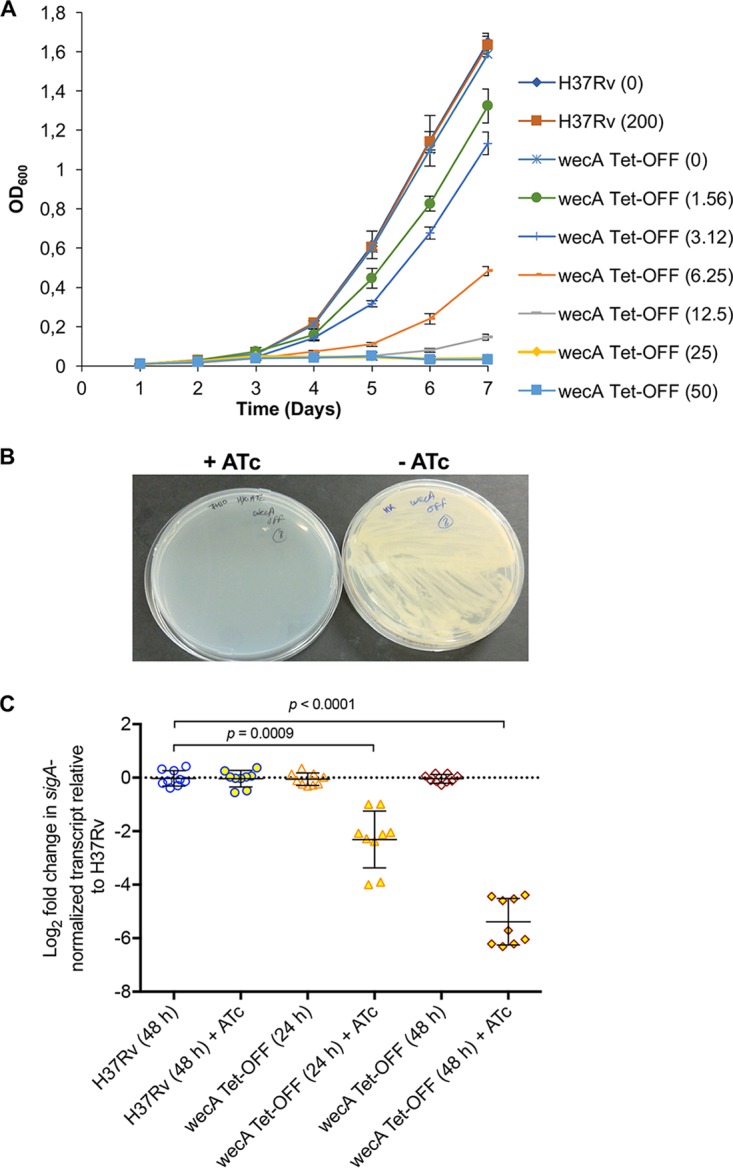
ATc dose-dependent growth of wecA conditional mutant of M. tuberculosis. (A) Growth of wecA Tet-Off was monitored in standard Middlebrook 7H9 liquid broth supplemented with the indicated concentrations of ATc (in parentheses; ng/ml), using the H37Rv and wecA-SCO strains as controls. Data are means ± SD of the results from three independent experiments. (B) Growth of the wecA Tet-Off mutant is suppressed in the presence of ATc on agar. (C) Silencing of wecA transcript to ATc treatment. Logarithmic-phase cultures of the wecA Tet-Off or H37Rv strains were treated with ATc for 24 and 48 h, as indicated, and the concentrations of wecA transcript relative to sigA were determined by ddPCR, as described in Materials and Methods. All of the data generated by QuantaSoft software included the 95% confidence interval.
Transcriptional silencing of wecA is bactericidal in M. tuberculosis in vitro and ex vivo.
Having confirmed the essentiality of wecA, we then assessed the impact of wecA silencing on the viability of M. tuberculosis. The wecA Tet-Off mutant was grown in standard 7H9 broth, with or without ATc, and samples collected over 7 days were plated on standard 7H10 agar containing the appropriate antibiotics to score CFU as an indicator of viability (Fig. 4A). In the absence of ATc, the wecA Tet-Off mutant showed growth comparable to that of the H37Rv control, and its ATc-dependent growth phenotype was stable over the course of the experiment, i.e., no CFU were observed on agar containing ATc (data not shown). In contrast, ATc treatment resulted in a progressive loss of viability, as evidenced by an ∼2-log10 decline in CFU over 7 days. The effect of wecA silencing on the viability of M. tuberculosis ex vivo was then assessed by infecting THP-1-derived macrophages with wecA Tet-Off or H37Rv. Infected macrophages were maintained in either standard RPMI medium or RPMI medium containing ATc, and M. tuberculosis viability was assessed by CFU enumeration over a period of 7 days (Fig. 4B). In the absence of ATc, the wecA Tet-Off mutant grew as well as H37Rv in THP-1 cells. ATc-induced silencing of wecA resulted in a slow decline in the viability of intracellular M. tuberculosis over the 7-day time course: the bacterial load remained constant until day 3 and thereafter showed a gradual decline, dropping by ∼0.8 log10 CFU over 7 days. Together, these results establish WecA as a bactericidal target in M. tuberculosis.
FIG 4.
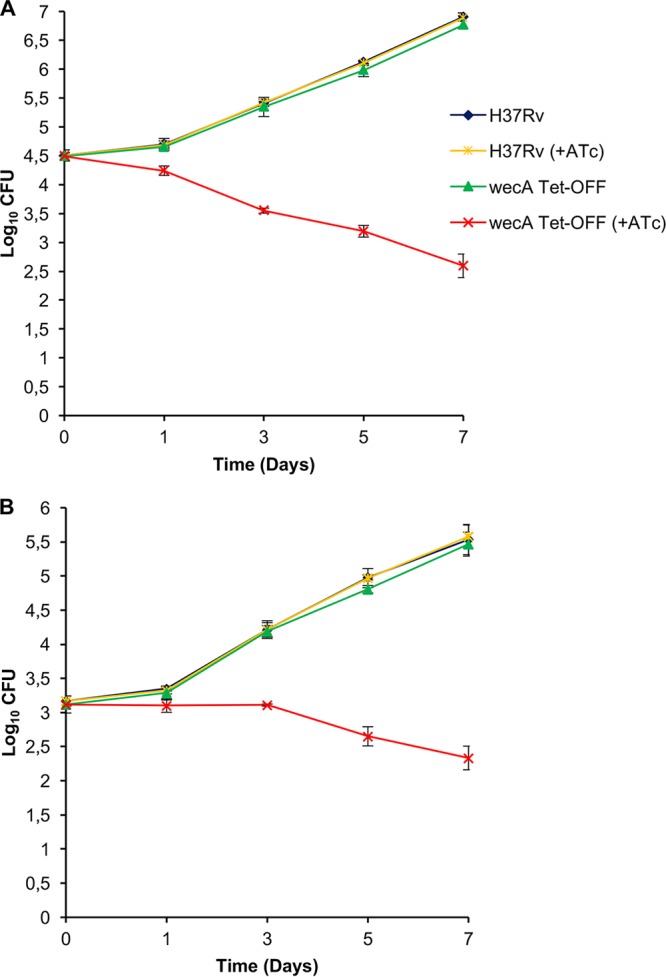
WecA is essential for the growth and survival of M. tuberculosis in both in vitro and intracellular environment. (A) The effect of wecA silencing on the viability of M. tuberculosis grown in vitro was assessed as described in Materials and Methods. The wecA Tet-Off and H37Rv strains were grown in the presence or absence of ATc (200 ng/ml), and the effect of silencing on viability was assessed by plating serial dilutions at the indicated times on 7H10 agar. (B) THP-1 cells were infected as described in Materials and Methods and grown in standard RPMI medium in the absence or presence of ATc (400 ng/ml), and the effect of wecA silencing on M. tuberculosis viability was assessed by plating serial dilutions as described in Materials and Methods. The data represent the mean ± SD from three biological replicates.
Optimization of radiometric assays for WecA and translocase I using crude mycobacterial membranes and commercial substrates.
Evidence of a shift in the targets of caprazamycin derivatives in B. subtilis from MraY (translocase I) to TagO (a WecA orthologue) upon chemical modification of the tested compounds (5) prompted us to develop simple radiometric assays using the same enzyme source and commercially available substrates to allow side-by-side evaluation of the effects of inhibitors on both WecA and translocase I activities in mycobacteria. We chose to use the nonpathogenic strains M. smegmatis mc2155 and M. tuberculosis H37Ra as model organisms for this purpose. Comparisons of WecA protein sequences between the enzymes from M. tuberculosis H37Rv and M. tuberculosis H37Ra or M. smegmatis mc2155 revealed 99.8% and 84.7% identical amino acid residues (Fig. S2); the same evaluations of translocase I protein sequences yielded 100% identity between M. tuberculosis H37Rv and M. tuberculosis H37Ra and 87.2% identity between M. tuberculosis H37Rv and M. smegmatis mc2155 (Fig. S4). Therefore, we considered the model organisms to be suitable surrogates. Our initial attempt to assay translocase I activity was based on the WecA assay (described above), using buffer A with membranes prepared from M. smegmatis or M. tuberculosis and UDP-[14C]-MurNAc-pentapeptide as the substrate. However, simply replacing UDP-[14C]-GlcNAc with UDP-[14C]-MurNAc-pentapeptide in the reaction mixture did not result in the production of lipid I. We thus modified the reaction conditions for translocase I according to the procedure described for E. coli membranes as the enzyme source (33). The reaction was thus performed in buffer C, which resulted in efficient lipid I production (Fig. 5). Contrary to previously published procedures where the reaction was stopped by the addition of pyridinium acetate and the products were extracted by repeated butanol washes (5, 34, 35), we proceeded exactly as stated above to obtain the WecA reaction products, i.e., by biphasic Folch wash. This led to the clean separation of lipid I product in the organic phase from the water-soluble substrates (Fig. 5).
FIG 5.

Optimization of WecA and translocase 1 assays. Organic and water phases were analyzed by TLC on silica-gel plates developed in 2-propanol–concentrated NH4OH–H2O (6:3:1), followed by autoradiography. To evaluate the activities of mycobacterial WecA and translocase 1, different buffer compositions were tested. (A) Buffer A (50 mM MOPS [pH 7.9], 10 mM MgCl2, 5 mM 2-mercaptoethanol); (B) buffer B (50 mM Tris-HCl [pH 8.0], 40 mM MgCl2, 0.5 mM EDTA, 50 mM sucrose, 5 mM 2-mercaptoethanol, 0.5% CHAPS, 50 μM undecaprenyl-P); (C) buffer C (100 mM Tris-HCl [pH 7.5], 30 mM MgCl2, 0.15% Triton X-100, 50 μM undecaprenyl-P, 100 μg/ml phosphatidyl glycerol).
Subsequently, we modified the reaction mixtures for measuring WecA activity by using buffer B (33), which increased efficiency of the WecA reaction by 60% for M. smegmatis and by about 80% for M. tuberculosis (Fig. 5).
Evaluation of potential dual activities of selected inhibitors against WecA and translocase I.
For evaluation of the optimized WecA and translocase I assays, we selected a panel of inhibitors of these enzymes: capuramycins SQ641 and SQ997, tunicamycin, and X-J99620886 (Fig. 6A). The chemically modified natural compound SQ641 is an analogue of capuramycin SQ997 with a lipophilic decanoyl side chain attached to the sugar moiety (36). SQ641 kills M. tuberculosis faster than other common anti-TB drugs and shows strong synergy with ethambutol, streptomycin, and SQ109 (37). The compound X-J799620886 was discovered in an old Sanofi-Aventis program, and it differs from commercially available tunicamycin only in the length of a lipophilic side chain.
FIG 6.

Effect of selected translocase I inhibitors on mycobacterial WecA and translocase I activities. Selectivity of tested compounds (200 μM) for WecA and translocase I activity was analyzed by radiometric assays using membrane fraction isolated from M. smegmatis mc2155 or M. tuberculosis H37Ra. Twenty percent of the lipid extract was loaded on a silica-gel TLC plate, developed in 2-propanol–concentrated NH4OH–H2O (6:3:1), and exposed to autoradiography film for 4 days. (A) Chemical structures of the compounds tested in this study. (B) Products of the reaction mixtures (GL1 or lipid I) after visualization by autoradiography.
In the initial screening, we tested all four compounds at a concentration of 200 μM, which caused severe inhibition of both WecA and translocase I activities in the membranes prepared from M. smegmatis and M. tuberculosis (Fig. 6B). To exclude nonspecific inhibitory effects, we also examined galactan polymerization and synthesis of the mycobacterial mannose-containing glycolipids phosphatidylinositol mannosides (PIMs) and polyprenyl-phospho-mannoses (PPMs). Polyprenylphosphate-containing substrates are utilized in these reactions similarly to WecA- and translocase I-catalyzed reactions. The reactions were performed as described with recombinant GlfT1 and GlfT2 enzymes produced in E. coli (8) or with mycobacterial membranes for monitoring PIM and PPM synthesis (15). Neither SQ641 nor X-J99620886, as representative inhibitors from our selection of compounds, inhibited any of the tested enzymes at 200 μM (Fig. S5). Thus, the observed robust inhibition of WecA and translocase I was likely due to specific effects of the tested drugs on these targets. However, there was a clear difference in these effects: while residual WecA activity was observed in the reaction mixtures with SQ641 and SQ997, the other two inhibitors, X-J99620886 and tunicamycin, appeared to completely abolish the reaction. In contrast, higher residual activity was found in the translocase I reaction for the X-J99620886 and tunicamycin inhibitors than for SQ641 and SQ997 (Fig. 6B).
These findings were confirmed in dose experiments performed with SQ641 and X-J99620886 in WecA and translocase I assays using membranes from M. tuberculosis H37Ra. The results showed that SQ641 indeed preferentially inhibited translocase I (50% inhibitory concentration [IC50], 0.165 μM) compared to WecA (IC50, 136 μM). Conversely, X-J99620886 potently inhibited both activities, with IC50s of 0.01427 μM for WecA and 13.32 μM for translocase I, indicating a preference for WecA in this case (Fig. S6). Overall, the IC50s suggested that both compounds are very efficient inhibitors of WecA and translocase I activity in mycobacterial membranes. However, each of these compounds has a strong preference toward one of the tested enzymes: SQ641 for translocase I and X-J99620886 for WecA. These results confirm that the assays can successfully be used to resolve the selectivity of such dual-activity inhibitors.
Tunicamycin and X-J99620886 act on WecA in M. tuberculosis.
The wecA Tet-Off mutant provided a tool to investigate the target selectivity of biochemically confirmed WecA inhibitors in whole M. tuberculosis cells using a checkerboard assay, in which the concentrations of ATc and a WecA inhibitor were varied in a two-dimensional array (Fig. 7). An ∼16-fold reduction in the MIC90 of tunicamycin (from 0.5 μg/ml to 0.03 μg/ml) was observed concomitant with the silencing of wecA (Fig. 7A). In contrast, wecA silencing had no effect on M. tuberculosis susceptibility to the standard TB drugs isoniazid or rifampin, whose mechanisms of action are unrelated to WecA inhibition (Fig. S8). The selective hypersensitization observed supports the notion that WecA is a target of tunicamycin in M. tuberculosis. The wecA Tet-Off mutant was also hypersensitive to the compound X-J99620886, as evidenced by a 4- to 8-fold shift in MIC90 (from 0.12 μg/ml to 0.03 to 0.015 μg/ml) upon wecA silencing (Fig. 7B). These results implicate WecA as a target of X-J99620886. In contrast, however, there was no significant change in the MIC of SQ641 against wecA-silenced M. tuberculosis (Fig. 7C), consistent with its considerably weaker activity against WecA in the enzyme assay.
FIG 7.
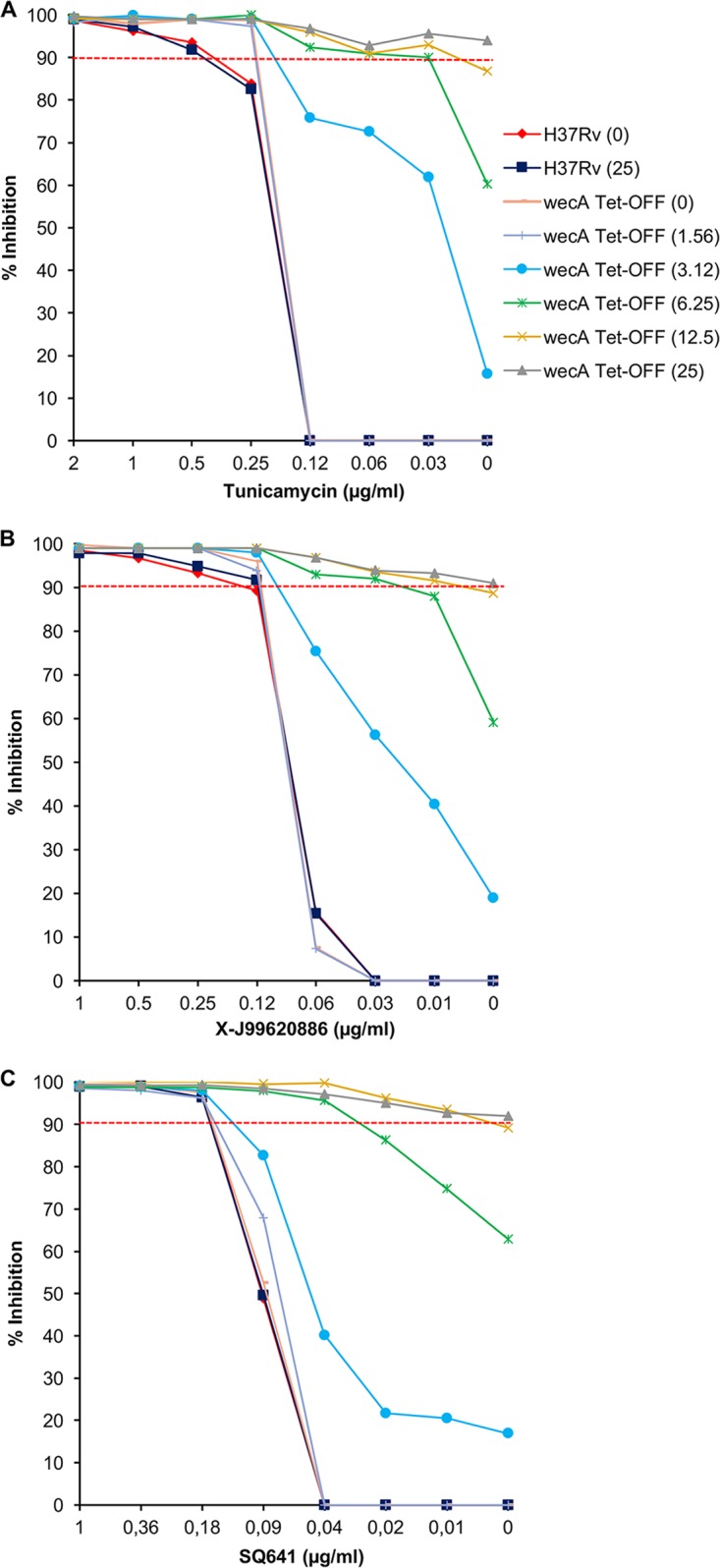
Silencing of wecA in M. tuberculosis H37Rv confers hypersensitivity to WecA inhibitors. A two-dimensional array of serial dilutions of ATc and WecA inhibitors were added to the cultures for determining the susceptibility of H37Rv and wecA Tet-Off strains to the WecA inhibitors. (A) Tunicamycin; (B) X-J99620886; (C) SQ641. Bacterial viability was assessed by the alamarBlue assay, as described in Materials and Methods. The values in parentheses represent ATc concentration in ng/ml.
DISCUSSION
Mycobacterial phosphoglycosyltransferase WecA is emerging as a novel and highly attractive target for the identification of new TB drugs, as a result of the progression of caprazamycin derivative CPZEN-45 as a preclinical candidate based on its favorable properties: aqueous solubility, toxicity profile (38), substantial in vitro activity against M. tuberculosis, and efficacy in mouse models of TB (http://www.newtbdrugs.org/pipeline/compound/cpzen-45). In this report, we demonstrate for the first time the enzymatic activity of the WecA orthologue from M. smegmatis and provide evidence in support of the findings of Jin et al. (9) that MSMEG_4947 and Rv1302 have WecA functions in mycobacteria. Our efforts to obtain these proteins by overexpression in E. coli or M. smegmatis failed. To date, the only successful WecA purification was described for an orthologue from the thermophilic bacterium T. maritima (23), although a recent attempt to replicate this procedure did not succeed in producing pure enzyme (39). Although we were unable to purify mycobacterial WecA to homogeneity, we could, however, demonstrate WecA activity in the partially purified orthologue from M. thermoresistibile, which can serve as a starting point for efforts toward the structural characterization of WecA from mycobacteria. Several recent reports on detailed structures of translocase I (MraY) (40–42), which belongs to the same family of polyprenyl phosphate–N-acetyl-hexosamine-1-phosphate transferases, hold promise for similar success with WecA enzymes.
MraY is an intensely investigated target for several classes of nucleoside antibiotics, including liposidomycins, mureidomycins, muraymycins, capuramycins, tunicamycins, and caprazamycins, among others (43, 44). Despite substantial effort, none of these compounds has yet progressed to human clinical trials (45). Structural information on MraY enzymes cocrystallized with muraymycin and tunicamycin that leads to a better understanding of the mechanisms of action and its inhibition is expected to aid in the development of new drug candidates not only for translocase I, but also for its paralogues, including WecA (41, 42). Given the similarities between WecA and translocase I, the design of compounds targeting both enzymes would be a particularly attractive option. In this context, a number of uridine-containing natural antibiotics have already been shown to inhibit both enzymes, albeit differentially (35, 46). This was further confirmed by our experiments with the capuramycin and tunicamycin derivatives SQ641 and X-J99620886, each of which varied in their inhibitory activities against WecA and translocase I by a factor of 103 (Fig. S6), in agreement with published data (46). However, a recent report by Mitachi et al. (47) describes the synthesis of muraymycin D1 and its amides, which inhibit WecA and translocase I of M. tuberculosis with much closer IC50s: muraymycin D1 and muraymycin D1 amide exhibited ∼60-fold higher activity against translocase I than against WecA, while muraymycin D1 diamide reached IC50s of 0.07 μM and 0.0096 μM for WecA and translocase I, respectively, reflecting an ∼7-fold difference in inhibitory activity on these two enzymes. This seminal work opens up an attractive possibility for a dual mechanism of action of the selected nucleoside antibiotic-derived inhibitors.
Since dual activity of an inhibitor is a highly favorable property (45, 48, 49), we developed simple assays to evaluate the inhibition of both mycobacterial WecA and translocase I. While numerous methods were reported for monitoring WecA and/or translocase I activities in different bacteria (33, 50, 51), to the best of our knowledge, a parallel evaluation of inhibitory activities of antibiotics on mycobacterial WecA and translocase I was reported for the first time only recently (47). Similar to our and numerous other assays, Mitachi et al. (47) employed crude mycobacterial membranes as a source of the tested enzymes. The activities of muraymycin D1 on WecA and translocase I were determined by fairly complicated fluorescence-based assays (51, 52). The value of these fluorescent assays is the potential to be converted to high-throughput formats. The radiometric assays described in the present paper are more suitable for low- or medium-throughput screening, although their transformation to a high-throughput format similar to that described by Hyland and Anderson (33) can be foreseen. Based on their reliability, yet simplicity, we envision these assays as being useful for a rapid evaluation of the inhibitory properties of natural product derivatives undergoing structural optimization. Efforts focused to improve the properties (such as bioavailability) of original natural compounds, along with novel chemistry allowing the development of procedures for total synthesis of the selected natural compounds, are documented extensively in the literature (44, 47, 53, 54). Reliable and simple assays for translocase I and WecA could thus help reveal possible target switching (5) and prioritize the natural product derivatives that exert comparable dual inhibition of WecA and translocase I enzymes in mycobacteria.
Target validation is a key step in the drug discovery process (55). In this study, we genetically validated WecA as a bactericidal drug target in M. tuberculosis. The wecA cKD mutant enabled us to assess the selectivity of molecules with demonstrated activity against the WecA enzyme in M. tuberculosis by target-based whole-cell screening. The hypersensitivity of wecA-silenced M. tuberculosis to tunicamycin and X-J99620886 suggested that WecA is a target for both compounds. These results, however, do not preclude translocase I as another tunicamycin target in M. tuberculosis. The hypersensitization of the wecA knockdown to tunicamycin was quite modest compared to the other target/drug couples assessed by the same methodology (29, 56), which may argue in favor of another target(s) for tunicamycin in addition to WecA. The recently published structure of MraY with bound tunicamycin (42) is consistent with this notion and could also be the case for other WecA inhibitors, such as CPZEN-45. In contrast, the capuramycin derivative SQ641, which showed potent WecA inhibition in the enzyme assay, does not appear to act on this target in whole cells, as evidenced using the wecA cKD mutant.
Natural products represent promising new leads for TB drug development, as demonstrated by several successful examples over the past few years (57–60). On the basis of the genetic validation of WecA as a bactericidal target and its pharmacological validation as an enzyme inhibited by several classes of natural nucleoside antibiotics, this enzyme represents an attractive target for the development of new medicines against TB.
MATERIALS AND METHODS
Cloning of wecA and its expression in M. smegmatis.
The Rv1302 gene from M. tuberculosis H37Rv and the MSMEG_4947 gene from M. smegmatis mc2155 were amplified using oligonucleotide primers Rv1302-pJAM-Fwd and Rv1302-pJAM-Rev or MSMEG_4947-pJAM-Fwd and MSMEG_4947-pJAM-Rev, containing BamHI and XbaI restriction sites (Table S1). The PCR fragments were digested and ligated into the similarly digested pJAM2 vector. The DNA sequences of the inserts were confirmed by sequencing and recombinant pJAM2 plasmids carrying Rv1302 from M. tuberculosis H37Rv or MSMEG_4947 from M. smegmatis mc2155 were transformed into M. smegmatis mc2155. Cells were grown in MM63 medium [5 mM (NH4)2SO4, 10 mM KH2PO4,18 mM FeSO4·7H2O (pH 7.0)] supplemented with 1 mM MgSO4, 0.025% (vol/vol) tyloxapol, 0.2% (wt/vol) succinate, and kanamycin (20 μg/ml) until the optical density at 600 nm (OD600) reached ∼0.5 (mid-log phase). Induction was initiated by the addition of acetamide at a final concentration of 0.2% (wt/vol). The expression of Rv1302 and MSMEG_4947 was tested at 37°C and 30°C. Optimal expression was achieved at 37°C after 4 h of induction for Rv1302 and 6 h of induction for MSMEG_4947. The resulting MSMEG_4947 product is a truncated version lacking the N-terminal 16 amino acids.
Cloning of wecA and its expression in E. coli.
For the production of recombinant His-tagged mycobacterial WecA protein in E. coli, three homologs of the mycobacterial wecA gene were selected: Rv1302 from M. tuberculosis H37Rv, MSMEG_4947 from M. smegmatis mc2155, and wecA from M. thermoresistibile ATCC 19527. The primers used for amplification of these genes are listed in the Table S1. The PCR fragments cloned into pET28a (N-terminal His tag) and pET29a (C-terminal His tag) plasmids were digested with NdeI and HindIII enzymes. Constructs based on pET-SUMO plasmids carrying Rv1302, MSMEG_4947, or wecA from M. thermoresistibile were prepared according to the user manual of the Champion pET SUMO expression system (Thermo Fisher Scientific). The prepared recombinant plasmids were then transformed into different expression strains, E. coli BL21(DE3), E. coli BL21(DE3) pLysS, E. coli C41(DE3), and E. coli C43(DE3), in which optimal expression was investigated at different time points during incubation at 37°C, 25°C, or 18°C and after induction with 1 mM or 0.4 mM isopropyl-β-d-thiogalactopyranoside (IPTG). The presence of recombinant mycobacterial WecA protein was analyzed in cell lysates and supernatants (15,000 × g, 20 min) of the lysates by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Western blotting.
Production, solubilization, and purification of WecA M. thermoresistibile.
One liter of Luria-Bertani (LB) medium supplemented with kanamycin (20 μg/ml) was inoculated (1:100) with a fresh overnight preculture of E. coli C43(DE3)/pET28a-wecAMth grown in the same medium at 37°C, and the culture was incubated at the same temperature with shaking at 130 rpm until the OD600 reached ∼0.5. The cultures where then kept at 4°C for 1 to 2 h and the OD600 measured again (∼0.6 to 0.7). Recombinant protein expression was induced with 0.4 mM IPTG by incubation at 25°C for 17 h. The cells were collected by centrifugation (3,500 × g for 20 min at 4°C), washed twice with 25 mM Tris-HCl (pH 7.5), and stored at −20°C before use. Approximately 4 g (wet weight) of E. coli C43(DE3)/pET28a-wecAMth cell pellet was suspended in 16 ml of buffer PA (25 mM Tris-HCl [pH 7.5], 300 mM NaCl, 10 mM MgCl2, 10% glycerol, 2 mM β-mercaptoethanol, 1 mM phenylmethylsulfonyl fluoride [PMSF]), and the cells were disrupted by sonication in 15-s pulses with 40-s cooling intervals between pulses, for a total of 20 cycles. The cell lysate was then centrifuged at 15,000 × g for 20 min at 4°C, and the supernatant was subsequently centrifuged at 100,000 × g for 1 h at 4°C. The membrane fraction was homogenized in 4 ml of buffer PA supplemented with 136 mM CHAPS (8% [wt/vol]), and proteins were solubilized by gentle rocking at 4°C for 2 h. The mixture was then centrifuged at 100,000 × g for 1 h at 4°C. The concentration of detergent was reduced by repeated filtration steps using an Amicon Ultra 15-ml centrifugal filter unit (10K molecular weight cutoff [MWCO]; Merck Millipore; 3,500 × g, 15 min, 4°C per one spin) and buffer PA. The sample was finally recovered in buffer PB (25 mM Tris-HCl [pH 7.5], 300 mM NaCl, 10 mM MgCl2, 10% glycerol, 2 mM β-mercaptoethanol, 0.1 mM PMSF, 0.5% CHAPS). Binding of His-tagged WecA from M. thermoresistibile was performed using a Talon cobalt resin (Clontech) with 2 h of incubation at 4°C. The mixture was then centrifuged at 90 × g for 3 min at 4°C to separate nonbound proteins and subsequently washed with 3 ml (two column volumes) of buffer PB. The resin was then suspended in 3 ml of buffer PB and transferred to a Poly-Prep chromatography column (Bio-Rad). Elution of the proteins from the affinity resin was performed with stepwise imidazole gradient (25 mM, 50 mM, 150 mM, 300 mM, and 1 M imidazole in buffer PB). Fractions containing His-tagged WecA protein were pooled and desalted with an Amicon Ultra 4-ml centrifugal filter (10K MWCO; Merck Millipore) by washing with buffer PB. For the final purification of WecA from M. thermoresistibile, rechromatography with Talon cobalt resin was performed in the same manner. Protein concentration was determined with a bicinchoninic acid (BCA) assay (Pierce BCA protein assay kit; Thermo Scientific).
Protein identification by mass spectrometry.
Following protein separation by SDS-PAGE and Coomassie brilliant blue staining, the gel pieces of interest were destained, reduced, and alkylated using dithiothreitol and iodoacetamide. The proteins were digested using sequencing-grade modified trypsin (Promega) overnight at 37°C. Extracted peptides were dried and dissolved in 0.1% trifluoroacetic acid and 2% acetonitrile (ACN). The samples were loaded onto a trap column (Acclaim PepMap100 C18, 75 μm by 20 mm; Dionex, CA, USA) and separated with a C18 column (Acclaim PepMap C18, 75 μm by 150 mm; Dionex) on an Ultimate 3000 RSLCnano system (Dionex) in a linear 25 min gradient (3 to 43% B) and flow rate of 300 nl/min. Two mobile phases were used: 0.1% (vol/vol) FA (A) and 80% (vol/vol) ACN with 0.1% FA (B). Eluted peptides were sprayed directly into an Orbitrap Elite mass spectrometer (Thermo Scientific, MA, USA), and spectral data sets were collected in the data-dependent mode using Top15 strategy for the selection of precursor ions for the higher-energy collisional dissociation (HCD) fragmentation (61). Each of the samples was analyzed in two technical replicates. The obtained data sets were processed by MaxQuant version 1.5.3.30 (62), with a built-in Andromeda search engine using carbamidomethylation (C) as a permanent modification and oxidation (M) as a variable modification. The search was performed against the E. coli protein database (UniProt), and the expected recombinant WecA sequence (M. thermoresistibile) included in a separate Fasta file.
For analysis of the proteins in solution, the samples were supplemented with 1 M Tris-HCl (pH 7.8) to final concentration of 25 mM, reduced in the presence of 5 mM dithiothreitol (30 min, 60°C), and alkylated by the addition of 15 mM iodoacetamide (20 min, room temperature, in the dark). The alkylation reaction was quenched by additional 5 mM dithiothreitol. Two micrograms of modified sequencing-grade trypsin (Promega) was added to the protein mixture, and the samples were incubated overnight at 37°C. The reaction mixture was acidified by the addition of 0.5% trifluoroacetic acid, and the peptides were purified by microtip C18 solid-phase extraction (SPE) and dried in the vacuum centrifuge. The samples were analyzed in technical triplicates by liquid chromatography-mass spectrometry (LC-MS), as described above, with the exception of longer separation column (50 cm) and longer gradient (240 min).
Construction of a wecA cKD mutant of M. tuberculosis.
To construct a mutant of M. tuberculosis in which the wecA promoter was replaced by the Tet-regulated promoter pmyc1tetO (63), a suicide plasmid carrying an amplicon spanning the ribosomal-binding site (RBS) (20 bp) and 322-bp 5′-terminal region of wecA, cloned as a SphI/NotI fragment in pSE100 (64), was electroporated into M. tuberculosis H37Rv and the transformants selected on Middlebrook 7H10 agar supplemented with oleic acid-albumin-dextrose-catalase (OADC) and hygromycin (Hyg). Individual colonies were grown to mid-log phase in Middlebrook 7H9 broth supplemented with OADC and Hyg. The site specificity of homologous recombination in the putative single-crossover (SCO) recombinants was confirmed by PCR using the primers listed in Table S1. Modulation of wecA expression using ATc was achieved by electroporation of pGMCK-OX38-T28 (65) into the SCO recombinant, wecA-SCO, to generate conditional mutants in the Tet-Off configuration (wecA Tet-Off). Integration into the chromosome was facilitated by codelivery of an additional suicide vector, pGA-OXP15-intL5, transiently expressing integrase (65). The transformants were selected on Middlebrook 7H10 supplemented with OADC, Hyg, and kanamycin (Km), in the presence and absence of ATc (200 ng/ml).
Gene expression analysis by droplet digital PCR.
Total RNA extraction from M. tuberculosis cultures, cDNA synthesis, designing of Primers/TaqMan minor groove binder (MGB) probes and ddPCR were performed as described previously (30).
Analysis of cidality caused by wecA silencing in vitro and in vivo.
To test the wecA-silencing effect on viability of M. tuberculosis, the wecA Tet-Off mutant and H37Rv strains were grown at 37°C in standard Middlebrook 7H9 broth to mid-exponential phase. An inoculum (∼5 × 104 CFU/ml) was added to standard Middlebrook 7H9 broth either with or without ATc (at 200 ng/ml, when used). Viability was assessed by plating serial dilutions at days 0, 1, 3, 5, and 7 on standard 7H10 agar containing the appropriate required antibiotics, in the presence (200 ng/ml) or absence of ATc. Aliquots withdrawn for CFU enumeration were washed with 1 ml of 7H9 broth to remove residual ATc and resuspended in 1 ml of fresh 7H9 broth prior to plating. Plates were incubated for 4 weeks before scoring CFU. For an evaluation of the survival of wecA-silenced M. tuberculosis in THP-1-derived macrophages, the experiment was performed as described previously (56, 66).
Drug susceptibility testing against M. tuberculosis strains.
An alamarBlue fluorescence-based broth microdilution assay was used to assess the MICs of compounds against M. tuberculosis strains, as described previously (30, 56).
Preparation of crude enzyme fractions from mycobacteria.
For enzyme experiments, mycobacteria were grown as follows: M. smegmatis mc2155 in Nutrient broth (Merck), M. smegmatis mc2155/pJAM2 and M. smegmatis mc2155/pJAM2-msmeg_4947 in MM63 medium (described above), and M. tuberculosis H37Ra in standard Middlebrook 7H9 broth (BD) supplemented with 10% albumin-dextrose-catalase (ADC) enrichment, 0.2% glycerol and 0.05% Tween 80. Enzymatic fractions were prepared as described previously (6), with slight modifications. The cell pellets (4-g to 10-g batches) were suspended in buffer A (50 mM morpholinepropanesulfonic acid [MOPS] [pH 7.9], 10 mM MgCl2, 5 mM 2-mercaptoethanol) or buffer D (20 mM Tris-HCl [pH 7.5], 10 mM MgCl2, 5 mM 2-mercaptoethanol) at a ratio 1 g of cells per 5 ml of buffer and disintegrated by sonication (30-s pulses with 90-s cooling intervals, 20 cycles) at 4°C. The cell lysates were fractionated to obtain the membrane fraction and cell envelope (P60) fraction as described previously (6). The final membrane and cell envelope fractions were resuspended in appropriate buffers at ratios of 0.3 ml of buffer/10 g of cells and 0.8 ml of buffer/5 g of cells, respectively.
Enzyme assays.
The basic WecA assay was performed as described previously (6), with minor modifications. The reaction mixtures contained 50 mM MOPS (pH 7.9), 10 mM MgCl2, 5 mM 2-mercaptoethanol, and 0.25 μCi of UDP-[14C]GlcNAc (specific activity, 300 mCi/mmol; American Radiolabeled Chemicals) in a final volume of 80 μl. The reactions were initiated by the addition of membrane fraction (∼300 μg protein) to the mixture. After incubation at 37°C for 1 h, reactions were stopped by the addition of 1.5 ml of CHCl3-CH3OH (2:1) followed by the addition of 170 μl of water to reach a final ratio of CHCl3-CH3OH-H2O of 4:2:1. The upper water phase and bottom organic phase were separated, and the organic phase containing radiolabeled lipids was dried under the stream of nitrogen. To remove residual UDP-[14C]GlcNAc, the organic phase was subjected to a repeated biphasic wash with CHCl3-CH3OH-H2O (4:2:1). The final dried organic phase was dissolved in 50 μl of CHCl3-CH3OH-concentrated NH4OH-H2O (65:25:0.5:3.6). Lipids from organic phase were loaded on aluminum-coated silica 60 F254 plate (Merck) and analyzed by TLC in CHCl3-CH3OH-concentrated NH4OH-H2O (65:25:0.5:3.6) followed by autoradiography using Kodak Bio-Max maximum-resolution (MR) films. Incorporation of the [14C]-GlcNAc from the water-soluble UDP-[14C]GlcNAc substrate to glycolipids was quantified by scintillation counting.
For a determination of both WecA and translocase I activities and the effects of the selected inhibitors, mycobacterial membranes were prepared in buffer D. The WecA assay was performed in the final volume of 50 μl containing 50 mM Tris-HCl (pH 8.0), 40 mM MgCl2, 0.5 mM EDTA, 50 mM sucrose, 5 mM 2-mercaptoethanol, 0.5% CHAPS, 50 μM undecaprenyl-P (buffer B), and 0.25 μCi of UDP-[14C]GlcNAc. The translocase I assay was performed in a final volume of 50 μl of containing 100 mM Tris-HCl (pH 7.5), 30 mM MgCl2, 0.15% Triton X-100, 50 μM undecaprenyl-P, 100 μg/ml phosphatidyl glycerol (buffer C), and 0.05 μCi of UDP-MurNAc-[14C]pentapeptide (prepared as described in supplemental Materials and Methods and in Fig. S7, or commercially obtained from the BacWAN facility, University of Warwick, Coventry, UK). Reactions were initiated by the addition of membrane fractions (180 to 200 μg of protein) isolated from M. smegmatis mc2155 or M. tuberculosis H37Ra. The reaction mixtures contained 0 to 200 μM inhibitors, which were added from dimethyl sulfoxide (DMSO) stocks; the final concentration of DMSO was 2%. Following 1 h of incubation at 37°C, the reaction mixtures were processed as described above. TLC was developed in 2-propanol–concentrated NH4OH–H2O (6:3:1). Incorporation of the [14C] label from water-soluble substrates to glycolipids was quantified by scintillation counting, and these data were used for IC50 determination. IC50s were calculated using GraphPad Prism 6 software from a log-dose versus response curve.
The same conditions were used for establishing the activity of purified WecA from M. thermoresistibile except for the source of the enzyme: the crude membrane fraction was replaced by 20 μg of proteins from the sample after purification, and the 0.5 mM EDTA was omitted from the reaction buffer. The reaction was carried out in the final volume of 80 μl.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the European Community's Seventh Framework Programme (grant 260872, More Medicines for Tuberculosis); the Slovak Research and Development Agency (contracts DO7RP-0015-11 and APVV-15-0515 to K.M.); the Ministry of Education, Science, Research and Sport of the Slovak Republic (grant VEGA 1/0441/15 to K.M.); the Howard Hughes Medical Institute (Senior International Research Scholar's grant to V.M.); the National Research Foundation of South Africa (to V.M.); and the South African Medical Research Council (to V.M.). Part of this contribution was enabled as the result of the implementation of the project “Technical infrastructure for biomedical research,” grant ITMS 26230120008, supported by the Research & Development Operational Programme funded by the ERDF (to P.B.). The funders had no role in the study design, data collection, and interpretation, or the decision to submit the work for publication.
We acknowledge Brigitte Gicquel from the Institut Pasteur, Paris, France, for providing us the pJAM2 plasmid; Vara Vissa and Dean Crick from Colorado State University, Fort Collins, CO, for pVV2, pVV16, and MurF expression plasmids; Todd Lowary from the University of Alberta for the GlfT2 expression plasmid; Jozef Nosek from Comenius University in Bratislava for help with sequence identification; and Jana Korduláková for discussions and critically reading the manuscript.
SQ641 intellectual property (IP) is owned by Sequella, Inc., and C.A.N. owns stock in Sequella.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.01310-17.
REFERENCES
- 1.WHO. 2016. Global tuberculosis report 2016. World Health Organization, Geneva, Switzerland: http://apps.who.int/iris/bitstream/10665/250441/1/9789241565394-eng.pdf?ua=1. [Google Scholar]
- 2.Zumla A, Nahid P, Cole ST. 2013. Advances in the development of new tuberculosis drugs and treatment regimens. Nat Rev Drug Discov 12:388–404. doi: 10.1038/nrd4001. [DOI] [PubMed] [Google Scholar]
- 3.Makarov V, Lechartier B, Zhang M, Neres J, van der Sar AM, Raadsen SA, Hartkoorn RC, Ryabova OB, Vocat A, Decosterd LA, Widmer N, Buclin T, Bitter W, Andries K, Pojer F, Dyson PJ, Cole ST. 2014. Towards a new combination therapy for tuberculosis with next generation benzothiazinones. EMBO Mol Med 6:372–383. doi: 10.1002/emmm.201303575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jackson M, McNeil MR, Brennan PJ. 2013. Progress in targeting cell envelope biogenesis in Mycobacterium tuberculosis. Future Microbiol 8:855–875. doi: 10.2217/fmb.13.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ishizaki Y, Hayashi C, Inoue K, Igarashi M, Takahashi Y, Pujari V, Crick DC, Brennan PJ, Nomoto A. 2013. Inhibition of the first step in synthesis of the mycobacterial cell wall core, catalyzed by the GlcNAc-1-phosphate transferase WecA, by the novel caprazamycin derivative CPZEN-45. J Biol Chem 288:30309–30319. doi: 10.1074/jbc.M113.492173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mikušová K, Mikuš M, Besra GS, Hancock I, Brennan PJ. 1996. Biosynthesis of the linkage region of the mycobacterial cell wall. J Biol Chem 271:7820–7828. doi: 10.1074/jbc.271.13.7820. [DOI] [PubMed] [Google Scholar]
- 7.Mills JA, Motichka K, Jucker M, Wu HP, Uhlik BC, Stern RJ, Scherman MS, Vissa VD, Pan F, Kundu M, Ma YF, McNeil M. 2004. Inactivation of the mycobacterial rhamnosyltransferase, which is needed for the formation of the arabinogalactan-peptidoglycan linker, leads to irreversible loss of viability. J Biol Chem 279:43540–43546. doi: 10.1074/jbc.M407782200. [DOI] [PubMed] [Google Scholar]
- 8.Beláňová M, Dianišková P, Brennan PJ, Completo GC, Rose NL, Lowary TL, Mikušová K. 2008. Galactosyl transferases in mycobacterial cell wall synthesis. J Bacteriol 190:1141–1145. doi: 10.1128/JB.01326-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jin Y, Xin Y, Zhang W, Ma Y. 2010. Mycobacterium tuberculosis Rv1302 and Mycobacterium smegmatis MSMEG_4947 have WecA function and MSMEG_4947 is required for the growth of M. smegmatis. FEMS Microbiol Lett 310:54–61. doi: 10.1111/j.1574-6968.2010.02045.x. [DOI] [PubMed] [Google Scholar]
- 10.Igarashi M, Nakagawa N, Doi N, Hattori S, Naganawa H, Hamada M. 2003. Caprazamycin B, a novel anti-tuberculosis antibiotic, from Streptomyces sp. J Antibiot (Tokyo) 56:580–583. doi: 10.7164/antibiotics.56.580. [DOI] [PubMed] [Google Scholar]
- 11.Takahashi Y, Igarashi M, Miyake T, Soutome H, Ishikawa K, Komatsuki Y, Koyama Y, Nakagawa N, Hattori S, Inoue K, Doi N, Akamatsu Y. 2013. Novel semisynthetic antibiotics from caprazamycins A-G: caprazene derivatives and their antibacterial activity. J Antibiot (Tokyo) 66:171–178. doi: 10.1038/ja.2013.9. [DOI] [PubMed] [Google Scholar]
- 12.Ikeda M, Wachi M, Jung HK, Ishino F, Matsuhashi M. 1991. The Escherichia coli mraY gene encoding UDP-N-acetylmuramoyl-pentapeptide: undecaprenyl-phosphate phospho-N-acetylmuramoyl-pentapeptide transferase. J Bacteriol 173:1021–1026. doi: 10.1128/jb.173.3.1021-1026.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mikušová K, Yagi T, Stern R, McNeil MR, Besra GS, Crick DC, Brennan PJ. 2000. Biosynthesis of the galactan component of the mycobacterial cell wall. J Biol Chem 275:33890–33897. doi: 10.1074/jbc.M006875200. [DOI] [PubMed] [Google Scholar]
- 14.Mikušová K, Beláňová M, Korduláková J, Honda K, McNeil MR, Mahapatra S, Crick DC, Brennan PJ. 2006. Identification of a novel galactosyl transferase involved in biosynthesis of the mycobacterial cell wall. J Bacteriol 188:6592–6598. doi: 10.1128/JB.00489-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Korduláková J, Gilleron M, Mikušová K, Puzo G, Brennan PJ, Gicquel B, Jackson M. 2002. Definition of the first mannosylation step in phosphatidylinositol mannoside synthesis. PimA is essential for growth of mycobacteria. J Biol Chem 277:31335–31344. [DOI] [PubMed] [Google Scholar]
- 16.Korduláková J, Gilleron M, Puzo G, Brennan PJ, Gicquel B, Mikušová K, Jackson M. 2003. Identification of the required acyltransferase step in the biosynthesis of the phosphatidylinositol mannosides of Mycobacterium species. J Biol Chem 278:36285–36295. doi: 10.1074/jbc.M303639200. [DOI] [PubMed] [Google Scholar]
- 17.Dhiman RK, Schulbach MC, Mahapatra S, Baulard AR, Vissa V, Brennan PJ, Crick DC. 2004. Identification of a novel class of omega,E,E-farnesyl diphosphate synthase from Mycobacterium tuberculosis. J Lipid Res 45:1140–1147. doi: 10.1194/jlr.M400047-JLR200. [DOI] [PubMed] [Google Scholar]
- 18.Jackson M, Crick DC, Brennan PJ. 2000. Phosphatidylinositol is an essential phospholipid of mycobacteria. J Biol Chem 275:30092–30099. doi: 10.1074/jbc.M004658200. [DOI] [PubMed] [Google Scholar]
- 19.Triccas JA, Parish T, Britton WJ, Gicquel B. 1998. An inducible expression system permitting the efficient purification of a recombinant antigen from Mycobacterium smegmatis. FEMS Microbiol Lett 167:151–156. doi: 10.1111/j.1574-6968.1998.tb13221.x. [DOI] [PubMed] [Google Scholar]
- 20.Amer AO, Valvano MA. 2002. Conserved aspartic acids are essential for the enzymic activity of the WecA protein initiating the biosynthesis of O-specific lipopolysaccharide and enterobacterial common antigen in Escherichia coli. Microbiology 148:571–582. doi: 10.1099/00221287-148-2-571. [DOI] [PubMed] [Google Scholar]
- 21.Lehrer J, Vigeant KA, Tatar LD, Valvano MA. 2007. Functional characterization and membrane topology of Escherichia coli WecA, a sugar-phosphate transferase initiating the biosynthesis of enterobacterial common antigen and O-antigen lipopolysaccharide. J Bacteriol 189:2618–2628. doi: 10.1128/JB.01905-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rath A, Glibowicka M, Nadeau VG, Chen G, Deber CM. 2009. Detergent binding explains anomalous SDS-PAGE migration of membrane proteins. Proc Natl Acad Sci U S A 106:1760–1765. doi: 10.1073/pnas.0813167106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Al-Dabbagh B, Mengin-Lecreulx D, Bouhss A. 2008. Purification and characterization of the bacterial UDP-GlcNAc:undecaprenyl-phosphate GlcNAc-1-phosphate transferase WecA. J Bacteriol 190:7141–7146. doi: 10.1128/JB.00676-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Svetlíková Z, Baráth P, Jackson M, Korduláková J, Mikušová K. 2014. Purification and characterization of the acyltransferase involved in biosynthesis of the major mycobacterial cell envelope glycolipid–monoacylated phosphatidylinositol dimannoside. Protein Expr Purif 100:33–39. doi: 10.1016/j.pep.2014.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Albesa-Jove D, Svetlíková Z, Tersa M, Sancho-Vaello E, Carreras-Gonzalez A, Bonnet P, Arrasate P, Eguskiza A, Angala SK, Cifuente JO, Korduláková J, Jackson M, Mikušová K, Guerin ME. 2016. Structural basis for selective recognition of acyl chains by the membrane-associated acyltransferase PatA. Nat Commun 7:10906. doi: 10.1038/ncomms10906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Edwards TE, Liao R, Phan I, Myler PJ, Grundner C. 2012. Mycobacterium thermoresistibile as a source of thermostable orthologs of Mycobacterium tuberculosis proteins. Protein Sci 21:1093–1096. doi: 10.1002/pro.2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Griffin JE, Gawronski JD, Dejesus MA, Ioerger TR, Akerley BJ, Sassetti CM. 2011. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog 7:e1002251. doi: 10.1371/journal.ppat.1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.DeJesus MA, Gerrick ER, Xu W, Park SW, Long JE, Boutte CC, Rubin EJ, Schnappinger D, Ehrt S, Fortune SM, Sassetti CM, Ioerger TR. 2017. Comprehensive essentiality analysis of the Mycobacterium tuberculosis genome via saturating transposon mutagenesis. mBio 8(1):e02133-16. doi: 10.1128/mBio.02133-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abrahams GL, Kumar A, Savvi S, Hung AW, Wen S, Abell C, Barry CE III, Sherman DR, Boshoff HI, Mizrahi V. 2012. Pathway-selective sensitization of Mycobacterium tuberculosis for target-based whole-cell screening. Chem Biol 19:844–854. doi: 10.1016/j.chembiol.2012.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Singh V, Brecik M, Mukherjee R, Evans JC, Svetlíková Z, Blaško J, Surade S, Blackburn J, Warner DF, Mikušová K, Mizrahi V. 2015. The complex mechanism of antimycobacterial action of 5-fluorouracil. Chem Biol 22:63–75. doi: 10.1016/j.chembiol.2014.11.006. [DOI] [PubMed] [Google Scholar]
- 31.Singh V, Dhar N, Pató J, Kolly GS, Korduláková J, Forbak M, Evans JC, Szekely R, Rybniker J, Palčeková Z, Zemanová J, Santi I, Signorino-Gelo F, Rodrigues L, Vocat A, Covarrubias AS, Rengifo MG, Johnsson K, Mowbray S, Buechler J, Delorme V, Brodin P, Knott GW, Ainsa JA, Warner DF, Kéri G, Mikušová K, McKinney JD, Cole ST, Mizrahi V, Hartkoorn RC. 2017. Identification of aminopyrimidine-sulfonamides as potent modulators of Wag31-mediated cell elongation in mycobacteria. Mol Microbiol 103:13–25. doi: 10.1111/mmi.13535. [DOI] [PubMed] [Google Scholar]
- 32.Evans JC, Trujillo C, Wang Z, Eoh H, Ehrt S, Schnappinger D, Boshoff HI, Rhee KY, Barry CE III, Mizrahi V. 2016. Validation of CoaBC as a bactericidal target in the coenzyme A pathway of Mycobacterium tuberculosis. ACS Infect Dis 2:958–968. doi: 10.1021/acsinfecdis.6b00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hyland SA, Anderson MS. 2003. A high-throughput solid-phase extraction assay capable of measuring diverse polyprenyl phosphate: sugar-1-phosphate transferases as exemplified by the WecA, MraY, and MurG proteins. Anal Biochem 317:156–165. doi: 10.1016/S0003-2697(03)00088-5. [DOI] [PubMed] [Google Scholar]
- 34.Brandish PE, Burnham MK, Lonsdale JT, Southgate R, Inukai M, Bugg TD. 1996. Slow binding inhibition of phospho-N-acetylmuramyl-pentapeptide-translocase (Escherichia coli) by mureidomycin A. J Biol Chem 271:7609–7614. doi: 10.1074/jbc.271.13.7609. [DOI] [PubMed] [Google Scholar]
- 35.Inukai M, Isono F, Takatsuki A. 1993. Selective inhibition of the bacterial translocase reaction in peptidoglycan synthesis by mureidomycins. Antimicrob Agents Chemother 37:980–983. doi: 10.1128/AAC.37.5.980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Koga T, Fukuoka T, Doi N, Harasaki T, Inoue H, Hotoda H, Kakuta M, Muramatsu Y, Yamamura N, Hoshi M, Hirota T. 2004. Activity of capuramycin analogues against Mycobacterium tuberculosis, Mycobacterium avium and Mycobacterium intracellulare in vitro and in vivo. J Antimicrob Chemother 54:755–760. doi: 10.1093/jac/dkh417. [DOI] [PubMed] [Google Scholar]
- 37.Reddy VM, Einck L, Nacy CA. 2008. In vitro antimycobacterial activities of capuramycin analogues. Antimicrob Agents Chemother 52:719–721. doi: 10.1128/AAC.01469-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Salomon JJ, Galeron P, Schulte N, Morow PR, Severynse-Stevens D, Huwer H, Daum N, Lehr CM, Hickey AJ, Ehrhardt C. 2013. Biopharmaceutical in vitro characterization of CPZEN-45, a drug candidate for inhalation therapy of tuberculosis. Ther Deliv 4:915–923. doi: 10.4155/tde.13.62. [DOI] [PubMed] [Google Scholar]
- 39.Das D, Walvoort MT, Lukose V, Imperiali B. 2016. A rapid and efficient luminescence-based method for assaying phosphoglycosyltransferase enzymes. Sci Rep 6:33412. doi: 10.1038/srep33412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chung BC, Zhao J, Gillespie RA, Kwon DY, Guan Z, Hong J, Zhou P, Lee SY. 2013. Crystal structure of MraY, an essential membrane enzyme for bacterial cell wall synthesis. Science 341:1012–1016. doi: 10.1126/science.1236501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chung BC, Mashalidis EH, Tanino T, Kim M, Matsuda A, Hong J, Ichikawa S, Lee SY. 2016. Structural insights into inhibition of lipid I production in bacterial cell wall synthesis. Nature 533:557–560. doi: 10.1038/nature17636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hakulinen JK, Hering J, Branden G, Chen H, Snijder A, Ek M, Johansson P. 2017. MraY-antibiotic complex reveals details of tunicamycin mode of action. Nat Chem Biol 13:265–267. doi: 10.1038/nchembio.2270. [DOI] [PubMed] [Google Scholar]
- 43.Bugg TD, Lloyd AJ, Roper DI. 2006. Phospho-MurNAc-pentapeptide translocase (MraY) as a target for antibacterial agents and antibacterial proteins. Infect Disord Drug Targets 6:85–106. doi: 10.2174/187152606784112128. [DOI] [PubMed] [Google Scholar]
- 44.Ichikawa S, Yamaguchi M, Matsuda A. 2015. Antibacterial nucleoside natural products inhibiting phospho-murnac-pentapeptide translocase; chemistry and structure-activity relationship. Curr Med Chem 22:3951–3979. doi: 10.2174/0929867322666150818103502. [DOI] [PubMed] [Google Scholar]
- 45.Silver LL. 2013. Viable screening targets related to the bacterial cell wall. Ann N Y Acad Sci 1277:29–53. doi: 10.1111/nyas.12006. [DOI] [PubMed] [Google Scholar]
- 46.Walvoort MT, Lukose V, Imperiali B. 2016. A modular approach to phosphoglycosyltransferase inhibitors inspired by nucleoside antibiotics. Chemistry 22:3856–3864. doi: 10.1002/chem.201503986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mitachi K, Aleiwi BA, Schneider CM, Siricilla S, Kurosu M. 2016. Stereocontrolled total synthesis of muraymycin D1 having a dual mode of action against Mycobacterium tuberculosis. J Am Chem Soc 138:12975–12980. doi: 10.1021/jacs.6b07395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Silver LL. 2011. Challenges of antibacterial discovery. Clin Microbiol Rev 24:71–109. doi: 10.1128/CMR.00030-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Okano A, Isley NA, Boger DL. 2017. Peripheral modifications of [Psi[CH2NH]Tpg4]vancomycin with added synergistic mechanisms of action provide durable and potent antibiotics. Proc Natl Acad Sci U S A 114:E5052–E5061. doi: 10.1073/pnas.1704125114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu Y, Breukink E. 2016. The membrane steps of bacterial cell wall synthesis as antibiotic targets. Antibiotics (Basel) 5:28. doi: 10.3390/antibiotics5030028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mitachi K, Siricilla S, Yang D, Kong Y, Skorupinska-Tudek K, Swiezewska E, Franzblau SG, Kurosu M. 2016. Fluorescence-based assay for polyprenyl phosphate-GlcNAc-1-phosphate transferase (WecA) and identification of novel antimycobacterial WecA inhibitors. Anal Biochem 512:78–90. doi: 10.1016/j.ab.2016.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Siricilla S, Mitachi K, Skorupinska-Tudek K, Swiezewska E, Kurosu M. 2014. Biosynthesis of a water-soluble lipid I analogue and a convenient assay for translocase I. Anal Biochem 461:36–45. doi: 10.1016/j.ab.2014.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Siricilla S, Mitachi K, Wan B, Franzblau SG, Kurosu M. 2015. Discovery of a capuramycin analog that kills nonreplicating Mycobacterium tuberculosis and its synergistic effects with translocase I inhibitors. J Antibiot (Tokyo) 68:271–278. doi: 10.1038/ja.2014.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nakamura H, Yoshida T, Tsukano C, Takemoto Y. 2016. Synthesis of CPZEN-45: construction of the 1,4-diazepin-2-one core by the Cu-catalyzed intramolecular amidation of a vinyl iodide. Org Lett 18:2300–2303. doi: 10.1021/acs.orglett.6b00943. [DOI] [PubMed] [Google Scholar]
- 55.Singh V, Mizrahi V. 2017. Identification and validation of novel drug targets in Mycobacterium tuberculosis. Drug Discov Today 22:503–509. doi: 10.1016/j.drudis.2016.09.010. [DOI] [PubMed] [Google Scholar]
- 56.Singh V, Donini S, Pacitto A, Sala C, Hartkoorn RC, Dhar N, Keri G, Ascher DB, Mondesert G, Vocat A, Lupien A, Sommer R, Vermet H, Lagrange S, Buechler J, Warner DF, McKinney JD, Pato J, Cole ST, Blundell TL, Rizzi M, Mizrahi V. 2017. The inosine monophosphate dehydrogenase, GuaB2, is a vulnerable new bactericidal drug target for tuberculosis. ACS Infect Dis 3:5–17. doi: 10.1021/acsinfecdis.6b00102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee RE, Hurdle JG, Liu J, Bruhn DF, Matt T, Scherman MS, Vaddady PK, Zheng Z, Qi J, Akbergenov R, Das S, Madhura DB, Rathi C, Trivedi A, Villellas C, Lee RB, Rakesh Waidyarachchi SL, Sun D, McNeil MR, Ainsa JA, Boshoff HI, Gonzalez-Juarrero M, Meibohm B, Bottger EC, Lenaerts AJ. 2014. Spectinamides: a new class of semisynthetic antituberculosis agents that overcome native drug efflux. Nat Med 20:152–158. doi: 10.1038/nm.3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kling A, Lukat P, Almeida DV, Bauer A, Fontaine E, Sordello S, Zaburannyi N, Herrmann J, Wenzel SC, Konig C, Ammerman NC, Barrio MB, Borchers K, Bordon-Pallier F, Bronstrup M, Courtemanche G, Gerlitz M, Geslin M, Hammann P, Heinz DW, Hoffmann H, Klieber S, Kohlmann M, Kurz M, Lair C, Matter H, Nuermberger E, Tyagi S, Fraisse L, Grosset JH, Lagrange S, Muller R. 2015. Antibiotics. Targeting DnaN for tuberculosis therapy using novel griselimycins. Science 348:1106–1112. [DOI] [PubMed] [Google Scholar]
- 59.Ling LL, Schneider T, Peoples AJ, Spoering AL, Engels I, Conlon BP, Mueller A, Schaberle TF, Hughes DE, Epstein S, Jones M, Lazarides L, Steadman VA, Cohen DR, Felix CR, Fetterman KA, Millett WP, Nitti AG, Zullo AM, Chen C, Lewis K. 2015. A new antibiotic kills pathogens without detectable resistance. Nature 517:455–459. doi: 10.1038/nature14098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tran AT, Watson EE, Pujari V, Conroy T, Dowman LJ, Giltrap AM, Pang A, Wong WR, Linington RG, Mahapatra S, Saunders J, Charman SA, West NP, Bugg TD, Tod J, Dowson CG, Roper DI, Crick DC, Britton WJ, Payne RJ. 2017. Sansanmycin natural product analogues as potent and selective anti-mycobacterials that inhibit lipid I biosynthesis. Nat Commun 8:14414. doi: 10.1038/ncomms14414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Michalski A, Damoc E, Lange O, Denisov E, Nolting D, Muller M, Viner R, Schwartz J, Remes P, Belford M, Dunyach JJ, Cox J, Horning S, Mann M, Makarov A. 2012. Ultra high resolution linear ion trap Orbitrap mass spectrometer (Orbitrap Elite) facilitates top down LC MS/MS and versatile peptide fragmentation modes. Mol Cell Proteomics 11:O111.013698. doi: 10.1074/mcp.O111.013698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cox J, Mann M. 2008. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 63.Ehrt S, Guo XV, Hickey CM, Ryou M, Monteleone M, Riley LW, Schnappinger D. 2005. Controlling gene expression in mycobacteria with anhydrotetracycline and Tet repressor. Nucleic Acids Res 33:e21. doi: 10.1093/nar/gni013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Guo XV, Monteleone M, Klotzsche M, Kamionka A, Hillen W, Braunstein M, Ehrt S, Schnappinger D. 2007. Silencing Mycobacterium smegmatis by using tetracycline repressors. J Bacteriol 189:4614–4623. doi: 10.1128/JB.00216-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Klotzsche M, Ehrt S, Schnappinger D. 2009. Improved tetracycline repressors for gene silencing in mycobacteria. Nucleic Acids Res 37:1778–1788. doi: 10.1093/nar/gkp015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Manganelli R, Voskuil MI, Schoolnik GK, Smith I. 2001. The Mycobacterium tuberculosis ECF sigma factor sigma E: role in global gene expression and survival in macrophages. Mol Microbiol 41:423–437. doi: 10.1046/j.1365-2958.2001.02525.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.